
Abstract
In this chapter, we will report the most recent investigations on modification of starch and their application as reinforcing filler in rubber composites. First, we will give a brief introduction on the characteristics of native starch, e.g., chemical structure, amylose content, morphology and crystallinity. Then, a brief review on different treatments used for starch modification, including gelatinization, plasticization, nanoparticles fabrication, and chemical grafting will be carried out. Finally, in order to prepare high performance starch reinforced rubber composites, three primary strategies reported so far, including (1) the addition of coupling agent, (2) modification of starch and (3) modification of rubber matrix will be highlighted.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
6.1 Introduction of Starch
Starch is one of the most abundant natural resources in nature, which can be widely derived from various bio-masses, such as corn, wheat, potato, cassava, rice and so on. In 2000, total 48.5 million tons of starches were produced all over the world. The main resources of starch are corn (81.24%), wheat (8.45%), potatoes (5.36%) and others (5.15%). The value of the output is worth €15 billion each year [1]. In 2006, the global market of starch was significantly increased to 2.4 billion tons [2]. According to a recent report, the global markets sales of starches and derivatives rose to $51.2 billion in 2012, and are expected to reach $77.4 billion by 2018 [3]. The rapid market growth promoted the researchers to look for new properties and application of starch.
Chemically, starch is a natural carbohydrate polymer, which consists of a large number of glucose units linked by glycosidic bonds. Its basic formula can be written as: (C6H10O5)n. The structure of starch is still under investigation due to its complexity [4]. It has been universally accepted that starch mainly contains two micro-structures: amylose and amylopectin, as shown in Fig. 6.1. Amylose, a linear molecular structure linked via α-(1-4) glycosidic bonds, is composed of 100–1000 glucose units. Its molecular weight is estimated to 20,000–225,000 g/mol. Amylopectin is a highly branched molecular structure consisting of short (1-4) α-d-glucan chains linked via α-(1-6) glycosidic bonds. Amylopectin has a much greater molecular weight than amylose, which is estimated to 200,000–1,000,000 g/mol. Depending on their botanic origin, the intrinsic properties of starch, such as amylose content, particle size, granule shape, crystallinity, crystalline type and gelatinization temperature are different. Table 6.1 summarizes the intrinsic properties of different types of starch.

Two micro-structures of starch: amylose and amylopectin
-
(1)
Amylose content. Normal starches have amylose content ranging from 15 to 30% and amylopectin content ranging from 70 to 85%. However, some starches have very high or extremely low amylose contents. For example, wrinkled pea starch [11] and amylomaize [12] have high amylose contents of 70 and 75%, respectively; whereas waxy maize starch [13] has extremely low amylose content of 1%. The content of amylose in starch was usually determined using two methods, such as the iodine affinity of solution of a defatted starch and the blue color derived from the amylose complex of iodine [8].
-
(2)
Average particle size and shape. The particle size of native starches ranges from 1 to 100 μm depending on their botanic origin. Potato starch granules have an oval-shaped form and an average particle size of 48 μm. Cassava starches are oval, round and truncated granules and have an average particle size of 33 μm. Wheat starch granules are disc shape and have an average particle size of 20 μm. Corn starches granules are a mixture of spherical and polyhedral shaped and have an average particle size of 15 μm. Rice starch granules show polyhedral-shaped form and have an average particle size of 6 μm. In general, the particle size increases as the following order: rice starch, corn starch, wheat starch, cassava starch, potato starch.
-
(3)
Crystallinity and crystalline type. Native starches are evidenced to be semi-crystalline natural polymers by X-ray diffraction (XRD) pattern. Their crystallinity ranges from 15 to 45% depending on their depending on their botanic origin. In native starch, the main component of amorphous regions is considered to be amylose. It was reported that the crystallinity of native starches was decreased as the increase of the amylose content [14]. The main component of the crystalline region is considered to be the amylopectin lamellae. The branched structure and high molecular weight of amylopectin strongly restrict the movement of starch chains and therefore cause the starch chains to be oriented [15]. From the XRD pattern, native starches are classified into three types: A, B and C types. For A-type starches, such as rice, corn and wheat starches, water molecules are located between each double helix. For B-type starches, such as cassava and potato starches, water molecules are located at the central cavity formed by six double helices [5]. For C-type starches, such as pea starches, water molecules are located both between each double helix and at the central cavity formed by six double helices. Therefore, C-type starches are considered as a mixture of A-type and B-type starches.
In order to further to understand the structure and intrinsic properties of starch, we took corn starch as an example and characterized it using Digital Camera, Scanning Electron Microscope (SEM), Attenuated Total Reflection-Fourier Transform Infrared spectroscopy (ATR-FTIR), Thermogravimetric Analysis (TGA), X-Ray Diffraction (XRD) and Nuclear Magnetic Resonance (NMR), as shown in Fig. 6.2.

Characterization of corn starch: a digital photo, b SEM microphoto, c ATR spectrum, d 1H-NMR spectrum, e XRD pattern and f TGA and DTG curves
Figure 6.2a shows that the native corn starch is a white powder. Figure 6.2b confirms that the corn starch has a mixture of spherical and polyhedral-shaped form. The particle size of corn starch is less than 20 μm. Figure 6.2c shows the ATR spectrum of corn starch. It can be seen that a wide and strong absorption peak appeared at 3313 cm−1 is attributed the O–H stretching. And its width is directly related to the formation of hydrogen bonding between two hydroxyl groups. Two absorption peaks at 2822 and 2932 cm−1 are ascribed to the asymmetric stretching of C–H in –CH3 or –CH2– groups. One absorption peak at 1640 cm−1 was due to adsorbed water. Two absorption peaks at 1440 and 1340 cm−1 are assigned to the angular deformation of C–H. Finally, the C–O absorption peaks could be found at 998 and 1077 cm−1 and C–O–C absorption peak could be found at 1149 cm−1. Figure 6.1d shows 1H-NMR spectrum of corn starch. The peak at 3.3 ppm is attributed to the hydrogen atom in the CH2–O groups. Two peaks at 3.6, 3.8 and 4.5 ppm are due to the hydrogen atoms in the CH–O group. Three peaks at 5.1, 5.5 and 5.6 ppm are assigned to the hydrogen atoms of the hydroxyl groups –OH. Figure 6.2e shows the XRD pattern of corn starch. Five diffraction peaks appearing in 15.1, 17.2, 18.0, 19.9 and 23.0 ppm can be observed, indicating that it is A-type semi-crystalline structure. Figure 6.2f shows the TGA and DTG curves of corn starch. Native corn starch shows characteristic three decomposition stages. The first stage occurring from 50 to 150 °C is attributed to the moisture evaporation. The second stage occurring from 239 to 353 °C is the major weight loss stage (62%), which is due to the decomposition of starch. From the DTG curve of corn starch, we can see the maximum decomposition temperature is about 312 °C. The third stage occurring from 353 to 600 °C is attributed to formation and evaporation of some volatile compounds. The char residue is 17% at 600 °C.
Starch granules tend to absorb the moisture at humid condition due to the strongly hydrophilic characteristics. Its moisture content has a significant influence on the morphology, thermal, mechanical properties and so on. Liu et al. [16] reported that the decomposition temperature of starch decreased with increasing moisture content. Zeleznak et al. [17] found that as the moisture content of starch increased, the melting temperature and the glass temperature decreased proportionally. Mali et al. [18] indicated that the increase in the moisture content led to both decrease in tensile strength and Young’s modulus of starch films. Vanderberg et al. [19] suggested that moisture could act as a plasticizer for starch. They explained that the addition of water improved the mobility of starch polymer chains.
Starch is also known as a completely biodegradable material, which can be broken down in natural environment by several microorganisms or bacteria, such as Bacillus amyloliquefaciens, Bacillus licheniformis, Aspergillus oryzae, Aspergillus niger, Bacillus subtilis, Malted barley and Bacillus acidopullulyticus [20]. The action of microorganisms for degradation of starch is different. For example, Bacillus amyloliquefaciens, Bacillus licheniformis and Aspergillus oryzae are mainly involved in cleaving α-1,4-oligosaccharide links to produce α-dextrin, maltose and oligosaccharides. Aspergillus niger is involved in cleaving α-1,4 and α-1,6-oligosaccharide to produce β-glucose. It is worth noting that these degraded dextrin, oligosaccharides and maltose can participate in catabolism to generate carbon dioxide and water. These produced carbon dioxide and water will recycle into starch through photo-synthesis process, again. Therefore, starch is also a renewable and biodegradable natural resource.
6.2 Modification of Starch
In recent years, starch-based products, such as starch-based coating, adhesives, flocculants, super absorbent, plastics and rubber biocomposites have been attracted a great attention from both academic and industrial researchers [21–26]. However, starch itself exhibits large particle size, high moisture absorption behavior, high hydrophilic behavior and high re-crystallization behavior. These above bottlenecks significantly limited the application of starch. Therefore, several methods, such as gelatinization, plasticization, hydrolysis and chemical grafting have been employed to modify the starch.
6.2.1 Gelatinization of Starch
Gelatinization generally consists of three stages. At the first stage, the starch granules in the water suspension irreversibly absorb water and swell under the continuously mechanical stirring and heat. At the second stage, when the temperature is over its gelatinization point, the intermolecular hydrogen bonds between starch molecules are significantly broken down. At the last stage, once the hydrogen bonds are broken, water molecules highly penetrate into the starch molecules, resulting in much greater swelling and the destruction of crystallites of starch [15]. After gelatinization, the branched amylopectin double helices are dissociated, as shown in Fig. 6.3, therefore and the crystalline structure of starch usually disappears.

Schematic illustration of starch gelatinization
Different types of starch have different gelatinization temperature. As shown in Table 6.1, the gelatinization temperatures are 61–18, 62–72, 52–63, 49–73 and 59–68 °C for rice, corn, wheat, cassava and potato starch, respectively. The pre-treatment process, such as milling affects the gelatinization temperature. After milling, the damaged starch can absorb water faster and therefore decreases the gelatinization temperature.
