
Abstract
Breast cancer (BC) is the most frequently diagnosed cancer in women worldwide. A range of microenvironmental and systemic factors coordinately influence BC development and progression. A variety of cell membrane receptors translate the message from these factors into the modulation of complex, context-dependent intracellular signaling cascades, often regulating critical cellular processes like growth, proliferation, movement, and survival. Deregulation of this intricate signaling system at the receptor, or the other downstream levels, influences, and can sometimes drive, tumor development and progression. This chapter focuses on the two major classes of cell surface receptors and their signaling cascades, namely, the receptor tyrosine kinases (RTKs) and the G protein-coupled receptors (GPCRs), which are most often deregulated in BC.
Conflict of interest: The authors declare no conflict of interest.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction
Breast cancer (BC) is the most frequently diagnosed cancer in women worldwide, with 1.7 million new cases diagnosed in 2012 [1]. In the United States alone, 231,840 new cases and 40,290 related deaths are expected to be seen in 2015 [2]. This heterogeneous disease is classified into several molecular subtypes, depending on the presence of specific cell surface receptors, including the estrogen receptor (ER), the progesterone receptor (PR), and the human epidermal growth factor 2 receptor (HER2) [3]. Luminal A BCs are ER+ and/or PR+ but HER2−. These are the most commonly seen BCs and have the best prognosis. Luminal B BCs are ER+ and/or PR+ and sometimes HER2+. These tumors have a higher proliferative index and are more aggressive. HER2+ BCs are ER− and PR− but HER2+. This subtype usually presents at a younger age with a poorer tumor grade and lymph node involvement, but the prognosis has improved dramatically since the clinical implementation of Herceptin, an anti-HER2 antibody. About 20% of BCs are triple negative (TNBC) or basal-like, that is, they are ER−, PR−, and HER2−. These tumors are often aggressive, have poorer prognosis, and lack any targeted therapies.
Research has focused extensively on the role of cell surface receptors like HER2 in the pathobiology of BC. There are numerous families of cell surface receptors, like receptor tyrosine kinases (RTKs), one example being HER2, and G protein-coupled receptors, that sense extracellular cues and transmit them into intracellular messages that regulate cell growth, proliferation, survival, migration, and differentiation. These receptors are often deregulated in BC and lead to tumor growth and metastasis. This chapter will focus on the identification of the receptors most often deregulated in BC, the common signaling pathways they activate, and the crosstalk that links them to one another.
2 RTKs and Their Downstream Signaling Targets
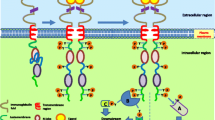
RTKs are cell surface receptors found on a diversity of cell types. All RTKs comprise an N-terminal extracellular ligand-binding domain, a single-pass transmembrane domain, and a C-terminal tyrosine kinase domain. Ligand binding induces a conformational change leading to the receptor homo- or heterodimerization and the consequent autophosphorylation of a series of tyrosine residues in the C-terminal tail. The phosphorylated tyrosines then act as docking sites for the SRC homology 2 (SH2) and phosphotyrosine-binding (PTB) domain-containing proteins, many of which are shared by the different RTKs. The RTK signaling program converges on the two major signaling pathways, namely, the phosphoinositide 3-kinase-protein kinase B/AKT (PI3K-PKB/AKT) and the rat sarcoma-mitogen-activated protein kinase/ERK (Ras-MAPK/ERK), that go on to regulate critical cellular processes like cell growth, proliferation, differentiation, migration, and apoptosis (Fig. 1.1).

RTK and GPCR signaling networks
One of the SH2 domain-containing effectors of RTKs is the regulatory subunit of the class I PI3K (p85), which when bound to the activated RTK or one of its tyrosine phosphorylated adaptors relieves its inhibition of the p110 catalytic subunit of PI3K, thereby leading to its activation [4]. PI3K p110 then phosphorylates a resident membrane lipid, phosphatidylinositol 4,5-bisphosphate (PIP2), to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), a major lipid second messenger [5]. The phosphatase and tensin homolog deleted on chromosome 10 (PTEN) counteracts the action of PI3K and converts PIP3 back to PIP2 [6, 7]. PIP3 recruits the pleckstrin homology (PH) domain-containing proteins to the membrane, of which there are more than 250 in the human genome, including AKT and the 3-phosphoinositide-dependent protein kinase 1 (PDK1) [8]. AKT is phosphorylated by PDK1 on threonine 308 (T308) and consequently by the mammalian target of rapamycin complex 2 (mTORC2) on serine 473 (S473), leading to its full activation [9, 10]. AKT then activates the mammalian target of rapamycin complex 1 (mTORC1) through two distinct pathways. AKT phosphorylates and suppresses the GTPase-activating protein (GAP) activity of the tuberous sclerosis complex 2 (TSC2) toward the Ras homolog enriched in the brain (Rheb) [11,12,13]. On the other hand, AKT phosphorylates and inhibits proline-rich AKT substrate of 40 KDa (PRAS40), which is implicated in the regulation of mTORC1 [14, 15]. mTORC1 regulates cell growth by controlling mRNA translation, via direct phosphorylation of the S6 kinase (p70S6K) and the 4E binding proteins (4EBPs) [16, 17]. AKT also phosphorylates the forkhead box O transcription factors (FOXOs), which results in their nuclear exclusion and proteasomal degradation, thus releasing cells from the FOXO-mediated cell cycle arrest [18,19,20]. The deactivation of FOXO, along with another target of AKT, the B-cell lymphoma 2 (BCL2)-associated agonist of cell death (BAD), coordinately represses apoptosis [21]. Finally, the AKT-mediated inhibition of glycogen synthase kinase 3 (GSK3) inhibits nuclear export and proteasomal degradation of cyclin D1, thus leading to its nuclear accumulation and induction of cell proliferation [22]. Thus, the PI3K-AKT pathway mainly regulates the cellular growth, proliferation, and survival programs in cells. Other than RTK deregulation, activating mutations in PI3K or deletion of PTEN is often found in BCs and further drives the oncogenic program in cells [23].
