
Abstract
The prospects of using organic materials in electronics and optoelectronics applications have attracted scientists and technologists since the 1970s. This field has become known as molecular electronics. Some successes have already been achieved, for example, the liquid-crystal display, organic light-emitting displays, and photoreceptors in electrophotography. Other products such as organic photovoltaic devices, chemical sensors and plastic transistors are developing fast. There is also a keen interest in exploiting technologies at the molecular scale that might eventually replace silicon devices. This chapter provides some of the background physics and chemistry to the interdisciplinary subject of molecular electronics. A review of some of the possible application areas for organic materials is presented and some speculation is provided regarding future directions.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Molecular electronics is concerned with the exploitation of organic and biological materials in electronics and optoelectronics [51.1, 51.2, 51.3]. The subject, as it has matured over the last 40 years, can broadly be divided into two themes. The first area, molecular materials for electronics, has its origins in materials science and concerns the development of devices that utilize the unique macroscopic properties of organic compounds. The most successful commercial product to date is the liquid-crystal display (GlossaryTerm
LCD
). However, following many years of research, devices such as organic light-emitting displays, organic photovoltaic (GlossaryTermPV
) devices, organic transistors, pyroelectric detectors for infrared imaging, and chemical and biochemical sensors are beginning to make a technological impact. The Nobel prize in chemistry for 2000 was awarded to three scientists working in this area: Alan Heeger, Alan MacDiarmid, and Hideki Shirakawa, who have made significant contributions to the development of electrically conductive polymers. More challenging is molecular-scale electronics. Here, the focus is on the behavior of individual organic molecules or groups of molecules. Topics such as molecular switches [51.3, 51.4, 51.5, 51.6, 51.7, 51.8, 51.9], molecular memories [51.10, 51.11, 51.12, 51.13], molecular rectifiers [51.14], negative differential-resistance junctions [51.15], deoxyribonucleic acid (DNA) electronics [51.16], and molecular manufacturing [51.17, 51.18] have all been described in the literature. It is much too early to say which, if any, of these could find their way into the commercial arena.This chapter provides an introduction to the interdisciplinary world of molecular electronics. In the first instance, the physics background to semiconductive organic compounds is outlined. A review of the available materials is presented and some of the possible device applications are described. There are currently a limited number of ways in which organic molecules can be deposited and manipulated on surfaces to form solid films, which can then be used in device structures. The most popular methods are outlined, and methods to pattern the films are described. The prospects for molecular-scale electronics are contrasted with the progress of the inorganic semiconductor industry. Finally, a selection of the ongoing work on molecular-scale devices is described and some speculation about future developments is given.
1 Electrically Conductive Organic Compounds
Metallic and semiconductive behavior is not restricted to inorganic materials. Figure 51.1 shows that the room-temperature conductivity values for organic polymers can extend over much of the spectrum of electrical conductivity, from insulating to semiconducting, and even metallic, behavior [51.19]. The physical explanation can be found in the nature of the chemical bonds that hold solids together.

Room-temperature conductivity values of conductive polymers compared to other materials. (After [51.19], with permission from Wiley)
1.1 Orbitals and Chemical Bonding
Carbon-based materials are unique in many respects. This is due to the many possible configurations of the electronic states of a carbon atom. Carbon has an atomic number of six and a valency of four. Its electron configuration is 1s2, 2s2, 2p2, that is, the inner 1s shell is filled and the four valence electrons available for bonding are distributed two each in the 2s orbital and the 2p orbitals. As the 2s orbital is spherically symmetrical (Fig. 51.2) it can form a bond in any direction. In contrast, the 2p orbitals are directed along mutually orthogonal axes and will tend to form bonds in these directions. When two or more of the valence electrons of carbon are involved in bonding with other atoms, the bonding can be explained by the construction of hybrid orbitals by mathematically combining the 2s and 2p orbitals. In the simplest case, the carbon 2s orbital hybridizes with a single p orbital. Taking the sum and difference of the two orbitals gives two sp hybrids; two p orbitals then remain. The sp orbitals are constructed from equal amounts of s and p orbitals; they are linear and 180∘ apart.

(a) 2s orbital. (b) 2py, 2px, and 2pz orbitals
Other combinations of orbitals lead to different hybrids. For example, from the 2s orbital and two 2p orbitals (e. g., a 2px and a 2py), three equivalent sp2 hybrids may be constructed. Each orbital is 33.3% s and 66.7% p. The three hybrids lie in the xy plane (the same plane defined by the two p orbitals), directed 120∘ from each other, and the remaining p orbital is perpendicular to the sp2 plane. Four sp3 hybrids may be derived from an s orbital and three p orbitals. These are directed to the corners of a tetrahedron with an angle of 109.5∘ between the bonds; each orbital is 25% s and 75% p.
A chemical bond can also be formed from a mixture of the above hybrid orbitals, for example, it is possible to have a hybridized orbital that is 23% s and 77% p. Thus, sp, sp2 and sp3 hybrids must be considered as limiting cases. Electrons in s orbitals have a lower energy than electrons in p orbitals. Therefore, bonds with more s character tend to be stronger.
Carbon forms four bonds in most compounds, resulting from its four valence electrons. In ethane, C2H6, the C–H bonds are all approximately C(sp3)–H(s), while the C–C bond is approximately C(sp3)–C(sp3). However, in the ethylene molecule, C2H4, each of the two carbons is attached to just three atoms while in acetylene, C2H2, each carbon atom is only attached to one carbon. In these compounds, the two carbon atoms are bound together by double (CH2 = CH2) or triple (CH ≡ CH) bonds, which involve sp2 and sp hybrids, respectively. These bonds can be considered to have two distinct components. For example, in ethylene, two sp2 hybrids on each carbon bond with the hydrogens. A third sp2 hybrid on each carbon forms a C(sp2)–C(sp2) single bond, leaving a p orbital left-over on each carbon. This orbital lies perpendicular to the plane of the six atoms. The two p orbitals are parallel to each other and have regions of overlap above and below the molecular plane. This type of bond, consisting of two separate sausage-like electron clouds lying above and below the planes of the carbon nuclei, is called a pi (π) bond. In contrast, the bond formed by the head-on overlap of the two carbon sp2 orbitals is known as a sigma (σ) bond and the electron cloud is densest at the midpoint between the carbon nuclei. The π bond has little electron density in the region between the carbon atoms and is much weaker than the σ bond. Figure 51.3 illustrates sigma and pi bonding. Carbon compounds that contain π bonds are said to be unsaturated, meaning that the carbon atoms, while having formed the requisite number of bonds, are not fully saturated in terms of their number of potential neighbors. Saturated carbon molecules, on the other hand, contain only single bonds.

(a) Sigma (σ) and (b) pi (π) bonding
1.2 Band Theory
The application of quantum mechanics to the electrons in the bonds of inorganic semiconductors led to the development of band theory, one of the great success stories of modern physics. Whenever two identical atoms are brought close together, the electron orbitals overlap and the energy level associated with each electron in the separated atoms is split into two new levels, with one above and one below the original level. A rigorous, quantum-mechanical description of the bonding reveals that the total number of orbitals must be conserved, that is, the total number of molecular orbitals must be the same as the number of atomic orbitals that went into their formation. Consider, for example, the formation of a hydrogen molecule from two separated atoms. When the two atoms approach each other so that their 1s orbitals overlap, two new orbitals (σ bonds ) are formed around the atoms, symmetric with respect to the interatomic axis. In one orbital, the bonding orbital, the electron has a lower energy than that in the isolated atomic orbital and in the other, the antibonding orbital, an electron has a higher energy. In the hydrogen molecule, the electron pair normally resides in the bonding orbital. The energy of the electrons in this orbital is lowered relative to that in the atomic orbitals, which is why the atoms remain bound together.
In an extended solid, many atoms can interact and many similar splittings of energy levels occur. For a solid containing approximately 1026 atoms (Avogadro’s number) each energy level splits, but the energies between these split levels are very small and continuous ranges or bands of energy are formed. Two such important bands are the valence band and the conduction band, analogous to the bonding and antibonding levels of the two-atom model. The energy gap, or band gap, between them is a forbidden energy range for electrons. Electrical conduction takes place by electrons moving under the influence of an applied electric field in the conduction band and/or holes moving in the valence band. Holes are really vacancies in a band but, for convenience, they may be regarded as positively charged carriers. For an electron, or hole, to gain energy from an applied electric field, and therefore for conductivity to occur, the charge carrier must be able to move into an unoccupied higher energy state. If the carrier cannot be accelerated by the field, then it cannot contribute to the electrical conduction. In a metal, the various energy bands overlap to provide a single energy band that is only partly full of electrons. An insulator has a full valence band and a relatively large energy separation (> 5 eV) to the higher conduction band. Most semiconductors possess the band structure of an insulator, but a forbidden energy gap of only 0.1–3 eV, so that carriers may be produced in the conduction and/or valence bands by optical or thermal means, or by doping with impurities.
An important feature of the band model is that the electrons are delocalized or spread over the lattice. The strength of the interaction between the overlapping orbitals determines the extent of delocalization that is possible for a given system. For many polymeric organic materials, the molecular orbitals responsible for bonding the carbon atoms of the chain together are the sp3 hybridized σ bonds, which do not give rise to extensive overlapping. The resulting band gap is large, as the electrons involved in the bonding are strongly localized on the carbon atoms and cannot contribute to the conduction process. This is why a simple saturated polymer such as polyethylene , ( CH2)n, is an electrical insulator.
A significant increase in the degree of electron delocalization may be found in unsaturated polymers, that is, those containing double and triple carbon–carbon bonds. If each carbon atom along the chain has only one other atom, for example, hydrogen, attached to it, the spare electron in a pz-orbital of the carbon atom overlaps with those of carbon atoms on either side forming delocalized molecular orbitals of π-symmetry. For a simple lattice of length L = Na, where N is the total number of atoms and a is the spacing between them, it can be shown that the total number of electron states in the lowest energy band is equal to N. This result is true for every energy band in the system and applies to three-dimensional (3-D) lattices. Allowing for the two spin orientations of an electron, the Pauli exclusion principle requires that there will be room for two electrons per cell of the lattice in an energy band. If each atom contributes one bonding electron, the valence band will be only half filled.
It might therefore be expected that a linear polymer backbone consisting of many strongly interacting coplanar pz orbitals, each of which contributes one electron to the resultant continuous π electron system, would behave as a one-dimensional (1-D) metal with a half-filled band. In chemical terms, this is a conjugated chain and may be represented by a system of alternating single and double bonds. It turns out that, for 1-D systems, such a chain can more efficiently lower its energy by introducing bond alternation (alternating short and long bonds). This limits the extent of electronic delocalization that can take place along the backbone. The effect is to open an energy gap in the electronic structure of the polymer. All conjugated polymers are semiconductors, with band gaps more than about 1.5 eV, rather than metals (N.B. the band gap in silicon is 1.1 eV at room temperature). Figure 51.4 shows the chemical π bonding and the electronic band structure of polyacetylene.