Some factors, such as water content, stirring and pressure may affect the gelatinization process. Wang et al. [27] investigated the effect of water content on the gelatinization of starch using experimentation and computer simulation. They observed that a minimum ratio of 14 water molecules to 1 anhydrous glucose unit was required to complete gelatinization. The minimum water content for completing gelatinization was defined as 63%. Garcia-Alonso et al. [28] studied the effect of magnetic stirring on the gelatinization of starch. They suggested that magnetic stirring during gelatinization led to form homogeneous gels and more standardized products. Herh and Kokini [29] demonstrated that starch exhibited very sensitive characteristic to the changes in the pressure. The conversion of gelatinized starch as well as gelatinization temperature increased as the increase in the pressure.
6.2.2 Plasticization of Starch
Native starch exhibits very high melting temperature (220–240 °C), which is higher than its decomposition temperature (220 °C), therefore, it is easily degraded before melting during the thermal processing. In order to apply starch in the polymer blends, plasticizer is usually used. The addition of plasticizer into starch leads to the decrease of its melting temperature and thus improves the processing property. The plasticized starch is commonly known as thermoplastic starch (TPS). Figure 6.4 illustrates the plasticization process of starch.

Schematic illustration of starch plasticization
By now, the use of various plasticizers, such as water, glycol, glycerol, sorbitol, critic acid, formamide, ethanolamide, urea, and acetamide to plasticize native starch have been reported. Table 6.2 summaries the common plasticizers used for the preparation of TPS. Water is considered as the most common plasticizer for the preparation of TPS. The addition of water causes a remarkable reduction in the melt viscosity of starch-based products [65, 66]. However, due to the poor mechanical performances resulted from water-plasticized starch [34], the water is often used together with the other plasticizers, such as glycerol [30–32], formamide [34] and glycol [36]. Wang et al. [34] found that when a mixture of water and formamide was used, the plasticization of starch as well as the fluidity of TPS/poly(lactic acid) blends were improved dramatically. The addition of formamide also increased the mechanical properties of TPS/poly(lactic acid) blends. Polyols, such as glycol, glycerol and sorbitol have significant effect on the plasticization of starch. The rigid starch granules have very high Young’s modulus due to the strong hydrogen bonds among the starch molecules. The addition of polyhydric alcohols can decrease the interaction among starch molecules. Thus, polyols can also act as plasticizers, which favor the movement of starch macromolecular chains, resulting in a reduction in the melting viscosity and an increase in the elongation. Yu et al. [35] reported that the addition of polyhydric alcohols, such as glycol, glycerol and hexylene glycol decreased the mechanical properties, apparent viscosity and crystalline structure of starch. Forssell et al. [39] investigated the phase and glass transition behavior of starch-glycerol-water mixtures. They found that the phase separation could easily occur, and when the water content was not too high, glycerol could inhibit amylopectin crystallization. Rodriguez-Gonzalez et al. [44] investigated the effect of glycerol content on the rheological and thermal properties of TPS. They found that as glycerol content increased from 29 to 40%, the elastic and loss modulus decreased gradually, meanwhile, the glass temperature Tg decreased from −45 to −56 °C. It is worth noting that the combined use of different plasticizers can optimize the performances of TPS [30–32, 34, 36, 47, 52, 55, 57, 59, 63]. Shi et al. [47] investigated the effect of critic acid on the glycerol-plasticized thermoplastic starch (GTPS). They found that compared with traditional GTPS, the novel critic acid-glycerol-co-plasticized thermoplastic starch (CGTPS) exhibited some new characteristics, such as partial esterification, low molecular weight and strong interaction between critic acid and starch. Teixeira et al. [52] comparatively studied the performances of glycerol-plasticized TPS/cellulose nanofibrills nanocomposites (TPSG) between glycerol-sorbitol-co-plasticized TPS/cellulose nanofibrills nanocomposites (TPSGS). The results indicated that compared with TPSG, TPSGS showed higher storage modulus and tensile strength. They explained that due to the higher molecular weight as well as the higher -OH content of sorbitol, the restriction effect of sorbitol on the nanocomposites was more prominent, resulting in higher stiffness.
One of the major bottlenecks for polyols-plasticized TPS is its retrogradation characteristic. The retrogradation occurs after polyols-plasticized TPS is stored for a period of time [67]. Kazuo et al. [68] found that urea had the retrogradation resistant ability for TPS. Therefore, to avoid the occurrence of retrogradation, various amides, such as formamide, ethanolamide, urea and acetamide have been studied in recent years. Ma et al. used the urea/formamide [59, 69] or urea/enthanolamide [63] as plasticizers for preparation of TPS. The results showed that the addition of urea ameliorated the retrogradation and mechanical properties of TPS. They also comparatively investigated three different amide plasticizers, such as formamide, acetamide and urea for TPS with the traditional plasticizer: glycerol as reference [58, 64]. The results showed that urea had the greatest ability to form the hydrogen bonds with starch, while polyols had the weakest ability to form the hydrogen bonds with starch. In addition, they confirmed that amide groups had the ability to suppress the retrogradation of TPS.
6.2.3 Preparation of Starch Nanoparticles
Particle size is one of the most crucial parameters for fillers to reinforce polymer matrix. Generally, fillers with particle size greater than 10 μm deteriorate the physical properties of polymer matrix rather than reinforcing; fillers with particle size from 1 to 10 μm primarily act as diluents and have no reinforcing capability in polymer matrix; fillers with particle size from 10 to 100 nm acts as reinforcing fillers, which significantly improve the performances of polymer compounds. Therefore, the performances of starch/polymer composites are strongly dependent on the particle size of starch. Recently, several methods, such as high-pressure homogenization [70, 71], precipitation [72, 73], micro-emulsion [74–76] and acid hydrolysis [77–88] have been developed to prepare the starch nanoparticles (SNPs).
Liu et al. [70] used high-pressure homogenization method (under a pressure of 207 MPa) to produce SNPs. After several run through a micro-fluidizer, the particle size of starch was reduced from micro- (3–6 μm) to nanometer (10–20 nm) without affecting its crystal structure and thermal stability. Ma and Yu [62] used ethanol as precipitant to produce SNPs through precipitation method and then modified the as-received SNPs with critic acid. The critic acid-modified SNPs ranged in particle size from 50 to 100 nm and exhibited good interaction with glycerol plasticized-pea starch matrix. On the base of precipitation method, the micro-emulsion method was developed. In the micro-emulsion method, surfactants, such as Span 80 [71, 74, 77], CTAB [76], Tween 80 [71, 76] were added into starch solution to form micro-emulsion before precipitation. Compared with the precipitation method, the SNPs prepared by the micro-emulsion method exhibited much smaller in the particle size [76].
Acid hydrolysis is one of the most popular methods used for the preparation of SNPs. The SNPs derived from starch granules through hydrolysis method are particularly called as “starch nanocrystals (SNCs)”. Native starch is a semi-crystalline polymer, including the amorphous and crystalline phases. The amorphous phases can dissolve by the treatment of acid hydrolysis; however, the water-insoluble crystalline lamellae can’t dissolve. Thus, SNCs refer to the remained crystalline lamellae. Jayakody and Hoover [77] indicated that the acid hydrolysis of starch included two steps. The first step is the hydrolysis of amorphous regions, which is affected by the amount of lipid complexed amylose chains, particle size, amylose content and pores on the surface. The second step is the hydrolysis of crystalline regions, which is affected by the mode of distribution of α (1-6) branches between the amorphous and the crystalline regions, amylopectin content and degree of packing of the double helices with the crystalline. Wang et al. [78] reported the differences in physical and chemical properties of acid hydrolyzed corn, potato and rice starch under the same hydrolysis conditions. They suggested that the inherent characteristics of starch played crucial roles in the properties of hydrolyzed starch. The yield and morphology of SNCs prepared by acid hydrolysis are dependent on the particle size, amylose/amylopectin ratio, crystalline type and hydrolysis conditions [77–81]. The effect of several hydrolysis conditions, such as temperature [82, 83], time [80, 82–84], acid type [85–87] and acid concentration [82, 83, 88] on the yield and morphology of SNCs has been extensively investigated. Angellier et al. [82] optimized the hydrolysis conditions for the preparation of SNCs using a response surface methodology. The conditions were optimized as following: (1) concentration: 3.16 M H2SO4, (2) time: 5 days, (3) temperature: 40 °C and (4) stirring speed; 100 rpm. Jayakody and Hoover [77] reported that the acid hydrolysis rate was very fast in the first 8 days. For example, with the acid hydrolysis (8 days, 2.2 N HCl, 35 °C), the waxy maize, normal maize, amylomaize V, amylomaize VII, waxy maize, oat and rice starches were hydrolyzed to the extent of 68.1, 61.1, 32.6, 28.5, 64.4 and 62.0%, respectively; with the acid hydrolysis (15 days, 2.2 N HCl, 35 °C), waxy maize, normal maize, amylomaize V, amylomaize VII, waxy maize, rice and oat starches were hydrolyzed to the extent of 77.3, 73.4, 37.0, 32.3, 75.3, and 72.9%, respectively. Singh and Ali [86] investigated the effect of different acids, such as HCl, HNO3, H2SO4 and H3PO4 on the starch hydrolysis. They indicated that HCl had the most effect on the starch hydrolysis, while H3PO4 had the weakest effect on the starch hydrolysis. Wang et al. [88] found that as the concentration of hydrochloric acid increased, the hydrolysis rate was increased. More recently, LeCorre reported [89] the significant effect of enzymatic pre-treatment on the acid hydrolysis of starch. They observed that the enzymatic pre-treatment significantly reduced the acid hydrolysis duration. For example, the duration of regular final yield of 15% SNCs was shortened from 5 days to 45 h.
6.2.4 Chemical Grafting of Starch
Starch has a large number of reactive hydroxyl groups on the backbones; therefore it is possible to modify its surface characteristic through a grafting copolymerization. It is found that vinyl monomers show highly reactive to be grafted onto the starch backbones. Various starch-graft-vinyl polymers have been synthesized, for example starch-g-poly(methyl methacrylate) [90–96], starch-g-polystyrene [97–102], starch-g-poly(butyl acrylate) [103–105], starch-g-poly(vinyl alcohol) [106–109], starch-g-poly(methacrylic acid) [109–112], starch-g-poly(acrylic acid) [113–118], starch-g-poly(acrylonitrile) [119–126], starch-g-poly(methacrylonitrile) [127–131], starch-g-poly(vinyl acetate) [132–135], starch-g-poly(acrylamide) [24, 136, 137], and starch-g-poly(styrene-maleic anhydride) [138]. Starch-graft-vinyl polymers are mainly synthesized by two methods: conventional redox-initiated method and irradiation-initiated method.