The Ras-ERK pathway is the other major signaling network that is modulated by the RTKs. The Src homology 2 domain-containing (SHC) and the growth factor receptor bound 2 (GRB2) are the main adaptor proteins that link the activated RTKs to the Ras-ERK pathway [24]. The RTKs interact with and activate SHC directly, which then recruits GRB2 to the cell membrane. Alternatively, GRB2 can also interact with RTKs directly or through another adaptor protein, like one of the insulin receptor substrates (IRSs) [25, 26]. GRB2 associates with the son of sevenless (SOS), which then recruits and activates Ras, by acting as a guanine nucleotide exchange factor (GEF), converting the GDP-bound Ras into the active GTP-bound form [27]. In a sequential manner, Ras activates Raf, which phosphorylates and activates MEK, which in turn phosphorylates and activates ERK [28, 29]. ERK is a serine-threonine kinase that has hundreds of cytoplasmic and nuclear targets. For example, ERK translocate to the nucleus and activates transcription factors like Ets-like gene 1 (Elk1) and c-Myc [30, 31]. In the nucleus, ERK also activates the mitogen- and stress-activated protein kinases (MSKs), which activate transcription factors like the cyclic AMP-responsive element-binding protein (CREB) and the activating transcription factor 1 (ATF1) [32]. In the cytoplasm, ERK phosphorylates the p90 ribosomal S6 kinases (RSKs), which inhibit apoptosis by phosphorylating BAD, but also translocate to the nucleus and activate transcription factors like CREB and c-Fos [33,34,35,36]. ERK-mediated phosphorylation of the MAPK-interacting kinases (MNKs) induces mRNA translation through phosphorylation of the eukaryotic initiation factor 4E (eIF4E) [37, 38]. The Ras-ERK pathway thus controls diverse cellular processes including cell growth, proliferation, differentiation, migration, and apoptosis.
In addition to the parallel activation immediately downstream of the receptors, coordination between the PI3K-AKT and the Ras-ERK signaling pathways can also be achieved by the interaction of activated Ras with the PI3K p110 catalytic subunit, independently of p85, leading to the PI3K-AKT pathway activation (Fig. 1.2) [39]. Like AKT, ERK and RSK can also phosphorylate and inhibit TSC2, leading to mTORC1 activation [40, 41]. The two pathways are also subject to the multiple levels of feedback inhibition. MEK promotes the membrane localization of PTEN, which downregulates the PI3K-AKT signaling pathway, while AKT can phosphorylate and inhibit Raf [42,43,44]. Furthermore, mTORC1, S6K, and ERK can downregulate both pathways by phosphorylating RTK substrates, like IRS1, on multiple inhibitory serine residues [45,46,47]. Thus, multiple feedback loops and crosstalk between the PI3K-AKT and the Ras-ERK pathways orchestrate the dynamic and intricate, context-dependent effects of multiple growth factors through their cognate RTKs.

Crosstalk between the PI3K-AKT and the Ras-ERK signaling pathways. Green lines indicate activation and red lines indicate inhibition. Solid black lines indicate a direct interaction, while dashed black lines indicate an indirect interaction
3 RTKs Often Deregulated in BC
The deregulation of RTK signaling plays an important role in the pathophysiology of many cancers, including BC [48]. Several mechanisms lead to the deregulation of RTK signaling, including RTK gene amplifications, activating mutations, protein overexpression, ligand overexpression or hyperactivation, and crosstalk with other cellular signaling components. Members of the ERBB family, MET, the insulin receptor (INSR), and the insulin-like growth factor receptor (IGF1R) are RTKs that are most often deregulated in BC.
