
Summary
Time-resolved electron paramagnetic resonance (TREPR) methods often play an important role in characterizing artificial photosynthetic systems. The radical pairs and triplet states generated in such systems are spin polarized because of the initial correlation of the electron spins and the spin selectivity of the electronic relaxation and electron transfer. The polarization makes the TREPR signals of the states fundamentally different from those of equilibrium systems and makes it possible to extract information about the geometry of the radical pairs and about the pathway and kinetics of electronic relaxation and electron transfer. In this chapter, we give an overview of the different types of TREPR experiments that can be performed on artificial photosynthetic complexes and the different polarization patterns that are observed. This is followed by a summary of recent results on a several selected systems, which illustrate the strengths and weakness of the technique.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
18.1 Introduction
18.1.1 Transient Paramagnetic Species in Natural Photosynthesis
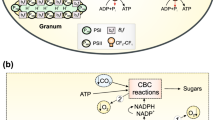
In oxygenic photosynthesis , the absorption of light leads to electron transfer through several proteins embedded in the thylakoid membrane (Fig. 18.1). This process results in the oxidation of water in the lumen and the reduction of NADP in the stroma. The initial steps take place in Photosystems I and II in which a series of sequential radical pairs are generated as the electron is transferred along the electron transfer chain. These radical pairs have been studied extensively using time-resolved electron paramagnetic resonance (TREPR) spectroscopy (Möbius 1997; van der Est 2001, 2009; Bittl and Zech 2001; Stehlik 2006; Thurnauer et al. 2004). A key feature of natural photosynthetic systems is the extremely high quantum yield of charge separation, which is achieved by optimizing the properties of the cofactors such that the rates of forward electron transfer are orders of magnitude faster than competing processes such as charge recombination or intersystem crossing. As a consequence of the fast electron transfer , the radical pairs are produced in spin states that are non-eigenstates of the spin system and the population distribution is far from equilibrium. This has a profound effect on the TREPR signals observed from photosynthetic reaction centers and allows details of the structure and dynamics of the system to be determined. TREPR methods and their application to natural photosynthesis have been summarized in detail in a number of review articles and book chapters (Möbius 1997; van der Est 2001, 2009; Bittl and Zech 2001; Stehlik 2006; Thurnauer et al. 2004).

Schematic diagram of the electron transport chain in oxygenic photosynthesis . The structures of the proteins have been generated from the following protein databank files using the program molmol (Koradi et al. 1996): Photosytem I 1JB0 (Jordan et al. 2001); Photosystem II 2AXT (Loll et al. 2005) Cytochrome b 6 f 1VF5(Kurisu et al. 2003); Plastocyanin 1JXD(Bertini et al. 2001); Ferredoxin 1FXI(Tsukihara et al. 1990); Ferredoxin-NADP+ reductase (FNR) 1FNB(Bruns and Karplus 1995)
The goal of artificial photosynthesis is to mimic the features of the natural systems and TREPR methods have been used widely to study artificial photosynthetic complexes. Despite the goal of mimicking the natural systems, the artificial complexes often display significant differences, which have important consequences for their time-resolved EPR spectra (Forbes et al. 2013).
18.1.2 Differences Between Natural and Artificial Photosynthesis
The term artificial photosynthesis refers to a wide range of complexes all of which mimic natural photosynthesis to some degree. These systems can be broadly grouped into the two classes shown in Fig. 18.2 based on whether the catalysis is homogeneous or heterogeneous. In photocells (Fig. 18.2a) a photosensitizer injects electrons into the conduction band of a semiconductor and activates heterogeneous catalysis at the electrode surface. In homogeneous biomimetic systems (Fig. 18.2b), synthetic analogues of the components of the photosynthetic reaction centers are coupled together to act as a shuttle for electrons between two catalysts that should perform oxidative and reductive chemistry in solution. The arrangement, shown in Fig. 18.2b, is only one of many possible schemes of this type and the position of the chromophore in the electron transfer chain, the nature of the bridges, donors and acceptors, the addition of antenna chromophores and many other factors can be varied. The vast majority of TREPR studies involve the characterization of such complexes, normally in the absence the redox catalysts, and thus, the primary focus of this chapter will be in this area.

Different possible schemes for artificial photosynthesis . (a). Photocell in which a photo-anode is used to split water and drive the reduction of protons at the cathode. (b). Donor-acceptor triad coupled to two catalysts to drive the oxidation of water and reduction of protons
In natural photosynthesis , the protein plays an essential role in tuning the properties of the cofactors and high quantum efficiency electron transfer has been achieved primarily through optimization of the protein-cofactor interactions (Allen and Williams 2014). In artificial donor-acceptor (D-A) complexes, the solvent and bridging groups between the donor and acceptor play the role of the protein. Because it is difficult to achieve the same level of control found in the natural photosystems, intersystem crossing in the donor and back electron transfer often compete with forward electron transfer and the lifetime of the final charge-separated state is usually shorter. The covalently bound bridging groups also generally provide stronger electronic coupling between the electron donors and acceptors than is found in a protein matrix in which the cofactors are non-covalently bound. In addition, partially ordered solvents are sometimes used to stabilize the charge separation. All of these factors lead to a wider variety of paramagnetic states and TREPR spectra for synthetic donor acceptor complexes compared to the natural photosystems.
In the following section, an overview of the basic principles of TREPR will be given, followed by a description of the spectra of the different species observed in artificial photosynthetic systems.
18.2 Time-Resolved EPR Methods
18.2.1 Transient EPR
The most straightforward TREPR experiment is transient EPR. A detailed description of the method can be found in Forbes et al. (2013) and only a brief overview is given here. As shown in Fig. 18.3, a laser flash with a pulse length of typically 10 ns or less is applied to the sample while it is irradiated with continuous microwaves in a static magnetic field. The microwave absorption is measured as a function of time giving a transient response. The magnetic field is then stepped over a range of values and a transient is collected at each field position. The resulting collection of transients forms a time/magnetic field dataset and transient EPR spectra can be generated by plotting the signal amplitude in a chosen time window as a function of the magnetic field.

Transient EPR. Continuous microwaves are used to monitor the EPR signal response of the sample to a short laser flash at a fixed magnetic field strength. The field is stepped over a region to create a time/field dataset from which transient EPR spectra can be extracted
If lock-in detection with 100 kHz field modulation is used, the response time is on the order of 100 μs. However, with so-called direct detection, the response time can be reduced to several tens of nanoseconds, and is limited by the Fourier broadening at short times. The faster response time comes at the cost of much lower sensitivity but this loss in sensitivity is compensated for by the strong spin polarization observed at short times. The polarization arises from the spin selectivity of the photoreactions and gives rise to both absorptive (A) or emissive (E) signal contributions. An important feature of TREPR studies is the analysis of the patterns of the absorptive and emissive polarization. In most spectroscopic experiments the absorption coefficient, the concentration and the temperature determine the intensity. However, for TREPR spectra the pathway by which the paramagnetic state was generated is the most important factor. Thus, the same state can have different spectra if it can be generated by different mechanisms . For example, singlet and triplet electron transfer can be distinguished by the polarization pattern of the resulting radical pair.
18.2.2 Pulsed EPR
Pulsed EPR techniques can also be used to measure light-induced paramagnetic species (Schweiger and Jeschke 2001). Such experiments are technically more challenging than transient EPR measurements but they provide additional information and allow the spin system to be manipulated by shifting population between the spin states. They can be grouped into two general types illustrated in Fig. 18.4. In the field-swept echo experiment (Fig. 18.4a) a two-pulse sequence is used to generate a spin echo (other echo sequences can also be used) and the height of the echo is recorded over a range of magnetic field positions to give a spectrum. In systems undergoing photochemical reactions, the delay after the laser flash at which the microwave pulses are applied can be varied to obtain the spectrum at different times during the reaction. In addition, the delay between the microwave pulses can be chosen to suppress or emphasize signal contributions from different species. To obtain a field-swept echo spectrum corresponding to the allowed single-quantum transitions of the spin system, the excitation bandwidths of the pulses should be small compared to the width of the spectrum. For molecular triplet states this is essentially always the case. However, for weakly coupled radical pairs, which have narrow spectral widths, the excitation bandwidth must be kept small.

Pulsed EPR experiments. (a) Field Swept Echo. After a laser flash, which generates a paramagnetic species, the two microwave pulses generate an echo. The field-swept echo spectrum is generated by measuring the echo amplitude as a function of the magnetic field. (b) Out-of-phase echo modulation. For a singlet-born radical pair the echo is phase shifted by 90° and is the amplitude of the echo is modulated strongly as a function of the spacing between the pulses by the spin-spin coupling
Echo modulation measurements are the other main class of pulse EPR experiments. They are illustrated in Fig. 18.4b using the out-of-phase echo modulation of a light-induced radical pair as an example. In an echo modulation experiment the height of the echo is measured as a function of the spacing between the pulses at a fixed magnetic field. The height of the echo is modulated by the weak interactions of the spin system. In the sequence shown in Fig. 18.4b only one pulse spacing is varied and hence a single modulation decay curve is obtained. For more complex pulse sequences multiple delays can be varied giving multidimensional modulation datasets.
In the case of a weakly coupled singlet-born radical pair, the echo is phase-shifted by 90° compared to that of a stable radical and shows deep modulations due to the coupling between the two spins. The values of the coupling constants can be obtained either by fitting calculated modulation curves to the experimental data or from the positions of the features in the Fourier transform of the modulation curve.
18.3 Quasi-Static Polarization Patterns
The EPR signals from a photoreaction depend on both the kinetics of the reaction and the spin dynamics of the light-induced paramagnetic species. In general, the analysis of TREPR data requires that the time dependent spin density matrix of the system be calculated (Schweiger and Jeschke 2001). However, if the microwave field is weak (i.e. in the linear response regime) and the signals are measured after the decay of any coherence effects (i.e. if off-diagonal elements of the density matrix can be ignored), the spin polarized transient EPR spectrum or field-swept echo spectrum can be calculated from the transitions between energy levels of the static spin Hamiltonian of the system.
In the light-induced radical pairs generated by electron transfer in D-A complexes, the strength of the spin-spin coupling has a strong effect on the observed TREPR spectra. There are two components to the coupling and they depend on the structure of the complex and the distance between the electrons. The dipolar coupling falls off with r −3 and the exchange coupling between the electrons can be written as:
where r 0 is the van der Waals radius and the attenuation factor β depends on the bridge between the donor and acceptor (Schubert et al. 2015). Thus, within the range of distances found in D-A complexes large variations in the strength of the coupling occur. For a system of two coupled S = 1/2 spins, there are two distinct regimes. If the spin-spin coupling is much larger than the difference of the Larmor precession frequencies, the eigenstates can be separated into singlet and triplet manifolds and the spin system is strongly coupled. In contrast, if the coupling is small compared to the difference in the precession frequencies, the spin system is weakly coupled.
18.3.1 Spin-Polarized, Weakly-Coupled Radical Pairs
Weakly coupled radical pairs generated from a singlet precursor have been described extensively in the literature because their importance in photosynthetic reaction centers (Kandrashkin et al. 1998; Norris et al. 1990; Closs et al. 1987; Hore et al. 1987; Stehlik et al. 1989; Angerhofer and Bittl 1996; Kandrashkin and van der Est 2001, 2007; Kamlowski et al. 1998; Savitsky et al. 2007, 2013). The electron spin polarization in such systems can be described using the spin correlated radical pair (SCRP) model (Forbes et al. 2013; Kandrashkin et al. 1998; Closs et al. 1987; Hore et al. 1987; Stehlik et al. 1989; Buckley et al. 1987). As shown in Fig. 18.5, there are four spin energy levels two of which are pure triplet states and two which have both singlet and triplet character. If the radical pair is formed from a pure singlet state only the energy levels with singlet character are populated and thus there are two absorptive and two emissive transitions. Electron transfer from a triplet precursor, also gives two absorptive and two emissive transitions but the pattern of emission and absorption is different. For a fixed orientation of the radical pair in the magnetic field, the spectrum (Fig. 18.5, bottom) consists of two antiphase doublets. The splitting of the doublets is determined by the spin-spin coupling and each of the doublets is centered at the effective g-value of the respective radicals. Both the splitting and the positions of the lines are orientation dependent.