In the conventional redox-initiated method, various organic/inorganic redox initiators, such as hydrogen peroxide [90, 108], potassium pervanadate [91], benzoyl peroxide [93, 102, 133, 138], 2,2-azobisisobutyronitrile [94], ceric ammonium nitrate [95, 104, 110, 113, 116, 120–123, 128, 129, 131, 134, 139], manganic pyrophosphate [96, 124–126], potassium persulphate [99, 101, 102, 109, 130], ammonium persulfate [105, 137], ferrous ammonium sulfate [108] and potassium permanganate [111, 112, 117], have been reported to synthesize starch-graft-vinyl polymers. Among these redox initiators, ceric ammonium nitrate is mostly preferred. Compared with the other initiators, ceric ammonium nitrate possesses some advantages, such as the simple mechanism of single electron transfer, formation of free radicals on the back bone polymer itself, production of pure graft copolymer and ease of application at an ambient temperature in aqueous medium [139]. A proposed grafting mechanism between methyl methacrylate and starch via ceric ammonium nitrate-initiated method is illustrated in Fig. 6.5. As shown in Fig. 6.5, when the initiator ceric ammonium nitrate was added in an inert atmosphere of nitrogen, starch macro radicals were produced by donating hydrogen atoms, as illustrated in reaction (i). In the presence of monomer MMA, these reactive starch radicals were easily added to the double bond in monomer MMA, producing starch-grafted MMA free radicals, as illustrated in reaction (ii). Reaction (iii) shows that many more MMA molecules were successively added to continuously propagate the reactive chain. Finally, this grafting copolymerization might be terminated by a combination of two reactive starch grafted MMA chains, as illustrated in reaction (iv).

A proposed grafting mechanism initiated by ceric ammonium nitrate [26]
Recently, irradiation-initiated methods, such as microwave, electron beam, UV and gamma-ray irradiation have been attracted considerable attention. Compared with the conventional redox-initiated method, irradiation-initiated method possesses some advantages, such as high percentage grafting, easy controlled, high conversion, energy saving, high grafting rate. Microwave irradiation is the most promising method for the synthesis of starch-graft-vinyl polymers. Because the free radicals are produced by means of microwave photos, steric hindrance can be completely avoided, resulting in much higher percentage grafting and higher conversion compared with redox-initiated method [140]. In addition, the grafting copolymerization can be completed in minutes compared with hours or days using the conventional redox-initiated method [141].
The grafting copolymerization of vinyl monomers onto starch backbone can be evidenced using XRD, FTIR, NMR, TGA, DSC and SEM observations. The grating parameters, such as grafting percentage (GP) and grafting efficiency (GE) are also employed to describe the extent of grafting copolymerization. GP and GE are calculated by the following equations:
where W1 is the weight of grafted polymers in the starch-graft-vinyl polymers, which can be obtained through soxhlet extraction followed by acid hydrolysis treatment; W2 is the weight of starch in the starch-graft-vinyl polymers; and W3 is the initial weight of monomer.
The synthesis conditions, such as pre-treatment, reaction time, temperature, monomer/starch ratio, initiator type and concentration have significant influence on the values of GP and GE. Athawale and Lele [131] synthesized the maize starch-g-poly(methacrylonitrile) copolymers using ceric ammonium nitrite as an initiator. The effect of reaction time, temperature, monomer concentration and initiator concentration on the GP and GE were investigated. The optimum conditions were determined as following: reaction time: 3 h; temperature: 35 °C, initiator concentration: 0.002 mol/l and monomer concentration: 0.755 mol/l. Cho and Lee [99] synthesized the starch-g-polystyrene copolymers by emulsion polymerization, using potassium persulfate as an initiator, sodium dodecylbenzenesulfonate as an emulsifier and tetraethylthiuram disulfide as a chain transfer agent. The influences of pre-heated, reaction time, initiator concentration, emulsifier concentration, chain transfer agent concentration on the GP were investigated. The results showed that the values of GP were increased as the reaction time, initiator concentration and emulsifier concentration increased, while decreased as the chain transfer agent concentration increased. In addition, they observed that when the starch was pre-heated in the water at 80 °C, higher GP values were achieved.
In the starch-graft-vinyl polymers, the component of grafted vinyl polymers acts as plasticizer, therefore the grafting of vinyl polymers affects the crystalline ability, thermal properties and morphology of starch. Gao et al. [96] found that after the starch was grafted with methyl methacrylate; four sharp crystal peaks observed from XRD pattern were merged into a smooth peak, suggesting that the crystal phase of starch was destroyed. Wang et al. [142] observed that starch-g-poly(butyl acrylate-co-acrylonitrile) copolymers exhibited much higher decomposition temperature compared with native starch, indicating the improved thermal stability. Jyothi [143] indicated that the glass temperature of modified starch was lower than neat starch, because the grafted polymers may act as internal plasticizers, which favored the movement of starch molecular chains. Li et al. [26] found the surface of starch-g-poly(methyl methacrylate) copolymers was covered by a layer of PMMA due to the heterogeneous copolymerization system. And as the GP increased, the granules of modified starch were gradually deformed, resulting in fragmentation or the formation of a deep groove in the central core region, indicating the developed plasticity. Therefore, the properties of starch-graft-vinyl polymers could be tailored with the GP value properly.
The modified starch copolymers have wide potential applications, such as reinforcing fillers in rubber industry, biodegradable matrix in plastic industry, packaging films in food industry, flooding agents in the oil drilling industry, adhesives in tape industry, coatings in wallpaper industry, flocculants in water treatment industry, superabsorbent polymers in baby diaper industry and bio-compatible materials in the medical industry. Owing to its good bio-compatibility as well as proper bio-degradability, starch copolymers are also considered as one of the most promising materials in the bone tissue engineering.
6.3 Starch Reinforced Rubber Composites
In the past two decades, researchers have made great efforts on the utilization of various natural resources to develop the sustainability of industrial products. Natural resources have merits of low cost, abundance, lightweight, renewability, biodegradability and environmental friendliness. Recently, the application of starch as reinforcing fillers in the rubber compounds is of great interest.
6.3.1 Native Starch Reinforced Rubber Composites
The potential use of native starch as reinforcing filler in rubber matrix has been investigated from a mechanical point of view. Owing to the hydrophilic surface characteristic, large particle size and high melting point, native starch is very difficult to uniformly disperse in the rubber matrix. Therefore, the incorporation of native starch into rubber matrix generally leads to the deterioration of mechanical properties of the composites. Kiing et al. [144] used the native sago starch to reinforce natural rubber. They found the tensile strength of the non-vulcanized native sago starch/natural rubber (NR) blends decreased from 18.74 to 2.41 MPa as the loading of native sago starch increased from 0 to 20%. Afiq et al. [145] also used native sago starch to reinforce NR. They found that the tensile strength of the vulcanized native sago starch/NR composites also decreased from 22.79 to 15.00 MPa as the loading of native sago starch increased from 0 to 25 phr. Khalaf et al. [146] reported the use of native maize starch to reinforce NR. They found that the tensile strength of the vulcanized native maize starch/NR composites decreased from approximately 24 to 2 MPa as the loading of native maize starch increased from 0 to 60 phr. Recently, to obtain uniform dispersion of starch in rubber matrix, a novel dispersion technique—latex compounding method was reported [147]. In this method, starch granules were gelatinized in aqueous solution at 90 °C to obtain the starch paste. Then the starch paste was compounded with rubber latex, followed by immediately co-coagulating the mixture using CaCl2 solution. By virtue of latex compounding method, the particle of starch was significantly reduced to less than 1 μm and the dispersion state of starch was improved; therefore, the starch/rubber composites prepared by latex compounding method showed superior mechanical properties compared to the composites prepared by solid compounding method.
Figure 6.6 shows mechanical properties of corn starch/styrene-butadiene rubber (SBR) composites prepared by solid and latex compounding method. It can be seen that the tensile strength, elongation and 300% modulus of neat SBR were 2.9 MPa, 636% and 1.2 MPa, respectively. The incorporation of 10 phr of starch into SBR matrix by solid compounding method decreased the tensile strength and elongation to 1.8 MPa and 415%, respectively; while slightly increased the 300% modulus. Further incorporation of starch from 10 to 40 phr into SBR matrix had no obvious effect on the tensile strength and 300% modulus, while caused a continuous decrease in the elongation, which might be due to the large particle size of starch. On the contrary, the incorporation of 10 phr starch into SBR matrix by latex compounding method increased the tensile strength, elongation and 300% modulus up to 3.1 MPa, 836% and 1.5 MPa, respectively. Further incorporation of starch had a positive effect on the mechanical properties. For example, as the starch concentration increased from 10 to 40 phr, the 300% modulus was successively increased from 1.5 to 3.1 MPa; the tensile strength was increased from 3.1 to 4.4 MPa. The maximum tensile strength (4.9 MPa) could be achieved when 30 phr of starch was incorporated. The improvement in the mechanical performances is attributed to the reduction in the starch particle size as well as the improved dispersion state, as evidenced in Fig. 6.7.

Mechanical properties of starch/SBR composites prepared by solid and latex compounding methods

FE-SEM micrographs of starch (20 phr)/SBR composites prepared by two methods: solid compounding method, scale bar: a 100 μm and b 10 μm; latex compounding method, scale bar: c 100 μm and d 10 μm
Figure 6.7 shows the FE-SEM micrographs of starch/SBR biocomposites prepared by solid and latex compounding method. For the composites prepared by solid compounding method, a large number of starch granules appear on the tensile-fractured surface, as shown in Fig. 6.7a, b. Most of the corn starch granules have larger particle size (20 μm). Additionally, a large number of voids could be observed on or near the boundaries of starch granules, which indicated the poor interfacial compatibility between hydrophilic starch and hydrophobic SBR matrix. For the composites prepared by latex compounding method, the shape of starch granules was strongly deformed after gelatinization and the particle size of starch granules was decreased, as shown in Fig. 6.7c, d. FE-SEM observations confirmed the reduction in the particle size of corn starch and the improvement in the dispersion of corn starch in SBR matrix, which caused a remarkable development in the mechanical properties.