(a) Chemical π bonding in polyacetylene. (b) Electronic band structure showing the normally empty π∗ band (conduction band) and the normally filled π band (valence band)
1.3 Electrical Conductivity
In polyacetylene, the valence band, or π band, is normally filled with electrons while the conduction band, or π∗ band is normally empty. Like silicon, the conductivity of polyacetylene can be changed by the addition of impurity atoms. However, the term doping is a misnomer as it tends to imply the use of minute quantities, parts per million or less, of impurities introduced into a crystal lattice. In the case of conductive polymers, typically 1–50% by weight of chemically oxidizing (electron withdrawing) or reducing (electron donating) agents are used to alter physically the number of π-electrons on the polymer backbone, leaving oppositely charged counterions alongside the polymer chain. These processes are redox chemistry. For example, the halogen-doping process that transforms polyacetylene to a good conductor is oxidation (or p-doping)
Reductive doping (n-doping) is also possible, for example, using an alkali metal
In both cases, the doped polymer is a salt. The counterions, I −3 or Na+, are fixed in position while the charges on the polymer backbone are mobile and contribute to the conductivity. The doping effect can be achieved because a π electron can be removed (or added) without destroying the σ backbone of the polymer so that the charged polymer remains intact. The increase in conductivity can be as much as 11 orders of magnitude.
The electrical properties of semiconductive organic polymers are not directly comparable to those of silicon. An important material parameter is the mobility of the charge carriers μ. This determines the (additional) velocity that a charge carrier (an electron or hole) acquires because of an applied electric field, and is defined by
where vd is the drift velocity of the carrier and E is the electric field. The mobility may be further related to the electrical conductivity σ by the expression
where n is the density of charge carriers and \(|q|\) is the magnitude of their charge (charge on an electron \(={\mathrm{1.6\times 10^{-19}}}\) C). The carrier mobility provides an indication of how quickly the carriers react to the field (i. e., the frequency response of the material). The greater the degree of electron delocalization, the larger the width of the bands (in energy terms) and the higher the mobility of the carriers within the band. For inorganic semiconductors such as silicon or gallium arsenide, the 3-D crystallographic structure provides for extensive carrier delocalization throughout the solid, resulting in a relatively high mobility μ.
Electrical conduction in polymers not only requires carrier transport along the polymer chains but also some kind of transfer, or hopping, between these chains, which tend to lie tangled up like a plate of spaghetti. The charge-carrier mobilities in organic polymers are therefore quite low, making it difficult to produce very high-speed electronic computational devices that are competitive with those based on silicon and gallium arsenide. However, some improvement in the carrier mobility can be achieved by both increasing the degree of order of the polymer chains [51.20, 51.21] and by improving the purity of the material. Table 51.1 contrasts the room-temperature carrier mobility values for Si, GaAs, and a number of different conductive organic compounds [51.20, 51.21, 51.22, 51.23, 51.24, 51.25, 51.26]. The exceptionally high value for graphene (\({\mathrm{2\times 10^{5}}}\,{\mathrm{cm^{2}/Vs}}\)) should be noted. However, this two-dimensional (2-D) material has a zero band gap, which is an obstacle to its use in certain electronic devices, such as field effect transistors. The values of the other organic materials are low in comparison with those of the inorganic semiconductors, but the field-effect mobilities of \(> {\mathrm{10}}\,{\mathrm{cm^{2}/Vs}}\) achieved recently make such conjugated small molecules and polymers useful for industrial application. In addition, other features make them more attractive for certain types of electronic devices, as indicated in the later sections.
2 Materials
Many conductive polymers have been synthesized to provide certain electronic features (e. g., band gap and electron affinity). The monomer repeat units are often based on five- or six-membered (benzene) carbon ring systems, including polypyrrole, polythiophene (and various other polythiophene derivatives), polyphenelenevinylene, and polyaniline [51.27]. The chemical structure of some of these materials is shown in Fig. 51.5. Such polymers generally show lower electrical conductivity than polyacetylene, however; they can have the advantage of high stability and processability.

Chemical structures of conductive polymers: (a) polypyrrole, (b) polyparaphenylene, (c) polyphenylenevinylene, (d) polythiophene
Conductive polymers represent only one category of organic electrical conductors. Another important class are the charge-transfer compounds [51.28, 51.29]. These are formed from a variety of molecules, primarily aromatic compounds (i. e., based on benzene) which can behave as electron donors (d) and electron acceptors (a). Complete transfer of an electron from a donor to an acceptor molecule results in a system that is electrically insulating (e. g., the transfer of a valence electron in a Na atom to a Cl atom, forming the compound NaCl). However, if the ratio of the number of donor molecules to the number of acceptor molecules differs from 1 : 1, for example, the stoichiometry is 1 : 2 or 2 : 3, or if there is incomplete transfer of an electron from a donor to an acceptor (say, six electrons in every ten are transferred), then partially filled electron energy bands can be formed and electron conduction is possible.
Charge-transfer interactions are strongest between donor molecules of low ionization potential and acceptor molecules of high electron affinity, provided that the donor and acceptor molecules have similar symmetry and are able to approach closely [51.29]. Well-known donor and acceptor molecules are tetrathiafulvalene (TTF) and tetracyanoquinodimethane (TCNQ) (Fig. 51.6a). The latter compound is a very strong acceptor forming first the radical anion and then the dianion (Fig. 51.6b). The stability of the semireduced radical ion, TCNQ\({}^{-\bullet}\), with respect to the neutral molecule mainly arises from the change from the relatively unstable quinoid structure to the aromatic one, allowing extensive delocalization of the π electrons over the carbon skeleton. As a consequence, TCNQ not only forms typical charge-transfer complexes but is also able to form true radical-ion salts, incurring complete one-electron transfer. Thus, on addition of lithium iodide to a solution of TCNQ, the simple lithium TCNQ salt is formed
Following removal of the free iodine precipitate, the TCNQ salt may be crystallized. The crystals show an electronic conductivity of about \({\mathrm{10^{-5}}}\,{\mathrm{S/cm}}\).

(a) Chemical structures of the charge-transfer compounds tetrathiafulvalene (TTF) and tetracyanoquinodimethane (TCNQ). (b) Oxidation and reduction of TCNQ
A 1 : 1 TCNQ : TTF salt exhibits a high room-temperature conductivity (\({\mathrm{5\times 10^{2}}}\,{\mathrm{S/cm}}\)) and metallic behavior is observed as the temperature is reduced to 54 K. The molecules in such compounds are arranged in segregated stacks, in which the donors and acceptors form separate donor stacks (ddddd…) and acceptor stacks (aaaaa…). The molecules are stacked in such a way that the π bonds on successive molecules can overlap to form bands. This overlap is different from the p-orbital overlap forming the π bands in conjugated polymers. The crystalline packing in these, and other, charge-transfer compounds generally leads to carrier mobility values that are higher than for semiconductive polymers.
Other electroactive compounds that may find application in molecular electronics are based on forms of pure carbon. Graphite consists of vast carbon sheets stacked one on top of another like a sheaf of papers. In pure graphite, these layers are about 0.335 nm apart, but they can be separated further by intercalating various molecules or by using adhesive tape, namely Scotch tape [51.30]. Finally, only one graphite sheet, graphene, can be obtained [51.30]. The bonding between the carbon atoms in the planes is mainly sp2 hybridizations consisting of a network of single and double bonds. As noted earlier, graphene is a zero band gap material. In case of multilayer graphene, weak interactions between the delocalized electron orbitals hold adjacent sheets together. The delocalized electron system in the planes results in semiconductive electrical behavior.
Under certain conditions, carbon forms regular clusters of 60, 70, 84, etc. atoms [51.31, 51.32]. A C60 cluster, shown in Fig. 51.7 , is composed of 20 hexagons and 12 pentagons and resembles a football. The diameter of the ball is about 1 nm. As with graphite, each carbon atom in C60 is bonded to three other carbon atoms. Thus, C60 can be considered as a rolled up layer of a single graphite sheet. The term buckministerfullerene was given originally to the C60 molecule because of the resemblance to the geodesic domes designed and built by Richard Buckminster Fuller. However, this term (or fullerene or buckyball) is used quite generally to describe C60 and related compounds. For example, a molecule with the formula C70 can be formed by inserting an extra ring of hexagons around the equator of the sphere, producing an elongated shell more like a rugby ball.