Energy level diagram and stick spectrum of a singlet-born weakly coupled radical pair
The observed spectrum also depends on the motion of the donor-acceptor complex . Several different situations arise depending on the solvent used and the temperature. At low temperature in frozen solution, the system can be treated as being in the rigid limit, while in liquid or nematic solution, rapid motion occurs that leads to averaging of the interactions. In the rigid limit, the sum over all possible orientations of the radical pair in the magnetic field must be taken. In natural photosynthetic reactions centers, the tumbling of the protein complexes is slow even in liquid solution so that the rigid limit applies to both frozen and liquid solution samples. In contrast, in donor-acceptor mimics, the rigid limit only applies below the freezing point of the solvent.
In the experimental spectrum, the lines are normally broadened by unresolved hyperfine couplings and if the inhomogenous linewidth is larger than the spin-spin coupling the antiphase doublet for each radical of a singlet-born radical pair is given by (Kandrashkin and van der Est 2001, 2007):
where i refers to the two radicals and the total spectrum is I total = I donor + I acceptor . Δω i is the inhomogeneous linewidth of each radical, ω eff = ħ −1 g eff βB 0 is the resonance frequency and ω 0 is the microwave frequency. The orientation dependences of d(θ,ϕ) and g eff (θ,ϕ) makes the polarization pattern dependent on the relative orientations of the principal axes of the two g-tensors and the vector from one radical to the other that defines the dipolar coupling. Thus, the spectra can be used to determine the geometry of the radical pair. However, if the exchange coupling is negligible, additional information is required to obtain a unique solution for the geometry (Kandrashkin and van der Est 2001, 2007). This is because the spin-spin coupling influences only the amplitude of the antiphase doublet described by Eq. 18.2 and not its shape. As a result, the absolute amplitude of the spectrum must be known if the spin-spin coupling and geometry is to be determined uniquely. Determination of the geometry also requires that the nature of the precursor (singlet, triplet or mixed singlet-triplet) be known. In the natural photosystems, the highly efficient forward electron transfer ensures that the initial state of the first observable radical pair is a pure singlet. In artificial donor-acceptor complexes, triplet electron transfer and/or significant singlet-triplet mixing can occur.
Figure 18.6 illustrates the sensitivity of radical pair powder spectra to geometry of the radical pair and the nature of the precursor. The black spectra (a, d) are for a singlet precursor and the red and blue spectra (b, c, e, f) for a triplet precursor with different population distributions of the spin states. The spectra on the (a–c) were calculated with the dipolar coupling axis parallel to the x-axis of the g-tensor of radical 1 and in the spectra on the right (d–f) the g-tensor has been rotated by 90° so that the y-axis is parallel to the dipolar coupling axis. For the singlet precursor spectra, (a, d) the sign of the polarization on the low-field end of the spectrum is sensitive to the orientation of the dipolar coupling vector relative to the x-axis of the g-tensor of radical 1. For a triplet precursor, however, the polarization pattern depends on the spin selectivity of the intersystem crossing by which the triplet state is formed (spin selective intersystem crossing is discussed in Sect. 18.3.4) and either sign (absorptive or emissive) is possible on the low field end of the spectrum. A characteristic feature of triplet state formation by intersystem crossing is that it also creates net polarization of the spin system, which is generally emissive (Salikhov et al. 1984). Thus, the spectra of radical pairs generated from a triplet precursor typically have net emissive polarization as shown in spectra c, d, e and f in Fig. 18.6

Calculated spin-polarized EPR spectra of a weakly coupled radical pair in the rigid limit. The spectra illustrate the effect of the relative orientation of the donor and acceptor and the influence of the precursor spin state. a, b, c: dipolar-coupling axis parallel to the x–axis of radical 1. d, e, f: Dipolar-coupling axis parallel to the y–axis of radical 1. a, d: singlet precursor. b, e: triplet precursor populated according to \( {S}_{\Omega, z}^2-\frac{1}{3}{\overrightarrow{S}}^2 \)in the molecular frame. c, f: triplet precursor populated according to \( {S}_{\Omega, x}^2-{S}_{\Omega, y}^2 \) in the molecular frame. Principal g-values of the two radicals of the radical pair: radical 1 gxx = 2.014, gyy = 2.008, gzz = 2.002; radical 2 gxx = gyy = gzz = 2.002. Dipolar coupling constant = −0.6 mT. Exchange coupling = 0
When the tumbling of the molecules is fast, e.g. liquid solution at room temperature, the orientation dependent terms are replaced by their average values. An important consequence of this is that the spectrum no longer depends on the dipolar coupling since its average value is zero. As a result the spectra no longer depend on the internal geometry of the radical pair. Moreover, if the exchange coupling is negligible and the radical pair is formed from a singlet precursor the absorptive and emissive lines cancel each other completely and no spectrum is observed. However, in most donor acceptor complexes the exchange coupling is sufficiently large that this situation does not arise but the radical pair signals are sometimes weak because the cancellation.
Liquid crystalline solvents in the nematic phase are often used for studying donor-acceptor complexes because the solvent dynamics stabilize the radical pair states (Hasharoni and Levanon 1995; Wiederrecht et al. 1999a, 1997.) In such solvents, the motion of the molecules is rapid but they have a non-zero average alignment relative to a direction known as the director. In the absence of any external fields the director varies randomly and there is no macroscopic alignment of the solvent. However, in the magnetic field of an EPR spectrometer, the director becomes aligned and in most nematic phases is parallel to the magnetic field. When a solute is dissolved in a nematic phase it also becomes partially ordered largely as a result of short-range interactions with the solvent (van der Est et al. 1987; Burnell and De Lange 1998). The ordering of a solute molecule is described by its order matrix, the elements of which are given by:
where i and j refer to a set of axes fixed in the molecule and Z is the direction of the magnetic field. Determining the principal axes of the order matrix in a molecule of low symmetry is not trivial but since the ordering is the result of short-range interactions, the principal z-axis of rod-like molecules is along the rod axis and for extended planar molecules it is perpendicular to the plane.
For a radical pair in a rapidly tumbling donor-acceptor complex in a nematic liquid crystal, the dipolar coupling averages to:
where θ is the angle between the vector and the magnetic field and the angled brackets mean the weighted average over all orientations. The term in angled brackets is the order parameter of the dipolar coupling vector. The g-values of the radicals are also averaged:
where S ii , i = x,y,z are the order parameters of the principal axes of the g-tensor. The order parameters are difficult to predict accurately for donor-acceptor complexes but approximate values can be estimated from the structure . In general the bridging groups define the long axis of the molecule (Fig. 18.2b) and the direction of the dipolar coupling. Since the ordering is determined by short-range interactions, orientations of the molecule with its long axis parallel to the director and the magnetic field are more probable than orientations with the long axis perpendicular to the field. Thus, the order parameter of the dipolar-coupling axis is generally positive.
Several calculated spectra of rapidly tumbling radical pairs are shown in Fig. 18.7. On the left, the motion is assumed to be isotropic, while on the right anisotropic motion as would be observed in a nematic liquid crystal has been assumed. The exchange coupling J has been taken to be negative. In the case of isotropic motion, the negative sign of J results in A/E polarization for each of the antiphase doublets when the precursor is a pure singlet state (a). For a triplet prescursor the sign of the multiplet polarization depends on the orientation of the complex in the magnetic field. For rapid motion, the multiplet polarization averages to zero and only the emissive polarization remains (b). If the exchange coupling is negligible (c) no spectrum is observed as discussed above. In an anisotropic solvent the spectra show significant differences. If the exchange and dipolar coupling constants have the same sign, then the splitting of the lines in the antiphase doublet, 2(J–d), can be of opposite sign in isotropic and anisotropic solvents, which results in an inversion of the sign of the polarization. With a triplet precursor the multiplet polarization does not average to zero in an anisotropic solvent and an E/A/E/A pattern with net emission is obtained for the choice of couplings used (e). Similarly, when the exchange coupling is negligible (f) the spectrum does not average to zero because the average dipolar coupling is non-zero.