6.3.2 Modified Starch Reinforced Rubber Composites
It is well known that the interfacial interaction between filler and rubber matrix, particle size, and dispersion state of filler in the rubber matrix are three crucial parameters that strongly affect the mechanical properties of filler/polymer composites. By virtue of latex compounding method, although the dispersion state of starch was improved, the mechanical properties of the composites were still low due to the poor interfacial interaction between native starch and rubber matrix. Great efforts have been made by researchers to enhance the interfacial interaction between starch and rubber matrix. These efforts can be classified into three strategies: (1) modification of starch, (2) modification of rubber matrix, and (3) addition of coupling agent. Table 6.3 gives a summary of the strategies used for improving the performances of starch/rubber compounds in recent years.
Addition of coupling agent is the most common method used to enhance the interfacial interaction between the hydrophobic rubber matrix and the hydrophilic filler, such as graphene oxide, carbon nanotubes, nano-clay, silica, halloysite nanotubes, cellulose, and starch. Coupling agent usually contains two functional groups with different reactivity. One functional group can be attached to the hydrophilic filler via the formation of chemical bonds, such as covalent bond and hydrogen bond. The other functional group reacts with rubber matrix. Therefore, coupling agent usually acts as a “bridge” linking two incompatible phases and provides stronger reinforcing effect. Figure 6.8 shows the molecular formulas of the most common coupling agents used to enhance the interfacial interaction between the hydrophobic rubber matrix and the hydrophilic filler, including maleic anhydride (MA), 4,4-methylene bis(phenyl isocyanate) (MDI), vinyltrimethoxysilane (VTMS), bis(triethoxysilylpropyl)tetrasulfide (TESPT), 3-aminopropyltriethoxysilane (APTES), N-2-(aminoethyl)-3-aminopropyltrimethoxysilane (AEAPTMS), 3-mercaptopropyltrimethoxysilane (MPTS), and 3-glycidoxypropyl trimethoxysilane (GPTMS).

Molecular formulas of the most common coupling agents
The addition of coupling agent to enhance the performance of starch/rubber composites could be dated back to 1997. Goodyear Tire Rubber Company [148] developed novel reinforcing filler called BioTRED to partially replace the conventional carbon black and silica. The BioTRED was prepared from the plasticization of starch using poly(ethylene vinyl alcohol). To enhance the interfacial interaction between the plasticized starch and rubber matrix, a coupling agent TESPT was added. With the addition of TESPT, the plasticized starch can be beneficially used as a partial replacement of the carbon black reinforcement. Carvalho et al. [149] prepared the thermoplastic starch/NR blends. They observed that the plasticizer glycerol contributed to the plasticization of starch and to the improvement of the starch-NR interface. Khalaf et al. [146] reported the potential use of MA and glycidyl methacrylate (GMA) as compatibilizers for maize starch/NR vulcanizates. Unfortunately, the incorporation of MA deteriorated the tensile strength of maize starch/NR vulcanizates. However, maize starch/NR vulcanizates with 1 phr of GMA showed an improvement in the tensile strength. In addition, it was observed that the addition of GMA accelerated the vulcanization rate of maize starch/NR vulcanizates. Wu et al. [150–152] reported the synergistic effect of RF and coupling agent AEAPTMS on the reinforcement of starch/SBR composites. Significant reinforcement can be achieved by co-modification of starch by RF and AEAPTMS. For example, with addition of 30 phr of RF/AEAPTMS-modified starch, the tensile strength and tear strength of SBR composites were 18.0 MPa and 45.6 kN/m, respectively. Li et al. [153] comparatively investigated the effectiveness of different coupling agents, including TESPT, APTES, MPTS and MDI on the PMMA-modified starch/SBR interfaces. Among these coupling agents, MDI contributed to the most significant improvement in the 300% modulus, while TESPT had little influence on the 300% modulus. The reaction mechanism of MDI in the PMMA-modified starch/SBR interfaces was illustrated in Fig. 6.9. MDI played as a crosslinking point in the PMMA-modified starch/SBR interfaces. On the one hand, the strong carbamate bonds formed between the isocyanate group of MDI and the hydroxyl groups of starch. On the other hand, there was a strong—adhesion between the benzene group of MDI and the benzene group of SBR. It is worth noting that the carbamate bonds formed between MDI and starch are much stronger than the hydrogen bonds formed between silane and starch. Therefore, MDI exhibited the superior reinforcement to other silane coupling agents, resulting in the highest values both in tensile strength and 300% modulus.

Proposed reaction mechanism among starch-MDI-SBR [153]
The second strategy is modification of starch. Through surface grafting of different reactive functional groups or polymers, modified starch with desired properties could be hand-tailored to be compatible with different types of rubber matrix. In 1968, Buchanan et al. [154] firstly used the modified starch—starch xanthate to reinforce SBR and NBR composites by a co-precipitation process. They indicated that starch xanthate had potential reinforcing ability for both SBR and NBR composites. However, the reinforcing ability of zinc starch xanthate was still limited. Wu et al. [150–152] used resorcinol formaldehyde (RF) to modified corn starch and investigated the reinforcing capacity of RF-modified starch in the SBR matrix. With increasing the RF content from 0 to 1.2 phr, the tensile strength increased from 2.7 to 11.1 MPa for 10 phr starch filled SBR composites. They postulated that the benzene group of RF oligomers in the RF-modified starch is compatible with SBR, causing the significant improvement in the tensile strength. Wang et al. [155] modified the cassava starch with carbon disulfide through esterification and then investigated the effect of esterification on the morphology, thermal and mechanical properties of starch/NR composites. The results showed that the esterified starch uniformly dispersed in the NR matrix and the thermal and mechanical properties were superior. Liu et al. [105] chemically modified the corn starch with surface grafting poly (butyl acrylate) (PBA) via radical polymerization using APS as initiator and then investigated the effect of surface grafting of PBA on the dispersion state and mechanical properties of NR composites. With increasing the PBA-modified starch content, the tensile strength increased from 26 MPa for neat NR to a maximum 32 MPa with 15 phr PBA-modified, and then decreased. The PBA graft chains acted as a multifunctional agent—dispersion agent, plasticizer and compatibilizer. The presence of PBA prevented the starch particles from agglomerates, reduced the crystallization of starch, and enhanced the interfacial interaction between starch and NR matrix. Li et al. [156] synthesized three types of modified corn starch—starch-graft-poly (methyl methacrylate), starch-graft-poly(butyl acrylate), and starch-graft-polystyrene via emulsion polymerization using KPS as initiator, and then compared the reinforcing capacities of three modified starches in SBR matrix. They found that starch-graft-poly (methyl methacrylate) exhibited the best reinforcing capacity in SBR matrix compared to the other two. A possible reinforcing mechanism is presented in Fig. 6.10. As shown in Fig. 6.8, the vinyl grafts not only protected the starch from aggregation, but also physically entangled with SBR chains, causing the significant improvement in the mechanical properties. The concentrations of MMA monomer used for grafting polymerization and starch were optimized [157]. When the concentration of MMA and starch was 10 and 30 phr, respectively; the best tensile strength (10.8 MPa) could be achieved.

Reinforcing mechanism of modified starch in the SBR matrix [156]
Starch nanocrystals were prepared from the native starch by acidic hydrolysis method. The obtained starch nanocrystals have a three-dimensional nano-scale. Compared with native starch, the starch nanocrystals have extremely large surface area, leading to the stronger interfacial interaction.
The starch nanocrystals could act as effective reinforcing filler and gas barrier agent in rubber matrix. Angellier et al. [158, 159] observed that starch nanocrystals displayed great reinforcing effect in a non-vulcanized NR matrix. When 10, 20 and 30 wt% of starch nanocrystals were incorporated into NR matrix, the relaxed storage modulus at 50 °C increased 10, 75 and 200% compared with neat NR, respectively. Moreover, a continuous decrease in water vapor permeability from 3.41 to 1.88 × 1010 g/(m.s.Pa) was observed as the increase of the starch nanocrystals from 0 to 30 wt%. The influence of potato starch nanocrystals on the mechanical and swelling behavior of non-vulcanized NR nanocomposites has been studied by Rajisha et al. [160]. The tensile strength increased from 2.6 to 13.8 MPa as the potato starch nanocrystals content increased from 0 to 20 wt%. The reinforcing capacity of starch nanocrystals in the non-vulcanized NR matrix was ascribed to the formation of a strong three-dimensional network via hydrogen bonding between starch nanoparticle clusters. The influence of moisture content of starch nanocrystals on the tensile properties of non-vulcanized NR nanocomposites was also studied [158]. It was found that the moisture content does not affect the elongation, however, as the moisture content increased from 0 to 98%, the tensile strength and tensile modulus gradually decreased from 14.4/26.5 to 2.7/5.7 MPa, respectively. The reinforcing mechanism of starch nanocrystals in the non-vulcanized NR matrix was further confirmed by Mele et al. [161], who applied two models—Kraus model and Maier and Goritz model to predict the Payne effect. They revealed that the phenomena of adsorption/desorption or slippage of NR chains on the surface of starch nanocrystals influenced the nonlinear viscoelastic properties of starch nanocrystals/non-vulcanized NR nanocomposites. LeCorre et al. [162] evaluated the influence of the botanic origin of starch nanocrystals on the performances of non-vulcanized NR nanocomposites. They suggested that the amylose content is an important factor determining the performances of starch nanocrystals/non-vulcanized NR nanocomposites. The higher the amylose content of native starch granules used for preparing starch nanocrystals, the lower the water uptake and reinforcing capacity. Bouthegourd et al. [163] investigated the effect of starch nanocrystals loading on the dispersion and electrical properties of non-vulcanized NR nanocomposites. They found that when the starch nanocrystals loading was greater than 15% w/w, a tendency for agglomeration was observed, which led to a decrease in the electrical resistivity. Valodkar and Thakore [164] prepared the starch nanoparticles from waxy maize starch by 3.16 M sulfuric acid hydrolysis, and then used 1,4-hexamethylene diisocyanate and acetic anhydride to modify the starch nanoparticles. The obtained isocyanated and acetylated starch nanoparticles displayed good compatibility with NR matrix, causing a significant improvement in the mechanical properties of vulcanized NR composites. Between these two modified starch nanoparticles, isocyanated starch nanoparticles exhibited superior reinforcing capacity than acetylated starch nanoparticles.