Chemical structure of C60 (top) and three classes of single-wall carbon nanotube (SWNT ) (a) (10,10) armchair SWNT, (b) (12,7) chiral SWNT, and (c) (15,0) zigzag SWNT
In addition to the spherical-shaped fullerenes, it is possible to synthesize tubular variations – carbon nanotubes [51.32, 51.33]. Such tubes are comprised of graphite-like sheets curled into a cylinder. Each tube may contain several cylinders nested inside each other. The tubes are capped at the end by cones or faceted hemispheres. Because of their very small diameters (down to around 0.7 nm), carbon nanotubes are prototype 1-D nanostructures. An important feature of a carbon nanotube is the orientation of the six-membered carbon ring in the honeycomb lattice relative to the axis of the nanotube. Three examples of single-wall carbon nanotubes (GlossaryTerm
SWCN
s) are shown in Fig. 51.7. The primary classification of a carbon nanotube is as either being chiral or achiral. An achiral nanotube is one whose mirror image has an identical structure to the original. There are only two cases of achiral nanotubes: armchair and zigzag (these names arise from the shape of the cross-sectional ring). Chiral nanotubes exhibit a spiral symmetry whose mirror image cannot be superimposed on the original structure. Carbon nanotubes are characterized by the chiral index (n, m), where the integers n and m specify each carbon nanotube uniquely [51.33]. An armchair nanotube corresponds to the special case n = m, while for a zigzag nanotube m = 0. All other (n, m) indexes correspond to chiral nanotubes. The electronic structure of a SWCN is either metallic or semiconducting, depending on its diameter and chirality.At low temperature, a SWNT is a quantum wire in which the electrons in the wire move without being scattered. Resistance measurements for various nanotube samples show that there are metallic and semiconducting nanotubes [51.33]. Carbon nanotubes can also be doped either by electron donors or electron acceptors [51.34]. After reaction with the host materials, the dopants are intercalated in the intershell spaces of the multiwalled nanotubes, and, in the case of single-walled nanotubes, either in between the individual tubes or inside the tubes.
The above confirms carbon’s uniqueness as an electronic material. It can be a good conductor in the form of graphite, an insulator in the form of diamond, or a flexible polymer (conductive or insulating) when reacted with hydrogen and other species. Carbon differs from other group IV elements, such as Si and Ge, which exhibit sp3 hybridization. Carbon does not have any inner atomic orbitals except for the spherical 1s orbital, and the absence of nearby inner orbitals facilitates hybridizations involving only the valence (outer) s and p orbitals. The fact that sp and sp2 hybridizations do not readily occur in Si and Ge might be related to the absence of organic materials made from these elements.
3 Plastic Electronics
3.1 Diodes and Transistors
Since the discovery of semiconducting behavior in organic materials, there has been a considerable research effort aimed at exploiting these properties in electronic and optoelectronic devices. The term plastic electronics refers to electronic devices incorporating polymeric organic compounds (although this term is often used more widely to include devices incorporating other semiconducting organic materials). Organic semiconductors can have significant advantages over their inorganic counterparts. For example, thin layers of conjugated small molecules and polymers can easily be made by low-cost methods such as spin coating. High-temperature deposition from vapour reactants is generally needed for inorganic semiconductors. Synthetic organic chemistry also offers the possibility of designing new materials with different band gaps. As noted in Sect. 51.1.3, the mobilities of the charge carriers in organic field-effect transistors (GlossaryTerm
FET
s) have exceeded 10 cm2 ∕ Vs. Therefore, such performance and the simple fabrication techniques for conjugated small molecules and polymers have attracted much industrial attention for the fabrication of organic transistor applications such as data storage and thin-film device arrays to address LCDs [51.20, 51.21, 51.22, 51.24, 51.25, 51.35, 51.36, 51.37].Semiconducting organic compounds have been used in a similar fashion to inorganic semiconductors (e. g., Si and GaAs) in metal/semiconductor/metal structures. A diode, or rectifying device, can be made by sandwiching a semiconductor between metals of different work functions. In the ideal case, an n-type semiconductor should make an Ohmic contact to a low work-function metal and a rectifying Schottky barrier to a high work-function metal [51.38]. One example is that of a semiconductive organic film sandwiched between aluminum and indium-tin-oxide electrodes [51.39]. This device also exhibits PV behavior and is a basic structure of organic PVs.
Organic materials have been used as the semiconducting layer in FET devices [51.22, 51.40, 51.41, 51.42]. These are three-terminal structures: a voltage applied to a metallic gate affects the electric current flowing between the source and drain electrodes. For transistor operation, charge must be injected easily from the source electrode into the organic semiconductor and the carrier mobility should be high enough to allow useful quantities of source–drain current to flow. The organic semiconductor and other materials with which it is in contact must also withstand the operating conditions without thermal, electrochemical, or photochemical degradation. Two performance parameters to be optimized in organic FETs are the field-effect mobility and the on/off ratio [51.41].
The operating characteristics of organic transistors and integrated circuits have improved markedly over recent years. This has been brought about by both improvements in the material synthesis and in the thin-film processing techniques [51.20, 51.21, 51.24, 51.25, 51.43, 51.44]. State-of-the-art organic FETs possess characteristics exceeding those of devices prepared from hydrogenated amorphous silicon, with mobilities around 1 cm2 ∕ Vs and on/off ratios greater than 106. As organic FETs are moving closer to applications, their reliability under realistic atmospheric as well as electrical operating conditions has come under more intense scrutiny.
A number of groups have also demonstrated transistor devices incorporating carbon nanotubes [51.45, 51.46, 51.47]. However, some key issues need to be addressed before nanotubes can be exploited fully in such applications. These include the reproducible fabrication of low-resistance electrical contacts and the accurate control of nanotube growth parameters [51.48]. The nanotube transistor devices realized experimentally have typical dimensions in the micron range. The real promise of carbon nanotube devices, however, lies in the possibility of nanoscale devices. Transistors incorporating graphenes have also been extensively studied because of their high mobility (up to 200000 cm2 ∕ Vs) [51.26, 51.30].
Thin-film transistors based on organic semiconductors are likely to form key components of plastic circuitry for use as display drivers in portable computers and pagers, and as memory elements in transaction cards and identification tags.
3.2 Organic Light-Emitting Structures
Reports of light emission from organic materials upon the application of an electric field (electroluminescence) have been around for many years. However, there has been an upsurge in interest following the initial report of organic light-emitting devices (GlossaryTerm
OLED
s) incorporating the conjugated small molecules (tris(8-hydroxyquinolinato)aluminum, Alq3 (Fig. 51.9) [51.49] or the conjugated polymer polyphenylenevinylene (PPV (Fig. 51.5)) [51.50]). The simplest OLED is an electroluminescent compound sandwiched between metals of high and low work function, as depicted in Fig. 51.8. The anode electrode is normally indium tin oxide (GlossaryTermITO
) as this material is semitransparent, allowing the light out of the device. On application of a voltage, electrons are injected from the low work function electrode into the lowest unoccupied molecular orbital (GlossaryTermLUMO
) level (conduction or π∗ band in the case of an organic compound possessing a delocalized electron system) of the organic compound and holes from the high work-function electrode into the highest occupied molecular orbital (GlossaryTermHOMO
) level (valence or π band in the case of an organic compound possessing a delocalized electron system). The recombination of these oppositely charged carriers then results in the emission of light. Work is focused on the use of low molecular-weight organic molecules and polymers and there is considerable industrial interest in the application of such materials to various display technologies [51.51, 51.52, 51.53, 51.54]. It is estimated that the global market for OLED displays will increase to $16 billion by 2020 [51.55]. Organic light-emitting devices offer a number of advantages over LCDs and the other technologies that currently dominate the flat-panel display market: OLEDs do not require backlighting and they can be made thinner and lighter. Furthermore, OLED displays provide higher contrast and truer colors, higher brightness, wider viewing angles, better temperature stability and faster response times than LCDs [51.51].
Schematic energy band structure of an organic light-emitting device (OLED). The recombination of electrons and holes results in the emission of light of frequency ν and energy hν

