
Abstract
Neurogenesis plays an indispensable role in the formation of the nervous system during development. The discovery that the adult brain still maintains neurogenic niches that allow the continued production of new cells after birth has changed the field of neuroscience. It has also opened a new venue of opportunities for the treatment of central nervous system disorders related to neuronal loss. This chapter has reviewed the studies showing that genetic or pharmacological manipulation of cannabinoid receptors (CB1 and CB2) or the enzymes responsible for endocannabinoid metabolism modify/regulate cell proliferation and neurogenesis during development and in the adult brain. A better characterization of the mechanisms involved in these effects could contribute to the development of new therapeutic alternatives to neurodegenerative and psychiatric disorders.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Cannabinoids
The term cannabinoid was first used to describe a class of substances with similar chemical structures extracted from the plant Cannabis sativa . More than 100 cannabinoids have been identified in this plant, including Δ9-tetrahydrocannabinol (THC) , the one responsible for its main psychological effects, and cannabidiol (CBD) , the major non-psychotomimetic compound [1]. The observation that the activity of psychoactive cannabinoids was intrinsically related to its chemical structure [2] raised the hypothesis that cannabinoid receptors would be present in the organism. In the late 1980s, the endocannabinoid (ECB) system started to be described with the identification of a specific receptor for THC in the central nervous system (CNS, [3]) that was subsequently cloned and named cannabinoid CB1 receptor [4].
CB1 receptors are now considered the most abundant metabotropic receptor in the mammals’ CNS and are also present in peripheral tissues. The CB1 receptors are widely expressed in presynaptic terminals , where they regulate the release of several neurotransmitters (e.g., GABAe, glutamate, serotonin, acetylcholine, dopamine) [5, 6]. A second cannabinoid receptor, named CB2 , was described in 1993 by Munro and colleagues [7]. Although initially thought to be expressed mainly in cells of the hematopoietic and immune systems, more recent studies have challenged this notion demonstrating that CB2 receptors may be expressed in neurons and is present in microglia and neural stem cells [8–10]. Of note, despite their different localization and, apparently, functions, both CB1 and CB2 receptors are coupled to a Gi/o protein [11].
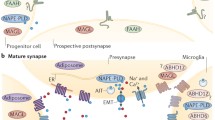
In addition to CB1 and CB2 receptors, their endogenous ligands (termed endocannabinoids) were also isolated in mammals. The most extensively investigated are those derived from arachidonic acid, arachidonoyl ethanolamide (anandamide-AEA), and 2-arachidonoyl glycerol (2-AG) , which are degraded by specific enzymes (Fig. 6.1, [12, 13]). AEA and 2-AG can also interact with other receptors such as proliferator-activated receptors (PPAR- α and γ). Moreover, AEA interacts with GPR55 and the Transient Receptor Potential Vanilloid Type 1 (TRPV1) [14].

Schematic representation of the endocannabinoid system in the brain. (?) Putative expression of CB2 receptor in neurons. *Microglial cells express CB1(constitutive) and CB2 (activated state) receptors. Endocannabinoids are produced in astrocytes, microglia, and neurons
Cannabinoids decrease neurotransmitter release by inhibiting calcium (Ca2+) and activating potassium channels [15]. They also affect short-term neuronal activity by reducing the depolarization-induced suppression of inhibition (DSI), mainly in GABAergic synapses, and the depolarization-induced suppression of excitation (DSE), in synapses that release glutamate and the neuropeptide cholecystokinin [16–18]. Moreover, cannabinoids display neuroprotective actions, being involved in the control of glutamate-induced excitotoxicity [19], and are critical regulators of neurodevelopment and adult neurogenesis [20].
In this chapter, we summarize the main pieces of evidence indicating that cannabinoid signaling on neural stem/progenitor cells affects their proliferation, maturation, and survival. These effects can modify CNS functions, being a potential new avenue for the development of novel therapeutic strategies for neurodegenerative and psychiatric disorders .
6.2 The Neurodevelopmental Role of the Endocannabinoid System
An extensive literature has addressed the consequences of developmental exposure to phytocannabinoids , mostly THC, and also to potent synthetic cannabinoid agonists. These studies have demonstrated that exposure of the immature nervous system to THC , in perinatal stages and/or the adolescence, is associated to numerous behavioral alterations [21]. Experimental evidence indicates that the developing brain is more sensitive to exogenous cannabinoid-induced plastic adaptations. These findings prompted the search of the neurobiological substrate of phytocannabinoid actions.
6.2.1 Expression of the Endocannabinoid System
The ECB system is present and functional since early stages of development, including the primordium of the nervous system, as well as in the restricted neurogenic areas of the adult brain (the hippocampal subgranular zone-SGZ and subventricular zone-SVZ). Along neuronal differentiation, CB1 and CB2 receptors show opposite patterns of expression, being increased and decreased, respectively [10, 22]. CB1 receptors are expressed, although at low levels, in neuroepithelial progenitor cells from early embryonic stages, and their levels increase along neural differentiation [20]. In addition, CB1 is enriched in white matter areas in embryonic stages, until the acquisition of its final expression pattern in the adult nervous system [23]. In vivo, CB1 receptor levels are associated with higher expression of differentiation markers of various neuronal lineages. CB1 receptor activity is more prominent in differentiated pyramidal projection neurons, interneurons, or cholinergic neurons than in their respective undifferentiated progenitor cells [20]. Little is known about the mechanisms controlling CB1 receptor expression during neurodevelopment. CB1 is induced during neuronal differentiation by neurotrophins such as brain-derived neurotrophic factor (BDNF) [24]. In mature GABAergic interneurons, CB1 expression is controlled by the 67-kDa isoform GABA -synthesizing enzyme glutamate decarboxylase [25] and in striatal neurons is regulated by the transcription factor REST via RE1 sites [26].
The CB1 receptor regulation by ECBS during development is poorly understood. 2-AG and AEA can be synthesized on-demand by surrounding differentiated neurons in response to neuronal activity. In addition, ECB can be produced in a paracrine/autocrine manner by neural progenitors (NPs) [27, 28]. The extracellular or intrinsic mechanisms responsible for ECB production in active neurogenic niches are not entirely understood. NPs produce and release the two major ECB compounds, namely, AEA and 2-AG, in response to increased intracellular Ca2+ concentration, and the ECB tone contributes to basal and stimulus-induced NP proliferation via CB1 receptors [27, 29, 30]. 2-AG levels in neurogenic niches are precisely regulated by diacylglycerol lipase (DAGL) and monoacylglycerol lipase (MAGL) activity. Ablation of DAGLα, but not of the β isoform, interferes with hippocampal and SVZ-derived neurogenesis [31] and pharmacological inhibition of DAGL activity in NP cultures reduces cell proliferation [32]. NPs express FAAH , the primary enzyme involved in AEA degradation, and its genetic ablation or pharmacological inhibition promote NP proliferation [27, 33].