Calculated spin-polarized EPR spectra of a weakly coupled radical pair undergoing rapid motion. The spectra illustrate the influence of the precursor spin state and the effect of partial ordering when the exchange and dipolar coupling constants are of the same sign. Left: isotropic motional averaging. Right: anisotropic averaging such that the dipolar-coupling axis has an order parameter of 0.4. The magnetic parameters and geometry of the radical pair are the same as in Fig. 18.6 except that J = −0.17 mT has been assumed for spectra a, b, d and e. Spectra a and d singlet precursor; b and e triplet precursor with populated according to \( {S}_{\Omega, z}^2-\frac{1}{3}{\overrightarrow{S}}^2 \); c and f exchange coupling J = 0, singlet precursor
18.3.2 Strongly-Coupled Radical Pairs
In donor-acceptor dyads it is common for the spin-spin coupling to be larger than the difference of the resonance frequencies of the two radicals. Under these conditions, the radical pair is strongly coupled and its spin states are nearly pure singlet and triplet states. Since the singlet state is EPR silent, the spectrum of such a radical pair is that of the triplet state, which gives an antiphase doublet. The sign of the polarization pattern and the amount of net polarization depend on how the radical pair was formed and what type of solvent is used (Fig. 18.8). In an isotropic solvent, the dipolar coupling is averaged to zero but an antiphase doublet is still observed (Fig. 18.8a, b). The reason for this is that the resonance positions of the two transitions in a triplet state are not identical and the difference between them is approximately \( {\left({\omega}_{eff}^D-{\omega}_{eff}^A\right)}^2/4 J \) (van der Est and Poddutoori 2013). For a molecular triplet state , the exchange coupling J is large and this difference is negligible but for a radical pair, the value of J can be sufficiently small that the two peaks are observable despite the fact that the dipolar coupling is averaged. In a liquid crystalline environment, the average dipolar coupling is no longer zero and the separation between the lines is determined by d eff (Fig. 18.8c, d) In either case, if the separation is much smaller than inhomogeneous broadening of the lines, the observed splitting of the antiphase doublet is determined by the linewidth, Δω, of the two overlapping lines and the intensity of the resulting pattern is determined by the separation, \( {\left({\omega}_{eff}^D-{\omega}_{eff}^A\right)}^2/4 J \) or 2d eff . Since the g-factors of most organic radicals do not differ strongly from the free electron value, \( \left({\omega}_{eff}^D-{\omega}_{eff}^A\right) \) is small and the separation of the lines in an isotropic solvent is typically much smaller than in a liquid crystal and hence the intensity is much weaker. Often, in an isotropic solvent the observed splitting is determined by the linewidth, while in a nematic liquid crystal it is due to the dipolar coupling as shown in Fig. 18.8. The sign of the dipolar-coupling constant in a radical pair is negative, and hence the sign of the pattern E/A or A/E, can be used to determine the population distribution. An E/A pattern (Fig. 18.8a, c) implies that the T 0 level of the radical pair is preferentially populated. An A/E pattern (Fig. 18.8b, d) implies excess population in T + and T − , which is only possible from triplet electron transfer .

Calculated spectra of a strongly coupled radical pair. a, b: isotropic solution c, d liquid crystalline solution with S = 0.6 for the dipolar coupling axis. a, c singlet precursor or triplet precursor with excess population in T 0. b, d: triplet precursor with excess population in T +/T −. Parameters: g 1 = 2.0023, g 2 = 2.0030, J = −2.0 mT, D = −1.5 mT, Δω = 0.5 mT
18.3.3 Sequential Radical Pairs
In D-A triads, a series of radical pairs can be produced by any of the following reactions, depending on where the chromophore is located in the electron transfer chain:
Here, I refers to a chromophore in the middle of the triad. In all cases, two sequential radical pairs are produced and if the lifetime of the first radical pair is long enough to allow singlet-triplet mixing to occur, the spin polarized TREPR spectrum of the second radical pair is affected. The mechanism of singlet-triplet mixing is illustrated in Fig. 18.9. Initially, the spin system is in a pure singlet state, in which the two spin vectors are antiparallel to one another. However, because the two spins in the radical pair are in different environments their precession frequencies differ. Thus, spin 2 will precess with a frequency (ω 1–ω 2) in a frame of reference rotating with the precession frequency of spin 1. As a result of the different precession frequencies, the spin system oscillates between the S and T 0 states.

Schematic representation of singlet-triplet mixing
The effect of singlet-triplet mixing on the spin polarization in sequential electron transfer has been described theoretically in detail (Kandrashkin et al. 1998, 2002; Norris et al. 1990; Hore 1996; Wang et al. 1992; Tang et al. 1996) and involves calculating the evolution of the density matrix. This is a rather involved process since the time at which the electron hops from one acceptor to the next is a statistical process and the integral over the ensemble must be taken. However, the polarization is usually observed at times that are long compared to the electron transfer lifetime and the decay of coherence effects. Under these conditions, Kandrashkin et al. (1998, 2002, 2007) were able derive analytical expressions for the spin polarization, which are extremely useful for understanding the observed polarization patterns. If the electron transfer is initiated from a singlet state, the singlet triplet mixing in each radical pair in the series generates additional polarization in the subsequent radical pairs. The polarization of a given radical pair can be broken down into three contributions: (i) singlet polarization, i.e. the polarization that would be seen without any singlet triplet mixing, (ii) a contribution that arises from the singlet-triplet mixing due differences in the g-values of the two radicals (iii) polarization associated with inhomogenous broadening in cases in which only one of the two unpaired electrons is transferred. These three contributions are illustrated in Fig. 18.10, spectra a, b and c, respectively. The relative intensities of these three contributions can be written as functions of the electron transfer rates and magnetic parameters of the radical pairs. The singlet contribution (spectrum a) is proportional to:
where the subscript 2 refers to the secondary radical pair. The additional polarization arising from singlet-triplet mixing due to the difference in the g-values of the radicals in the precursor (spectrum b) is given by:
where k 1 is the decay rate of the precursor, which is usually dominated by the forward electron transfer rate. The contribution from the inhomogeous line broadening (spectrum c) is:
The spectra on the right of Fig. 18.10 show how the polarization pattern of the secondary radical pair changes as the lifetime and magnetic parameters of the precursor change. The observed changes are primarily a result of changes in the contribution described by Eq. 18.8. In Fig. 18.10d, the lifetime of the precursor has been varied. In accordance with Eq. 18.8, the net polarization of the two radicals increases as the lifetime is increased from 1 ns (Fig. 18.10d, black spectrum) to 3 ns (Fig. 18.10d, blue spectrum). In Fig. 18.10e, the spin-spin coupling in the precursor has been varied. The sign of the net polarization depends on the sign of the coupling in the precursor and in the observed radical pair. In the spectra in Fig. 18.10 the dipolar coupling has been set to zero and the value of J in the secondary pair is positive. When the value of J in precursor is also positive (Fig. 18.10e, blue spectrum) the radical with resonances at lower field shows net emission and the higher field antiphase doublet shows net absorption. When the sign of J in the precursor is negative (Fig. 18.10e, red and black spectra) the pattern is reversed and the low field antiphase doublet shows net absorption. The effect of the difference in the g-values of the radicals in the precursor is shown in Fig. 18.10f. If the two radicals in the precursor have the same g-factor then no net polarization is observed in the secondary radical pair (Fig. 18.10f, red spectrum). When the sign of the g-factor difference is the same in the precursor and secondary radical pair, and J is negative in the precursor and positive in the secondary radical pair (Fig. 18.10f, black spectrum), the low field doublet has net absorption and the high field doublet has net emission. When the sign of the g-factor difference is opposite the sign of the net polarization in each doublet is reversed (Fig. 18.10f, blue spectrum). Thus, with sufficient information about the g-factors of the radicals and the sign of the coupling it is possible to deduce, the approximate lifetime of the precursor from such spectra.

The effect of ST-mixing on the TREPR spectra of the secondary radical pair in sequential electron transfer . a: singlet contribution b: additional polarization due the g-factor difference in the precursor radical pair; c: additional polarization resulting from inhomogenous hyperfine broadening in the precursor; d: Influence of the precursor lifetime, black τ = 1 ns, black τ = 2 ns, blue τ = 3 ns; e: Influence of the spin-spin coupling in the precursor, black J = −1.0 mT, red J = −0.5 mT, blue J = 0.5 mT; f: Influence of the g-factor difference in the precursor, black g donor – g acceptor = −0.0027, red g donor – g acceptor = 0.0, blue g donor – g acceptor = 0.0027
18.3.4 Triplet States
In both natural and artificial photosynthetic systems, molecular triplet states can also be formed, either by intersystem crossing from the excited singlet state of a chromophore or by charge recombination. Because both of these processes are spin selective, the resulting triplet state is spin polarized. However, the selectivity of the two processes is different and hence the TREPR spectra differ (Budil and Thurnauer 1991; Thurnauer 1979). This can be a very useful tool in characterizing the photo-physics of D-A complexes, especially when used in combination with transient optical methods.
Figure 18.11 illustrates the two different pathways by which a molecular triplet state can be formed and how they result in different population distributions in the spin sublevels. Charge recombination is shown in Fig. 18.11a and intersystem crossing in Fig. 18.11b. In both cases, singlet-triplet interconversion must occur but the mechanism is different. As discussed above, when the charge-separated state D + A − is formed, mixing between S and T 0 occurs as the spins precess and as a result recombination to the triplet state can occur. Because the singlet-triplet mixing in the radical pair is only between S and T 0, only the T 0 sublevel of the triplet state is populated.