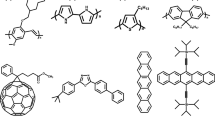
Representative chemical structures of green (a) fluorescent, (b) phosphorescent, and (c) TADF emitters
Many techniques have been used in attempts to optimize the performance of OLEDs. For example, a thin inorganic insulating layer such as LiF, or an organic monolayer may be inserted between the cathode and the emissive material [51.56, 51.57, 51.58]. A thin insertion layer of poly(ethylene imine) (PEI) [51.54, 51.58] between a metal oxide such as ITO or ZnO as a bottom cathode and an emitting layer greatly improves the performance of OLEDs. Such inverted OLEDs, or GlossaryTerm
iOLEDS
, possess the configuration of substrate/cathode/PEI/emitting layer/anode (the configuration of conventional OLEDs shown in Fig. 51.8 is substrate/anode/emitting layer/cathode). In recent years, iOLEDs have attracted much attention because the absence of air-sensitive electron injection materials such as LiF, Ca, and Cs results in a long device lifetime [51.59, 51.60]. Stringent encapsulation of flexible/stretchable devices on flexible substrates, such as a plastic film, is rather difficult and hence the development of iOLEDs is essential for future flexible/stretchable displays.Electron- and hole-transporting layers can also be introduced between the cathode and the emitting layer and between the anode and the emitting layer, respectively, to improve and balance the injection of charge carriers [51.61].
When an electron and a hole recombine to form an excited molecular state in an organic material, the spins of the electrons in the excited and ground levels can either point in the same direction (triplet state) or in the opposite directions (singlet state). For quantum-mechanical reasons, 75% of recombination events are associated with triplet states which, in most cases, do not emit photons when they decay to the ground state. Hence, the production of emission from the triplet state of organic materials is a further means to improve the device efficiency [51.62, 51.63]. Indeed, the internal quantum efficiency, which is defined as the ratio of the number of photons that can be extracted for electroluminescence to the injected current (i. e., the number of injected carriers), is 25% in fluorescent emitters. In contrast, the internal quantum efficiency of OLEDs using phosphorescent materials is 100% [51.64]. However, the design of the phosphorescent materials is greatly limited because the heavy atom effect (spin–orbital interaction) must be induced, for example, by using rare metals, to realize highly efficient radiative transition from the triplet excited state to the ground state. The internal quantum efficiency of 100% has also been achieved using the up-conversion of triplet excitons to singlet excitons by thermal energy, known as thermally activated delayed fluorescence (GlossaryTerm
TADF
) [51.65]. TADF emitters can include ubiquitous atoms such as C, H, and N without using rare metals. Recent studies have revealed that highly efficient delayed fluorescence can be achieved by optimizing the molecular design [51.62]. Representative fluorescent, phosphorescent, and TADF emitters are shown in Fig. 51.9.Table 51.2 shows some important parameters of polymer-based OLEDs with different color outputs [51.54]. There is also a keen interest in developing white-light organic displays. For example, a large-area white-light-emitting OLED could provide a solid-state light source that might compete with conventional lighting technologies. Different methods of making an intrinsically white-emitting OLED by blending emissive species, either in single or multiple layers have been demonstrated [51.66, 51.67, 51.68, 51.69, 51.70].
3.3 Photovoltaic Devices
Concerns over global climate change, local air pollution, and resource depletion are making PVs an increasingly attractive method of energy supply. The current technology is based on single-crystal silicon solar cells. These have developed since the 1940s and now possess conversion efficiencies of around 15% for commercial devices (although figures of around 25% are reported in the laboratory) [51.71]. However, the technology is more expensive than conventional power generation, and there is much research on alternative materials. Photovoltaics using organic compounds, such as polymers or dyes, offer the possibility of large-scale manufacture at low temperature coupled with low cost. Until the end of the twentieth century, little progress had been made and energy conversion efficiencies of up to only about 1% were achieved [51.72]. However, the availability of new conductive organic materials and different PV designs has significantly improved on this figure. To 2015, several laboratories have reported conversion efficiencies of around 10% [51.71, 51.73, 51.74, 51.75, 51.76].
An organic solar cell device is very similar in structure to the OLED described in the previous section. If the incoming photons have energy greater than the band gap of the polymer (or greater than the HOMO–LUMO separation in the case of organic molecular materials) then the light will be absorbed, creating electrons and holes. In an inorganic PV cell, these electrons and holes would be generated within, or close to, a depletion region in the semiconductor and they would be free to migrate to opposite electrodes, where they can do useful work in an external electrical load. However, in the organic material the electrons and holes are bound together in excitons. An immediate problem in organic PV cells is to split these excitons. This can be conveniently done at an interface, the simplest being the junction between the electrodes and the organic material. Under open-circuit conditions, holes are collected at the high work-function electrode (e. g., ITO) and electrons at the low work-function electrode (e. g., Al). Generally, the open-circuit output voltage of the PV device depends on the work function difference between the electrodes. However, in a p-n junction, the maximum available voltage is determined by the difference of the quasi Fermi levels of the two charge carriers, that is, n-doped semiconductor energy level and p-doped semiconductor energy level, respectively. In organic PV, the open-circuit voltage is found to be linearly dependent on the HOMO level of the electron-donating material (p-type semiconductor quasi Fermi level) and the LUMO level of the electron-accepting material (n-type semiconductor quasi Fermi level) [51.77, 51.78, 51.79]. Improvements in the efficiency of the exciton-splitting process can be achieved using organic compounds incorporating electron-donating and electron-accepting species. By creating interfaces of differing electron affinities, it is possible to enhance the probability of electron transfer between the molecules. The creation of the interfaces is carried out by blending electron-donating and electron-accepting species, to produce a bulk heterojunction [51.80, 51.81, 51.82]. This exhibits a donor–acceptor phase separation on a 10–20 nm length scale. In such a nanoscale interpenetrating network, each interface is within a distance less than the exciton diffusion length from the absorbing site. The bulk heterojunction concept significantly increases the interfacial area between the donor and acceptor phases and results in improved efficiency solar cells.
An alternative approach to organic PVs exploits a dye-sensitized solar cell , or Grätzel cell [51.83] . Here, the incoming photons are absorbed by molecules of a dye on a semiconductor surface with subsequent energy and electron transfer to the semiconductor. An electron is returned to the oxidized dye via an electrolyte. The efficiency of such devices is 11.9% [51.71, 51.84].
4 Molecular-Scale Electronics
4.1 Moore’s Laws
The second strand to molecular electronics (molecular-scale electronics) recognizes the spectacular size reduction in the individual processing elements in integrated circuits over recent years. The first microprocessor chip manufactured in 1972 by Intel (8008) had a clock speed of 200 kHz and contained 3500 transistors. There are 1.4 billion transistors on the Pentium Core i7 chip (June, 2014), fabricated using 22 nm process technology, and operating at a clock speed of 3–4 GHz.
Moore’s law (or Moore’s first law) states that the functions per chip double every 1.5 years. This will probably describe developments over at least the next decade. The semiconductor industries have produced an international technology roadmap for the future of complementary metal oxide semiconductor (GlossaryTerm
CMOS
) technology [51.85]. Figure 51.10 shows the anticipated growth in the density of the transistors in both the microprocessor unit (GlossaryTermMPU
) and the dynamic random-access memory (GlossaryTermDRAM
) of a CMOS chip over the next decade. The prediction is for a 10 nm minimum feature size (gate length) for the MPU and 1011 transistors per cm2 in the case of the memory by the year 2022. These figures are regularly updated. This device density is remarkable and provides the means to store significant amounts of data (Table 51.3 [51.3]). However, molecular-scale electronics has the potential for further increases in device density. For example, using ≈ 1–3 nm organic molecules as the processing elements, 1013–1014 devices could be fitted into 1 cm2 [51.3].
Predicted feature size in CMOS devices . (After [51.85])
There are, however, a number of technological issues that will need to be overcome for the predictions for the CMOS-based roadmap to be realized [51.86, 51.87]. Not least are the materials limitations of the silicon/silicon dioxide system. For example, charge leakage becomes a problem when the insulating silicon dioxide layers are thinned to a few nanometer. The scaling of physical dimensions can also be limited by minute variations in size and spatial fluctuations in the doping concentration. Lithographic techniques will face enormous challenges to achieve the high controllability needed to eliminate variations from device to device. Heat dissipation is another key factor. A Pentium Core i7 chip with about 1 billion transistors operating at the current nanosecond rate can emit 60–90 W of heat. Financial issues are also problematic as ever more complex integrated circuits are produced. Intel’s Fab 32, a chip-fabrication facility (GlossaryTerm
FAB
) that opened in Chandler, AZ, USA, in August 2005, cost $3 billion to construct and equip. The cost of building a fab is expected to rise to over $10 billion by 2025 [51.88]. This significant increase in cost (Moore’s second law) is due to the extremely sophisticated tools that will be needed to form the increasingly small features of the devices. Molecular-scale technology will, of course, also need to address such problems – plus many more.4.2 Nanoscale Organic Films
Before organic molecules can be exploited in device architectures (i. e., those familiar from Si-based microelectronics) ways must be found to deposit them onto surfaces. Well-established methods of organic film deposition include electrodeposition, thermal evaporation, and spinning [51.89]. The Langmuir–Blodgett (LB) technique, self-assembly, and layer-by-layer electrostatic deposition are further means for producing layers of organic materials. These allow ultrathin-film assemblies of organic molecules to be engineered at the molecular level and are of particular relevance to molecular-scale electronics [51.90, 51.91].
Langmuir–Blodgett films are prepared by first depositing a small quantity of an amphiphilic compound (i. e., one containing both polar and nonpolar groups) dissolved in a volatile solvent onto the surface of purified water [51.91, 51.92]. The classical materials are long-chain fatty acids, such as n-octadecanoic acid (stearic acid). When the solvent has evaporated, the organic molecules may be organized into a floating two-dimensional crystal by compression on the water surface. As the area available to the organic molecules is reduced, the floating film will undergo several phase transformations. These are, to a first approximation, analogous to 3-D gas, liquid, and solid phases. The phase changes may readily be identified by monitoring the surface pressure as a function of the area occupied by the molecules in the film. This is the two-dimensional equivalent to the pressure-versus-volume isotherm for a gas. In the gaseous state the molecules are far enough apart on the water surface that they exert little force on one another. As the surface area of the monolayer is reduced, the hydrocarbon chains will begin to interact. The liquid state that is formed is generally called the expanded monolayer phase. The hydrocarbon chains of the molecules in such a film are in a random, rather than regular orientation, with their polar groups in contact with the subphase. As the molecular area is progressively reduced, condensed phases may appear. There may be more than one of these and the emergence of each condensed phase can be accompanied by constant-pressure regions of the isotherm, as observed in the cases of a gas condensing to a liquid and a liquid solidifying. In the condensed monolayer states, the molecules are closely packed and are oriented with their hydrocarbon chain pointing away from the water surface. The area per molecule in such a state will be similar to the cross-sectional area of the hydrocarbon chain, that is, \(\approx{\mathrm{0.19}}\,{\mathrm{nm^{2}/molecule}}\).
If the surface pressure is held constant in one of the condensed phases, then the film may be transferred from the water surface onto a suitable solid substrate simply by raising and lowering the latter through the monolayer–air interface. Figure 51.11 shows a schematic diagram of the equipment required for LB film deposition. In this technique, introduced by Langmuir and Blodgett [51.91] the floating condensed monolayer is transferred, like a carpet, as the substrate is raised and/or lowered through the air/monolayer interface. A number of different LB deposition modes are possible. The most commonly encountered situation is Y-type deposition, which refers to monolayer transfer on both the upward and downward movements of the substrate. Instances in which the floating monolayer is only transferred to the substrate as it is being inserted into the subphase, or only as it is being removed, are also observed. These deposition modes are called X-type and Z-type depositions, respectively.

System for the deposition of Langmuir–Blodgett films
Self-assembly is a much simpler process than that of LB deposition. Monomolecular layers are formed by the immersion of an appropriate substrate into a solution of the organic material [51.90]. The best known examples of self-assembled systems are organosilicon on hydroxylated surfaces (SiO2, Al2O3, glass etc.) and alkanethiols on gold, silver, and copper [51.93]. However, other combinations include:
-
Dialkyl sulfides on gold
-
Dialkyl disulfides on gold
-
Alcohols and amines on platinum
-
Carboxylic acids on aluminum oxide and silver
-
Phosphonic acids on aluminum oxide and ITO.
The self-assembly process is driven by the interactions between the head group of the self-assembling molecule and the substrate, resulting in a strong chemical bond between the head group and a specific surface site, for example, a covalent Si–O bond for alkyltrichlorosilanes on hydroxylated surfaces.
The combination of the self-assembly process with molecular recognition offers a powerful route to the development of nanoscale systems that may have technological applications as chemical sensing or switching devices. For instance, the complexation of a neutral or ionic guest at one site in a molecule may induce a change in the optical or redox properties of the system. Monolayers containing both an electroactive TTF unit and a metal-binding macrocycle assembled onto platinum have been shown to exhibit electrochemical recognition to Ag+ ions [51.94].
The self-assembly process, as described above, is usually restricted to the deposition of a single molecular layer on a solid substrate. However, chemical means can be exploited to build up multilayer organic films. A method pioneered by Sagiv is based on the successive absorption and reaction of appropriate molecules [51.95, 51.96]. The head groups react with the substrate to give a permanent chemical attachment and each subsequent layer is chemically attached to the one before in a very similar way to that used in systems for supported synthesis of proteins.
Another technique for building up thin films of organic molecules is driven by the ionic attraction between opposite charges in two different polyelectrolytes, the so-called layer-by-layer assembly technique [51.100, 51.97, 51.98, 51.99]. A solid substrate with a positively charged planar surface is immersed in a solution containing an anionic polyelectrolyte and a monolayer of polyanion is adsorbed (Fig. 51.12). Since the adsorption is carried out at relatively high concentrations of the polyelectrolyte, most of the ionic groups remain exposed to the interface with the solution and thus the surface charge is reversed. After rinsing in pure water, the substrate is immersed in a solution containing the cationic polyelectrolyte. Again, a monolayer is adsorbed but now the original surface charge is restored, resulting in the formation of a multilayer assembly of both polymers. It is possible to use a sensitive optical technique, such as surface plasmon resonance to monitor, in situ, the growth of such electrostatically assembled films [51.101]. The layer-by-layer method has been used to build up layers of conductive polymers, for example, partially doped polyaniline and a polystyrene polyanion [51.102]. Biocompatible surfaces consisting of alternate layers of charged polysaccharides and oppositely charged synthetic polymers can also be deposited in this way [51.103]. A related, but alternative, approach uses layer-by-layer adsorption driven by hydrogen-bonding interactions [51.104].

Deposition of layer-by-layer polyelectrolyte films by the sequential immersion of a solid substrate in solutions of the polyanions and polycations
The organization of the organic molecules in multilayer assemblies may be investigated by a number of analytical techniques including x-ray and neutron reflection, electron diffraction, and infrared spectroscopy [51.91]. Figure 51.13 shows an example of an atomic force micrograph of a 12-layer n-eicosanoic acid LB film deposited onto single-crystal silicon [51.105]. Lines of individual molecules are evident at the magnification shown. Figure 51.14a,b contrasts the molecular organization expected in an LB multilayer with that in electrostatically deposited layer-by-layer films. For the latter case, the polyelectrolyte chains within each layer will become entangled, and may even penetrate into the layers above and below, leading to a less-ordered film than that produced by LB deposition.