3.1 HER2
The amplification of the HER2 gene, a member of the ERBB family of RTKs, is seen in approximately 20% of BCs, and HER2 overexpression correlates with a worse BC prognosis [49,50,51]. In these patients, HER2 overexpression correlates with tumor size, grade, proliferative index, aneuploidy, lack of steroid hormone receptors, and metastatic disease. HER2 (also named ERBB2 or NEU), belongs to the ERBB family, with three additional members: the epidermal growth factor receptor (EGFR, also named ERBB1), ERBB3, and ERBB4, all of which have been shown to be overexpressed and/or hyperactivated in BC to varying degrees. For example, EGFR is often overexpressed in basal-like TNBC [52]. There are 11 ligands that activate this family of RTKs, and they can be subdivided into three groups [53]. The first includes the epidermal growth factor (EGF), the transforming growth factor α (TGFα), and amphiregulin, which bind specifically to EGFR. The second includes betacellulin, heparin-binding EGF (HB-EGF), and epiregulin, which bind EGFR and ERBB4. Neuregulins (NRGs) make up the third group of ligands and are further subdivided into two subgroups, based on the ability to activate ERBB3 and ERBB4 (NRG1 and NRG2) or ERBB4 alone (NRG3 and NRG4). All ligands exist as membrane-anchored precursors, often co-expressed and even overexpressed with the ERBBs in the same cancer cells. Metalloproteases, mainly of the a disintegrin and metalloprotease (ADAM) family, cleave the precursors, leading to ectodomain shedding and activation of ERBB signaling in an autocrine or paracrine fashion [54].
Like other prototypical RTKs, all ERBBs can form functional homo- or heterodimers, with the exception of HER2, which does not appear to bind a ligand, and ERBB3, which is impaired in the intrinsic kinase activity and thus cannot form functional homodimers [55, 56]. Though HER2 is not self-autonomous, its extracellular domain conformation mimics that of the ligand-bound receptor, thus allowing HER2 to form functional heterodimers with other ERBBs [57]. HER2 is in fact the preferred binding partner of other ERBBs, and intriguingly the HER2-ERBB3 heterodimer is the most mitogenic and transforming of all the receptor combinations [58,59,60,61]. The C terminus of each of the ERBBs is unique (11–25% identity) and is able to bind to a diversity of intracellular targets. All of the ERBB members activate the Ras-ERK signaling pathway by directly interacting with the adaptor proteins SHC and GRB2 [62]. The regulatory subunit of PI3K (p85) directly interacts with ERBB3 and ERBB4. ERBB3 has the most [6] binding sites for p85, while EGFR and HER2 lack them all together, thus the HER2-ERBB3 heterodimer is the most potent activator of the PI3K-AKT signaling pathway, promoting cell growth, proliferation, and survival [63, 64]. Alternatively, ERBBs can activate the PI3K pathway through Ras. Together with the multitude of ligands, the different combinations of receptor dimers, and the unique C-terminal tails, this family of RTKs is capable of regulating diverse cellular processes implicated in cell growth, proliferation, differentiation, migration, and apoptosis.
3.2 MET
The hepatocyte growth factor receptor or MET is another RTK that is overexpressed in about 20% of BCs, particularly in the basal-like TNBCs [65]. Hepatocyte growth factor (HGF) is the only known ligand of MET, and it is often co-expressed with its receptor in the same tumor cells, particularly in the leading edge of the tumor [66, 67]. The expression of both, the receptor and the ligand, correlates with tumor grade, proliferative index, metastatic disease, and poor prognosis [68,69,70,71,72,73,74]. The HGF-mediated activation of MET leads to activation of the Ras-ERK pathway through the direct interaction of SHC and GRB2 with the receptor or through the recruitment of an insulin-like substrate (IRS)-like adaptor, the GRB2-associated-binding protein 1 (GAB1). The p85 regulatory subunit of PI3K also interacts with MET directly or through GAB1 and leads to activation of the PI3K-AKT pathway [75].
3.3 INSR
The INSR is overexpressed in as many as 80% of BCs and is associated with poor survival [76, 77]. The INSR is encoded by a gene composed of 22 exons found on chromosome 19. From this single gene, two receptor isoforms, INSR-A and INSR-B, are expressed as a result of alternative splicing. These two isoforms differ in inclusion/exclusion of exon 11, a 36 bp region encoding a 12 amino acid peptide located at the C-terminal end of the INSR alpha subunit [78]. INSR-B represents the full-length isoform and is expressed in insulin-responsive tissues including the liver, muscle, and adipose tissue. Conversely, INSR-A is expressed from the spliced transcript that lacks exon 11 and plays a significant role in fetal development by regulating cell growth and proliferation [79, 80]. The INSR-A and INSR-B isoforms display unique ligand specificity and downstream signaling potential. INSR-A exhibits an almost twofold higher affinity for insulin as compared to INSR-B and has a much stronger affinity for the insulin-like growth factor II (IGFII) [81,82,83]. INSR-A is the prevailing isoform overexpressed in both BC cells in culture and patient tumors [77, 84]. Therefore, increased INSR-A expression may negatively impact BC development, particularly in the context of hyperinsulinemia, as in the cases of diabetes or obesity. Indeed, hyperinsulinemia is an adverse prognostic factor in BC that is associated with increased risk of recurrence or death [85]. INSR-A expression is also elevated beyond that of the related IGF1R in some BCs suggesting INSR-A plays a role in mediating the growth-promoting effects of IGF-II in breast tumorigenesis [84, 86].