Different possible pathways by which a molecular triplet state can be formed. (a): Radical pair recombination. (b): Intersystem crossing
In the case of intersystem crossing, the presence of spin-orbit coupling results in mixing of the singlet and triplet states. Because this interaction is governed by molecular symmetry, while the wave functions of the spin states are determined largely by the Zeeman interaction with the external field, the intersystem crossing rates to the three triplet sublevels differ and depend on the orientation of the molecule in the field. The probability of intersystem crossing to a given sublevels can be written as a linear combination of probabilities associated with each of the three principal axes in the molecule (Forbes et al. 2013; Levanon 1987):
The probabilities p x , p y and p z depend on the strength of the spin-orbit coupling in the molecular x, y and z directions. The coefficients c ix , c iy , and c iz depend on the orientation of the molecule and can be obtained by writing the triplet wavefunctions in terms of the zero-field wavefunctions:
Because, the absolute amplitude of the spin polarization is usually not known only the ratios of the probabilities can be determined. Moreover, since their sum equals one, there are only two independent probabilities. This description also does not take net polarization of the spin system into account.
An more elegant way to describe the population distribution is to use the traceless diagonal part of the density matrix Δρ, which represents the differences in the populations of the spin levels and can be expanded in terms of the matrix representations of the spin operators (Kandrashkin et al. 2006a). It may also be written as the sum of a multiplet polarization contribution Δρ M with equal amounts of absorption and emission and a net polarization contribution Δρ N with either pure emission or absorption. The mutliplet polarization is invariant to inversion and thus can be described by the even powers of the spin operators (Salikhov et al. 1984). In the case of radical pair recombination the multiplet polarization can be written:
For intersystem crossing the multiplet polarization represents the differences in the intersystem crossing rates that arise from the traceless anisotropy of the spin-orbit coupling, and two contributions are needed (Kandrashkin et al. 2006a, b):
where θ and ϕ describe the orientation of the molecule in the magnetic field. The important difference between the two cases is that the polarization generated by radical pair recombination does not depend on the orientation of the molecule while the polarization generated during intersystem crossing does. Any measured polarization pattern can be reproduced as a linear combination of these contributions:
The parameters\( {\kappa}_M^{axial} \),\( {\kappa}_M^{non- axial} \) can be related to the probabilities p x , p y and p z such that \( {\kappa}_M^{axial} \) = 1.0, \( {\kappa}_M^{non- axial} \) = 0.0 corresponds to p x :p y :p z = 0:0:1; \( {\kappa}_M^{axial} \) = −1.0, \( {\kappa}_M^{non- axial} \) = 1.0 corresponds to p x :p y :p z = 0:1:0 and \( {\kappa}_M^{axial} \) = −1.0, \( {\kappa}_M^{non- axial} \) = −1.0 corresponds to p x :p y :p z = 0:0:1.
Figure 18.12 shows examples of these contributions to the polarization pattern for a free-base porphyrin. The spectra a and b correspond to \( \Delta {\rho}_M^{axial} \)and \( \Delta {\rho}_M^{non- axial} \), respectively and are the contributions to the multiplet polarization from intersystem crossing. Spectrum c is the pattern generated by radical pair recombination and spectrum d is the net polarization. Spectrum e, dashed line, is a weighted sum of spectra a, b and d that reproduces the experimental spectrum (solid line).

Spin polarization patterns for molecular triplet states . a: multiplet contribution \( \Delta {\rho}_M^{axial} \) (Eq. 18.8); b: multiplet contribution\( \Delta {\rho}_M^{non- axial} \) (Eq. 18.8); c: radical pair recombination contribution \( \Delta {\rho}_M^{RP} \) (Eq. 18.7); d: net polarization contribution proportional to –Sz; e simulation of the polarization pattern of a free-base porphyrin with κ axial : κ non − axial : κ net = − 1.0 : 0.47 : 0. solid line: experimental spectrum, dashed line: simulation. For all of the spectra D = 38.4 mT, E = 7.8 mT and g = 2.0023
18.4 Time Dependent Effects
The time traces in a TREPR experiment also contain a significant amount of information. However, extracting this information is more complicated than the analysis of the spin polarization patterns.
18.4.1 Electron Transfer
TREPR data can be used to determine electron transfer rates but the range of accessible lifetimes is limited. The theoretical lower limit of the time resolution is determined by Fourier broadening and for an experiment performed at 9 GHz (X-band) this limit is on the order of 1–10 ns. In practice, the bandwidth of the resonator and the detection results in a response time that is several tens of nanoseconds or more, depending on the instrument. The sensitivity of such instruments is usually not high enough to detect, the equilibrium Boltzmann polarization and hence the range of available lifetimes is also limited by T1 relaxation. Nonetheless, in favorable cases, lifetimes in the range of ~50 ns to several tens of microseconds are accessible. Usually, the decay of the TREPR signal of a radical pair is determined by a combination of charge recombination and spin relaxation and it is difficult to distinguish the two effects using the EPR data alone. Hence, in general it is necessary to use both optical and EPR methods to obtain a clear picture of the kinetics . The main advantage of TREPR methods for measuring kinetics is that in contrast to optical methods, only paramagnetic species are observed and there are no overlapping signals from diamagnetic excited states or the ground state. In addition, triplet states and radical pairs are easily distinguished and the spin selectivity of the electron transfer can be studied. An example of this is illustrated in Fig. 18.13. Electron transfer with predominantly singlet character generates a weakly coupled radical pair with the population distribution shown on the left. This distribution results in the polarization pattern shown under the energy level diagram. Charge recombination from states with singlet character is faster than from those with pure triplet character. Thus states Ψ2 and Ψ3 are depopulated relative to Ψ1 and Ψ4. As a result the polarization pattern becomes weaker and inverts. A time trace corresponding to the field position marked with an arrow under the spectrum on the left is shown on the right of Fig. 18.13. The initial population distribution results in a strong emissive signal at this field position. As the singlet recombination occurs the signal rapidly inverts and then decays slowly as the states with triplet character are depopulated. From such time traces, the singlet and triplet backreaction rates can be determined. In most cases, spin relaxation between the pure triplet states (Ψ1 and Ψ4) and the mixed singlet/triplet states (Ψ2 and Ψ3) is expected to be the rate-limiting step in triplet recombination.

Example of spin selective charge recombination of a radical pair and its effect on the associated TREPR signals
18.4.2 Quantum Beats
If a radical pair is generated on a time scale such that no significant singlet-triplet mixing occurs, its initial state is a pure singlet as shown in Fig. 18.9 and singlet-triplet mixing occurs due to the subsequent precession of the spins. This motion of the spin system modulates the intensity of the EPR transitions. The intensity of the transitions shown in Fig. 18.5 are determined by the product of the transition probability and the population difference. Because ψ 1 and ψ 4 are pure triplet sates, the transition probability is proportional to the triplet character of the mixed states ψ 2 and ψ 3 Thus, as the spin system oscillates between a pure singlet and pure triplet state the intensity of the EPR transitions oscillate. Such oscillations are referred to as quantum beats (Bittl and Kothe 1991; Salikhov et al. 1990) and they can be observed at short times following the laser flash. At longer time, they decay as the relative phase of the oscillations in different parts of the sample becomes random. Typically, phase relaxation times are on the order of 100 ns or less, which makes the quantum beats difficult to observe. Because they depend on the rate of singlet-triplet mixing they can be analyzed to obtain the magnetic parameters of the spin system. However, this information can also be obtained from the analysis of the spin polarization pattern, which is much easier to measure.
18.4.3 Out of Phase Echo Modulation
Early observation of the electron spin echo signals from photosynthetic samples showed that the echo was phase shifted by 90° compared to the echo from a stable radical (Thurnauer et al. 1979, 1982; Thurnaur and Norris 1980; Thurnauer and Clark 1984). The origin of the phase shift was later shown to be a result of the correlation between the electron spins and that the amplitude of the echo is modulated by the spin-spin interactions (Salikhov et al. 1992; Tang et al. 1994). This provides an elegant way of measuring the spin-spin coupling and it has been widely used to characterize the natural photosynthetic systems (Bittl and Zech 2001; Angerhofer and Bittl 1996; Savitsky et al. 2007, 2013; Bittl and Kothe 1991; Borovykh et al. 2002; Bittl and Zech 1997; Zech et al. 1996). For many artificial complexes the out-of-phase echo is not observed or the modulation decays too rapidly to determine the spin-spin coupling. The absence of an out-of-phase echo indicates a lack the correlation between the spins, which can occur as a result of dephasing of singlet-triplet mixing in the primary radical pair or when triplet electron transfer is the dominant mechanism . As a result there are far fewer reports of the use of out-of-phase echo modulation experiments on D-A complexes.
18.5 Recent Results
18.5.1 Early Results on Donor-Acceptor Complexes
The early results of TREPR experiments on donor-acceptor complexes for artificial photosynthesis are summarized in a number of review articles (Levanon and Möbius 1997; Savitsky and Moebius 2006; Gust et al. 2001; Wasielewski 1992, 2006; Verhoeven 2006). In these initial studies, the goal was primarily to mimic various aspects of the natural photosystems. Some of the first systems to be investigated were the porphyrin-quinone dyads and triads studied by Möbius and Kurreck (Lendzian et al. 1991; Hasharoni et al. 1993; Batchelor et al. 1995; Kay et al. 1995; Elger et al. 1998; Fuhs et al. 2000; Wiehe et al. 2001) that were designed to mimic the chlorophyll donor and quinone acceptors of photosynthetic reaction centers . While most of these complexes displayed light induced electron transfer from the porphyrin to the quinone, the TREPR spectra were very different from those of the natural systems because of differences in the strength of the spin-spin coupling, relaxation dynamics and electron transfer pathway. The widest array of D-A systems has been studied by Wasielewski and co-workers (Wiederrecht et al. 1999a, 1997, 1999b; Wasielewski 1992, 2006; Wasielewski et al. 1988, 1990, 1991, 1993, 1995; Hasharoni et al. 1995, 1996; Laukenmann et al. 1995; van der Est et al. 1996; Levanon et al. 1998; Heinen et al. 2002; Shaakov et al. 2003; Dance et al. 2006, 2008a, b; Jakob et al. 2006; Tauber et al. 2006; Ahrens et al. 2007; Mi et al. 2009; Carmieli et al. 2009; Giacobbe et al. 2009; Scott et al. 2009; Miura et al. 2010; Wilson et al. 2010; Miura and Wasielewski 2011; Ls et al. 2012; Colvin et al. 2012, 2013). Using the charge transfer bands of compounds such as 4-aminonaphthalene-1,8-dicarboximide and 3,5-dimethyl-4-(9-anthracenyl) julolidine to initiate electron transfer , efficient, long-lived charge separation could be achieved by attaching secondary donors such as aniline, tetrathiafulvalene (TTF) and tetramethylbenzo-bisdioxole (BDX) and secondary acceptors such naphthalenediimide (NDI) and pyromellitic diimide (PI). An important feature of these dyads and triads is that long-lived radical pair formation occurs in both liquid and frozen solution, which greatly facilitates TREPR measurements. The low reorganization energy of fullerene (C60) as an electron acceptor also promotes long-lived charge separation as first shown by Gust and Moore (Liddell et al. 1994; Carbonera et al. 1998). Using these systems it has been possible to reproduce all of the characteristic TREPR signatures seen in photosynthetic reaction centers and obtain important information about the electron transfer . In the following sections we highlight a number of recent examples and summarize the TREPR studies on organic bulk heterojunction solar cells .
18.5.2 Sequential Electron Transfer in Triads
As discussed in Sect 18.3.3, in a sequential electron transfer reaction the spin dynamics that occurs during the lifetime of the primary radical pair affects the polarization pattern of the subsequent radical pairs. We have investigated this phenomenon in a series of triads based on aluminum(III) porphyrin (AlPor) in which an acceptor (NDI or C60) and secondary donor (TTF) are attached on opposite faces of the porphyrin as shown in Fig. 18.14 (van der Est and Poddutoori 2013; Poddutoori et al. 2013, 2015). Excitation of the AlPor leads to transfer of the excited electron in the LUMO of the porphyrin to the LUMO of the acceptor as well as hole transfer from the HOMO of the porphyrin to the HOMO of the donor. This reaction creates the radical pair TTF•+NDI•– or TTF•+C60 •– depending on which acceptor is used.