Atomic force micrograph of a 12-layer, n-eicosanoic-acid Langmuir–Blodgett film deposited onto silicon. (From [51.105], with permission from ACS)

Molecular organization in (a) a Langmuir–Blodgett multilayer assembly and (b) a polyelectrolyte multilayer deposited by the electrostatic method
4.3 Patterning Technologies
The problem of connecting together the individual processing elements in any future molecular computer is challenging. Each one of the 1.4 billion transistors in the Pentium Core i7 chip is addressable and connected to a power supply. Organic molecules can be difficult to arrange on a surface or in a 3-D array such that each molecule is addressable. Planar inorganic materials are normally patterned using photolithography. Here, a surface is first covered with a light-sensitive photoresist, which is exposed to ultraviolet light through a contact mask. Either the exposed photoresist (positive resist) or the unexposed regions (negative resist) can then be developed to leave a positive or negative image of the mask on the surface. This approach is routinely used in the fabrication of devices based on inorganic semiconductors. However, difficulties can be encountered when used with organic films, as the photoresists themselves are based on organic compounds.
Brittain et al. [51.106] describe a series of soft lithographic methods that may be better suited to the patterning of organic layers. Pouring a liquid polymer, such as polydimethylsiloxane (PDMS) onto a master made from silicon forms a pattern-transfer element. The polymer is allowed to cure to form an elastomer, which can then be removed from the master. This replica can subsequently be used as a stamp to transfer chemical ink, such as a solution of an alkanethiol, to a surface.
Scanning microscopy methods offer a powerful means of manipulating molecules. Careful control of an atomic force microscope (GlossaryTerm
AFM
) tip can allow patterns to be drawn in an organic film [51.108, 51.109]. Such techniques can also be used to reposition molecules, such as the fullerene C60, on surfaces and to break up an individual molecule [51.110]. A further approach that has recently been developed is called dip-pen nanolithography (GlossaryTermDPN
) [51.107] (Fig. 51.15). This is able to deliver organic molecules in a positive printing mode. An AFM tip is used to write alkanethiols on a gold thin film in a manner analogous to that of a fountain pen. Molecules flow from the AFM tip to a solid substrate (paper) via capillary transport, making DPN a potentially useful tool for assembling nanoscale devices. Recent developments of DPN have included an overwriting capability that allows one nanostructure to be generated and the areas surrounding that nanostructure to be filled with a second type of ink [51.111]. Perhaps the greatest limitation in using scanning probe methodologies for ultrahigh-resolution nanolithography over large areas derives from the serial nature of most techniques. However, multiple-pen nanoplotters capable of performing parallel lithography have been reported [51.112, 51.113]. In addition, PDMS elastomeric tip arrays of up to 11 million pens that were made with a silicon master have been developed for low-cost, high-throughput patterning [51.114]. The DPN method has also been used to deposit magnetic nanostructures [51.115] and arrays of protein molecules [51.116].
Dip-pen patterning showing the transfer of an ink onto paper using the tip of an AFM. (After [51.107])
The need to combine large-area coatings with device patterning has resulted in the development of direct-write fabrication methods, such as ink-jet printing [51.117, 51.118, 51.119, 51.120]. Although ink-jet printhead droplet ejection can be achieved with thermal (bubble-jet) and piezoelectric modes of operation, the majority of published literature on ink-jet printing as a tool for manufacturing organic devices has been the result of using piezoelectric-actuated printers. Piezoelectric printhead technology is favored primarily because it applies no thermal load to the organic inks and is compatible with the printing of digital images. The combination of solution-processable emissive polymers with ink-jet printing offers some promise in the development of low-cost high-resolution displays [51.121]. The technique has also been applied to the manufacturing of all-polymer transistor circuits [51.122, 51.123]. Ultrafine printing with 1 μm resolution has recently been demonstrated using an ink-jet system with subfemtoliter ejection accuracy [51.124].
Liquid thin films exhibit a number of unique physical processes, such as dewetting, pinning, and capillary action, and these processes have been investigated as a potential tool of patterning organic micro- and nanostructures on large-area substrates by low-cost solution techniques. For example, the control of the dewetting behavior of liquid thin films using substrates having a prepatterned variation of surface energies or structures has been used to guide organic materials to desired surfaces [51.126, 51.127]. The capillary action of the liquid into narrow channels has also been utilized to manipulate and deposit organic materials [51.128, 51.129] and biomaterials [51.130, 51.131], in which PDMS stamps have been adopted to form hydrophobic channels. In several of these approaches, pattern length scale and feature sizes are limited to the lateral dimension of prepatterned templates. However, further investigation of the self-assembly of organic molecules and physical phenomena in liquid thin films enables us to downscale dimensions relative to the size of the templates. An example is shown in Fig. 51.16 [51.125, 51.132]. This method uses the convex surfaces of an elastomeric stamp as forming positive motifs. As the stamp is gently placed on a liquid thin film made of a dilute solution, capillary forces drive the solution to form menisci under the stamp instead of filling the grooves, as shown in Fig. 51.16a. Depending on the intermolecular and molecule–substrate interactions, organic molecules are self-assembled on the substrate into patterns of split nanowires or strings of nanodots (Fig. 51.16b). A capability of the method for nanopatterning has been demonstrated using a molecular magnet [51.125] and a molecular semiconductor [51.132].

(a) Schematics of stamp-assisted deposition process and (b) atomic force micrograph of dotted structures printed from a diluted solution of molecular magnets. (After [51.125], with permission from the ACS)
4.4 Molecular Device Architectures
The bottom-up approach to molecular electronics offers many intriguing prospects for manipulating materials on the nanometer scale, thereby providing opportunities to build up novel architectures with predetermined and unique physical and/or chemical properties. Two relatively simple examples of organic superlattice structures are the incorporation of an electric polarization into a multilayer array to form thin films exhibiting pyroelectric behavior [51.133] and the use of noncentrosymmetric layers for second-order nonlinear optical response, for example, second-harmonic generation [51.134]. In both instances, the multilayer thin films can be built up using the LB approach.
To realize functional nanoelectronic circuits, a number of workers have investigated the electrical characteristics of structures in which organic molecules are sandwiched between two metallic electrodes. Of particular interest is the possibility of observing molecular rectification using monolayer or multilayer films. This follows the prediction [51.135] that an asymmetric organic molecule containing a donor and an acceptor group separated by a short σ-bonded bridge, allowing quantum-mechanical tunneling, should exhibit diode characteristics. There have been many attempts to demonstrate this effect in the laboratory, particularly in organic thin films [51.136, 51.137, 51.138]. Asymmetric current-versus-voltage behavior has certainly been recorded for many metal/insulator/metal structures, although these results are often open to several interpretations as a result of the asymmetry of the electrode configuration.
In other cases, switching behavior has been reported [51.12, 51.13, 51.138, 51.139, 51.3, 51.4, 51.5, 51.6, 51.7, 51.8]. As with the work on molecular rectification, the origin of the switching is not always clear. For example, is this a property of the organic molecules, the metallic electrode or of the presence of any interfacial (e. g., oxide) layer (or a combination of these)? Bistable rotaxane molecules have been used as the basis of some of these studies; an example is shown in Fig. 51.17a. This molecule is amphiphilic and the ring component can move between the polar and nonpolar regions of the main part of the molecule. The molecules can be assembled on an electrode using the LB approach and a top electrode is then deposited to form the crossbar structure shown in Fig. 51.17b [51.5, 51.6, 51.7]. Alternatively, the molecules can be solution-cast between Pt source and drain electrodes (with gaps of 1–2 nm) in transistor structures [51.9]. In some of the experiments, the absence of switching using control compounds suggests that the rotaxane molecule itself is responsible for the bistability [51.7].

(a) Bistable rotaxane. (b) Schematic diagram of cross-wire structure for switching studies. (After [51.5])
Bistable switching intrinsic to a molecular phenomenon has also been reported in single molecular layers formed by the self-assembly approach [51.139, 51.140]. A switching behavior trigged by an external voltage in a substituted conjugated molecular wire of bipyridy-dinitro oligophenylene-ethynylene dithiol [51.139] has been confirmed by studies both on monolayers and isolated molecules using various two-terminal configurations, such as crossed wire electrodes, a scanning tunneling microscope and a pair of electrodes with a nanometer-sized gap [51.140]. The experimental and theoretical studies indicate that such voltage-triggered switching originates in the presence of the nitro groups and their dipole moment facilitates molecular rotation during voltage sweeps [51.140, 51.141]. However, the fabrication of monolayer devices sandwiched metallic electrodes often suffers from electrical shorts, due to structural defects and/or the penetration of metallic atoms in the organic layer. In a pioneering work, monolayer formation in nanopores patterned by lithography has been exploited in device fabrication [51.15]. A simple approach that uses conducting thin films such as poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) [51.142] and multilayer graphene [51.143] as protecting interlayers for the metal penetration has recently been developed for high-yield, large-area fabrication of monolayer device arrays without electrical shorts. This technique has also been used to the demonstrate switching behavior in monolayer devices comprising photochromic molecules [51.144] and transition metal complex molecules [51.145]. Such results augur well for the development of molecular-scale logic circuitry.
Nanoscale organic devices can also exploit charge storage on nanoparticles or at interfaces. One important metal oxide semiconductor (GlossaryTerm
MOS
) device is the flash memory [51.146]. This is similar in structure to a MOS field-effect transistor (GlossaryTermMOSFET
), except that it has two gate electrodes, one on top of the other. The top electrode forms the control gate, below which a floating gate is capacitively coupled to the control gate and the underlying silicon. The memory cell operation involves putting charge on the floating gate or removing it, corresponding to two logic levels. Nanoflash devices utilize single or multiple nanoparticles as the charge-storage elements. These are usually embedded in the gate oxide of a FET and located in close proximity (2–3 nm) to the transistor channel [51.147]. Figure 51.18 shows the characteristics of a memory device incorporating metallic nanoparticles deposited by the LB technique [51.10]. The device structure is shown schematically in the figure. The gold nanoparticles (Q-Au) were of nominal diameter 10 nm and passivated with an organic capping layer. Cadmium arachidate (CdAr2) LB layers were used to provide an insulating gate layer. Figure 51.18 also shows the normalized capacitance versus voltage (C–V) data, measured at 1 MHz and at a voltage scan rate of 40 mV ∕ s for three different device structures:-
Al/SiO2/p-Si
-
Al/20 LB layers CdAr2/SiO2/p-Si
-
Al/20 LB layers CdAr2/one LB layer Q–Au/p-Si.
The C–V curve for the reference Al/SiO2/Si sample reveals the usual accumulation/depletion/inversion characteristics associated with metal–insulator–semiconductor (GlossaryTerm
MIS
) structures, with a flat-band voltage of approximately −1 V. Negligible hysteresis was evident on reversing the voltage scan. The data for the Si/SiO2/CdAr2 structure also show clear accumulation, depletion, and inversion regions, again with no hysteresis on reversing the direction of the voltage scan. The most significant difference in the structures with and without the Q-Au nanoparticles is the relatively large hysteresis in the MIS structure containing the Q-Au layer. This was thought to be indicative of charge storage in the gold nanoparticles [51.10]. In a somewhat different approach, the same group has used a self-assembly technique to chemically attach gold nanoparticles to a SiO2 surface [51.148]. When incorporated into a transistor structure, the resulting device was shown to behave as a nonvolatile electrically erasable programmable read-only memory [51.149]. Alternatively, gold nanoparticles deposited by layer-by-layer assembly using polyions have been applied to the fabrication of nonvolatile memory based on organic transistors [51.150, 51.151]. Recent progress involves the use of a mixed self-assembled monolayer of aliphatic and C60-functionalized phosphonic acid molecules as a molecular charge storage dielectric layer, which allows low-voltage operation of the organic transistor memory [51.152].
(a) Metal–insulator–semiconductor (MIS) structure incorporating gold nanoparticles; a transmission electron micrograph of the nanoparticles is shown. (b) Normalized capacitance versus voltage characteristics for different MIS devices. (After [51.10], © 2003 ACS)
Molecular-scale electronics may also offer increased device densities by fabricating 3-D architectures. In 1989, the principle of a 3-D memory based on LB films was described [51.153]. The device requires a molecule with a central conjugated region of high electron affinity (for an n-type material, the electron affinity is the energy difference between the bottom of the conduction band and the vacuum level) surrounded by aliphatic substituents of low electron affinity, A multilayer structure, as shown in Fig. 51.19, could be used to store one N-bit word, the presence or absence of charge on the n-th layer representing a 0 or 1 of the n-th bit. The LB film could be assembled on the gate of an FET and, on application of an electric field, transport of bits across the layers may be detected as induced charge on the gate.