The role of extracellular signaling cues promoting ECB production is solely known for 2-AG generation, whereas signals driving AEA levels remain elusive, as the expression pattern of NAPE-PLD (N-acyl phosphatidylethanolamine phospholipase D) and FAAH (fatty acid amide hydrolase) enzymes responsible for AEA synthesis and degradation, respectively, during brain development remains unknown. Fibroblast growth factor (FGF) in coordination with neural cell adhesion molecule increases 2-AG levels via DAGL coupled with PLCγ activation. Alternatively, NGF via TrkA enhances 2-AG production during neurite outgrowth of cholinergic neurons by controlling the levels of MAGL [14]. In NPs, the high expression levels of DAGLα have been shown to rapidly decrease along their differentiation into GABAergic neuronal cells [34], through a mechanism that relies on the regulation of the transcriptional regulator specificity protein 1. On the contrary, retinoic acid-induced neuronal-like differentiation of neuroblastoma cells increases first DAGLα expression and later DAGLβ [35].
A variety of neuroactive molecules acting via ionotropic and metabotropic receptors have the potential to engage ECB generation via increased Ca2+ levels or Gq-PLC activation. These responses may occur after neurotransmitter-mediated neuronal activity and are also associated with spontaneous neuronal activity during cortical development. However, the contribution of spontaneous neuronal activity (during brain development) or neuronal synaptic activity (in adult neurogenic niches) in NP cell fate regulation, via ECB production, remains unknown. In addition to CB1 receptors, CB2 receptor activity regulates NP cell proliferation, cell cycle maintenance, and neural differentiation [10, 32, 36]. Whereas CB2 receptor regulation clearly regulates stem/progenitor cell responses, its expression levels and the identity of neural cells expressing it remain obscure.
6.2.2 Cannabinoid Signaling Consequences in the Developing Brain
6.2.2.1 Proliferation
The first pieces of evidence for an active role of cannabinoid signaling in NP cells came from studies on the regulation of adult neurogenesis by pharmacological cannabinoid manipulation or genetic ablation of the CB1 receptor [20, 37]. These studies evidenced that ablation of CB1 receptor expression reduced hippocampal and SVZ NP cell proliferation in vivo. Likewise, CB1 receptor absence in vitro inhibits self-renewal and NP proliferation [27]. Recent findings suggest that the positive role of CB1 receptor signaling in adult neurogenesis is reminiscent of its role in NP proliferation and identity during cortical development Fig. 6.2a [38].

The endocannabinoid system exerts a regulatory role on neural cell fate at different levels. Cannabinoid signaling regulates (a) NP proliferation and identity of progenitor cells, (b) neuronal and glial differentiation, and (c) neuronal morphogenesis and migration
CB1 receptor signaling controls neural cell fate decisions during CNS development by regulating the expression of genes responsible for neural identity [39]. In differentiating neuroblasts, CB1 activation regulates the homeodomain containing transcription factor Pax6 post-translationally via PI3K/Akt-dependent phosphorylation, and this is in turn responsible for its positive actions in neurite outgrowth [40]. In addition, CB1 activation increases Pax6 expression in cortical progenitors, driving the expansion towards basal intermediate progenitors by inducing the expression of the transcription factor Tbr2/eomes [38].
6.2.2.2 Neuronal Differentiation and Morphogenesis
CB1 receptor signaling also affects neuronal differentiation acting in post-mitotic cells and, in an independent manner of its regulatory role, in undifferentiated NPs. CB1 signaling activates NP cell proliferation and pro-survival signaling pathways that contribute to the regulation of cell cycle maintenance and the switch between cell proliferation and differentiation/migration. On the other hand, post-mitotic conditional CB1 receptor ablation does not affect cortical progenitor expansion but only neuronal differentiation (Fig. 6.2b) [41]. CB1 regulates the balance between the expression of Ctip2 and Satb2, two transcriptional regulators that are involved in the decision switch of deep- versus upper-layer cortical neurons. Ctip2 drives deep-layer cortical neuronal identity and corticospinal connectivity, whereas Satb2 is involved in intracortical projection neurons selectively arising from upper cortical layers [42]. Deletion of CB1 during mouse cortical development lowered Ctip2 expression and generation of deep-layer V neurons, and this is reflected in the reduced ability for skilled motor activity of CB1-deficient mice [39].
Cannabinoid signaling also exerts a crucial regulatory role in axon guidance and morphogenesis (Fig. 6.2c) [14]. CB1 receptor located in axon growth cones of differentiating neurons induces its collapse in response to DAGL -derived 2-AG , [43]. A tight spatiotemporal regulation of 2-AG availability has been suggested accordingly to the differential subcellular localization of 2-AG metabolizing enzymes [44]. MAGL is enriched in tubulin-consolidating axon shafts while DAGL accumulates in actin-rich motile axon tips, thus generating a 2-AG gradient that triggers axonal growth cone collapse. In cortical and retinal neurons, CB1 regulates axonal growth cone by controlling the plasma membrane localization of the Dcc (deleted in colorectal cancer) receptor [45], whereas in GABAergic interneurons the monomeric G protein RhoA is involved [43]. CB1 receptor regulation of growth cone collapse and neurite retraction relies on its ability to regulate actomyosin cytoskeleton via RhoA/ROCK signaling and Rac1/WAVE complex [46, 47].
CB1 receptor regulation of growth cone dynamics is responsible for its role in the establishment of long-range subcortical projections. Ablation or pharmacological blockade of CB1 receptors in utero alters corticothalamic projections and induces axon fasciculation deficits [48]. The complementary expression pattern of DAGL in thalamocortical axons and of MAGL in corticothalamic and thalamocortical developing axons contribute to the generation of spatially restricted 2-AG pools. It has therefore been suggested a potential role for 2-AG as one of the molecules responsible for the timely developmental coordination between corticothalamic and thalamocortical projection “hand-shaking” [49]. The CB1 receptor thus exerts an acute/short-term regulation of growth cone signaling in neurite tips, as well as long-lasting changes in neurogenic gene expression that affect neuronal wiring and connectivity.