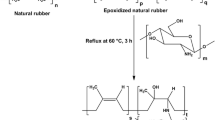
The last strategy is modification of rubber matrix. The introduction of polar groups, such as acrylonitrile, maleic anhydride and citraconic anhydride into nonpolar rubber chains can largely enhance the interfacial bonding between matrix and hydrophilic fillers, such as silica, clay, starch and cellulose. This method is very complex and relatively expensive. Therefore, the use of this method to prepare modified starch/rubber composites has rarely been reported. Rouilly et al. [165] chemically modified natural rubber latex with surface grafting of a cationic hydrophilic polymer—poly (dimethylaminoethyl methacrylate) (polyDMAEMA). They investigated the effect of polyDMAEMA modification on the mechanical properties and water sensitivity of NR/starch films. Owing to the formation of hydrogen bond between polyDMAEMA and starch, the polyDMAEMA-modified NR/starch films exhibited better elongation, toughness and water sensitivity than NR/starch films. Nakason et al. [166–168] synthesized two types of modified NR–NR-graft-poly(methyl methacrylate) (NR-g-PMMA) and maleated NR (MNR), and then compounded them with cassava starch. They observed that the curing time and tensile strength were decreased with the increase of cassava starch loading. Electron beam irradiation is one of the most effective radiation technologies used to improve the interfacial interaction in polymer blends. Senna et al. [169] modified the NR matrix using electron beam irradiation and then compounded the irradiated NR with starch. Unfortunately, the electron beam irradiation decreased the mechanical properties of NR/starch composites. Owing to the high cost, processing inconvenience and poor mechanical properties, it seems that modification of rubber matrix is not appropriate to prepare the high performance rubber/starch composites.
6.3.3 Starch/Carbon Black Hybrids Reinforced Rubber Composites
By now, various fillers, such as carbon black, silica, nano-clay, carbon nanotube, graphene, nano-CaCO3, starch, cellulose, chitin, protein, wood flour, rice bran have been used as filler to improve the overall performances of rubber composites. Carbon black and silica have been the main fillers to reinforce rubber composites in most industrial applications. The incorporation of carbon black significantly improved the tensile strength, modulus, and tear strength and abrasion resistance. Silica is hydrophilic; therefore its reinforcing capacity is not well as carbon black. To obtain better mechanical performances of silica/rubber composites, coupling agent is usually used. However, silica has its own advantages. For example, in tire treads, silica provided lower rolling resistance, leading to the improvement in the fuel consumption. The use of Nano-sized fillers, such as nano-clay, carbon nanotube, grapheme oxide, nano-CaCO3 to reinforce rubber composites has been also investigated. Owing to the “nano effects” and high aspect ratio, the incorporation of only a small amount of nano-sized fillers not only significantly improved the mechanical properties, but also other performances of rubber composites, such as gas permeability and fire retardancy. Very recently, natural fillers, such as starch, cellulose, chitin, protein, wood flour, rice bran are of great interest. It well known that natural fillers have merits of low cost, abundance, lightweight, renewability, biodegradability and environmental friendliness. Different types of fillers have their own advantages; therefore the utilization of hybrid filler to prepare rubber composites should bring the benefits from both fillers. It has been reported the utilization of silica/carbon black [170], clay/carbon black [171], graphene/carbon black [172], rice husk/carbon black [173], starch/carbon black [174, 175], layered double hydroxide/multiwalled carbon nanotube [176], graphene/multiwalled carbon nanotube [177], kaolin/silica [178], and carbon nanotube/silica [179] hybrids to improve the dispersion state, mechanical properties, vulcanization characteristic, electrical sensitivity, thermal stability, gas permeability, fire retardancy, and abrasive wear resistance of rubber composites.
There are few reports on the starch/other filler hybrids rubber composites. Wu et al. [174] used the starch to partially replace carbon black in SBR composites. They observed that compared with the solid compounding of starch particles into SBR compounds, the latex compounding of starch particles into SBR compounds improved the abrasion resistance of the starch/carbon black/SBR composites. However, the compounding of starch deteriorated the tensile strength of carbon black/SBR composites using both solid and latex compounding methods. Kim et al. [175] used the PMMA-modified starch to partially replace carbon black in NBR composites. They found that the synergistic effect of PMMA-modified starch and carbon black in the NBR composites. When the concentrations of PMMA-modified starch and carbon black were 25 and 25 phr, respectively, the optimal tensile strength (18.5 MPa) was obtained, which were much higher than the tensile strength (11.8 MPa) of PMMA-modified starch(50 phr)/NBR and the tensile strength (13.5 MPa) carbon black(50 phr)/NRB composites.
6.4 Summary and Future Challenges
Starch, as one the most abundant natural resources, can serve as promising candidate to reinforce rubber matrix, providing economic, renewable, and biodegradable advantages. To date, most of the investigations concentrated on how to maximally improve its reinforcing capacity. For example, in order to improve interfacial adhesion between starch and rubber matrix, some reinforcing mechanisms were proposed. However, there are lack of investigations on the vulcanization and biodegradability of starch/rubber composites. Li et al. [156] found that the incorporation of starch accelerated the vulcanization rate of SBR rubber, whereas the accelerating mechanism has not been explained properly. In addition, starch/rubber composites are considered as biodegradable materials, thus the research on their biodegradability is very essential in the future.
Starch nanocrystals can be derived from native starch granules. Owing to the three-dimensional nano scale, specific plate-like morphology and strong percolation network, starch nanocrystals possessed some advantages compared with native starch granules. For example, starch nanocrystals/rubber composites exhibited much lower gas permeability, better thermal stability and lower solvent absorption than native starch/rubber composites. Therefore, compared with native starch/rubber composites, starch nanocrystals/rubber composites are expected to have a wider range of application. However, the quick preparation of starch nanocrystals from native starch granules is one challenge. Currently, the starch nanocrystals were mainly prepared by acid hydrolysis, which took a long period. In addition, after hydrolysis, the separation of starch nanocrystals from acids is very difficult. Therefore, novel techniques should be developed to quickly prepare starch nanocrystals from native starch granule and to easily separate the starch nanocrystals from acids. In addition, the uniform dispersion of starch nanocrystals in the rubber matrix is another challenge.
References
Evaluation of the Community Policy for Starch and Starch Products. Commission of the European Communities (2002)
U. Marz, World market for starches/glucose, emphasizing Cassava. Report Code: FOD037A (2006)
U. Marz, Starch/glucose: global markets. Report Code: FOD037B (2013)
A. Buléon et al., Starch granules: structure and biosynthesis. Int. J. Biol. Macromol. 23(2), 85–112 (1998)
D. Le Corre, J. Bras, A. Dufresne, Starch nanoparticles: a review. Biomacromolecules 11(5), 1139–1153 (2010)
K. Sriroth et al., Cassava starch granule structure–function properties: influence of time and conditions at harvest on four cultivars of cassava starch. Carbohydr. Polym. 38(2), 161–170 (1999)
E. Nuwamanya et al., Crystalline and pasting properties of cassava starch are influenced by its molecular properties (2010)
J.N. BeMiller, R.L. Whistler (eds.), Starch: Chemistry and Technology (Academic Press, Massachusetts, 2009)
R.A. Moura, The effect of physical aging, starch particle size, and starch oxidation on thermal-mechanical properties of poly (lactic acid)/starch composites. ProQuest (2006)
A.L. Adejumo, A.F. Aderibigbe, S.K. Layokun, Cassava starch: production, physicochemical properties and hydrolysation: a review. Adv. Food Energy Secur. 2, 8–17 (2011)
R.M. McCready et al., Determination of starch and amylose in vegetables. Anal. Chem. 22(9), 1156–1158 (1950)
G.K. Adkins, C.T. Greenwood, Studies on starches of high amylose-content: Part VII. Observations on the potentiometric iodine-titration of amylomaize starch. Carbohydr. Res. 3(1), 81–88 (1966)