(a) Molecular memory holding N bits (top) compared to a conventional silicon memory holding 1 bit (bottom). (b) Electron energy versus perpendicular distance for molecular memory with no applied electric field. (After [51.153], with permission from Elsevier)
Recent technological advancements have allowed the fabrication of structures in which one or few molecules bridge metallic electrodes through chemical bonds, such as SH terminal anchor groups. Such metal–molecule–metal junctions (molecular junctions ) [51.154] have been extensively studied as potential building blocks for molecular integrated circuits. One of key technologies is the development of metallic electrodes with a few nanometers gap (nanogap electrode ) capable of bridging small molecules. Currently, nanogap electrodes formed by mechanical break and electromigration of a prepatterned notched gold nanowire are most widely used for the fabrication of molecular junctions in planer configurations (these approaches have been referred to as break junction techniques ) [51.155, 51.156]. The gap width of electrodes formed by the mechanical break junction technique can be tuned by bending a flexible substrate under in situ monitoring of tunneling current flowing in the gap, allowing forming a junction of a single molecule with electrodes reproducibly [51.155]. The electromigration break junction technique only provides a statistical formation of molecule–electrode junctions, but is able to form transistor structures using back-gate electrodes having a thin gate dielectric layer (e. g., Al2O3), as shown in Fig. 51.20 a [51.157]. A molecular transistor that utilizes the electrostatic gating of molecular orbitals for the modulation of current flow has recently been demonstrated in benzene dithiols covalently bonded to gold electrodes (Fig. 51.20b) [51.158]. The effects of different anchor groups (e. g., NC, NH3, and COOH), molecular length, molecular conformation and substituents on the electrical properties of molecular junctions have extensively been investigated by the break junction techniques including a scanning tunneling microscope-controlled break junction [51.159] toward the development of molecular devices with various electronic functions.

(a) Scanning electron micrograph of gold nanogap electrodes formed by the electromigration break junction technique on a gate dielectric layer comprising a 3 nm thick aluminum oxide. (After [51.157], Courtesy of Nature Publishing Group). (b) Current versus voltage characteristics of a molecular transistor based on benzene dithiols at different gate voltages. (After [51.158], Courtesy of Nature Publishing Group)
5 DNA Electronics
The study of the electronic behavior of organic compounds has led some scientists to work on the electrical properties of biological materials. DNA is arguably the most significant molecule in nature. It may also be an important material for molecular electronics applications. Reports into the electronic properties of DNA have already generated controversy in the literature [51.16, 51.160, 51.161, 51.162, 51.163, 51.164]. According to some, DNA is a molecular wire of very small resistance. Others, however, find that DNA behaves as an insulator. These seemingly contradictory findings can probably be explained by the different DNA sequences and experimental conditions used to monitor the conductivity [51.16].
The DNA strand in the double-helix arrangement consists of a long polymer backbone consisting of repeating sugar molecules and phosphate groups (Fig. 51.21). Each sugar group is attached to one of four bases, guanine (G), cytosine (C), adenine (A), and thymine (T). The chemical bonding is such that an A base only ever pairs with a T base, while a G always pairs with a C. Some of the electron orbitals belonging to the bases overlap quite well with each other along the long axis of the DNA. This provides a path for electron transfer along the molecule, in a similar fashion to the 1-D conduction seen in conjugated polymers (Sect. 51.1.2).