In postnatal stages, cannabinoid receptor activity regulates astroglial and oligodendroglial differentiation (Fig. 6.2b). CB1 receptor activity increases astroglial differentiation and GFAP expression in the developing cortex [50]. In oligodendrocyte progenitor cells CB1 and CB2 activation promotes the expression of Olig-2 in a PI3K/Akt/mTORC1-dependent manner [51], and their activation by 2AG or WIN55,212-2 administration favors white matter recovery and oligodendrocyte differentiation [52, 53].
Noteworthy, ECB signaling in oligodendrocytes via CB2 receptors can contribute to neuron axon pathfinding by modulating Slit/Robo signaling in corticothalamic neurons expressing CB1 receptor [54].
6.3 Pathological Implications of Cannabinoid Signaling in the Developing Brain
The neurodevelopmental role of the ECB system and its ability to regulate neural cell fate has important implications in regard to its potential contribution to neurodevelopmental disorders. Likewise, exposure to plant-derived cannabinoids, cannabinergic drugs interacting with the ECB system (i.e., modulators of ECB synthesis and degradation), or pollutants interfering with the ECB system can induce functional alterations in the adult progeny. Extensive literature exists regarding the consequences of cannabinoid-exposure during adolescence indicating that this is a critical period of susceptibility to deleterious actions produced by these compounds [21]. Less is known about the consequences of prenatal cannabinoid administration or embryonic manipulation of cannabinoid signaling [54, 55]. Cannabinoid-induced alterations of the nervous system development have been demonstrated in different experimental models. In early embryonic chick development, administration of a THC analogue disrupts neurogenesis and affects brain, somite and spinal cord primordium development, indicating that the ECB system is active in early cell fate decisions of neural tube progenitor cells [56]. In pregnant rats, administration of WIN-55,212-2 during the gestational period induces changes in dorsal pallial migrating neuroblasts and marginal zone interneurons [57]. Unfortunately, the impact of WIN-55,212-2 treatment in the progeny’s brain was not investigated.
6.3.1 Neuronal Hyperexcitability and Epileptogenesis
Constitutive absence of CB1 receptors in null mice results in increased seizure susceptibility that is mostly attributed to the lack of the neuromodulatory role of presynaptic CB1 receptors [58]. In addition, the neurodevelopmental alterations associated with the loss of CB1 receptors in early stages, i.e., during embryonic development when synaptic activity is still absent or emerging, can shed new light on the cellular mechanisms responsible for epileptogenesis and the appropriate balance of excitation/inhibition (E/I) . Alterations of neurogenesis and changes of excitatory and inhibitory neuronal cell populations are, therefore, essential for coordinated activity. Considering the evidence that the ECB via CB1 receptors regulates both excitatory projection neuron specification and GABAergic interneuron morphogenesis and local microcircuits, these alterations can contribute to the higher susceptibility and severity to seizures as a consequence of CB1 signaling manipulation. In agreement, embryonic THC administration exerts a deleterious impact in deep-cortical layer projection neurons and increases seizure susceptibility via CB1 receptors [59]. In this study, the impact of THC in interneurons and particularly in CCK basket cells was not investigated, but selective neuronal lineage rescue of CB1 receptor expression [60] revealed that CB1 receptors expressed in projection neurons and the GABAergic lineage contribute to seizure susceptibility. Likewise, prenatal THC administration, by interfering with cytoskeleton stability via c-Jun N-terminal kinase and Superior Cervical Ganglion 10/stathmin-2 protein levels, decreases Schaffer collateral-induced long-term depression and perisomatic basket cell surrounding pyramidal cell somata [61]. Interference with the correct generation of different neuronal subpopulations can be responsible for embryonic THC-induced E/I unbalance. In addition to CB1 receptor regulation of neuronal differentiation, cannabinoid signaling actions in neuronal migration can contribute to developmental epileptogenesis. Genetic ablation of CB1 receptors during cortical development exerts a radial migration blockade that results in ectopic projection neurons resembling subcortical band heterotopias (Díaz-Alonso, de Salas-Quiroga, Galve-Roperh, personal communication). Noteworthy, transient CB1 receptor knockdown restricted to embryonic stages exerts long-lasting migration blockade that persists in the adulthood and induces increased seizure susceptibility. The promigratory role of CB1 receptors during brain development (Fig. 6.2c) is in agreement with the described role of the ECB system regulating neuroblasts migration in the adult rostral migratory stream [62]. These findings support the notion that cannabinoid signaling controls the appropriate E/I balance by additional mechanisms to the canonical CB1 receptor neuromodulation.
6.3.2 Neuropsychiatric Disorders
Experimental evidence described herein reveals that defective ECB signaling or developmental exposure to phytocannabinoids can induce alterations in neuronal number, specification and functional properties, or morphological changes that may be responsible not only for seizure susceptibility but also for neuropsychiatric actions of cannabinoid signaling. The neurobiological substrate responsible for the emotional, social interaction, and cognitive changes induced by phytocannabinoid consumption or by an unbalanced ECB signaling during brain development remains largely unknown [54, 55]. In agreement with previous evidence of CB1 regulation of CCK development, a recent study showed that embryonic THC administration correlated with selective changes in the development of CCK basket cells, but not other interneuron populations. Embryonic THC administration compromised feedforward and feedback inhibition in the progeny [63]. The persistent inhibitory deficits in the adult progeny was associated with deficient social interaction, but not increased anxiety, as reported in many studies where THC was administered in the adolescent period [21]. The impact of THC in CCK development raises the hypothesis of a potential interaction between cannabinoid signaling and autism. Noteworthy, autism-related mutations of neuroligin 3 are associated with changes in CB1 constitutive activity [64]. THC administration during adolescence, but not later, interferes with GABA maturation and functionality in the prefrontal cortex, highlighting the importance of developmental actions in cannabinoid effects [65]. On the other hand, CB1 receptor blockade in the adult can counteract several phenotypic markers of the Fragile X model (based on the loss of fragile X mental retardation protein FRMP) [66]. The consequences of manipulating CB1 receptor signaling during brain development in autism models remain to be investigated. Furthermore, the role of CB1 in interneuron developmental changes underlying the pathogenesis of schizophrenia constitutes an expanding field of research [67].