On the cell surface, the INSR exists as a heterotetrameric protein comprised of two extracellular alpha subunits and two transmembrane beta subunits. The beta subunit of the receptor possesses tyrosine kinase activity, which is stimulated upon binding of the ligand to the alpha subunit [87, 88]. Upon activation, INSR phosphorylates a number of substrates including IRS1–4, SHC, and GAB1 [89]. IRSs and GAB1 recruit the p85 regulatory subunit of PI3K, leading to the PI3K-AKT pathway activation. The Ras-ERK pathway is activated by the recruitment of GRB2-SOS complex by SHC or IRSs [26, 90]. INSR-B regulates the metabolic effects of insulin mainly through the PI3K-AKT pathway, while INSR-A activates the mitogenic program through both, the Ras-ERK and the PI3K-AKT pathways [91, 92]. Consequently, inhibition of INSR-A is actively being explored as a therapeutic option in breast and other cancers with clinical trials focusing on testing small molecule inhibitors and monoclonal antibodies directed against key components of these signaling networks [93]. Systemic modification of receptor ligands represents another strategy for targeting INSR signaling in cancer. For example, reduction in circulating insulin levels via administration of the antidiabetic drug metformin is being explored as a treatment option for cancers associated with obesity and hyperinsulinemia, especially BC. Indeed, administration of metformin to early-stage, nondiabetic BC patients led to reductions in circulating insulin and cancer cell proliferation, as well as suppressed INSR activity as indicated by reductions in AKT and ERK signaling [94].
3.4 IGF1R
Close to 50% of human breast tumors express the activated form of IGF1R, and gene expression signatures consistent with IGF1R activation are associated with poor outcome in BC patients [95, 96]. The IGF1R is homologous to the INSR but exhibits preferential binding to IGFI and IGFII over insulin. It is also a heterotetrameric protein complex consisting of two extraceulluar alpha subunits and two transmembrane beta subunits but plays a more significant role in the regulation of mitogenic signaling. The IGF1R shares numerous binding partners and effector proteins with the INSR, including IRSs and SHC adaptors, and is known to stimulate cell growth and proliferation via activation of the PI3K-AKT and Ras-ERK signaling pathways [93, 97]. A second IGF receptor, namely, IGF2R, is also commonly expressed by numerous cells; however, it lacks catalytic activity and is not involved in intracellular signaling [98]. Instead, IGF2R exhibits a high affinity for IGFII and is thought to sequester the growth factor from stimulating IGF1R [97, 99]. As a result, IGF2R may exhibit tumor suppressor properties by decreasing the bioactivity of IGFII and indirectly modulating signaling by IGF1R.
Due to their homology and strong structural similarities, the INSR and IGF1R have the ability to form hybrid receptors composed of one hemireceptor of each type. In addition, the two INSR isoforms can also combine to form hybrids, generating the potential for multiple insulin and IGF-sensitive receptors (INSR-A, INSR-B, INSR-A/B, IGF1R, INSR/IGF1R) to be expressed by a single cell. Hybrid receptors appear to bind IGFI with a higher affinity than insulin, and they exhibit different ligand specificities depending on the INSR isoform present. For example, INSR-A/IGF1R hybrids bind IGFI, IGFII, and insulin, while INSR-B/IGF1R hybrids typically bind IGF1 [91]. Since cancer cells frequently express high levels of both INSR and IGF1R, it is not surprising that they also overexpress hybrid receptors. Indeed, human breast tumors express high levels of hybrid receptors, and most of the effects of IGF1 are believed to be mediated by INSR-A/IGF1R hybrids. Furthermore, BC cells are known to secrete IGFII, creating the potential for autocrine stimulation of tumor cell growth and proliferation via activation of INSR-A, IGF1R, and INSR/IGF1R hybrid receptors [84, 100]. Consequently, human BC cells are highly sensitive to the growth-promoting effects of insulin and IGFs, and INSR/IGF1R expression may be a key event in tumor development and growth.
4 GPCRs and Their Downstream Signaling Targets
The G protein-coupled receptors (GPCRs) are the largest group of cell surface receptors that regulate cell motility, growth, proliferation, differentiation, and survival. The discovery of the Mas oncogene, a GPCR, in 1986 provided the first direct link between GPCRs and their role in cellular transformation [101]. Since then, many GPCRs where shown to be overexpressed or mutated in a diversity of cancers, including BC.
GPCRs are seven-pass transmembrane domain-containing receptors with an intracellular C-terminal tail that interacts with the heterotrimeric G proteins [102]. Upon ligand binding, the receptor undergoes a conformational change that allows it to act as a GEF, converting the GDP-bound G protein α subunit to the GTP-bound, active form. This causes the G protein α subunit to dissociate from the βγ subunits, initiating a multitude of signaling cascades. There are numerous G protein subtypes, each with unique signaling abilities. For example, the Gα12/13 activates several Rho GEFs leading to activation of Rho, a small GTPase that regulates cytoskeletal dynamics and cell motility, largely implicated in cancer metastasis. Gαq/11 activates the phospholipase C beta (PLCβ), which initiates the calcium and diacylglycerol (DAG) signaling cascades that regulate cell motility, proliferation, and gene expression. The GPCR signaling also crosstalks to the PI3K-AKT and the RAS-ERK pathways. The DAG-activated protein kinase C (PKC) phosphorylates and activates Raf, thereby leading to the activation of ERK [103]. The Gβγ subunits bind directly to PI3Kγ and activate the PI3K-AKT signaling pathway [104].