Structure of axial aluminum(III) porphyrin-based D-A triads
The transient EPR spectra of the triads are shown in Fig. 18.15. In both cases, the spectrum consists of two antiphase doublets with an E/A/E/A polarization pattern. The low field doublet is due to TTF•+, while the high-field doublet arises from the reduced acceptor, either NDI•– or C60 •–. The antiphase doublets are not symmetric and in each doublet one of the two peaks is considerably stronger than the other. With NDI as the acceptor (Fig. 18.15, left) the low-field doublet due to TTF•+ shows net emissive polarization and the NDI•– doublet at higher field has net absorption. With C60 as the acceptor, the situation is reversed (Fig. 18.15, right). The E/A/E/A pattern is consistent with singlet electron transfer and negative spin-spin coupling. However, pure singlet electron transfer leads to symmetric antiphase doublets as shown in Fig. 18.10. The net polarization of each doublet is due to singlet-triplet mixing during the lifetime of the precursors. The simulations of the spectra (red curves) take this mixing into account (van der Est and Poddutoori 2013; Poddutoori et al. 2013, 2015). The different sign of the net polarization in the two cases can be easily understood as the result of a change in the sign of the g-factor difference in the precursor state. As shown in Eq. 18.7, the sign of the additional polarization generated by singlet-triplet mixing depends on the sign of g D –g A in the precursor state. For the radical pair AlPor•+NDI•– the g-factor difference is negative, while for TTF•+AlPor•– and AlPor•+C60 •– it is positive. Thus, the data are consistent with the following electron transfer sequences in the two triads:
and

Room temperature transient EPR spectra of two axial triads from the series shown in Fig. 18.14
In the latter case, the EPR data are also consistent with electron transfer to C60 being the initial step, however, fluorescence quenching data suggest that electron donation by TTF occurs first. In principle, the lifetime of the precursor state can be estimated from the net polarization of the antiphase doublets, however, this requires that the strength of the spin-spin coupling in the precursor is known. For the spectra shown in Fig. 18.15, a lifetime of about 1 ns is obtained if the spin-spin coupling is assumed to be ~2 mT but this combination of values for the lifetime and coupling is not unique.
18.5.3 Quantum Beats
As discussed in Sect. 18.4.2, the strong correlation between the electrons when a radical pair is generated from a pure singlet state produces coherence effects in the TREPR signals. These quantum beat oscillations are not easy to observe because of the rapid de-phasing of the spins. This is particularly true when the measurements must be performed at room temperature. In most D-A complexes charge separation that is long lived enough to be observed by TREPR requires stabilization of the radical pair states by solvent reorganization. As a result, the radical pairs cannot be observed at low temperature and hence there are very few reports of quantum beat measurements. However, there are some complexes in which this phenomenon can be studied (Laukenmann et al. 1995; Krzyaniak et al. 2015). The first observation of quantum beat oscillations in a D-A complex was reported by Kothe, Norris, Wasielewski and co-workers (Laukenmann et al. 1995). More recently, a new optical method of indirect observation of the singlet-triplet coherence has reported by Wasielewski and co-workers (Kobr et al. 2012; Krzyaniak et al. 2015). They designed a complex in which two electron-transfer steps can be driven by two short laser pulses at different wavelengths. The first pulse generates the primary radical pair, which is then irradiated to drive the secondary electron transfer and create the secondary radical pair. In a first study it was shown that the secondary radical pair is spin-correlated and generated initially in a singlet state (Kobr et al. 2012). In a very recent report it was then shown that by varying the delay between the two laser flashes changes in the TREPR spectrum of the secondary radical pair are observed that are consistent with coherent singlet-triplet mixing in the primary radical pair.
18.5.4 Echo Modulations
The values of the spin-spin couplings are crucial for understanding the relationship between molecular structure and the efficiency of electron transfer . This is because the exchange coupling can be directly related to the electronic coupling between the donor and acceptor and the dipolar coupling provides information about the distance between the separated electrons. However, as discussed in Sect. 18.3.1, if the coupling is smaller than the inhomogeneous linewidth it cannot be obtained unambiguously from the TREPR spectrum. However, the out-of-phase echo experiment described in Sect. 18.2.2 provides an elegant and accurate way of the determining the couplings. Again, however, this experiment is difficult to perform at room temperature because of short T2 relaxation. Carmieli et al. (2009) recently reported out-of-phase echo modulation results on the complexes shown in Fig. 18.16, which can be measured at low temperature. In complexes 1 and 2 excitation of the charge transfer band between 6ANI and PI and subsequent donation by BDX or TTF results in a long-lived radical pair. In complex 3, the charge transfer band between DMJ and An is excited and the charge separation is stabilized by electron transfer to NI.

Structures of donor acceptor complexes for out-of-phase echo modulation experiments (Reprinted with permission from (Carmieli et al. 2009). Copyright (2009) American Chemical Society)
The out-of-phase echo modulation curves for the three complexes and their Fourier transforms are shown in Fig. 18.17. It is immediately apparent that the modulation frequency for complex 3 is much lower than for complexes 1 and 2 and that this results in a narrower Fourier transform spectrum. This lower frequency is the result of a significantly larger distance between the spins in complex 3. Closer inspection of the curves for complexes 1 and 2 shows that there are subtle differences between them despite the fact that the complexes are virtually identical. In 2 the perpendicular component of the coupling is smaller than in 1 but the parallel component is larger and the sign of the ν ∥ and \( {\nu}_{\parallel}^{\prime } \) signal components are opposite in 1 and 2. These differences are a result of a difference in the sign of J in the two complexes. This is a surprising result, given that the two complexes differ only in the nature of the donor. Although the origin of the difference remains unclear, the results provide a good demonstration of the use of the out-of-phase echo modulation to accurately determine the dipolar and exchange contributions to the coupling.

18.5.5 Polymer-Fullerene Blends
TREPR methods have also been applied to the study of charge separation in organic bulk heterojunction solar cells . These systems consist of blends of a conductive polymer, usually a substituted polythiophene, and an electron acceptor usually a substituted fullerene.
Light excitation of the polymer leads to charge separation at the polymer/fullerene junction with good yield and a long lifetime and if the heterojunction is sandwiched between two electrodes of a closed circuit a photocurrent is generated (Sariciftci et al. 1992). For such systems, the factors that allow the Coulomb attraction between the separated charges to be overcome is not well understood and this has motivated a number of TREPR studies (Da Ros et al. 1999; Pasimeni et al. 2001a, b; Behrends et al. 2012; Kobori et al. 2013; Miura et al. 2013; Kraffert et al. 2014; Lukina et al. 2015; Niklas et al. 2015). The structures of some of the fullerenes and polythiophenes that have been used in these studies are shown in Fig. 18.18. Pasimeni and co-workers (Da Ros et al. 1999; Pasimeni et al. 2001a, b) reported the first TREPR spectra on a blend of N-mTEGFP and ST6 (Figs. 18.18 and 18.19). The E/A/E/A polarization is indicative of a spin correlated radical pair. The authors were able to simulate the spectrum (Fig. 18.19 dotted curve) assuming a singlet-born radical pair with a dipolar coupling constant of D = −121 ± 4 μT, which corresponds to a distance of 28.4 ± 0.3 Å between the radicals. Because a distribution of geometries for the radical pair is expected, a random distribution of orientations of the dipolar coupling vector relative to the g-tensors was taken and the principal axes of the g-tensors of the two radicals were assumed to be collinear. No out-of-phase echo was detectable for these samples (Pasimeni et al. 2001b). Behrends et al. (2012; Kraffert et al. 2014) obtained a virtually identical spectrum from a blend of PCBM and P3HT (Fig. 18.18) and showed that it could be simulated using purely isotropic spin-spin coupling and argued that it is not possible to obtain a unique value for the coupling. As discussed in Sect. 18.3.1, the geometry and spin-spin coupling can only be determined if the absolute intensity of the spectrum is known and this is generally not the case for TREPR data. They also showed that the polarization decays to a purely absorptive pattern at late times that was the same as that observed by steady-state EPR, suggesting that it was due to separated polarons.

Structures of several polythiophenes and fullerene derivates used in organic bulk heterojunction solar cells