6.4 Adult Neurogenesis
At the beginning of the twentieth century, independent researchers reported what they believed to be the first description of mitotic figures in the adult nervous system of mammals [68]. However, this finding was not recognized because of the accepted dogma based on Santiago Ramon y Cajal’s view that, reflecting the limitations of the techniques available at that time, it was impossible to identify dividing neurons in the adult brain [69].
For more than 100 years, evidence of adult neurogenesis was denied, as the accepted view was that this process could only happen during embryonic periods, stopping just after birth. In the early 1960s, Joseph Altman, a scientist of the Massachusetts Institute of Technology, using tritiated thymidine administered intraperitoneally in adult rats, reported that “a proliferative region of granule cells was identified in the dentate gyrus of the hippocampus” [70, 71]. Almost 15 years later, Dr. Michael Kaplan presented additional evidence that new neurons are added in specific regions of the young and adult rat brain, including the neocortex, hippocampal formation, and olfactory bulb [72–74]. However, it was the work of [75], which reported that new neurons are indeed generated in the hippocampus of adult humans that established one of the most exciting recent fields in neuroscience: adult neurogenesis.
Adult neurogenesis is a complex process that evolves from the initial division of precursor cells until the effective differentiation and generation of a new functional and integrated neuron. In the words of Dr. G. Kempermann: “Neurogenesis is a process, not an event.”. It can be more precisely defined as an in vivo process that involves cell division, survival (not all cells that divide will survive), migration, differentiation, and maturation [76–78]. Neural proliferative capacity has been reported in different brain regions, such as the hypothalamus and the cell layers surrounding the third ventricle [79]. However, the best characterized neurogenic areas in the adult brain are the SVZ of the lateral walls of the lateral ventricle and SGZ of the dentate gyrus (DG) of the hippocampal formation [80]. Both regions have a resident population of neural stem/progenitor cells that can originate neurons, astrocytes, and oligodendrocytes [81].
Despite the half-century of research separating the initial findings of Altman from our current knowledge, the particular function/physiological role of adult neurogenesis, as well as the key regulators of this process, remain under debate. So far, it seems to be a consensus that experience modulates neurogenesis in the adult brain either positively or negatively. Voluntary exercise or enrichment environment enhances proliferation in neurogenic niches [82]. Conversely, chronic stress exposure decreases neurogenesis. However, due the different neurobiological nature of the two main neurogenic niches, it is reasonable to infer that neurogenesis in SVZ and SGZ might be recruited differently and consequently exerts distinct or complementary roles on brain functions [77].
In the SVZ, neurogenesis is regulated by the olfactory experience of the animals [83, 84]. Odor exposure can increase the survival of newborn neurons and improve memory in a learned odor discrimination task, suggesting that neurogenesis in the olfactory bulb is recruited during learning and memory processes related to olfactory stimulation [85]. However, due to the relevance of the hippocampus for several brain functions and its implication on the genesis of neuropsychiatric disorders, much closer attention has been paid to SGZ than SVZ neurogenesis [86, 87].
Hippocampal neurogenesis is proposed to be important for at least some forms of learning and memory. Positive associations between them have been replicated by independent groups in rodents and humans [88–90]. For example, voluntary running and exposure to enriched environments improve learning and memory process with a concomitant increase in cell proliferation and survival of new DG generated neurons [82, 91, 92].
In addition, decreased adult hippocampal neurogenesis has been associated with psychiatric disorders such as anxiety, schizophrenia, and mood disorders. Stressful experiences that can precipitate symptoms of anxiety and mood disorders down-regulate hippocampal neurogenesis [33, 93, 94]. Snyder et al. [95] showed that impaired SGZ, but not SVZ, neurogenic capacity facilitates stress-induced depressive-like symptoms and disrupt the essential negative feedback of hippocampus in hypothalamic-pituitary-adrenal (HPA) axis [95]. Adult hippocampal neurogenesis has also been implicated in the mechanism of pattern separation [96, 97]). Pattern separation is a complex concept that involves CA3 region as an associative network between a spatial location and a situation or an object that allows completion of memory during recall [98]. It has been hypothesized that this event is highly regulated by new neurons formed in the DG . In addition, several authors have demonstrated that neurogenesis is relevant for the perception of an event as stressful or not [99]. In the light of psychiatric conditions that involve an initial exposure to a traumatic event, such as posttraumatic stress disorder, the intact capacity of DG to produce new neurons has been associated with a poor ability of fear discrimination and overgeneralization (Besnard and Sahay 2015).
Of note, drugs used in the clinical practice for the treatment of psychiatric disorders , such as antidepressants or lithium, normalize or even facilitate hippocampal neurogenesis [94, 100]. Moreover, compounds with therapeutic potential for psychiatric conditions, such as cannabinoids, also impacts positively in adult hippocampal neurogenesis [33, 101].
6.4.1 Cannabinoids and Adult Neurogenesis
Several independent groups around the world have demonstrated the importance of the ECB system in the modulation of different steps required for neurogenesis: cell proliferation, differentiation, maturation, and survival (Fig. 6.3, [37, 86]). Indeed, activation of CB receptors regulates intracellular pathways involved in cell proliferation, differentiation, survival, and the integration of new cells in already established circuitries, such as the MEK/ERK/CREB and PI3K/Akt/mTOR and BDNF production [14, 37]. Also, voluntary exercise seems to increase adult hippocampal neurogenesis through a facilitation of CB1 -mediated neurotransmission. Finally, a positive association between cannabinoid-induced neurogenesis and behavioral improvement has been observed in animal models of anxiety, psychosis, depression, and memory impairment (as further discussed in item 1.5 of this chapter). Chronic (10 days), but not the acute administration of HU-210, a synthetic cannabinoid, induces neurogenesis in mice. A very similar picture is found after repeated administration of WIN55,212-2, a CB1/CB2 agonist [27, 32, 101, 102].