O. Paredes-Lopez, Molecular Biotechnology for Plant Food Production (CRC Press, Boca Raton, 1999)
N.W.H. Cheetham, L. Tao, Variation in crystalline type with amylose content in maize starch granules: an X-ray powder diffraction study. Carbohydr. Polym. 36(4), 277–284 (1998)
H. Liu, F. Xie, L. Yu, L. Chen, L. Li, Thermal processing of starch-based polymers. Prog. Polym. Sci. 34, 1348–1368 (2009)
X. Liu, L. Yu, H. Liu, L. Chen, L. Lin, In situ thermal decomposition of starch with constant moisture in a sealed system. Polym. Degrad. Stab. 93, 260–262 (2008)
K.J. Zeleznak, R.C. Hoseney, The glass transtion in starch. Cereal Chem. 64, 121–124 (1987)
S. Mali, L.S. Sakanaka, F. Yamashita, M.V.E. Grossmann, Water sorption and mechanical properties of cassava starch films and their relation to plasticizing effect. Carbohydr. Polym. 60, 283–289 (2005)
C. Van der Berg, Food water relationships: progress and integration, comments and thoughts. H. Levine, L. Slade (eds.), Water Relationships in Foods (Plenum Press, New York, 1991), pp. 21–28
M.A. Garcia, M.N. Martino, N.E. Zaritzky, Starch-based coatings: effect on refrigerated strawberry (Fragaria ananassa) quality. J. Sci. Food Agric. 76, 411–420 (1998)
S.H. Imam, S.H. Gordon, L. Mao, L. Chen, Environmentally friendly wood adhesive from a renewable plant polymer: characteristics and optimization. Polym. Degrad. Stab. 73, 529–533 (2001)
Y. Wei, F. Cheng, H. Zheng, Synthesis and flocculating properties of cationic starch derivatives. Carbohydr. Polym. 74, 673–679 (2008)
J. Wu, Y. Wei, J. Lin, S. Lin, Study on starch-graft-acrylamide/mineral powder super absorbent composite. Polymer 44, 6513–6520 (2003)
B.R. Pant, H.-J. Jeon, H.H. Song, Radiation cross-linked carboxymethylated starch and iron removal capacity in aqueous solution. Macromol. Res. 19, 307–312 (2011)
M.-C. Li, J.K. Lee, U.R. Cho, Synthesis, characterization, and enzymatic degradation of starch-grafted poly(methyl methacrylate) copolymer films. J. Appl. Polym. Sci. 125, 405–414 (2012)
S.S. Wang, W.C. Chiang, B. Zhao, X.G. Zheng, I.H. Kim, Experimental analysis and computer simulation of starch-water interactions during phase transition. J. Food Sci. 56, 121–124 (1991)
A. Garcia-Alonso, A. Jimenez-Escrig, N. Martin-Carroon, L. Bravoa, F. Saura-Calixto, Assessment of some parameters involved in the gelatinization and retrogration of starch. Food Chem. 66, 181–187 (1999)
P.K. Herh, J.L. Kokini, The effect of pressure on the gelatinization of starch using small amplitude oscillatory measurements under pressure, in Proceeding of Institute of Food Technologies 51st Annual Meeting (1990)
J.J.G. Van Soest, K. Benes, D. De Wit, The influence of starch molecular mass on the properties of extruded thermoplastic starch. Polymer 37, 3543–3552 (1996)
J.J.G. Van Soest, R.C. Bezemer, D. De Wit, J.F.G. Vliegenthart, Influence of glycerol on the melting of potato starch. Ind. Crops Prod. 5, 1–9 (1996)
J.J.G. Van Soest, D.B. Borger, Structure and properties of compression-molded thermoplastic starch materials from normal and high-amylose maize starches. J. Appl. Polym. Sci. 64, 631–644 (1997)
K. Dean, L. Yu, D.Y. Wu, Preparation and characterization of melt-extruded thermoplastic starch/clay nanocomposites. Compos. Sci. Technol. 67, 413–421 (2007)
N. Wang, J. Yu, P.R. Chang, X. Ma, Influence of formamide and water on the properties of thermoplasticstarch/poly(lactic acid) blends. Carbohydr. Polym. 71, 109–118 (2008)
J. Yu, J. Gao, T. Lin, Biodegradable thermoplastic starch. J. Appl. Polym. Sci. 62, 1491–1494 (1996)
D. Lourdin, L. Coignard, H. Bizot, P. Colonna, Influence of equilibrium relative humidity and plasticizer concentration on the water content and glass transition of starch materials. Polymer 38, 5401–5406 (1997)
A.L.M. Smits, M. Wubbenhorst, P.H. Kruiskamp, J.J.G. van Soest, J.F.G. Vliegenthart, J. Van Turnhout, Structure evolution in amylopectin/ethylene glycol mixtures by Hbond formation and phase separation studied with dielectric relaxation spectroscopy. J. Phys. Chem. B 105, 5630–5636 (2001)
A.L. Da Róz, A.J.F. Carvalho, A. Gandini, A.A.S. Curvelo, The effect of plasticizers on thermoplastic starch compositions obtained by melt processing. Carbohydr. Polym. 63, 412–417 (2006)
P.M. Forssell, J.M. Mikkilä, G.K. Moates, Roger Parker, Phase and glass transition behaviour of concentrated barley starch-glycerol-water mixtures, a model for thermoplastic starch. Carbohydr. Polym. 34, 275–282 (1997)
A.A.S. Curvelo, A.J.F. de Carvalho, J.A.M. Agnelli, A thermoplastic starch–cellulosic fibers composites: preliminary results. Carbohydr. Polym. 45, 183–188 (2001)
Z. Liu, X.S. Yi, Y. Feng, Effects of glycerin and glycerol monstearate on performance of thermoplastic starch. J. Mater. Sci. 36, 1809–1815 (2001)
H.-M. Park, X. Li, C.-Z. Jin, C.-Y. Park, W.-J. Cho, C.-S. Ha, Preparation and properties of biodegradable thermoplastic starch/clay hybrids. Macromol. Mater. Eng. 287, 553–558 (2002)
H.-M. Park, W.-K. Li, C.-Y. Park, W.-J. Cho, C.-S. Ha, Environmentally friendly polymer hybrids Part I mechanical, thermal, and barrier properties of thermoplastic starch/clay nanocomposites. J. Mater. Sci. 38, 909–915 (2003)
F.J. Rodriguez-Gonzalez, B.A. Ramsay, B.D. Favis, Rheological and thermal properties of thermoplastic starch with high glycerol content. Carbohydr. Polym. 58, 139–147 (2004)
B. Chen, J.R.G. Evans, Thermoplastic starch–clay nanocomposites and their characteristics. Carbohydr. Polym. 61, 455–463 (2005)
M.A. Huneault, H. Li, Morphology and properties of compatibilized polylactide/thermoplastic starch blends. Polymer 48, 270–280 (2007)
R. Shi, Z. Zhang, Q. Liu, Y. Han, L. Zhang, D. Chen, W. Tian, Characterization of citric acid/glycerol co-plasticized thermoplastic starch prepared by melt blending. Carbohydr. Polym. 69, 748–755 (2007)
M.-F. Huang, J.-G. Yu, X.-F. Ma, Studies on the properties of montmorillonite-reinforced thermoplastic starch composites. Polymer 45, 7017–7026 (2007)
L. Wang, R. Shogren, C. Carriere, Preparation and properties of thermoplastic starch–polyester laminate sheets by coextrusion. Polym. Eng. Sci. 40, 499–506 (2000)
A.P. Mathew, A. Dufresne, Morphological investigation of nanocomposites from sorbitol plasticized starch and tunicin whiskers. Biomacromolecules 3, 609–617 (2002)
K. Krogars, J. Heinamaki, M. Karjalainen, A. Niskanen, M. Leskela, J. Yliruusi, Enhanced stability of rubbery amylose-rich maize starch films plasticized with a combination of sorbitol and glycerol. Int. J. Pharm. 251, 205–208 (2003)
E.D.M. Teixeira, D. Pasquini, A.A.S. Curvelo, E. Corradini, M.N. Belgacem, A. Dufresne, Cassava bagasse cellulose nanofibrils reinforced thermoplastic cassava starch. Carbohydr. Polym. 78, 422–431 (2009)
N. Wang, J.G. Yu, X.F. Ma, Y. Wu, The influence of citric acid on the properties of thermoplastic starch/linear low-density polyethylene blends. Carbohydr. Polym. 67, 446–453 (2007)
N. Wang, X. Zhang, N. Han, S. Bai, Effect of citric acid and processing on the performance of thermoplastic starch/montmorillonite nanocomposites. Carbohydr. Polym. 76, 68–73 (2009)
N. Wang, J. Yu, P.R. Chang, X. Ma, Influence of citric acid on the properties of glycerol-plasticized dry starch (DTPS) and DTPS/Poly(lactic acid) blends. Starch/Starke 59, 409–417 (2007)
X. Ma, P.R. Chang, J. Yu, M. Stumborg, Properties of biodegradable citric acid-modified granular starch/thermoplastic pea starch composites. Carbohydr. Polym. 75, 1–8 (2009)
J. Yu, N. Wang, X. Ma, The effects of citric acid on the properties of thermoplastic starch plasticized by glycerol. Starch/Starke 59, 494–504 (2005)
X. Ma, J. Yu, The plastcizers containing amide groups for thermoplastic starch. Carbohydr. Polym. 57, 197–203 (2004)
X. Ma, J. Yu, F. Jin, Urea and formamide as a mixed plasticizer for thermoplastic starch. Polym. Int. 53, 1780–1785 (2004)
X. Ma, J. Yu, Formamide as the plasticizer for thermoplastic starch. J. Appl. Polym. Sci. 93, 1769–1773 (2004)
X. Ma, J. Yu, H.F. Kennedy, Studies on the properties of natural fibers-reinforced thermoplastic starch composites. Carbohydr. Polym. 62, 19–24 (2005)
X. Ma, J. Yu, Studies on the properties of formamide plasticized thermoplastic starch. Acta Polym. Sin. 2, 240–245 (2004)
X. Ma, J, Yu, J. Wan, Urea and ethanolamide as a mixed plasticizer for thermoplastic starch. Carbohydr. Polym. 64, 267–273 (2006)
X. Ma, J. Yu, The effect of plasticizers containing amide groups on the properties of thermoplastic starch. Starch/starke 56, 545–551 (2004)
P.S. Walia, J.W. Lawton, R.L. Shogren, F.C. Felker, Effect of moisture level on the morphology and melt flow behavior of thermoplastic starch/poly(hydroxyl ester ether) blends. Polymer 41, 8083–8093 (2000)
J.L. Wilett, M.M. Millard, B.K. Jasberg, Extrusion of waxy maize starch: melt rheology and molecular weight degradation of amylopectin. Polymer 38, 5983–5989 (1997)