DNA double helix showing the position of the four bases: guanine (G), cytosine (C), adenine (A), and thymine (T)
Theoretical and experimental work now suggests that a hole (i. e., a positive charge) is more stable on a G–C base pair than on an A–T base pair [51.16]. The energy difference between these two pairs is substantially larger than the thermal energy of the charge carrier. Under these conditions, a hole will localize on a particular G–C base pair. Because the A–T base pairs have a higher energy, they act as a barrier to hole transfer. However, if the distance between two G–C base pairs is small enough, the hole can tunnel quantum-mechanically from one pair to the next. In this way, charge carriers are able to shuttle along a single DNA molecule over a distance of a few nanometers.
Recent experimental and theoretical results suggest that native DNA strands consisting of the specific sequence is a wide band-gap semiconductor [51.165, 51.166, 51.167]. However, the conductivity of DNA is still controversial, especially in experimental work, because it depends on conditions such as trapped water and counter ions in the DNA strands. Such factors may allow a different channel for conduction and also influence on the geometrical arrangements of base pairs DNA. In addition, modification of DNA with metals and ions can change the electronic behavior. For the realization of DNA electronics, it is imperative to predict the electronic properties theoretically as well as to control the experimental and useable conditions of the DNA.
DNA chips exploit the fact that short strands of DNA will bind to other segments of DNA that have complementary sequences, and can therefore be used to probe whether certain genetic codes are present in a given specimen of DNA. Microfabricated chips with many parallel DNA probes are becoming widespread in analytical and medical applications. Currently, the chips are read out optically, but further miniaturization might require new read-out schemes, possibly involving the electron-transfer properties of DNA.
Computations by chemical or biological reactions overcome the problem of parallelism and interconnections in a classical system. If a string of DNA can be put together in the right sequence, it can be used to solve combinational problems. The calculations are performed in test tubes filled with strands of DNA. Gene sequencing is used to obtain the result. For example, Adleman [51.168] calculated the traveling-salesman problem to demonstrate the capabilities of DNA computing. DNA computing on parallel problems potentially provides 1014 millions of instructions per second (GlossaryTerm
MIPS
) and uses less energy and space than conventional supercomputers. While CMOS supercomputers operate 109 operations per Joule, a DNA computer could perform about 1019 operations per Joule. Data could potentially be stored on DNA in a density of approximately 1 bit ∕ nm3 while existing storage media such as DRAMs require 1012 nm3 to store 1 bit.The fact that a DNA strand binds specifically with other strand having a complementary sequence is relevant to the formation of electronic nano-structures. This is so-called DNA origami in a literature. Using DNA strands having various designed sequences, many architectures such as 2-D sheets [51.169], various shapes and patterns [51.170], and 3-D structures [51.171], were formed by self-assembly. These structures are useful as scaffolds for other materials to implement an electronic function [51.172].
The differences in the electronic properties of the nucleotides have been expected to be usable for identifying the sequence of DNA. Since a DNA strand was shown to translocate through nanopores fabricated on solid state films, many efforts have been made to sequence DNA using electronic measurements [51.173, 51.174, 51.175]. The difference in tunneling current through the nucleotides was measured with small electrodes made by break-junction [51.176], which suggests the possibility of DNA sequencing electronically.
6 Conclusions
Organic compounds possess a wide range of fascinating physical and chemical properties that make them attractive candidates for exploitation in electronic and optoelectronic devices. It is not, however, anticipated that these materials will displace silicon in the foreseeable future as the dominant material for fast signal processing. It is much more likely that organic materials will find use in other niche areas of electronics, where silicon and other inorganic semiconductors cannot compete. Examples already exist, such as liquid-crystal displays, photoreceptors in electrophotography, and certain chemical sensors. Organic light-emitting structures are likely to make a major impact in the marketplace as display and lighting devices.
Over the first decades of the twenty-first century, classical CMOS technology will come up against a number of technological barriers. The bottom-up approach to molecular electronics provides an alternative and attractive way forward and, as such, it is currently an area of exciting interdisciplinary activity. However, the challenges in fabricating molecular switches and connecting them together are still formidable. Living systems use a different approach; these assemble themselves naturally from molecules and are extremely energetically efficient when compared with man-made computational devices. More radical approaches to materials fabrication and device design, exploiting self-organization, may be needed to realize fully the potential offered by molecular-scale electronics.
References
M.C. Petty: Molecular Electronics: From Principles to Practice (Wiley, Chichester 2008)
T.H. Richardson (Ed.): Functional Organic and Polymeric Materials (Wiley, Chichester 2000)
J.M. Tour: Molecular Electronics (World Scientific, New Jersey 2003)
P.E. Kornilovitch, A.M. Bratkovsky, R.S. Williams: Phys. Rev. B 66, 245413 (2002)
Y. Chen, G.-Y. Jung, D.A.A. Ohlberg, X. Li, D.R. Stewart, J.O. Jeppesen, K.A. Nielsen, J.F. Stoddart, R.S. Williams: Nanotechnology 14, 462 (2003)
Y. Chen, D.A.A. Ohlberg, X. Li, D.R. Stewart, R.S. Williams, J.O. Jeppesen, K.A. Nielsen, J.F. Stoddart, D.L. Olynick, E. Anderson: Appl. Phys. Lett. 82, 1610 (2003)
M.R. Diehl, D.W. Steuerman, H.-R. Tseng, S.A. Vignon, A. Star, P.C. Celestre, J.F. Stoddart, J.R. Heath: Chem. Phys. Chem. 4, 1335 (2003)
L. Ma, S. Pyo, J. Ouyang, Q. Xu, Y. Yang: Appl. Phys. Lett. 82, 1419 (2003)
H. Yu, Y. Luo, K. Beverly, J.F. Stoddart, H.-R. Tseng, J.R. Heath: Angew. Chem. 42, 5706 (2003)
S. Paul, C. Pearson, A. Molloy, M.A. Cousins, M. Green, S. Kolliopoulou, P. Dimitrakis, P. Normand, D. Tsoukalas, M.C. Petty: Nano. Lett. 3, 533 (2003)
J.M. Tour, L. Cheng, D.P. Nackashi, Y. Yao, A.K. Flatt, S.K. St. Angelo, T.E. Mallouk, P.D. Franzen: J. Am. Chem. Soc. 125, 13279 (2003)
L.D. Bozano, B.W. Kean, V.R. Deline, J.R. Salem, J.C. Scott: Appl. Phys. Lett. 84, 607 (2004)
J. Ouyang, C.-W. Chu, C.R. Szmanda, L. Ma, Y. Yang: Nature Mater. 3, 918 (2004)
R.M. Metzger: J. Solid State Chem. 168, 696 (2002)
J. Chen, W. Wang, M.A. Reed, A.M. Rawlett, D.W. Price, J.M. Tour: Appl. Phys. Lett. 77, 1224 (2000)
C. Dekker, M.A. Ratner: Phys. World 14, 29 (2001)
K.E. Drexler: Nanosystems: Molecular Machinery, Manufacturing and Computation (Wiley, New York 1992)
C. Nicolini (Ed.): Molecular Manufacturing (Plenum, New York 1996)
S. Roth: One-Dimensional Metals (VCH, Weinheim 1995)
M. Ikawa, T. Yamada, H. Matsui, H. Minemawari, J. Tsutsumi, Y. Horii, M. Chikamatsu, R. Azumi, R. Kumai, T. Hasegawa: Nat. Commun. 3, 1176 (2012)
H.-R. Tseng, H. Phan, C. Luo, M. Wang, L.A. Perez, S.N. Patel, L. Ying, E.J. Kramer, T.-Q. Nguyen, G.C. Bazan, A.J. Heeger: Adv. Mater. 26, 2993 (2014)
C.D. Dimitrakopoulos, D.J. Mascaro: IBM J. Res. Devel. 45, 11 (2001)
M.R. Bryce, M.C. Petty: Nature 374, 771 (1995)
M. Abe, T. Mori, I. Osaka, K. Sugimoto, K. Takimiya: Chem. Mater. 27, 5049 (2015)
H. Minemawari, T. Yamada, H. Matsui, J. Tsutsumi, S. Haas, R. Chiba, R. Kumai, T. Hasegawa: Nature 475, 364 (2011)
K.S. Novoselov, V.I. Fal’ko, L. Colombo, P.R. Gellert, M.G. Schwab, K. Kim: Nature 490, 192 (2012)
W.J. Feast, J. Tsibouklis, K.L. Pouwer, L. Groenendaal, E.W. Meijer: Polymer 37, 5017 (1996)
J.R. Ferraro, J.M. Williams: Introduction to Synthetic Electrical Conductors (Academic, Orlando 1987)
J.D. Wright: Molecular Crystals (Cambridge Univ. Press, Cambridge 1995)
K.S. Novoselov: Nobel Lecture, Graphene: Materials in the Flatland, https://www.nobelprize.org/nobel_prizes/physics/laureates/2010/novoselov_lecture_OnlinePDF.pdf (2010)
H.W. Kroto, D.R.M. Walton (Eds.): The Fullerenes (Cambridge Univ. Press, Cambridge 1993)
G. Timp (Ed.): Nanotechnology (Springer, Berlin, Heidelberg 1998)
R. Saito, G. Dresselhaus, M.S. Dresselhaus: Physical Properties of Carbon Nanotubes (Imperial College, London 1998)
L. Duclaux: Carbon 40, 1751 (2002)
F. Li, A. Nathan, Y. Wu, B.S. Ong: Organic Thin Film Transistor Integration: A Hybrid Approach (Wiley, Weinheim 2011)
Plastic Logic: A basic display, http://www.plasticlogic.com/ (2016)
S.R. Forrest: Nature 428, 911 (2004)
E.H. Rhoderick: Metal–Semiconductor Contacts (Clarendon, Oxford 1978)
Y.L. Hua, M.C. Petty, G.G. Roberts, M.M. Ahmad, M. Hanack, M. Rein: Thin Solid Films 149, 161 (1987)
G. Horowitz: Adv. Mater. 2, 286 (1990)
H.E. Katz: J. Mater. Chem. 7, 369 (1997)
S. Scheinert, G. Paasch: Phys. Status Solidi (a) 201, 1263 (2004)
E.C.P. Smits, S.G.J. Mathijssen, P.A. van Hal, S. Setayesh, T.C.T. Geuns, K.A.H.A. Mutsaers, E. Cantatore, H.J. Wondergem, O. Werzer, R. Resel, M. Kemerink, S. Kirchmeyer, A.M. Muzafarov, S.A. Ponomarenko, B. de Boer, P.W.M. Blom, D.M. de Leeuw: Nature 455, 956 (2008)
H. Kawaguchi, T. Someya, T. Sekitani, T. Sakurai: IEEE Solid-State Circuits 40, 177 (2005)
M.S. Fuhrer, B.M. Kim, T. Durkop, T. Brintlinger: Nano. Lett. 2, 755 (2002)
A. Javey, Q. Wang, A. Ural, Y.M. Li, H.J. Dai: Nano. Lett. 2, 929 (2002)
F. Léonard, J. Tersoff: Phys. Rev. Lett. 88, 258302 (2002)
K. Tsukagoshi, N. Yoneya, S. Uryu, Y. Aoyagi, A. Kanda, Y. Ootuka, B.W. Alphenaar: Physica B 323, 107 (2002)
C.W. Tang, S.A. VanSlyke: Appl. Phys. Lett. 51, 913 (1987)
J.H. Burroughes, D.D.C. Bradley, A.R. Brown, R.N. Marks, K. Mackay, R.H. Friend, P.L. Burns, A.B. Holmes: Nature 347, 359 (1990)
K. Müllen, U. Scherf (Eds.): Organic Light Emitting Devices: Synthesis, Properties and Applications (Wiley, Weinheim 2006)
T. Tsujimura: OLED Display Fundamentals and Applications (Wiley, Hoboken 2012)
S. Schols: Device Architecture and Materials for Organic Light-Emitting Devices (Springer, Dordrecht 2011)
Cambridge Display Technology: OLED Display Technology, http://www.cdtltd.co.uk/ (2016)
K. Ghaffarzadeh, N. Bardsley: OLED Lighting Opportunities 2015–2025: Forecasts, Technologies, Players (IDTechEx, Cambridge 2015)
L.S. Hung, C.W. Tang, M.G. Mason: Appl. Phys. Lett. 70, 152 (1997)