Complex modulation of the endocannabinoid system during the process of adult hippocampal neurogenesis . Blue arrows facilitation of the formation of new cells/new neurons, N.A data not available or inconclusive. Based on in vivo studies
The two main compounds of the plant Cannabis sativa , THC and CBD , also affect adult hippocampal neurogenesis. Repeated treatment with CBD for 15-days prevented β-amyloid-induced neurotoxicity via activation of the proliferator-activated receptor-γ (PAAR-γ), suggesting a mechanism for CBD neuroprotective effects [103]. Wolf et al. [30] suggested that chronic treatment with CBD (42 days) decreases cell proliferation but stimulates cell survival. These responses were mediated by CB1 receptors, as CBD effects were absent in CB1 receptor knockout mice. Also, repeated CBD (30 mg/kg) treatment for 14 days prevented a stress-induced decrease in cell survival and differentiation in mice. In non-stressed mice, CBD increased the number of double-labeled BrdU/NeuN cells in the dentate gyrus [33]. These results were associated with increased levels of AEA, but not 2-AG , in the hippocampus of mice treated with CBD [33]. On the other hand, THC, a partial CB1 receptor agonist, decreased proliferation and, at the same time, spatial memory [30].
The participation of ECB in the modulation of neurogenesis has also been investigated. For example, hippocampal cell proliferation is increased in FAAH deficient mice and in animals treated with URB597, an FAAH inhibitor [27]. On the other hand, the ECB uptake inhibitor, AM404, reversed the trimethylthiazoline (TMT)-induced decrease of neurogenesis [104]. Finally, the genetic ablation of the enzyme responsible for 2-AG synthesis reduced cell proliferation, the number of doublecortin (a neuroblast marker) positive cells, and decreased the survival of newborn cells in the DG [31, 105].
The facilitation of CB2 signaling also influences adult neurogenesis. Repeated administration of HU-308, a CB2 receptor agonist, during 5 days, induces neural precursor cells proliferation in the DG. This effect seems to recruit Akt/mTORC1 pathway [36]. In the opposite way, the administration of CB2 inverse agonist (JTE907) or antagonists (SR144528 or AM630) reduces cell proliferation and the number of BrdU labeled cells in the SVZ and SGZ [10, 32, 36]. The involvement of CB2 receptors in these results was confirmed by the failure of a CB2 agonist to induce any change in neurogenesis in animals deficient for this receptor [10, 36].
In the case of studies using pharmacological and genetic regulation of CB1 receptors, the results are controversial. CB1-deficient mice exhibit low rates of proliferation, astrogliogenesis, and neurogenesis in the DG and SVZ [27]. Also, repeated administration of the CB1 antagonists/inverse agonists, SR141716A, and AM251, decreased neurogenesis in some studies [106]. Other groups, nevertheless, suggested that these drugs facilitate neurogenesis [30, 104, 107]. Interestingly, the effects of some of these cannabinergic drugs were preserved in CB1 but not in TRPV1-deficient mice [107]. These discrepancies may be related to the use of different animal species, strain or gender, cannabinergic drugs , and doses employed. In addition, contradictory results may be the consequence of different BrdU-administration schedule, and time-point of analysis, which may induce alternative interpretations. For example, Wolf et al. [30] found increased cell proliferation 1 and 24 h after treatment with AM251, but a decrease in cell maturation 48 h and 7 days later.
6.4.2 Neurogenesis, Cannabinoids, and Neuropsychiatric/Neurodegenerative Disorders: What’s the Correlation?
Considering that the ECB system modulates adult neurogenesis and that this process is impaired in neuropsychiatric and neurodegenerative disorders, it is plausible that cannabinoids may induce beneficial or detrimental effects in the brain and influence behavior by controlling newly generated neuron-induced plasticity. Cannabinoids are effective in modulating neurogenesis in various animal models of depression, anxiety disorders, Alzheimer’s disease , and cerebral ischemia. Some of these studies are not only based on associative results, but suggest causality, once the direct ablation of hippocampal neurogenesis by different methods prevented the therapeutic effects induced by distinct cannabinoids tested.
Acute treatment with AM404, an ECB uptake inhibitor, reversed the trimethylthiazoline-induced decrease of hippocampal cell proliferation and promoted anxiolytic-like effect [104]. In the same sense, sub-chronic treatment with the CB1/CB2 agonist HU210 induced anxiolytic- and antidepressant-like effects accompanied by an increase in neurogenesis [101]. Although a controversial finding, authors suggested that neurogenesis ablation through hippocampal X-ray irradiation prevented HU210-induced behavioral responses [101]. In agreement, repeated injections of CBD reversed the anxiogenic-like responses and the neurogenesis impairment produced by chronic stress in a CB1-dependent manner [33]. These effects were completely lost after ganciclovir administration to transgenic mice that express thymidine kinase under the control of the GFAP promoter, a method used to ablate only adult dividing precursor cells. In accordance, a recent study showed that the enhancement of 2-AG -induced neurotransmission by the MAGL inhibitor, JZL184, also prevented the anxiogenic- and pro-depressive-like effects, as well as the decrease in neurogenesis, induced by chronic stress [108]. Strengthening this hypothesis, the antidepressant-like effect produced by a single injection of the CB1 antagonist SR141716A was lost after sub-chronic administration of the drug, probably due to the reduction in neurogenesis observed in these animals [106].
Several studies in the literature show that (1) neurogenesis is altered in some neurodegenerative diseases, and (2) cannabinoids can improve behavioral responses, as memory impairment, and brain damage, in animal models of these disorders. For example, Esposito et al. [103] showed that chronic administration of CBD in rats that previously received β-amyloid injection in the hippocampus, an animal model of Alzheimer’s disease , decreases reactive gliosis, neuronal damage and facilitates adult hippocampal neurogenesis through PPAR-γ receptors. Also, cannabinoids can ameliorate age-related reduction in neurogenesis, suggesting that these compounds could replenish damaged/death neurons during neurodegeneration [32, 102]. In the middle cerebral artery occlusion rat model , widely used to evaluate cerebral ischemic injury, daily injections of oleoylethanolamide, a monounsaturated analog of anandamide, improved the spatial cognitive impairment concomitant to an increase in BDNF and hippocampal neurogenesis [109]. Also, CB2 receptor regulation counteracts alcohol-induced decline in neurogenesis [110].
Taken together, these pieces of evidence suggest that cannabinoids could exert anxiolytic- and antidepressant-like effects as well as neuroprotection through an enhancement of adult neurogenesis. New studies using cannabinergic drugs that modulate the ECB tone in long-term studies of animal models of mood, cognitive, or neurodegenerative disorders are urgently needed to clarify these important aspects.