J.J.G. Van Soest, N. Knooren, Influence of glycerol and water content on the structure and properties of extruded starch plastic sheets during aging. J. Appl. Polym. Sci. 64, 1411–1422 (1996)
O. Kazuo, A. Kenji, O. Shin, T. Yoshimura, S. Rengakuji, Y. Nakamura, I. Yamazaki, S. Murotani, C. Shimasaki, Effects of the noncyclic cyanamides on the retrogradation of waxy corn starch. Bull. Chem. Soc. Japan 73, 1283–1284 (2000)
X.F. Ma, J.G. Yu, J. Feng, A mixed plasticizer for preparation of thermoplastic starch. Chin. Chem. Lett. 15, 741–744 (2004)
D. Liu, Q. Wu, H. Chen, P.R. Chang, Transitional properties of starch colloid with particle size reduction from micro- to nanometer. J. Colloid Interface Sci. 339, 117–124 (2009)
A. Shi, D. Li, L. Wang, B. Lia, B. Adhikari, Preparation of starch-based nanoparticles through high-pressure homogenization and miniemulsion cross-linking: influence of various process parameters on particle size and stability. Carbohydr. Polym. 83, 1604–1610 (2011)
X. Ma, R. Jian, P.R. Chang, R. Yu, Fabrication and characterization of citric acid-modified starch nanoparticles/plasticized-starch composites. Biomacromolecules 9, 3314–3320 (2008)
Y. Tan, K. Xu, L. Li, C. Liu, C. Song, P. Wang, Fabrication of size-controlled starch-based nanospheres by nanoprecipitation. ACS Appl. Mater. Interfaces 1, 956–959 (2009)
S. Xiao, X. Liu, C. Tong, J. Liu, D. Tang, L. Zhao, Studies of poly-L-lysine-starch nanoparticle preparation and its application as gene carrier. Sci. China, Ser. B: Chem. 48, 162–9166 (2005)
D. Yu, S. Xiao, C. Tong, C. Lin, X. Liu, Dialdehyde starch nanoparticles: preparation and application in drug carrier. Chin. Sci. Bull. 52, 2913–2918 (2007)
S.F. Chin, S.C. Pang, S.H. Tay, Size controlled synthesis of starch nanoparticles by a simple nanoprecipitation method. Carbohydr. Polym. 86, 1817–1819 (2011)
L. Jayakody, R. Hoover, The effect of lintnerization on cereal starch granules. Food Res. Int. 35, 665–680 (2002)
Y.-J. Wang, V.-D. Truong, L. Wang, Structures and physicochemical properties of acid-thinned corn, potato and rice starches. Starch/Starke 53, 570–576 (2001)
D.L. Corre, J. Bras, A. Dufresne, Starch nanoparticles: a review. Biomacromolecules 11, 1139–1153 (2010)
D. LeCorre, J. Bras, A. Dufresne, Influence of botanic origin and amylose content on the morphology of starch nanocrystals. J. Nanopart. Res. 13, 7193–7208 (2011)
V. Singn, S.Z. Ali, Comparative acid modification of various starches. Starch/Starke 39, 402–405 (1987)
H. Angellier, L. Choisnard, S. Molina-Boisseau, P. Ozil, A. Dufresne, Optimization of the preparation of aqueous suspensions of waxy maize starch nanocrystals using a response surface methodology. Biomacromolecules 5, 1545–1551 (2004)
D.L. Corre, J. Bras, L. Choisnard, A. Dufresne, Optimization of the batch preparation of starch nanocrystals to reach daily time-scale. Starch/Starke 64, 489–496 (2012)
Y. Chen, C. Liu, R.P. Chang, X. Cao, D.P. Anderson, Bionanocomposites based on pea starch and cellulose nanowhiskers hydrolyzed from pea hull fibre: effect of hydrolysis time. Carbohydr. Polym. 76, 607–615 (2009)
V. Sigh, S.Z. Ali, Acid degradation of starch. The effect of acid and starch type. Carbohydr. Polym. 41, 191–195 (2000)
V. Sigh, S.Z. Ali, Properties of starches modified by different acids. Int. J. Food Prop. 11, 495–507 (2008)
M.B. Tasic, B.V. Konstantinovic, M.L. Lazic, V.B. Veljkovic, The acidhydrolysis of potato tuber mash in bioethanol production. Biochem. Eng. J. 43, 208–211 (2009)
Y.-J. Wang, V.-D. Truong, L. Wang, Structures and rheological properties of corn starch as affected by acid hydrolysis. Carbohydr. Polym. 52, 327–333 (2003)
D. LeCorre, E. Vahanian, A. Dufresne, J. Bras, Enzymatic pretreatment for preparing starch nanocrystals. Biomacromolecules 13, 132–137 (2012)
C.E. Brockway, Efficiency and frequency of grafting of methyl methacrylate to granular corn starch. J. Polym. Sci. part A: Gen. Pap. 2, 3721–3731 (1964)
P. Ghosh, S.K. Paul, Photograft copolymerization of methyl methacrylate on potato starch using potassium pervanadate as initiator. J. Macromol. Sci. part A Chem. 20, 261–269 (1983)
L. Nurmi, S. Holappa, N. Mikkonen, J. Seppala, Controlled grafting of acetylated starch by atom transfer radical polymerization of MMA. Eur. Polym. J. 43, 1372–1382 (2007)
V. Pimpan, P. Thothong, Synthesis of cassava starch-g-poly(methyl methacrylate) copolymers with benzoyl peroxide as an initiator. J. Appl. Polym. Sci. 101, 4083–4089 (2006)
B.N. Misra, R. Dogra, Grafting onto Starch. IV. Graft copolymerization of methyl methacrylate by use of AIBN as radical initiator. J. Macromol. Sci. Part A Chem. 14, 763–770 (1980)
F.E. Okieimen, O.B. Said, Studies on the graft copolymerization of methyl methacrylate onto starch. Acta Polym. 40, 708–710 (1989)
J.-P. Gao, R.-C. Tian, J.-G. Yu, M.-L. Duan, Graft copolymers of methy methacrylate onto canna starch using manganic pyrophosphate as an initiator. J. Appl. Polym. Sci. 53, 1091–1102 (1994)
G.F. Fanta, R.C. Burr, W.M. Doana, C.R. Russell, Graft polymerization of styrene onto starch by simultaneous cobalt-60 irradiation. J. Appl. Polym. Sci. 21, 425–433 (1977)
K. Kaewtatip, V. Tanrattanakul, Preparation of cassava starch grafted with polystyrene by suspension polymerization. Carbohydr. Polym. 73, 647–655 (2008)
C.G. Cho, K. Lee, Preparation of starch-g-polystyrene copolymer by emulsion polymerization. Carbohydr. Polym. 48, 125–130 (2002)
S. Kitkamjornwong, M. Sonsuk, S. Wittayapichet, P. Prasassarakich, P.-C. Vejjanukroh, Degradation of styrene-g-cassava starch filled polystyrene plastics. Polym. Degrad. Stab. 66, 323–335 (1999)
J.M. Fang, P.A. Fowler, C.A.S. Hill, Studies on the grafting of acryloylated potato starch with styrene. J. Appl. Polym. Sci. 96, 452–459 (2005)
R.A. De Graaf, L.P.B.M. Janssen, The production of a new partially biodegradable starch plastic by reactive extrusion. Polym. Eng. Sci. 40, 2086–2094 (2000)
P. Liu, Z. Su, Surface-initiated atom transfer radical polymerization (SI-ATRP) of n-butylacrylate from starch granules. Carbohydr. Polym. 62, 159–163 (2005)
M.B. Vazquez, I. Goni, M. Gurruchaga, M. Valero, G.M. Guzman, Graft polymerization of acrylic monomers onto starch fractions. IV. Effect of reaction time on the grafting of butyl acrylate onto amylose. J. Polym. Sci., Part A: Polym. Chem. 25, 719–725 (1987)
C. Liu, Y. Shao, D. Jia, Chemically modified starch reinforced natural rubber composites. Polymer 49, 2176–2181 (2008)
M. Zhai, F. Yoshhii, T. Kume, K. Hashim, Syntheses of PVA/starch grafted hydrogels by irradiation. Carbohydr. Polym. 50, 295–303 (2002)
M. Zhai, F. Yoshhii, K. Hashim, Radiation modification of starch-based plastic sheets. Carbohydr. Polym. 52, 311–317 (2003)
Z. Zhu, R. Zhui, Slow release behavior of starch-g-poly (vinylalcohol) matrix for 2,4,5-trichlorophenoxyacetic acid herbicide. Eur. Polym. J. 37, 1913–1919 (2001)
M.K. Beliakova, A.A. Aly, F.A. Abdel-Mohdy, Grafting of poly (methacrylic acid) on starch and poly (vinyl alcohol). Starch-Starke 56, 407–412 (2004)
V.D. Athawale, S.C. Rathi, Syntheses and characterization of starch-poly(methacrylic acid) graft copolymers. J. Appl. Polym. Sci. 66, 1399–1403 (1997)
M.I. Khalil, K.H. Mostafa, A. Hebeish, Synthesis of poly(methacrylic acid-)starch graft copolymers using Mn-IV-acid system. Starch-Starke 42, 107–111 (1990)
K.M. Mostafa, Graft polymerization of methacrylic acid on starch and hydrolyzed starches. Polym. Degrad. Stab. 50, 189–194 (1995)
V.D. Athawale, V. Lele, Graft copolymerization onto starch. II. Grafting of acrylic acid and preparation of it’s hydrogels. Carbohydr. Polym. 35, 21–27 (1998)
Q.-Z. Yan, W.-F. Zhang, G,-D, Lu, X,-T, Su, C.-C. Ge, Frontal copolymerization synthesis and property characterization of starch-graft-poly(acrylic acid) hydrogels. Chem. Eur. J. 11, 6609–6615 (2005)
S. Kiatkamjornwong, W. Chomsaksakul, M. Sonsukc, Radiation modification of water absorption of cassava starch by acrylic acid/acrylamide. Radiat. Phys. Chem. 59, 413–427 (2000)
E. Al, G. Guclu, T.B. Iyim, S. Emik, S. Ozgumus, Synthesis and properties of starch-graft-acrylic acid/Na-montmorillonite superabsorbent nanocomposite hydrogels. J. Appl. Polym. Sci. 109, 16–22 (2008)
K.M. Mostafa, Graft polymerization of acrylic acid onto starch using potassium permanganate acid (redox system). J. Appl. Polym. Sci. 56, 263–269 (1995)
V.D. Athawale, V. Lele, Recent trends in hydrogels based on starchgraft-acrylic acid: a review. Starch-Starke 53, 7–13 (2001)
S.E. Abdel-Aal, Y.H. Gad, A.M. Dessouki, Use of rice straw and radiation-modified maize starch/acrylonitrile in the treatment of wastewater. J. Hazard. Mater. 129, 204–215 (2006)
G.F. Fanta, R.C. Burr, C.R. Russell, C.E. Rist, Graft copolymers of starch. I. Copolymerization of gelatinized wheat starch with acrylonitrile. Fractionation of copolymer and effect of solvent on copolymer composition. J. Appl. Polym. Sci. 10, 929–937 (1966)