5 GPCRs Often Deregulated in BC
5.1 PAR1
The protease activated receptor 1 (PAR1) is a GPCR that is overexpressed in TNBC and correlates with metastatic disease and poor prognosis [105, 106]. The zinc-dependent matrix metalloprotease 1 (MMP1), thrombin, and other proteases cleave the extracellular domain of PAR1 exposing a new N terminus that binds to and activates the receptor [107, 108]. PAR1 then couples to multiple G proteins (Gαq/11, Gαi/o, Gα12/13) to regulate cell migration and proliferation, in part through the activation of Rho and ERK, respectively. PAR1 has been shown to be required and sufficient for the regulation of growth and invasion of BC cells in a mouse xenograft model [107, 109].
5.2 GPR161
The GPR161 is another GPCR that is overexpressed in TNBC and correlates with cancer relapse [110]. Overexpression of GPR161 in mammary epithelial cells transforms them via a yet unidentified mTORC1-dependent signaling pathway [110].
5.3 Wnt
The Wnt signaling pathway is hyperactivated in basal-like BCs and correlates with poor survival [111, 112]. The canonical Wnt signaling pathway results in nuclear accumulation of β-catenin, where it acts as a transcriptional coactivator for the T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors [113]. In the absence of the Wnt signal, β-catenin is sequestered in the cytoplasm by a destruction complex, containing GSK3β, which targets β-catenin for proteasomal degradation. Frizzled (FZD) is the GPCR for the Wnt family of ligands. When FZD is activated by the Wnt ligand, it acts together with the co-receptors, the low-density lipoprotein receptor-related protein 5 and 6 (LRP5/6), to disrupt the β-catenin destruction complex. This allows β-catenin to accumulate in the cytoplasm and translocate to the nucleus to activate its transcriptional program. The knockdown of FZD7 in TNBC cell lines reduces expression of β-catenin target genes, the transformation of these cells in vitro, and their ability to form tumors in vivo [114]. In addition, more than 40% of invasive breast tumors have a hypermethylation of the promoter, and therefore a strong downregulation of expression of the secreted frizzled-related proteins (sFRPs), the negative regulators of the Wnt signaling pathway [115, 116].
6 Crosstalk Between RTKs and GPCRs
The RTKs crosstalk with each other through multiple feedback and transactivation mechanisms. For example, MET can interact with and be transactivated by ERBBs, thus synergizing in the regulation of the downstream pathway components [117, 118]. Furthermore, RTK signaling often parallels or synergizes with GPCR signaling. The GPCRs can be upstream or downstream of the RTKs, and GPCRs are under the transcriptional regulation of RTKs and vice versa [119]. Furthermore, GPCRs and RTKs can transactivate each other. For example, GPCR activation regulates ectodomain shedding of the ERBB ligands. The PAR1 and the Wnt pathway have been shown to transactivate EGFR and HER2 in this manner [115,116,117,123]. Thus, GPCRs can activate the PI3K-AKT and the Ras-ERK pathways directly or through transactivation of the RTKs. In addition, EGFR-mediated activation of ERK induces nuclear translocation of the pyruvate kinase (PKM), which regulates β-catenin transcriptional activity [124]. The expression of β-catenin target genes can further be induced through the AKT- or RSK-dependent inhibition of GSK3β or the direct phosphorylation of β-catenin by AKT [120,121,127].
7 Other Receptors Deregulated in BC
The tumor microenvironment is a complex milieu of cell surface and secreted factors that affect BC development and progression. Tumor-associated fibroblasts (TAFs), endothelial cells, and inflammatory cells comprise the majority of the tumor microenvironment and express factors that affect tumor progression. Tumor cells express a number of non-RTK and/or non-GPCR receptors, the discussion of which is beyond the scope of this chapter, that sense signals from the microenvironment and often integrate them into the common pathways described above. For example, plexins, the receptors of semaphorins, originally described for their role in axon guidance, have now been implicated in BC metastasis, in part due to their ability to be transactivated by HER2 and MET and to activate Rho signaling [128, 129]. Tumor cells also express a number of cytokine receptors that interpret the pro-inflammatory signaling from leukocytes, tumor-associated macrophages (TAMs), TAFs, and autocrine loops. Cytokine receptors can activate several pro-survival and proliferation pathways but can also transactivate RTKs [130]. Lastly, integrins and cadherins, the cell adhesion mediators, are often deregulated in metastatic BC and play a central role in the epithelial-to-mesenchymal transition as well as in the activation of oncogenic signaling. Integrins can feed into both the PI3K-AKT and the Ras-ERK signaling pathways [131, 132]. Integrins also regulate ERBB expression at the mRNA translation level, as well as interact directly with ERBBs and regulate their tyrosine kinase activity [133, 134]. Further insight into the complexity of these intracellular crosstalk networks will aid in the identification of effective therapeutic targets and the mechanisms of therapeutic resistance.