6.5 Conclusions and Perspectives
In this chapter, we have presented evidence indicating that cannabinoids exert an important neurodevelopmental regulatory role on and mediate plastic events in the adult brain (Figs. 6.2 and 6.3). Important unanswered questions, however, remain. For example, is the modulation of neurogenesis by endocannabinoid signaling always positive, or can it be deleterious in some pathological conditions? What are the precise mechanisms by which cannabinoid regulate neurogenesis, neurodevelopment, and cell fate? What is the role of non-cannabinoid mediated mechanisms (e.g., TRPV1, GPR55, PPAR-γ receptors) in cannabinoid modulation of neurogenesis? What intracellular pathways are involved? These open questions indicate that we are only at the beginning of our journey. However, the results so far clearly support the perspective that new knowledge in this area could bring important contributions to the therapy of neuropsychiatric and neurodevelopmental disorders.
Abbreviations
- 2-AG:
-
2-arachidonoylglycerol
- AEA:
-
Anandamide
- Ca2+ :
-
Calcium
- CB1 :
-
Cannabinoid receptor type 1
- CB2 :
-
Cannabinoid receptor type 2
- CBD:
-
Cannabidiol
- DAGL:
-
Diacylglycerol lipase
- DG:
-
Dentate gyrus
- E/I:
-
Excitation/inhibition
- ECB:
-
Endocannabinoids
- MAGL:
-
Monoacylglycerol lipase
- NP:
-
Neural progenitor
- SGZ:
-
Subgranular zone
- SVZ:
-
Subventricular zone
- THC:
-
Δ9-tetrahydrocannabinol
References
Pertwee RG (2005) Pharmacological actions of cannabinoids. Handb Exp Pharmacol 168:1–51
Mechoulam R, Lander N, Varkony TH, Kimmel I, Becker O, Ben-Zvi Z et al (1980) Stereochemical requirements for cannabinoid activity. J Med Chem 23(10):1068–1072
Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, Howlett AC (1988) Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34(5):605–613
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346(6284):561–564
Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066
Katona I, Freund TF (2008) Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med 14(9):923–930
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature (London) 365:61–65
Lisboa SF, Gomes FV, Guimaraes FS, Campos AC (2016) Microglial cells as a link between cannabinoids and the immune hypothesis of psychiatric disorders. Front Neurol 7:5
Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA et al (2006) Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci 1074:514–536
Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzman M, Galve-Roperh I (2006) Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J 20(13):2405–2407
Howlett AC (2002) The cannabinoid receptors. Prostaglandins Other Lipid Mediat 68–69:619–631
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G et al (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258(5090):1946–1949
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR et al (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50(1):83–90
Maccarrone M, Guzman M, Mackie K, Doherty P, Harkany T (2014) Programming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci 15(12):786–801
Szabo B, Schlicker E (2005) Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol 168:327–365
Diana MA, Marty A (2004) Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br J Pharmacol 142(1):9–19
Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410(6828):588–592
Yoshida T, Hashimoto K, Zimmer A, Maejima T, Araishi K, Kano M (2002) The cannabinoid CB1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar Purkinje cells. J Neurosci 22(5):1690–1697
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A et al (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302(5642):84–88
Galve-Roperh I, Chiurchiu V, Diaz-Alonso J, Bari M, Guzman M, Maccarrone M (2013) Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res 52(4):633–650
Rubino T, Parolaro D (2016) The impact of exposure to cannabinoids in adolescence: insights from animal models. Biol Psychiatry 79(7):578–585
Begbie J, Doherty P, Graham A (2004) Cannabinoid receptor, CB1, expression follows neuronal differentiation in the early chick embryo. J Anat 205(3):213–218
Romero J, Garcia-Palomero E, Berrendero F, Garcia-Gil L, Hernandez ML, Ramos JA et al (1997) Atypical location of cannabinoid receptors in white matter areas during rat brain development. Synapse 26(3):317–323
Maison P, Walker DJ, Walsh FS, Williams G, Doherty P (2009) BDNF regulates neuronal sensitivity to endocannabinoids. Neurosci Lett 467(2):90–94
Eggan SM, Lazarus MS, Stoyak SR, Volk DW, Glausier JR, Huang ZJ et al (2012) Cortical glutamic acid decarboxylase 67 deficiency results in lower cannabinoid 1 receptor messenger RNA expression: implications for schizophrenia. Biol Psychiatry 71(2):114–119
Blazquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, Resel E et al (2011) Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain 134(Pt 1):119–136
Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B et al (2005) The endocannabinoid system drives neural progenitor proliferation. FASEB J 19(12):1704–1706
Butti E, Bacigaluppi M, Rossi S, Cambiaghi M, Bari M, Cebrian Silla A et al (2012) Subventricular zone neural progenitors protect striatal neurons from glutamatergic excitotoxicity. Brain 135(Pt 11):3320–3335
Rubio-Araiz A, Arevalo-Martin A, Gomez-Torres O, Navarro-Galve B, Garcia-Ovejero D, Suetterlin P et al (2008) The endocannabinoid system modulates a transient TNF pathway that induces neural stem cell proliferation. Mol Cell Neurosci 38(3):374–380
Wolf SA, Bick-Sander A, Fabel K, Leal-Galicia P, Tauber S, Ramirez-Rodriguez G et al (2010) Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun Signal 8:12
Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M et al (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30(6):2017–2024
Goncalves MB, Suetterlin P, Yip P, Molina-Holgado F, Walker DJ, Oudin MJ et al (2008) A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol Cell Neurosci 38(4):526–536
Campos AC, Ortega Z, Palazuelos J, Fogaca MV, Aguiar DC, Diaz-Alonso J et al (2013) The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: involvement of the endocannabinoid system. Int J Neuropsychopharmacol 16(6):1407–1419
Walker DJ, Suetterlin P, Reisenberg M, Williams G, Doherty P (2010) Down-regulation of diacylglycerol lipase-alpha during neural stem cell differentiation: identification of elements that regulate transcription. J Neurosci Res 88(4):735–745
Jung KM, Astarita G, Thongkham D, Piomelli D (2011) Diacylglycerol lipase-alpha and -beta control neurite outgrowth in neuro-2a cells through distinct molecular mechanisms. Mol Pharmacol 80(1):60–67