Transient EPR spectrum of a sexithiophene – fulleropyrrolidine blend. Solid line: experimental spectrum, Dashed line: simulation (Reprinted from (Pasimeni et al. 2001b), with permission from Elsevier)
Kobori and co-workers (Kobori et al. 2013; Miura et al. 2013) studied the same P3HT-PCBM blend and obtained similar time-dependent spectra. In contrast to Behrends et al. (2012; Kraffert et al. 2014) they argued that the absorptive spectrum observed at late time was broader than the steady-state spectrum and therefore assigned it to the coupled radical pair after relaxation of the initial spin polarization . They were able to reproduce the experimental spectra as a function of time using the stochastic Liouville equation to take the relaxation into account. To calculate the spectra, they assumed a single average geometry for the P3HT•+PCBM•– radical pair and estimated the dipolar coupling from the width of the spectrum. The exchange coupling and geometry were obtained by fitting to the experimental data. When the side chains on the polythiophene were varied and the polymer blend was annealed, different values for the exchange and dipolar couplings and geometries were obtained (Miura et al. 2013). From these data, the distance dependence of the exchange coupling was calculated and a value of β = 0.2 Å−1 for the attenuation factor of the electronic coupling was estimated.
Niklas et al. (2015) compared the TREPR spectra of the blends of PCBM with the three polythiophenes P3HT, PTB7 and PCDTBT shown in Fig. 18.18. These blends show similar TREPR spectra but differing degrees of net polarization of the two radicals occur. For the PTB7-containing blend a spin-polarized spectrum of the triplet state of PTB7 is observed in addition to the radical pair. The polarization pattern of the triplet state showed that it is due to radical pair recombination (see Sect. 18.3.4). The intensity of the triplet state increased substantially in experiments performed at 130 GHz as a result of the faster singlet-triplet mixing at higher magnetic field. Together the polarization patterns of the radical pairs and the appearance of the triplet state spectrum clearly indicate that a series of sequential radical pairs is formed and that the observed pair is the secondary pair.
In the analysis by Kobori et al. (2013) an average geometry was assumed but the width of the distribution was not evaluated, although the relaxation times they obtained suggest a relatively broad distribution. Out-of-phase echo studies provide a more accurate measure of the spin-spin coupling and the width of the distribution. Lukina et al. (2015) recently reported such experiments and found that although a light-induced out-of-phase echo from P3HT+PCBM− radical pair is observed it is not modulated. They rationalized the absence of modulation as a result of delocalization of the hole on the polythiophene chain resulting in a distribution of spin-spin couplings, which leads to damping of the echo modulation. The echo decay curve could be simulated with a simple model in which the hole on the polythiophene is distributed over a distance spanning about 4 nm.
18.6 Concluding Remarks
As can be seen from the examples presented here TREPR experiments provide important information both for the characterization and design of artificial photosynthetic D-A complexes. As for any spectroscopic technique, there are limitations. The most important of these for TREPR is the comparitively slow inherent response time which is on the order of 10 ns. However as discussed above, the spin dynamics occuring on a shorter time scale can have an influence on the observed data and it is possible to deduce properties of the states that cannot be observed directly. The organic bulk heterojunction studies illustrate how the complexity of the data analysis increases with the complexity of the systems. However, they also show that by combining data from various experiments it is possible to gain useful insights even in disordered amorphous systems.
References
Ahrens MJ, Kelley RF, Dance ZE, Wasielewski MR (2007) Photoinduced Charge Separation in Self-Assembled Cofacial Pentamers of Zinc-5, 10, 15, 20-Tetrakis (Perylenediimide) Porphyrin. Phys Chem Chem Phys 9 (12):1469–1478
Allen JP, Williams JC (2014) Energetics of Cofactors in Photosynthetic Complexes: Relationship Between Protein–Cofactor Interactions and Midpoint Potentials. In: Golbeck JH, van der Est A (eds) The Biophysics of Photosynthesis. Biophysics for the Life Sciences. Springer, New York, pp 275–295
Angerhofer A, Bittl R (1996) Radicals and Radical Pairs in Photosynthesis. Photochem Photobiol 63 (1):11–38
Batchelor SN, Sun L, Möbius K, Kurreck H (1995) Time-resolved EPR Studies of Covalently Linked Porphyrin—Crown Ether—Quinones, Dissolved in Liquid Crystals. Magn Reson Chem 33 (13):S28–S33
Behrends J, Sperlich A, Schnegg A, Biskup T, Teutloff C, Lips K, Dyakonov V, Bittl R (2012) Direct detection of photoinduced charge transfer complexes in polymer fullerene blends. Phys Rev B 85 (12):125206
Bertini I, Bryant DA, Ciurli S, Dikiy A, Fernández CO, Luchinat C, Safarov N, Vila AJ, Zhao J (2001) Backbone Dynamics of Plastocyanin in Both Oxidation States Solution Structure of the Reduced Form and Comparison with the Oxidized State. J Biol Chem 276 (50):47217–47226
Bittl R, Kothe G (1991) Transient EPR of Radical Pairs in Photosynthetic Reaction Centers: Prediction of Quantum Beats. Chem Phys Lett 177 (6):547–553
Bittl R, Zech SG (1997) Pulsed EPR Study of Spin-Coupled Radical Pairs in Photosynthetic Reaction Centers: Measurement of the Distance Between and in Photosystem I and between and in Bacterial Reaction Centers. J Phys Chem B 101 (8):1429–1436
Bittl R, Zech SG (2001) Pulsed EPR Spectroscopy on Short-Lived Intermediates in Photosystem I. Biochim Biophys Acta 1507 (1):194–211
Borovykh I, Kulik L, Dzuba S, Hoff A (2002) Out-of-phase Stimulated Electron Spin-Echo Appearing in the Evolution of Spin-Correlated Photosynthetic Triplet-Radical Pairs. J Phys Chem B 106 (46):12066–12071
Bruns CM, Karplus AP (1995) Refined Crystal Structure of Spinach Ferredoxin Reductase at 1.7 Å Resolution: Oxidized, Reduced and 2′-phospho-5′-AMP Bound States. J Mol Biol 247 (1):125–145
Buckley C, Hunter D, Hore P, McLauchlan K (1987) Electron Spin Resonance of Spin-Correlated Radical Pairs. Chem Phys Lett 135 (3):307–312
Budil DE, Thurnauer MC (1991) The Chlorophyll Triplet State as a Probe of Structure and Function in Photosynthesis. Biochim Biophys Acta 1057 (1):1–41
Burnell E, De Lange C (1998) Prediction from Molecular Shape of Solute Orientational Order in Liquid Crystals. Chem Rev 98 (6):2359–2388
Carbonera D, Di Valentin M, Corvaja C, Agostini G, Giacometti G, Liddell PA, Kuciauskas D, Moore AL, Moore TA, Gust D (1998) EPR Investigation of Photoinduced Radical Pair Formation and Decay to a Triplet State in a Carotene-Porphyrin-Fullerene Triad. J Am Chem Soc 120 (18):4398–4405
Carmieli R, Mi Q, Ricks AB, Giacobbe EM, Mickley SM, Wasielewski MR (2009) Direct Measurement of Photoinduced Charge Separation Distances in Donor− Acceptor Systems for Artificial Photosynthesis Using OOP-ESEEM. J Am Chem Soc 131 (24):8372–8373
Closs GL, Forbes MD, Norris JR (1987) Spin-Polarized Electron Paramagnetic Resonance Spectra of Radical Pairs in Micelles: Observation of Electron Spin-Spin Interactions. J Phys Chem 91 (13):3592–3599
Colvin MT, Ricks AB, Scott AM, Co DT, Wasielewski MR (2012) Intersystem Crossing Involving Strongly Spin Exchange-Coupled Radical Ion Pairs in Donor–bridge–Acceptor Molecules. J Phys Chem A 116 (8):1923–1930
Colvin MT, Carmieli R, Miura T, Richert S, Gardner DM, Smeigh AL, Dyar SM, Conron SM, Ratner MA, Wasielewski MR (2013) Electron Spin Polarization Transfer from Photogenerated Spin-Correlated Radical Pairs to a Stable Radical Observer Spin. J Phys Chem A 117 (25):5314–5325
Da Ros T, Prato M, Guldi D, Alessio E, Ruzzi M, Pasimeni L (1999) A noncovalently linked, dynamic fullerene porphyrin dyad. Efficient formation of long-lived charge separated states through complex dissociation. Chem Commun (7):635–636
Dance ZE, Mi Q, McCamant DW, Ahrens MJ, Ratner MA, Wasielewski MR (2006) Time-Resolved EPR Studies of Photogenerated Radical Ion Pairs Separated by p-Phenylene Oligomers and of Triplet States Resulting from Charge Recombination. J Phys Chem B 110 (50):25163–25173
Dance ZE, Ahrens MJ, Vega AM, Ricks AB, McCamant DW, Ratner MA, Wasielewski MR (2008a) Direct Observation of the Preference of Hole Transfer over Electron Transfer for Radical Ion Pair Recombination in Donor-Bridge-Acceptor Molecules. J Am Chem Soc 130 (3):830–832
Dance ZE, Mickley SM, Wilson TM, Ricks AB, Scott AM, Ratner MA, Wasielewski MR (2008b) Intersystem Crossing Mediated by Photoinduced Intramolecular Charge Transfer: Julolidine-Anthracene Molecules with Perpendicular π Systems. J Phys Chem A 112 (18):4194–4201
Elger G, Fuhs M, Müller P, Gersdorff J, Wiehe A, Kurreck H, Möbius K (1998) Time-resolved EPR Studies of Photoinduced Electron Transfer Reactions in Photosynthetic Model Porphyrin Quinone Triads. Mol Phys 95 (6):1309–1323
Forbes MDE, Jarocha LE, Sim S, Tarasov VF (2013) Time-Resolved Electron Paramagnetic Resonance Spectroscopy: History, Technique, and Application to Supramolecular and Macromolecular Chemistry. In: Ian HW, Nicholas HW (eds) Advances in Physical Organic Chemistry, vol Volume 47. Academic Press, pp 1–83