8 Outlook
BC is one of the most common cancers worldwide and the second leading cause of cancer-related death in women [135]. It is a heterogeneous disease with a complex molecular etiology. A great deal of research has focused on the mechanisms underlying BC development, growth, and progression. Dysregulated RTK signaling has been identified as a critical event in breast tumorigenesis. For example, HER2 and IR are overexpressed by 20 and 80% of BCs respectively, and mutation of PI3K, a key mediator of RTK signaling, is mutated in 35% of human breast tumors [77, 135, 136]. Identification of such oncogenic proteins has led to a deeper understanding of BC and allowed for the development of targeted therapies for the treatment of this disease. Nevertheless, additional research is required to characterize mechanisms of tumor initiation as well as therapeutic resistance. Indeed, crosstalk between different RTK pathways and the existence of signaling feedback mechanisms are poorly understood processes that play critical roles in BC development and resistance to therapy. In the future, fundamental studies focusing on these issues in vitro should be combined with clinical research and early phase clinical trials to further characterize the role of RTKs in BC and identify new targets for anticancer therapies.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M et al (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer J Int du Cancer 136(5):E359–E386
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65(1):5–29
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM (1998) Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol 18(3):1379–1387
Hawkins PT, Jackson TR, Stephens LR (1992) Platelet-derived growth factor stimulates synthesis of PtdIns(3,4,5)P3 by activating a PtdIns(4,5)P2 3-OH kinase. Nature 358(6382):157–159
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI et al (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275(5308):1943–1947
Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T et al (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95(1):29–39
Lemmon MA (2008) Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol 9(2):99–111
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB et al (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7(4):261–269
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307(5712):1098–1101
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4(9):648–657
Manning BD, Cantley LC (2003) Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 28(11):573–576
Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D et al (1994) rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem 269(23):16333–16339
Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ et al (2003) Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 278(12):10189–10194
Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E et al (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25(6):903–915
Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ et al (1997) Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277(5322):99–101
Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE (1992) Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 257(5072):973–977
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96(6):857–868
Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP (2003) FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem 278(38):35959–35967
Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ et al (2002) Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol 22(7):2025–2036
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y et al (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91(2):231–241
Diehl JA, Cheng M, Roussel MF, Sherr CJ (1998) Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12(22):3499–3511
Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X et al (2005) PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 65(7):2554–2559
Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G et al (1992) A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70(1):93–104
Sasaoka T, Rose DW, Jhun BH, Saltiel AR, Draznin B, Olefsky JM (1994) Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem 269(18):13689–13694
Skolnik EY, Batzer A, Li N, Lee CH, Lowenstein E, Mohammadi M et al (1993) The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science 260(5116):1953–1955
Sasaoka T, Draznin B, Leitner JW, Langlois WJ, Olefsky JM (1994) Shc is the predominant signaling molecule coupling insulin receptors to activation of guanine nucleotide releasing factor and p21ras-GTP formation. J Biol Chem 269(14):10734–10738
Moodie SA, Willumsen BM, Weber MJ, Wolfman A (1993) Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 260(5114):1658–1661
Warne PH, Viciana PR, Downward J (1993) Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 364(6435):352–355
Alvarez E, Northwood IC, Gonzalez FA, Latour DA, Seth A, Abate C et al (1991) Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J Biol Chem 266(23):15277–15285
Cruzalegui FH, Cano E, Treisman R (1999) ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 18(56):7948–7957
Deak M, Clifton AD, Lucocq LM, Alessi DR (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17(15):4426–4441
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286(5443):1358–1362
Zhao Y, Bjorbaek C, Moller DE (1996) Regulation and interaction of pp90(rsk) isoforms with mitogen-activated protein kinases. J Biol Chem 271(47):29773–29779
Sturgill TW, Ray LB, Erikson E, Maller JL (1988) Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 334(6184):715–718
Chen RH, Abate C, Blenis J (1993) Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A 90(23):10952–10956
Fukunaga R, Hunter T (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J 16(8):1921–1933
Waskiewicz AJ, Flynn A, Proud CG, Cooper JA (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 16(8):1909–1920
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ et al (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370(6490):527–532
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121(2):179–193
Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 101(37):13489–13494
Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K et al (1999) Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286(5445):1738–1741
Zimmermann S, Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286(5445):1741–1744
Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, Baccarini M (2013) MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell 50(1):43–55
De Fea K, Roth RA (1997) Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J Biol Chem 272(50):31400–31406
Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE et al (2001) A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci U S A 98(8):4640–4645
Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M et al (2004) Absence of S6 K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431(7005):200–205
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235(4785):177–182
Ross JS, Fletcher JA, Linette GP, Stec J, Clark E, Ayers M et al (2003) The Her-2/neu gene and protein in breast cancer 2003: biomarker and target of therapy. Oncologist 8(4):307–325
King CR, Kraus MH, Aaronson SA (1985) Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science 229(4717):974–976
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z et al (2004) Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res: An Official J Am Assoc Cancer Res 10(16):5367–5374
Tebbutt N, Pedersen MW, Johns TG (2013) Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer 13(9):663–673
Arteaga CL, Engelman JA (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25(3):282–303
Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL 3rd (1994) Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A 91(17):8132–8136
Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA (2010) ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A 107(17):7692–7697
Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr et al (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421(6924):756–760
Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA et al (1995) Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 10(9):1813–1821
Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L et al (1996) Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 15(10):2452–2467
Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A (1995) Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J 14(17):4267–4275
Graus-Porta D, Beerli RR, Daly JM, Hynes NE (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16(7):1647–1655
Prigent SA, Gullick WJ (1994) Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J 13(12):2831–2841
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE (2003) The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A 100(15):8933–8938
Soltoff SP, Carraway KL 3rd, Prigent SA, Gullick WG, Cantley LC (1994) ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol 14(6):3550–3558
Ho-Yen CM, Green AR, Rakha EA, Brentnall AR, Ellis IO, Kermorgant S et al (2014) C-Met in invasive breast cancer: is there a relationship with the basal-like subtype? Cancer 120(2):163–171
Tuck AB, Park M, Sterns EE, Boag A, Elliott BE (1996) Coexpression of hepatocyte growth factor and receptor (Met) in human breast carcinoma. Am J Pathol 148(1):225–232
Ma J, DeFrances MC, Zou C, Johnson C, Ferrell R, Zarnegar R (2009) Somatic mutation and functional polymorphism of a novel regulatory element in the HGF gene promoter causes its aberrant expression in human breast cancer. J Clin Invest 119(3):478–491
Chen HH, Su WC, Lin PW, Guo HR, Lee WY (2007) Hypoxia-inducible factor-1alpha correlates with MET and metastasis in node-negative breast cancer. Breast Cancer Res Treat 103(2):167–175
Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K et al (2005) C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer J Int du Cancer 113(4):678–682
Raghav KP, Wang W, Liu S, Chavez-MacGregor M, Meng X, Hortobagyi GN et al (2012) cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin Cancer Res: An Official J Am Assoc Cancer Res 18(8):2269–2277
Edakuni G, Sasatomi E, Satoh T, Tokunaga O, Miyazaki K (2001) Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int 51(3):172–178
Garcia S, Dales JP, Charafe-Jauffret E, Carpentier-Meunier S, Andrac-Meyer L, Jacquemier J et al (2007) Poor prognosis in breast carcinomas correlates with increased expression of targetable CD146 and c-Met and with proteomic basal-like phenotype. Hum Pathol 38(6):830–841
Garcia S, Dales JP, Charafe-Jauffret E, Carpentier-Meunier S, Andrac-Meyer L, Jacquemier J et al (2007) Overexpression of c-Met and of the transducers PI3K, FAK and JAK in breast carcinomas correlates with shorter survival and neoangiogenesis. Int J Oncol 31(1):49–58
Yamashita J, Ogawa M, Yamashita S, Nomura K, Kuramoto M, Saishoji T et al (1994) Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res 54(7):1630–1633
Gherardi E, Birchmeier W, Birchmeier C, Vande WG (2012) Targeting MET in cancer: rationale and progress. Nat Rev Cancer 12(2):89–103
Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL et al (2008) Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res 68(24):10238–10246
Mulligan AM, O’Malley FP, Ennis M, Fantus IG, Goodwin PJ (2007) Insulin receptor is an independent predictor of a favorable outcome in early stage breast cancer. Breast Cancer Res Treat 106(1):39–47
Seino S, Seino M, Nishi S, Bell GI (1989) Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A 86(1):114–118
Denley A, Wallace JC, Cosgrove LJ, Forbes BE (2003) The insulin receptor isoform exon 11- (IR-A) in cancer and other diseases: a review. Horm Metab Res = Hormon- und Stoffwechselforschung = Hormones et metabolisme 35(11–12):778–785
Moller DE, Yokota A, Caro JF, Flier JS (1989) Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol 3(8):1263–1269
Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A et al (1999) Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 19(5):3278–3288
Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, McClain DA (1990) Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J 9(8):2409–2413
Yamaguchi Y, Flier JS, Yokota A, Benecke H, Backer JM, Moller DE (1991) Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology 129(4):2058–2066
Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P et al (1999) Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 18(15):2471–2479
Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y et al (2002) Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol Off J Am Soc Clin Oncol 20(1):42–51
Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y et al (1999) Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res: An Official J Am Assoc Cancer Res 5(7):1935–1944
Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clauser E et al (1985) The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell 40(4):747–758
Kasuga M, Hedo JA, Yamada KM, Kahn CR (1982) The structure of insulin receptor and its subunits. Evidence for multiple nonreduced forms and a 210,000 possible proreceptor. J Biol Chem 257(17):10392–10399
Cohen P (2006) The twentieth century struggle to decipher insulin signalling. Nat Rev Mol Cell Biol 7(11):867–873
Pronk GJ, McGlade J, Pelicci G, Pawson T, Bos JL (1993) Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem 268(8):5748–5753
Belfiore A, Frasca F, Pandini G,Sciacca L, Vigneri R (2009) Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocrine Reviews 30(6):586–623
Poloz Y, Stambolic V (2015) Obesity and cancer, a case for insulin signaling. Cell Death Dis 6(12):e2037