Palazuelos J, Ortega Z, Diaz-Alonso J, Guzman M, Galve-Roperh I (2012) CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J Biol Chem 287(2):1198–1209
Prenderville JA, Kelly AM, Downer EJ (2015) The role of cannabinoids in adult neurogenesis. Br J Pharmacol 172(16):3950–3963
Díaz-Alonso J, Aguado T, de Salas-Quiroga A, Ortega Z, Guzman M, Galve-Roperh I (2015) CB1 cannabinoid receptor-dependent activation of mTORC1/Pax6 signaling drives Tbr2 expression and basal progenitor expansion in the developing mouse cortex. Cereb Cortex 25(9):2395–2408
Díaz-Alonso J, Aguado T, Wu CS, Palazuelos J, Hofmann C, Garcez P, et al. The CB(1) cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. J Neurosci 2012a;32(47):16651–65
Bromberg KD, Ma'ayan A, Neves SR, Iyengar R (2008) Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science 320(5878):903–909
Díaz-Alonso J, Guzman M, Galve-Roperh I. Endocannabinoids via CB(1) receptors act as neurogenic niche cues during cortical development. Philos Trans R Soc Lond B Biol Sci 2012b;367(1607):3229-41
Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD (2013) Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci 14(11):755–769
Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urban GM et al (2007) Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 316(5828):1212–1216
Keimpema E, Barabas K, Morozov YM, Tortoriello G, Torii M, Cameron G et al (2010) Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. J Neurosci 30(42):13992–14007
Argaw A, Duff G, Zabouri N, Cecyre B, Chaine N, Cherif H et al (2011) Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J Neurosci 31(4):1489–1499
Njoo C, Agarwal N, Lutz B, Kuner R (2015) The cannabinoid receptor CB1 interacts with the WAVE1 complex and plays a role in actin dynamics and structural plasticity in neurons. PLoS Biol 13(10):e1002286
Roland AB, Ricobaraza A, Carrel D, Jordan BM, Rico F, Simon A et al (2014) Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. Elife 3:e03159
Mulder J, Aguado T, Keimpema E, Barabas K, Ballester Rosado CJ, Nguyen L et al (2008) Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc Natl Acad Sci U S A 105(25):8760–8765
Wu CS, Zhu J, Wager-Miller J, Wang S, O'Leary D, Monory K et al (2010) Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci 32(5):693–706
Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B et al (2006) The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci 26(5):1551–1561
Gomez O, Sanchez-Rodriguez A, Le M, Sanchez-Caro C, Molina-Holgado F, Molina-Holgado E (2011) Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br J Pharmacol 163(7):1520–1532
Arevalo-Martin A, Garcia-Ovejero D, Molina-Holgado E (2010) The endocannabinoid 2-arachidonoylglycerol reduces lesion expansion and white matter damage after spinal cord injury. Neurobiol Dis 38(2):304–312
Tomas-Roig J, Wirths O, Salinas-Riester G, Havemann-Reinecke U (2016) The cannabinoid CB1/CB2 agonist WIN55212.2 promotes oligodendrocyte differentiation in vitro and neuroprotection during the cuprizone-induced central nervous system demyelination. CNS Neurosci Ther 22(5):387–395
Alpár A, Tortoriello G, Calvigioni D, Niphakis MJ, Milenkovic I, Bakker J et al (2014) Endocannabinoids modulate cortical development by configuring Slit2/Robo1 signalling. Nat Commun 5:4421
Di Marzo V, Stella N, Zimmer A (2015) Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci 16(1):30–42
Psychoyos D, Hungund B, Cooper T, Finnell RH (2008) A cannabinoid analogue of Delta9-tetrahydrocannabinol disrupts neural development in chick. Birth Defects Res B Dev Reprod Toxicol 83(5):477–488
Saez TM, Aronne MP, Caltana L, Brusco AH (2014) Prenatal exposure to the CB1 and CB2 cannabinoid receptor agonist WIN 55,212-2 alters migration of early-born glutamatergic neurons and GABAergic interneurons in the rat cerebral cortex. J Neurochem 129(4):637–648
Soltesz I, Alger BE, Kano M, Lee SH, Lovinger DM, Ohno-Shosaku T et al (2015) Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat Rev Neurosci 16(5):264–277
de Salas-Quiroga A, Diaz-Alonso J, Garcia-Rincon D, Remmers F, Vega D, Gomez-Canas M et al (2015) Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc Natl Acad Sci U S A 112(44):13693–13698
Ruehle S, Remmers F, Romo-Parra H, Massa F, Wickert M, Wortge S et al (2013) Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J Neurosci 33(25):10264–10277
Tortoriello G, Morris CV, Alpar A, Fuzik J, Shirran SL, Calvigioni D et al (2014) Miswiring the brain: Delta9-tetrahydrocannabinol disrupts cortical development by inducing an SCG10/stathmin-2 degradation pathway. EMBO J 33(7):668–685
Oudin MJ, Gajendra S, Williams G, Hobbs C, Lalli G, Doherty P (2011) Endocannabinoids regulate the migration of subventricular zone-derived neuroblasts in the postnatal brain. J Neurosci 31(11):4000–4011
Vargish GA, Pelkey KA, Yuan X, Chittajallu R, Collins D, Fang C et al (2016) Persistent inhibitory circuit defects and disrupted social behaviour following in utero exogenous cannabinoid exposure. Mol Psychiatry
Földy C, Malenka RC, Sudhof TC (2013) Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 78(3):498–509
Cass DK, Flores-Barrera E, Thomases DR, Vital WF, Caballero A, Tseng KY (2014) CB1 cannabinoid receptor stimulation during adolescence impairs the maturation of GABA function in the adult rat prefrontal cortex. Mol Psychiatry 19(5):536–543
Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, Agustin-Pavon C, Pastor A, Mato S et al (2013) Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19(5):603–607
Volk DW, Lewis DA (2016) The role of endocannabinoid signaling in cortical inhibitory neuron dysfunction in schizophrenia. Biol Psychiatry 79(7):595–603
Allen E (1912) The cessation of mitosis in the central nervous system of the albino rat. J Comp Neurol 19:547–568
Gross CG (2000) Neurogenesis in the adult brain: death of a dogma. Nat Rev Neurosci 1:67–73
Altman J (1963) Autoradiographic investigation of cell proliferation in the brains of rats and cats. Anat Rec 145:573–591
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124(3):319–335
Kaplan MS (1981) Neurogenesis in the 3-month-old rat visual cortex. J Comp Neurol 195(2):323–338
Kaplan MS (1983) Proliferation of subependymal cells in the adult primate CNS: differential uptake of DNA labelled precursors. J Hirnforsch 24(1):23–33
Kaplan MS (2001) Environment complexity stimulates visual cortex neurogenesis: death of a dogma and a research career. Trends Neurosci 24(10):617–620
Eriksson PS, Perfilieva E, BjörkEriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4(11):1313–1317