Fuhs M, Elger G, Mobius K, Osintsev A, Popov A, Kurreck H (2000) Multifrequency Time-Resolved EPR (9.5 GHz and 95GHz) on Covalently Linked Porphyrin-Quinone Model Systems for Photosynthetic Electron Transfer: Effect of Molecular Dynamics on Electron Spin Polarization. Mol Phys 98 (15):1025–1040
Giacobbe EM, Mi Q, Colvin MT, Cohen B, Ramanan C, Scott AM, Yeganeh S, Marks TJ, Ratner MA, Wasielewski MR (2009) Ultrafast Intersystem Crossing and Spin Dynamics of Photoexcited Perylene-3, 4: 9, 10-bis (dicarboximide) Covalently Linked to a Nitroxide Radical at Fixed Distances. J Am Chem Soc 131 (10):3700–3712
Gust D, Moore TA, Moore AL (2001) Mimicking Photosynthetic Solar Energy Transduction. Acc Chem Res 34 (1):40–48
Hasharoni K, Levanon H (1995) Attenuation of Intramolecular Electron Transfer Rates in Liquid Crystals. J Phys Chem 99 (14):4875–4878
Hasharoni K, Levanon H, von Gersdorff J, Kurreck H, Möbius K (1993) Photo-Induced Electron Transfer in Covalently Linked Donor–Acceptor Assemblies in Liquid Crystals. Time-Resolved Electron Paramagnetic Resonance. J Chem Phys 98 (4):2916–2926
Hasharoni K, Levanon H, Greenfield SR, Gosztola DJ, Svec WA, Wasielewski MR (1995) Mimicry of the Radical Pair and Triplet States in Photosynthetic Reaction Centers with a Synthetic Model. J Am Chem Soc 117 (30):8055–8056
Hasharoni K, Levanon H, Greenfield SR, Gosztola D, J, Svec WA, Wasielewski MR (1996) Radical Pair and Triplet State Dynamics of a Photosynthetic Reaction-Center Model Embedded in Isotropic Media and Liquid Crystals. J Am Chem Soc 118 (42):10228–10235
Heinen U, Berthold T, Kothe G, Stavitski E, Galili T, Levanon H, Wiederrecht G, Wasielewski MR (2002) High Time Resolution Q-band EPR Study of Sequential Electron Transfer in a Triad Oriented in a Liquid Crystal. J Phys Chem A 106 (10):1933–1937
Hore P (1996) Research Note Transfer of Spin Correlation Between Radical Pairs in the Initial Steps of Photosynthetic Energy Conversion. Mol Phys 89 (4):1195–1202
Hore P, Hunter D, McKie C, Hoff A (1987) Electron Paramagnetic Resonance of Spin-Correlated Radical Pairs in Photosynthetic Reactions. Chem Phys Lett 137 (6):495–500
Jakob M, Berg A, Stavitski E, Chernick ET, Weiss EA, Wasielewski MR, Levanon H (2006) Photoinduced Electron Transfer Through Hydrogen Bonds in a Rod-Like Donor–Acceptor Molecule: A Time-Resolved EPR Study. Chem Phys 324 (1):63–71
Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauss N (2001) Three-Dimensional Structure of Cyanobacterial Photosystem I at 2.5 Ångstrom Resolution. Nature 411 (6840):909–917
Kamlowski A, Zech SG, Fromme P, Bittl R, Lubitz W, Witt HT, Stehlik D (1998) The Radical Pair State in Photosystem I Single Crystals: Orientation Dependence of the Transient Spin-Polarized EPR Spectra. J Phys Chem B 102 (42):8266–8277
Kandrashkin Y, van der Est A (2001) A New Approach to Determining the Geometry of Weakly Coupled Radical Pairs From Their Electron Spin Polarization Patterns. Spectrochim Acta C 57 (8):1697–1709
Kandrashkin YE, van der Est A (2007) Time-resolved EPR spectroscopy of Photosynthetic Reaction Centers: from Theory to Experiment. Appl Magn Reson 31 (1–2):105–122
Kandrashkin YE, Salikhov K, van der Est A, Stehlik D (1998) Electron Spin Polarization in Consecutive Spin-Correlated Radical Pairs: Application to Short-Lived and Long-Lived Precursors in Type 1 Photosynthetic Reaction Centres. Appl Magn Reson 15 (3–4):417–447
Kandrashkin YE, Vollmann W, Stehlik D, Salikhov K, Van der Est A (2002) The Magnetic Field Dependence of the Electron Spin Polarization in Consecutive Spin Correlated Radical Pairs in Type I Photosynthetic Reaction Centres. Mol Phys 100 (9):1431–1443
Kandrashkin YE, Asano MS, van der Est A (2006a) Light-Induced Electron Spin Polarization in Vanadyl Octaethylporphyrin: I. Characterization of the Excited Quartet State. J Phys Chem A 110 (31):9607–9616
Kandrashkin Y, Poddutoori P, Van Der Est A (2006b) Novel Intramolecular Electron Transfer in Axial bis (terpyridoxy) phosphorus (V) Porphyrin Studied by Time-Resolved EPR Spectroscopy. Appl Magn Reson 30 (3–4):605–618
Kay C, Kurreck H, Batchelor S, Tian P, Schlüpmann J, Möbius K (1995) Photochemistry of a Butylene Linked Porphyrin-Quinone Donor-Acceptor System Studied by Time-Resolved and Steady-State EPR Spectroscopy. Appl Magn Reson 9 (4):459–480
Kobori Y, Noji R, Tsuganezawa S (2013) Initial Molecular Photocurrent: Nanostructure and Motion of Weakly Bound Charge-Separated State in Organic Photovoltaic Interface. J Phys Chem C 117 (4):1589–1599
Kobr L, Gardner DM, Smeigh AL, Dyar SM, Karlen SD, Carmieli R, Wasielewski MR (2012) Fast Photodriven Electron Spin Coherence Transfer: A Quantum Gate Based on a Spin Exchange J-Jump. J Am Chem Soc 134 (30):12430–12433
Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: A Program for Display and Analysis of Macromolecular Structures. J Mol Graphics 14 (1):51–55
Kraffert F, Steyrleuthner R, Albrecht S, Neher D, Scharber MC, Bittl R, Behrends J (2014) Charge Separation in PCPDTBT: PCBM Blends from an EPR Perspective. J Phys Chem C 118 (49):28482–28493
Krzyaniak MD, Kobr L, Rugg BK, Phelan BT, Margulies EA, Nelson JN, Young RM, Wasielewski MR (2015) Fast Photo-Driven Electron Spin Coherence Transfer: the Effect of Electron-Nuclear Hyperfine Coupling on Coherence Dephasing. J Mat Chem C
Kurisu G, Zhang HM, Smith JL, Cramer WA (2003) Structure of the Cytochrome b(6)f Complex of Oxygenic Photosynthesis: Tuning the Cavity. Science 302 (5647):1009–1014
Laukenmann K, Weber S, Kothe G, Oesterle C, Angerhofer A, Wasielewski MR, Svec WA, Norris JR (1995) Quantum Beats of the Radical Pair State in Photosynthetic Models Observed by Transient Electron Paramagnetic Resonance. J Phys Chem 99 (12):4324–4329
Lendzian F, Schlüpmann J, von Gersdorff J, Möbius K, Kurreck H (1991) Investigation of the Light-Induced Charge Transfer between Covalently Linked Porphyrin and Quinone Units by Time-Resolved EPR Spectroscopy. Angew Chem Int Edit 30 (11):1461–1463
Levanon H (1987) Spin Polarized Triplets Oriented in Liquid Crystals. Res Chem Intermed 8 (3):287–320
Levanon H, Möbius K (1997) Advanced EPR Spectroscopy on Electron Transfer Processes in Photosynthesis and Biomimetic Model Systems. Annu Rev Bioph Biom 26 (1):495–540
Levanon H, Galili T, Regev A, Wiederrecht GP, Svec WA, Wasielewski MR (1998) Determination of The Energy Levels of Radical Pair States in Photosynthetic Models Oriented in Liquid Crystals with Time-Resolved Electron Paramagnetic Resonance. J Am Chem Soc 120 (25):6366–6373
Liddell PA, Sumida JP, Macpherson AN, Noss L, Seely GR, Clark KN, Moore AL, Moore TA, Gust D (1994) Preparation and Photophysical Studies of Porphyrin-C60 Dyads. Photochem Photobiol 60 (6):537–541
Loll B, Kern J, Saenger W, Zouni A, Biesiadka J (2005) Towards Complete Cofactor Arrangement in the 3.0 Ångstrom Resolution Structure of Photosystem II. Nature 438 (7070):1040–1044
Lukina EA, Popov AA, Uvarov MN, Kulik LV (2015) Out-of-Phase Electron Spin Echo Studies of Light-Induced Charge-Transfer States in P3HT/PCBM Composite. J Phys Chem B
Mi Q, Ratner MA, Wasielewski MR (2009) Time-Resolved EPR Spectra of Spin-Correlated Radical Pairs: Spectral and Kinetic Modulation Resulting from Electron− Nuclear Hyperfine Interactions. J Phys Chem A 114 (1):162–171
Miura T, Wasielewski MR (2011) Manipulating Photogenerated Radical Ion Pair Lifetimes in Wirelike Molecules Using Microwave Pulses: Molecular Spintronic Gates. J Am Chem Soc 133 (9):2844–2847
Miura T, Carmieli R, Wasielewski MR (2010) Time-Resolved EPR Studies of Charge Recombination and Triplet-State Formation within Donor− Bridge− Acceptor Molecules Having Wire-Like Oligofluorene Bridges. J Phys Chem A 114 (18):5769–5778
Miura T, Aikawa M, Kobori Y (2013) Time-Resolved EPR Study of Electron–Hole Dissociations Influenced by Alkyl Side Chains at the Photovoltaic Polyalkylthiophene: PCBM Interface. J Phys Chem Lett 5 (1):30–35
Niklas J, Beaupré S, Leclerc M, Xu T, Yu L, Sperlich A, Dyakonov V, Poluektov OG (2015) Photoinduced Dynamics of Charge Separation: From Photosynthesis to Polymer–Fullerene Bulk Heterojunctions. J Phys Chem B 119 (24):7407–7416
Norris J, Morris A, Thurnauer M, Tang J (1990) A General Model of Electron Spin Polarization Arising from the Interactions within Radical Pairs. J Chem Phys 92 (7):4239–4249
Pasimeni L, Franco L, Ruzzi M, Mucci A, Schenetti L, Luo C, Guldi DM, Kordatos K, Prato M (2001a) Evidence of High Charge Mobility in Photoirradiated polythiophene–Fullerene Composites. J Mater Chem 11 (4):981–983
Pasimeni L, Ruzzi M, Prato M, Da Ros T, Barbarella G, Zambianchi M (2001b) Spin Correlated Radical Ion Pairs Generated by Photoinduced Electron Transfer in Composites of Sexithiophene/Fullerene Derivatives: A Transient EPR Study. Chem Phys 263 (1):83–94
Poddutoori PK, Zarrabi N, Moiseev AG, Gumbau-Brisa R, Vassiliev S, van der Est A (2013) Long-Lived Charge Separation in Novel Axial Donor–Porphyrin–Acceptor Triads Based on Tetrathiafulvalene, Aluminum (III) Porphyrin and Naphthalenediimide. Chem Eur J 19 (9):3148–3161
Poddutoori PK, Lim GN, Sandanayaka AS, Karr PA, Ito O, D'Souza F, Pilkington M, van der Est A (2015) Axially Assembled Photosynthetic Reaction Center Mimics Composed of Tetrathiafulvalene, Aluminum (iii) Porphyrin and Fullerene Entities. Nanoscale 7 (28):12151–12165