Pollak M (2012) The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12(3):159–169
Dowling RJ, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR et al (2015) Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res 17:32
Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG et al (2008) Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol Off J Am Soc Clin Oncol 26(25):4078–4085
Farabaugh SM, Boone DN, Lee AV (2015) Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front Endocrinol 6:59
Heidegger I, Massoner P, Sampson N, Klocker H (2015) The insulin-like growth factor (IGF) axis as an anticancer target in prostate cancer. Cancer Lett 367(2):113–121
Massoner P, Ladurner-Rennau M, Eder IE, Klocker H (2010) Insulin-like growth factors and insulin control a multifunctional signalling network of significant importance in cancer. Br J Cancer 103(10):1479–1484
De Souza AT, Hankins GR, Washington MK,Orton TC, Jirtle RL (1995) M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet 11(4):447–449
Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V et al (2002) A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab 87(1):245–254
Young D, Waitches G, Birchmeier C, Fasano O, Wigler M (1986) Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 45(5):711–719
Oldham WM, Hamm HE (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9(1):60–71
Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H et al (1993) Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature 364(6434):249–252
Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J et al (1997) The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89(1):105–114
Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y et al (1998) Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 4(8):909–914
Hernandez NA, Correa E, Avila EP, Vela TA, Perez VM (2009) PAR1 is selectively over expressed in high grade breast cancer patients: a cohort study. J Transl Med 7:47
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120(3):303–313
Vu TK, Hung DT, Wheaton VI, Coughlin SR (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64(6):1057–1068
Yang E, Cisowski J, Nguyen N, O’Callaghan K, Xu J, Agarwal A et al (2015) Dysregulated protease activated receptor 1 (PAR1) promotes metastatic phenotype in breast cancer through HMGA2. Oncogene 35(12):1529–1540
Feigin ME, Xue B, Hammell MC, Muthuswamy SK (2014) G-protein-coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proc Natl Acad Sci U S A 111(11):4191–4196
Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH (2010) Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol 176(6):2911–2920
Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y et al (2000) Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A 97(8):4262–4266
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13(1):11–26
Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC et al (2011) FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 30(43):4437–4446
Klopocki E, Kristiansen G, Wild PJ, Klaman I, Castanos-Velez E, Singer G et al (2004) Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int J Oncol 25(3):641–649
Veeck J, Geisler C, Noetzel E, Alkaya S, Hartmann A, Knuchel R et al (2008) Epigenetic inactivation of the secreted frizzled-related protein-5 (SFRP5) gene in human breast cancer is associated with unfavorable prognosis. Carcinogenesis 29(5):991–998
Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC (2000) Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem 275(12):8806–8811
Khoury H, Naujokas MA, Zuo D, Sangwan V, Frigault MM, Petkiewicz S et al (2005) HGF converts ErbB2/Neu epithelial morphogenesis to cell invasion. Mol Biol Cell 16(2):550–561
Garcia-Sainz JA, Romero-Avila MT, Medina LC (2010) Dissecting how receptor tyrosine kinases modulate G protein-coupled receptor function. Eur J Pharmacol 648(1–3):1–5
Daub H, Weiss FU, Wallasch C, Ullrich A (1996) Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379(6565):557–560
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C et al (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402(6764):884–888
Arora P, Cuevas BD, Russo A, Johnson GL, Trejo J (2008) Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 27(32):4434–4445
Schlange T, Matsuda Y, Lienhard S, Huber A, Hynes NE (2007) Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res 9(5):R63
Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W et al (2011) Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480(7375):118–122
Eldar-Finkelman H, Seger R, Vandenheede JR, Krebs EG (1995) Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3 T3 cells. J Biol Chem 270(3):987–990
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H et al (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282(15):11221–11229
He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA et al (2007) PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet 39(2):189–198
Swiercz JM, Worzfeld T, Offermanns S (2008) ErbB-2 and met reciprocally regulate cellular signaling via plexin-B1. J Biol Chem 283(4):1893–1901
Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D et al (2002) The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat Cell Biol 4(9):720–724
Qiu Y, Ravi L, Kung HJ (1998) Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature 393(6680):83–85
Dans M, Gagnoux-Palacios L, Blaikie P, Klein S, Mariotti A, Giancotti FG (2001) Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J Biol Chem 276(2):1494–1502
Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM (1997) Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell 91(7):949–960
Falcioni R, Antonini A, Nistico P, Di Stefano S, Crescenzi M, Natali PG et al (1997) Alpha 6 beta 4 and alpha 6 beta 1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp Cell Res 236(1):76–85
Yoon SO, Shin S, Lipscomb EA (2006) A novel mechanism for integrin-mediated ras activation in breast carcinoma cells: the alpha6beta4 integrin regulates ErbB2 translation and transactivates epidermal growth factor receptor/ErbB2 signaling. Cancer Res 66(5):2732–2739
Elster N, Collins DM, Toomey S, Crown J, Eustace AJ, Hennessy BT (2015) HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Res Treat 149(1):5–15
Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S et al (2004) The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 3(8):772–775
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Poloz, Y., Dowling, R.J.O., Stambolic, V. (2017). Fundamental Pathways in Breast Cancer 1: Signaling from the Membrane. In: Veronesi, U., Goldhirsch, A., Veronesi, P., Gentilini, O., Leonardi, M. (eds) Breast Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-48848-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-48848-6_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48846-2
Online ISBN: 978-3-319-48848-6
eBook Packages: MedicineMedicine (R0)