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11(5):339–350
Kempermann G (2008) The neurogenic reserve hypothesis: what is adult hippocampal neurogenesis good for? Trends Neurosci 31(4):163–169
Suh H, Deng W, Gage FH (2009) Signaling in adult neurogenesis. Annu Rev Cell Dev Biol 25:253–275
Chaker Z, George C, Petrovska M, Caron JB, Lacube P, Caille I et al (2016) Hypothalamic neurogenesis persists in the aging brain and is controlled by energy-sensing IGF-I pathway. Neurobiol Aging 41:64–72
Kempermann G, Jessberger S, Steiner B, Kronenberg G (2004) Milestones of neuronal development in the adult hippocampus. Trends Neurosci 27(8):447–452
Gage FH (2000) Mammalian neural stem cells. Science 287:1433–1438
Van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2:266–270
Lledo PM, Alonso M, Grubb MS (2006) Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci 7(3):179–193
Lledo PM, Saghatelyan A (2005) Integrating new neurons into the adult olfactory bulb: joining the network, life-death decisions, and the effects of sensory experience. Trends Neurosci 28(5):248–254
Alonso M, Viollet C, Gabellec MM, Meas-Yedid V, Olivo-Marin JC, Lledo PM (2006) Olfactory discrimination learning increases the survival of adult-born neurons in the olfactory bulb. J Neurosci 26(41):10508–10513
Fogaça MV, Galve-Roperh I, Guimaraes FS, Campos AC (2013) Cannabinoids, neurogenesis and antidepressant drugs: is there a link? Curr Neuropharmacol 11(3):263–275
Opendak M, Gould E (2015) Adult neurogenesis: a substrate for experience-dependent change. Trends Cogn Sci 19(3):151–161
Coras R, Siebzehnrubl FA, Pauli E, Huttner HB, Njunting M, Kobow K et al (2010) Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain 133(11):3359–3372
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999) Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2(3):260–265
Kempermann G, Gage FH (2002) Genetic determinants of adult hippocampal neurogenesis correlate with acquisition, but not probe trial performance, in the water maze task. Eur J Neurosci 16(1):129–136
Kee NJ, Preston E, Wojtowicz JM (2001) Enhanced neurogenesis after transient global ischemia in the dentate gyrus of the rat. Exp Brain Res 136(3):313–320
Tashiro A, Makino H, Gage FH (2007) Experience-specific functional modification of the dentate gyrus through adult neurogenesis: a critical period during an immature stage. J Neurosci 27(12):3252–3259
Czéh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M et al (2001) Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci U S A 98(22):12796–12801
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301(5634):805–809
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA (2011) Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476(7361):458–461
Besnard A, Sahay A (2016) Adult hippocampal neurogenesis, fear generalization, and stress. Neuropsychopharmacology 41(1):24–44
Sahay A, Wilson DA, Hen R (2011) Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron 70(4):582–588
Rolls ET (2013) The mechanisms for pattern completion and pattern separation in the hippocampus. Front Syst Neurosci 7:74
Egeland M, Zunszain PA, Pariante CM (2015) Molecular mechanisms in the regulation of adult neurogenesis during stress. Nat Rev Neurosci 16(4):189–200
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I et al (2009) Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62(4):479–493
Jiang W, Zhang Y, Xiao L, Van Cleemput J, Ji SP, Bai G et al (2005) Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J Clin Invest 115(11):3104–3116
Marchalant Y, Brothers HM, Wenk GL (2009) Cannabinoid agonist WIN-55,212-2 partially restores neurogenesis in the aged rat brain. Mol Psychiatry 14(12):1068–1069
Esposito G, Scuderi C, Valenza M, Togna GI, Latina V, De Filippis D et al (2011) Cannabidiol reduces Abeta-induced neuroinflammation and promotes hippocampal neurogenesis through PPARgamma involvement. PLoS One 6(12):e28668
Hill MN, Kambo JS, Sun JC, Gorzalka BB, Galea LA (2006) Endocannabinoids modulate stress-induced suppression of hippocampal cell proliferation and activation of defensive behaviours. Eur J Neurosci 24(7):1845–1849
Jenniches I, Ternes S, Albayram O, Otte DM, Bach K, Bindila L et al (2016) Anxiety, stress, and fear response in mice with reduced endocannabinoid levels. Biol Psychiatry 79(10):858–868
Lee S, Kim DH, Yoon SH, Ryu JH (2009) Sub-chronic administration of rimonabant causes loss of antidepressive activity and decreases doublecortin immunoreactivity in the mouse hippocampus. Neurosci Lett 467(2):111–116
Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO et al (2004) Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol 66(2):204–208
Zhang Z, Wang W, Zhong P, Liu SJ, Long JZ, Zhao L et al (2015) Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus 25(1):16–26
Yang LC, Guo H, Zhou H, Suo DQ, Li WJ, Zhou Y et al (2015) Chronic oleoylethanolamide treatment improves spatial cognitive deficits through enhancing hippocampal neurogenesis after transient focal cerebral ischemia. Biochem Pharmacol 94(4):270–281
Rivera P, Blanco E, Bindila L, Alen F, Vargas A, Rubio L et al (2015) Pharmacological activation of CB2 receptors counteracts the deleterious effect of ethanol on cell proliferation in the main neurogenic zones of the adult rat brain. Front Cell Neurosci 9:379
Acknowledgements
We would like to thank Franciele Scarante for her technical support on design and graphic art of Figs. 6.1 and 6.3, and members of our research groups for an inspiring scientific environment. ACC and FSG are recipients of FAPESP grants. IGR research is funded by PI15-00310, RTC-2015-3364-1, and S2011-BMD-2336 supported by the Instituto de Salud Carlos III, Mineco (Plan Estatal de I+D+i 2013-2016) and Comunidad de Madrid. Research was cofinanced by the European Development Regional Fund “A way to achieve Europe” (ERDF). JPL is a recipient of FPU (Ministerio de Educación) fellowship. MVF is recipient of a CAPES fellowship.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Campos, A.C., Paraíso-Luna, J., Fogaça, M.V., Guimarães, F.S., Galve-Roperh, I. (2017). Cannabinoids as Regulators of Neural Development and Adult Neurogenesis. In: Pébay, A., Wong, R. (eds) Lipidomics of Stem Cells. Stem Cell Biology and Regenerative Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-49343-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-49343-5_6
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-49342-8
Online ISBN: 978-3-319-49343-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)