Salikhov KM, Molin YN, Sagdeev R, Buchachenko A (1984) Spin Polarization and Magnetic Effects in Radical Reactions.
Salikhov K, Bock C, Stehlik D (1990) Time Development of Electron Spin Polarization in Magnetically Coupled, Spin Correlated Radical Pairs. Appl Magn Reson 1 (2):195–211
Salikhov K, Kandrashkin YE, Salikhov A (1992) Peculiarities of Free Induction and Primary Spin Echo Signals for Spin-Correlated Radical Pairs. Appl Magn Reson 3 (1):199–216
Sariciftci N, Smilowitz L, Heeger AJ, Wudl F (1992) Photoinduced Electron Transfer from a Conducting Polymer to Buckminsterfullerene. Science 258 (5087):1474–1476
Savitsky A, Moebius K (2006) Photochemical Reactions and Photoinduced Electron-Transfer Processes in Liquids, Frozen Solutions, and Proteins as Studied by Multifrequency Time-Resolved EPR Spectroscopy. Helv Chem Acta 89 (10):2544–2589
Savitsky A, Dubinskii A, Flores M, Lubitz W, Möbius K (2007) Orientation-Resolving Pulsed Electron Dipolar High-Field EPR Spectroscopy on Disordered Solids: I. Structure of Spin-Correlated Radical Pairs in Bacterial Photosynthetic Reaction Centers. J Phys Chem B 111 (22):6245–6262
Savitsky A, Niklas J, Golbeck J, Mobius K, Lubitz W (2013) Orientation Resolving Dipolar High-Field EPR Spectroscopy on Disordered Solids: II. Structure of Spin-Correlated Radical Pairs in Photosystem I. J Phys Chem B 117 (38):11184–11199
Schubert C, Margraf J, Clark T, Guldi D (2015) Molecular Wires–Impact of π-Conjugation and Implementation of Molecular Bottlenecks. Chem Soc Rev
Schweiger A, Jeschke G (2001) Principles of Pulse Electron Paramagnetic Resonance. Oxford University Press, Oxford
Scott AM, Miura T, Ricks AB, Dance ZE, Giacobbe EM, Colvin MT, Wasielewski MR (2009) Spin-Selective Charge Transport Pathways through p-Oligophenylene-Linked Donor− Bridge− Acceptor Molecules. J Am Chem Soc 131 (48):17655–17666
Shaakov S, Galili T, Stavitski E, Levanon H, Lukas A, Wasielewski MR (2003) Using Spin Dynamics of Covalently Linked Radical Ion Pairs to Probe the Impact of Structural and Energetic Changes on Charge Recombination. J Am Chem Soc 125 (21):6563–6572
Stehlik D (2006) Transient EPR Spectroscopy as Applied to Light-Induced Functional Intermediates Along the Electron Transfer Pathway in Photosystem I. In: Golbeck JH (ed) Photosystem I: The Light-Driven Plastocyanin: Ferredoxin Oxidoreductase. Advances in Photosynthesis and Respiration, vol 24. Springer, Dordrecht, The Netherlands, pp 361–386
Stehlik D, Möbius K (1997) New EPR Methods for Investigating Photoprocesses with Paramagnetic Intermediates. Annu Rev Phys Chem 48 (1):745–784
Stehlik D, Bock CH, Petersen J (1989) Anisotropic Electron Spin Polarization of Correlated Spin Pairs in Photosynthetic Reaction Centers. J Phys Chem 93 (4):1612–1619
Tang J, Thurnauer MC, Norris JR (1994) Electron Spin Echo Envelope Modulation Due to Exchange and Dipolar Interactions in a Spin-Correlated Radical Pair. Chem Phys Lett 219 (3):283–290
Tang J, Bondeson S, Thurnauer MC (1996) Effects of Sequential Electron Transfer on Electron Spin Polarized Transient EPR Spectra at High Fields. Chem Phys Lett 253 (3):293–298
Tauber MJ, Kelley RF, Giaimo JM, Rybtchinski B, Wasielewski MR (2006) Electron Hopping in π-Stacked Covalent and Self-Assembled Perylene Diimides Observed by ENDOR Spectroscopy. J Am Chem Soc 128 (6):1782–1783
Thurnauer MC (1979) ESR Study of the Photoexcited Triplet State in Photosynthetic Bacteria. Res Chem Intermed 3 (1):197–230
Thurnauer MC, Clark C (1984) Electron Spin Echo Envelope Modulation of The Transient EPR Signals Observed in Photosynthetic Algae and Chloroplasts. Photochem Photobiol 40 (3):381–386
Thurnauer M, Bowman M, Norris J (1979) Time-Resolved Electron Spin Echo Spectroscopy Applied to the Study of Photosynthesis. FEBS Lett 100 (2):309–312
Thurnauer M, Rutherford A, Norris J (1982) The Effect of Ambient Redox Potential on the Transient Electron Spin Echo Signals Observed in Chloroplasts and Photosynthetic Algae. Biochim Biophys Acta 682 (3):332–338
Thurnauer MC, Poluektov OG, Kothe G (2004) Time-Resolved High-Frequency and Multifrequency EPR Studies of Spin-Correlated Radical Pairs in Photosynthetic Reaction Center Proteins. In: Grinberg O, Berliner L (eds) Very High Frequency (VHF) ESR/EPR. Biological Magnetic Resonance, vol 24. Springer, New York, pp 165–206
Thurnaur MC, Norris JR (1980) An Electron Spin Echo Phase Shift Observed in Photosynthetic Algae: Possible Evidence for Dynamic Radical Pair Interactions. Chem Phys Lett 76 (3):557–561
Tsukihara T, Fukuyama K, Mizushima M, Harioka T, Kusunoki M, Katsube Y, Hase T, Matsubara H (1990) Structure of the [2Fe-2S] Ferredoxin I from the Blue-Green Alga Aphanothece sacrum at 2·2 Å Resolution. J Mol Biol 216 (2):399–410
van der Est A (2001) Light-Induced Spin Polarization in Type I Photosynthetic Reaction Centres. Biochim Biophys Acta 1507 (1):212–225
van der Est A (2009) Transient EPR: Using Spin Polarization in Sequential Radical Pairs to Study Electron Transfer in Photosynthesis. Photosynth Res 102 (2–3):335–347
van der Est A, Poddutoori PK (2013) Light-Induced Spin Polarization in Porphyrin-Based Donor–Acceptor Dyads and Triads. Appl Magn Reson 44 (1–2):301–318
van der Est A, Kok M, Burnell E (1987) Size and Shape Effects on the Orientation of Rigid Molecules in Nematic Liquid Crystals. Mol Phys 60 (2):397–413
van der Est A, Fuechsle G, Stehlik D, Wasielewski M (1996) X-and K-band Transient EPR of the Light Induced Radical Ion Pairs in Photosynthetic Model Systems. Berich Bunsen Gesell 100 (12):2081–2085
Verhoeven JW (2006) On the Role of Spin Correlation in the Formation, Decay, and Detection of Long-Lived, Intramolecular Charge-Transfer States. J Photoch Photobio C 7 (1):40–60
Wang Z, Tang J, Norris JR (1992) The Time Development of the Magnetic Moment of Correlated Radical Pairs. J Magn Reson 97 (2):322–334
Wasielewski MR (1992) Photoinduced Electron Transfer in Supramolecular Systems for Artificial Photosynthesis. Chem Rev 92 (3):435–461
Wasielewski MR (2006) Energy, Charge, and Spin Transport in Molecules and Self-Assembled Nanostructures Inspired by Photosynthesis. J Org Chem 71 (14):5051–5066
Wasielewski MR, Johnson DG, Svec WA, Kersey KM, Minsek DW (1988) Achieving High Quantum Yield Charge Separation in Porphyrin-Containing Donor-Acceptor Molecules at 10 K. J Am Chem Soc 110 (21):7219–7221
Wasielewski MR, Gaines III GL, O'Neil MP, Svec WA, Niemczyk MP (1990) Photoinduced Spin-Polarized Radical Ion Pair Formation in a Fixed-Distance Photosynthetic Model System at 5 K. J Am Chem Soc 112 (11):4559–4560
Wasielewski MR, Gaines III GL, O'neil MP, Svec WA, Niemczyk MP (1991) Spin-Polarized Radical Ion Pair Formation Resulting from Two-Step Electron Transfer from the Lowest Excited Singlet State of a Fixed-Distance Photosynthetic Model System at 5 K. Mol Cryst Liq Cryst 194 (1):201–207
Wasielewski MR, Gaines III GL, Wiederrecht GP, Svec WA, Niemczyk MP (1993) Biomimetic Modeling oF Photosynthetic Reaction Center Function: Long-Lived, Spin-Polarized Radical Ion Pair Formation in Chlorophyll-Porphyrin-Quinone Triads. J Am Chem Soc 115 (22):10442–10443
Wasielewski MR, Wiederrecht GP, Svec WA, Niemczyk MP (1995) Chlorin-Based Supramolecular Assemblies for Artificial Photosynthesis. Sol Energ Mat Sol C 38 (1):127–134
Wiederrecht GP, Svec WA, Wasielewski MR (1997) Differential Control of Intramolecular Charge Separation and Recombination Rates Using Nematic Liquid Crystal Solvents. J Am Chem Soc 119 (26):6199–6200
Wiederrecht GP, Svec WA, Wasielewski MR (1999a) Controlling the Adiabaticity of Electron-Transfer Reactions Using Nematic Liquid-Crystal Solvents. J Phys Chem B 103 (9):1386–1389
Wiederrecht GP, Svec WA, Wasielewski MR, Galili T, Levanon H (1999b) Triplet States with Unusual Spin Polarization Resulting from Radical Ion Pair Recombination at Short Distances. J Am Chem Soc 121 (33):7726–7727
Wiehe A, Senge MO, Schäfer A, Speck M, Tannert S, Kurreck H, Röder B (2001) Electron Donor–Acceptor Compounds: Exploiting the Triptycene Geometry for the Synthesis of Porphyrin Quinone Diads, Triads, and a Tetrad. Tetrahedron 57 (51):10089–10110
Wilson TM, Zeidan TA, Hariharan M, Lewis FD, Wasielewski MR (2010) Electron Hopping among Cofacially Stacked Perylenediimides Assembled by Using DNA Hairpins. Angew Chem Int Edit 49 (13):2385–2388
Zech SG, Lubitz W, Bittl R (1996) Pulsed EPR Experiments on Radical Pairs in Photosynthesis: Comparison of the Donor-Acceptor Distances In Photosystem I and Bacterial Reaction Centers. Berich Bunsen Gesell 100 (12):2041–2044
Acknowledgments
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Council Canada. AvdE wishes to thank Stefan Weber and the members of his research group and the members of FRIAS in Freiburg for their great hospitality during the writing of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
van der Est, A., Poddutoori, P.K. (2017). Time-Resolved EPR in Artificial Photosynthesis. In: Hou, H., Najafpour, M., Moore, G., Allakhverdiev, S. (eds) Photosynthesis: Structures, Mechanisms, and Applications. Springer, Cham. https://doi.org/10.1007/978-3-319-48873-8_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-48873-8_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48871-4
Online ISBN: 978-3-319-48873-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)