G.-Y. Jung, C. Pearson, L.E. Horsburgh, I.D.W. Samuel, A.P. Monkman, M.C. Petty: J. Phys. D: Appl. Phys. 33, 1029 (2000)
Y. Zhou, C. Fuentes-Hernandez, J. Shim, J. Meyer, A.J. Giordano, H. Li, P. Winget, T. Papadopoulos, H. Cheun, J. Kim, M. Fenoll, A. Dindar, W. Haske, E. Najafabadi, T.M. Khan, H. Sojoudi, S. Barlow, S. Graham, J.-L. Brédas, S.R. Marder, A. Kahn, B. Kippelen: Science 336, 327 (2012)
K. Morii, M. Ishida, T. Takashima, T. Shimoda, Q. Wang, M.K. Nazeeruddin, M. Grätzel: Appl. Phys. Lett. 89, 183510 (2006)
H. Fukagawa, K. Morii, M. Hasegawa, Y. Arimoto, T. Kamada, T. Shimizu, T. Yamamoto: Appl. Phys. Express 7, 082104 (2014)
C. Adachi, S. Tokito, T. Tsutsui, S. Saito: Jpn. J. Appl. Phys. 27, L269 (1988)
C. Adachi: Jpn. J. Appl. Phys. 53, 060101 (2014)
M. Ikai, S. Tokito, Y. Sakamoto, T. Suzuki, Y. Taga: Appl. Phys. Lett. 79, 156 (2001)
C. Adachi, M.A. Baldo, M.E. Thompson, S.R. Forrest: J. Appl. Phys. 90, 5048 (2001)
H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi: Nature 492, 234 (2012)
J. Kido, M. Kimura, K. Nagai: Science 267, 1332 (1995)
J. Kido, H. Shionoya, K. Nagai: Appl. Phys. Lett. 67, 2281 (1995)
J. Thompson, R.I.R. Blyth, M. Mazzeo, M. Anni, G. Gigli, R. Cingolani: Appl. Phys. Lett. 79, 560 (2001)
G.M. Farinola, R. Ragni: Chem. Soc. Rev. 40, 3467 (2011)
M.C. Gather, A. Köhnen, K. Meerholz: Adv. Mater. 23, 233 (2011)
M.A. Green, K. Emery, Y. Hishikawa, W. Warta, E.D. Dunlop: Prog. Photovolt: Res. Appl. 24, 3 (2016)
C.W. Tang: Appl. Phys. Lett. 48, 183 (1986)
V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya, H. Murata: Nat. Photonics 9, 403 (2015)
M. Saito, I. Osaka, Y. Suzuki, K. Takimiya, T. Okabe, S. Ikeda, T. Asano: Sci. Rep. 5, 14202 (2015)
L. Huo, T. Liu, X. Sun, Y. Cai, A.J. Heeger, Y. Sun: Adv. Mat. 27, 2938 (2015)
J. Subbiah, B. Purushothaman, M. Chen, T. Qin, M. Gao, D. Vak, F.H. Scholes, X. Chen, S.E. Watkins, G.J. Wilson, A.B. Holmes, W.W.H. Wong, D.J. Jones: Adv. Mat. 27, 702 (2015)
S. Günes, H. Neugebauer, N.S. Sariciftci: Chem. Rev. 107, 1324 (2007)
C.J. Brabec, A. Cravino, D. Meissner, N.S. Sariciftci, T. Fromherz, M. Minse, L. Sanchez, J.C. Hummelen: Adv. Funct. Mater. 11, 374 (2001)
M. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A.J. Heeger, C. Brabec: Adv. Mater. 18, 789 (2006)
M. Hiramoto, H. Fujiwara, M. Yokoyama: Appl. Phys. Lett. 58, 1062 (1991)
S. Morita, A.A. Zakhidov, K. Yoshino: Solid State Commun. 82, 2084 (1992)
G. Yu, J. Gao, J.C. Hummelen, F. Wudl, A.J. Heeger: Science 270, 1789 (1995)
B. O’Regan, M. Grätzel: Nature 353, 737 (1991)
S. Zhang, X. Yang, Y. Numata, L. Han: Environ. Sci. 6, 1443 (2013)
Semiconductor Industry Association: International Technology Roadmap for Semiconductors (ITRS), http://www.itrs2.net/ (2011)
D.J. Frank, R.H. Dennard, E. Nowak, P.M. Solomon, Y. Taur, H.-S.P. Wong: Proc. IEEE 89, 259 (2001)
J. Greer, A. Korkin, J. Lanbanowski (Eds.): Nano and Giga Challenges in Microelectronics (Elsevier, Amsterdam 2003)
IC Knowledge LLC: The Economics of the ITRS, http://www.icknowledge.com/ (2011)
M.C. Petty: Organic thin-film deposition techniques. In: Functional Organic and Polymeric Materials, ed. by T.H. Richardson (Wiley, Chichester 2000) p. 7
A. Ulman: Ultrathin Organic Films (Academic, San Diego 1991)
M.C. Petty: Langmuir–Blodgett Films (Cambridge Univ. Press, Cambridge 1996)
D.R. Talham: Chem. Rev. 104, 5479 (2004)
J.C. Love, L.A. Estroff, J.K. Kriebel, R.G. Nuzzo, G.M. Whitesides: Chem. Rev. 105, 1103 (2005)
A.J. Moore, L.M. Goldenberg, M.R. Bryce, M.C. Petty, A.P. Monkman, C. Marenco, J. Yarwood, M.J. Joyce, S.N. Port: Adv. Mater. 10, 395 (1998)
L. Netzer, J. Sagiv: J. Am. Chem. Soc. 105, 674 (1983)
L. Netzer, R. Iscovici, J. Sagiv: Thin Solid Films 99, 235 (1983)
G. Decher, J.D. Hong, J. Schmitt: Thin Solid Films 210/211, 831 (1992)
G. Decher, Y. Lvov, J. Schmitt: Thin Solid Films 244, 772 (1994)
G. Decher, J.B. Schlenoff (Eds.): Multilayer Thin Films (Wiley, Weinheim 2003)
K. Ariga, J.P. Hill, Q. Ji: Phys. Chem. Chem. Phys. 9, 2319 (2007)
C. Pearson, J. Nagel, M.C. Petty: J. Phys. D: Appl. Phys. 34, 285 (2001)
J.H. Cheung, W.B. Stockton, M.F. Rubner: Macromolecules 30, 2712 (1997)
Y. Lvov, M. Onda, K. Ariga, T. Kunitake: J. Biomater. Sci. Polym. Edn. 9, 345 (1998)
W.B. Stockton, M.F. Rubner: Macromolecules 30, 2717 (1997)
S.A. Evanson, J.P.S. Badyal, C. Pearson, M.C. Petty: J. Phys. Chem. 100, 11672 (1996)
S. Brittain, K. Paul, X.-M. Zhao, G. Whitesides: Phys. World 11, 31 (1998)
R.D. Piner, J. Zhu, F. Xu, S. Hong, C.A. Mirkin: Science 283, 661 (1999)
L.F. Chi, L.M. Eng, K. Graf, H. Fuchs: Langmuir 8, 2255 (1992)
A. Krämer, R.R. Fuierer, C.B. Gorman: Chem. Rev. 103, 4367 (2003)
J. Gimzewski: Phys. World 11, 25 (1998)
S. Hong, J. Zhu, C.A. Mirkin: Science 286, 523 (1999)
S. Hong, C.A. Mirkin: Science 288, 1808 (2000)
K. Salaita, Y. Wang, J. Fragala, R.A. Vega, C. Liu, C.A. Mirkin: Angew. Chem. Int. Ed. 45, 7220 (2006)
F. Huo, Z. Zheng, G. Zheng, L.R. Giam, H. Zhang, C.A. Mirkin: Science 321, 1658 (2008)
X.G. Liu, L. Fu, S.H. Hong, V.P. Dravid, C.A. Mirkin: Adv. Mater. 14, 231 (2002)
K.B. Lee, S.J. Park, C.A. Mirkin, J.C. Smith, M. Mrksich: Science 295, 1702 (2002)
S.P. Speakman, G.G. Rozenberg, K.J. Clay, W.I. Milne, A. Ille, I.A. Gardner, E. Bresler, J.H.G. Steinke: Organic Electron. 2, 65 (2001)
G. PerHin, B.T. Khuri-Yakub: Rev. Sci. Instrum. 73, 2193 (2002)
H. Sirringhaus, T. Shimoda (Eds.): MRS Bull. 28, 802 (2003)
M. Singh, H.M. Haverinen, P. Dhagat, G.E. Jabbour: Adv. Mater. 22, 673 (2010)
I.D. Rees, K.L. Robinson, A.B. Holmes, C.R. Towns, R. O’Dell: MRS Bull. 27, 451 (2002)
H. Sirringhaus, T. Kawase, R.H. Friend, T. Shimoda, M. Inbasekaran, W. Wu, E.P. Woo: Science 290, 2123 (2000)
A.C. Arias, S.E. Ready, R. Lujan, W.S. Wong, K.E. Paul, A. Salleo, M.L. Chabinyc, R. Apte, R.A. Street, Y. Wu, P. Liu, B. Ong: Appl. Phys. Lett. 85, 3304 (2004)
T. Sekitani, Y. Noguchi, U. Zschieschang, H. Klauk, T. Someya: Proc. Natl. Acad. Sci. USA 105, 4976 (2008)
M. Cavallini, F. Biscarini, J. Gomez-Segura, D. Ruiz, J. Veciana: Nano Lett. 3, 1527 (2003)
M. Böltau, S. Walheim, J. Mlynek, G. Krausch, U. Steiner: Nature 391, 877 (1998)
R. van Hameren, P. Schön, A.M. van Buul, J. Hoogboom, S.V. Lazarenko, J.W. Gerritsen, H. Engelkamp, P.C.M. Christianen, H.A. Heus, J.C. Maan, T. Rasing, S. Speller, A.E. Rowan, J.A.A.W. Elemans, R.J.M. Nolte: Science 314, 1433 (2006)
E. Kim, Y. Xia, G.M. Whitesides: Nature 376, 581 (1995)
F. Zhang, T. Nyberg, O. Inganäs: Nano Lett. 2, 1373 (2002)
R.S. Kane, S. Takayama, E. Ostuni, D.E. Ingber, G.M. Whitesides: Biomaterials 20, 2363 (1999)
E. Delamarche, D. Juncker, H. Schmid: Adv. Mater. 17, 2911 (2005)
M. Cavallini, F. Biscarini: Nano Lett. 3, 1269 (2003)
C.A. Jones, M.C. Petty, G.G. Roberts: IEEE Trans. Ultrasonics Ferroelec. Freq. Control 35, 736 (1988)
P.J. Skabara: Organic second-order non-linear optical materials. In: Functional Organic and Polymeric Materials, ed. by T.H. Richardson (Wiley, Chichester 2000) p. 295
A. Aviram, M.A. Ratner: Chem. Phys. Lett. 29, 277 (1974)
A.S. Martin, J.R. Sambles, G.J. Ashwell: Phys. Rev. Lett. 70, 218 (1993)
G.J. Ashwell, D.S. Gandolfo: J. Mater. Chem. 11, 246 (2001)
J.R. Heath, P.J. Kuekes, G.S. Snider, R.S. Williams: Science 280, 1716 (1998)
A.S. Blum, J.G. Kushmerick, D.P. Long, C.H. Patterson, J.C. Yang, J.C. Henderson, Y.X. Yao, J.M. Tour, R. Shashidhar, B.R. Ratna: Nat. Mater. 4, 167 (2005)
E. Lörtscher, J.W. Ciszek, J. Tour, H. Riel: Small 2, 973 (2006)
V. Meded, A. Bagrets, A. Arnold, F. Evers: Small 5, 2218 (2009)
H.B. Akkerman, P.W.M. Blom, D.M. de Leeuw, B. de Boer: Nature 441, 69 (2006)
G. Wang, Y. Kim, M. Choe, T.-W. Kim, T. Lee: Adv. Mater. 23, 755 (2011)
A.J. Kronemeijer, H.B. Akkerman, T. Kudernac, B.J. van Wees, B.L. Feringa, P.W.M. Blom, B. de Boer: Adv. Mater. 20, 1467 (2008)
J. Lee, H. Chang, S. Kim, G.S. Bang, H. Lee: Angew. Chem. Int. Ed. 48, 8501 (2009)
B.G. Streetman, S. Banjeree: Solid State Electronic Devices (Prentice Hall, New Jersey 2000)
R. Compañó (Ed.): Technology Roadmap for Nanoelectronics (Office for Official Publications of the European Communities, Luxembourg 2001)
S. Kolliopoulou, P. Dimitrakis, P. Normand, H.-L. Zhang, N. Cant, S.D. Evans, S. Paul, C. Pearson, A. Molloy, M.C. Petty: J. Appl. Phys. 94, 5234 (2003)
S. Kolliopoulou, P. Dimitrakis, P. Normand, H.-L. Zhang, N. Cant, S.D. Evans, S. Paul, C. Pearson, A. Molloy, M.C. Petty, D. Tsoukalas: Micro. Eng. 73/74, 725 (2004)
Z. Liu, F. Xue, Y. Su, Y.M. Lvov, K. Varahramyan: IEEE Trans. Nanotech. 5, 379 (2006)
S.-J. Kim, Y.-S. Park, S.-H. Lyu, J.-S. Lee: Appl. Phys. Lett. 96, 033302 (2010)
M. Burkhardt, A. Jedaa, M. Novak, A. Ebel, K. Voitchovsky, F. Stellacci, A. Hirsch, M. Halik: Adv. Mater. 22, 2525 (2010)
P.E. Burrows, K.J. Donovan, E.G. Wilson: Thin Solid Films 179, 129 (1989)
H. Song, M.A. Reed, T. Lee: Adv. Mater. 23, 1583 (2011)
M.A. Reed, C. Zhou, C.J. Muller, T.P. Burgin, J.M. Tour: Science 278, 252 (1997)
H. Park, A.K.L. Lim, A.P. Alivisatos, J. Park, P.L. McEuen: Appl. Phys. Lett. 75, 301 (1999)
W.J. Liang, M.P. Shores, M. Bockrath, J.R. Long, H. Park: Nature 417, 725 (2002)
H. Song, Y. Kim, Y.H. Jang, H. Jeong, M.A. Reed, T. Lee: Nature 462, 1039 (2009)
B. Xu, N.J. Tao: Science 301, 1221 (2003)
H.-W. Fink, C. Schöneberger: Nature 398, 407 (1999)
R.S. Phadke: Appl. Biochem. Biotechnol. 96, 269 (2001)
A. Rakitin, P. Aich, C. Papadopoulos, Y. Kobzar, A.S. Vedeneev, J.S. Lee, J.M. Xu: Phys. Rev. Lett. 86, 3670 (2001)
C.N.R. Rao, A.K. Cheetham: J. Mater. Chem. 11, 2887 (2001)
H. Tabata, L.T. Cai, J.H. Gu, S. Tanaka, Y. Otsuka, Y. Sacho, M. Taniguchi, T. Kawai: Synth. Met. 133/134, 469 (2003)
R.G. Endres, D.L. Cox, R.R.P. Singh: Rev. Mod. Phys. 76, 195 (2004)
M. Taniguchi, T. Kawai: Physica E 33, 1 (2006)
S.S. Mallajosyula, S.K. Pati: J. Phys. Chem. Lett. 1, 1881 (2010)
L. Adleman: Science 266, 1021 (1994)
E. Winfree, F. Liu, L.A. Wenzler, N.C. Seeman: Nature 394, 539 (1998)
P.W.K. Rothemund: Nature 440, 297 (2006)
W.M. Shih, J.D. Quispe, G.F. Joyce: Nature 427, 618 (2004)
H. Yan, S.H. Park, G. Finkelstein, J.H. Reif, T.H. LaBean: Science 26, 1882 (2003)
J. Li, D. Stein, C. McMullan, D. Branton, J.A. Golovchenko: Nature 412, 166 (2001)
P. Chen, J. Gu, E. Brandin, Y.-R. Kim, Q. Wang, D. Branton: Nano Lett. 4, 2293 (2004)
M. Zwolak, M.D. Ventra: Rev. Mod. Phys. 80, 141 (2008)
M. Tsutsui, M. Taniguchi, K. Yokota, T. Kawai: Nat. Nanotech. 5, 286 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Petty, M.C., Nagase, T., Suzuki, H., Naito, H. (2017). Molecular Electronics. In: Kasap, S., Capper, P. (eds) Springer Handbook of Electronic and Photonic Materials. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-319-48933-9_51
Download citation
DOI: https://doi.org/10.1007/978-3-319-48933-9_51
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48931-5
Online ISBN: 978-3-319-48933-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)