G.F. Fanta, R.C. Burr, C.R. Russell, C.E. Rist, Graft copolymers of starch. II. Copolymerization of gelatinized wheat starch with acrylonitrile: influence of reaction conditions on copolymer composition. J. Polym. Sci. Part B: Polym. Lett. 4, 765–769 (1966)
G.F. Fanta, R.C. Burr, C.R. Russell, C.E. Rist, Graft copolymers of starch. III. Copolymerization of gelatinized wheat starch with acrylonitrile. influence of chain modifiers on copolymer composition. J. Appl. Polym. Sci. 11, 457–463 (1967)
G.F. Fanta, R.C. Burr, C.R. Russell, C.E. Rist, Copolymers of starch and polyacrylonitrile: the dilution effect. J. Appl. Polym. Sci. 13, 133–140 (1969)
R. Mehrotra, B. Ranby, Graft copolymerization onto starch. I. Complexes of Mn3+ as initiators. J. Appl. Polym. Sci. 21, 1647–1654 (1977)
R. Mehrotra, B. Ranby, Graft copolymerization onto starch. II. Grafting of acrylonitrile to granular native potato starch by manganic pyrophosphate initiation. Effect of reaction conditions on grafting parameters. J. Appl. Polym. Sci. 21, 3407–3415 (1977)
R. Mehrotra, B. Ranby, Graft copolymerization onto starch. III. Grafting of acrylonitrile to gelatinized potato starch by manganic pyrophosphate initiation. J. Appl. Polym. Sci. 22, 2991–3001 (1978)
M. Tahan, A. Zikha, Alkali metal alkoxide derivatives of starch and dextrin as initiators of graft polymerization of methacrylonitrile. Eur. Polym. J. 5, 347–359 (1969)
B. Vazquez, I. Goni, M. Gurruchaga, M. Valero, G. Martin, Guzman, Synthesis and characterization of graft copolymers of methacrylonitrile/methacrylate mixtures onto amylomaize by the ceric ion method. J. Polym. Sci., Part A: Polym. Chem. 30, 1542–1548 (1992)
V.D. Athawale, V. Lele, Thermal studies on granular maize starch and its graft copolymers with vinyl monomers. Starch-Starke 52, 205–213 (2000)
K. Mostafa, M. Morsy, Modification of carbohydrate polymers via grafting of methacrylonitrile onto pregelled starch using potassium monopersulfate/Fe2+ redox pair. Polym. Int. 53, 885–890 (2004)
V.D. Athawale, V. Lele, Syntheses and characterisation of graft copolymers of maize starch and methacrylonitrile. Carbohydr. Polym. 41, 407–416 (2000)
G.F. Fanta, R.C. Burr, W.M. Doane, C.R. Russell, Graft polymerization of vinyl acetate onto starch. Saponification to starch–g–poly(vinyl alcohol). J. Appl. Polym. Sci. 23, 229–240 (1979)
B.N. Misra, R. Dogra, I. Kaur, D. Sood, Grafting onto starch. II. Graft copolymerization of vinyl acetate onto starch by radical initiator. J. Polym. Sci.: Polym. Chem. Edn. 18, 341–344 (1980)
G.F. Fanta, D. Trimnell, J.H. Salch, Graft polymerization of methyl acrylate–vinyl acetate mixtures onto starch. J. Appl. Polym. Sci. 49, 1679–1682 (1993)
J. Huang, H.A. Schols, Z. Jin, E. Sulmann, A.G.J. Voragen, Characterization of differently sized granule fractions of yellow pea, cowpea and chickpea starches after modification with acetic anhydride and vinylacetate. Carbohydr. Polym. 67, 11–20 (2007)
W.-C. Chan, C.-Y. Chiang, Flocculation of clay suspensions with water-insoluble starch grafting acrylamide/sodium allylsulfonated copolymer powder. J. Appl. Polym. Sci. 58, 1721–1726 (1995)
J. Zhang, A. Li, A. Wang, Study on superabsorbent composite. VI. Preparation, characterization and swelling behaviors of starch phosphate-graft-acrylamide/attapulgite superabsorbent composite. Carbohydr. Polym. 65, 150–158 (2006)
G.-X. Chen, H. Geng, L. Luo, B. Wu, Q.F. Li, Synthesis and properties of starch-grafted polystyrene-maleic anhydride copolymer. J. Appl. Polym. Sci. 126, E109–E115 (2012)
G. Mino, S. Kaizerman, A new method for the preparation of graft copolymers. Polymerization initiated by ceric ion redox systems. J. Appl. Polym. Sci. 31, 242–243 (1958)
G. Sen, R. Kumar, S. Ghosh, S. Pal, A novel polymeric flocculant based on polyacrylamide grafted carboxymethylstarch. Carbohydr. Polym. 77, 822–831 (2009)
Y. Wei, F. Cheng, H. Zheng, Synthesis and flocculating properties of cationic starch derivatives. Carbohydr. Polym. 74, 673–679 (2008)
C. Wang, X. Li, J. Chen, G. Fei, H. Wang, Q. Liu, Synthesis and characterization of polyacrylonitrile pregelled starch graft copolymers using ferrous sulfate-hydrogen peroxide redox initiation system as surface szing agent. J. Appl. Polym. Sci. 122, 2630–2638 (2011)
A.N. Jyothi, Starch graft copolymers: novel application in industry. Compos. Interfaces 17, 165–174 (2010)
S.C. Kiing, K. Dzulkefly, P.H. Yiu, Characterization of biodegradable polymer blends of acetylated and hydroxypropylated sago starch and natural rubber. J. Polym. Environ. 21, 995–1001 (2013)
M.M. Afiq, A.R. Azura, Effect of sago starch loadings on soil decomposition of Natural Rubber Latex (NRL) composite films mechanical properties. Int. Biodeterior. Biodegradation 85, 139–149 (2013)
A.I. Khalaf, E.M. Sadek, Compatibility study in natural rubber and maize starch blends. J. Appl. Polym. Sci. 125, 959–967 (2012)
Y.P. Wu, M.Q. Ji, Q. Qi, Y.Q. Wang, L.Q. Zhang, Preparation, structure and properties of starch/rubber composites prepared by co-coagulating rubber latex and starch paste. Macromol. Rapid Commun. 25, 565–571 (2004)
F.G. Corvasce, T.D. Linster, G. Thielen, U.S. Patent 5,672,639 (1997)
A.J.F. Carvalho, A.E. Job, N. Alves, A.A.S. Curvelo, A. Gandini, Thermoplastic starch/natural rubber blends. Carbohydr. Polym. 53, 95–99 (2003)
Y.P. Wu, Q. Qi, G.H. Liang, L.Q. Zhang, A strategy to prepare high performance starch/rubber composites: in situ modification during latex compounding process. Carbohydr. Polym. 65, 109–113 (2006)
H. Tang, Q. Qi, Y. Wu, G. Liang, L. Zhang, J. Ma, Reinforcement of elastomer by starch. Macromol. Mater. Eng. 291, 629–637 (2006)
Q. Qi, Y. Wu, M. Tian, G. Liang, L. Zhang, M. Jun, Modification of starch for high performance elastomer. Polymer 47, 3896–3903 (2006)
M.C. Li, X. Ge, U.R. Cho, Effectiveness of coupling agents in the poly (methyl methacrylate)-modified starch/styrene-butadiene rubber interfaces. Mater. Lett. 92, 132–135 (2013)
R.A. Buchanan, O.E. Weislogel, C.R. Russell, C.E. Rist, Starch in rubber. Zinc starch xanthate in latex masterbatching. Ind. Eng. Chem. Prod. Res. Dev. 7, 155–158 (1968)
Z.F. Wang, Z. Peng, S.D. Li, H. Lin, K.X. Zhang, X.D. She, X. Fu, The impact of esterification on the properties of starch/natural rubber composites. Compos. Sci. Technol. 69, 1797–1803 (2009)
M.C. Li, X. Ge, U.R. Cho, Emulsion grafting vinyl monomers onto starch for reinforcement of styrene-butadiene rubber. Macromol. Res. 21, 519–528 (2013)
M.C. Li, U.R. Cho, Mechanical performance, water absorption behavior and biodegradability of poly (methyl methacrylate)-modified starch/SBR biocomposites. Macromol. Res. 21, 793–800 (2013)
H. Angellier, S. Molina-Boisseau, A. Dufresne, Processing and structure properties of waxy-maize starch nanocrystal reinforced natural rubber. Macromolecules 38, 3783–3792 (2005)
H. Angellier, S. Molina-Boisseau, A. Dufresne, Mechanical properties of waxy-maize starch nanocrystal reinforced natural rubber. Macromolecules 38, 9161–9170 (2005)
K.R. Rajisha, H.J. Maria, L.A. Pothan, Z. Ahmad, S. Thomas, Preparation and characterization of potato starch nanocrystal reinforced natural rubber nanocomposites. Int. J. Biol. Macromol. 67, 147–153 (2014)
P. Mele, S. Molina-Boisseau, A. Dufresne, Reinforcing mechanisms of starch nanocrystals in a nonvulcanized natural rubber matrix. Biomacromocules 12, 1487–1493 (2011)
D. LeCorre, J, Bras, A. Dufresne, Influence of the botanic origin of starch nanocrystals on the morphological and mechanical properties of natural rubber nanocomposites. Macromolecular Mater. Eng. Doi:10.1002/mame.201100317
E. Bouthegourd, K.R. Rajisha, N. Kalarical, J.M. Saiter, S. Thomas, Natural rubber latex/potato starch nanocrystal nanocomposites: correlation morphology/electrical properties. Mater. Lett. 65, 3615–3617 (2011)
M. Valodkar, S. Thakore, Organically modified nanosized starch derivatives as excellent reinforcing agents for bionanocomposites. Carbohydr. Polym. 86, 1244–1251 (2011)
A. Rouilly, L. Rigal, R.G. Gillbert, Synthesis and properties of composites of starch and chemically modified natural rubber. Polymer 45, 7813–7820 (2004)
C. Nakason, A. Kaesaman, S. Homsin, S. Kiatkamjonwong, Rheological and curing behavior of reactive blending. I. Natural rubber-g-poly(methyl methacrylate)-cassava starch. J. Appl. Polym. Sci. 89, 1453–1463 (2003)
C. Nakason, A. Kaesaman, A. Rungvichaniwat, K. Eardrod, S. Kiatkamjonwong, Rheological and curing behavior of reactive blending. II. Maleated natural rubber-cassava starch. J. Appl. Polym. Sci. 81, 2803–2813 (2001)
C. Nakason, A. Kaesaman, K. Eardrod, Cure and mechanical properties of natural rubber-g-poly(methyl methacrylate)—cassava starch compounds. Mater. Lett. 59, 4020–4025 (2005)
M.M. Senna, R.M. Mohamed, A.N. Shehab-Eldin, S. El-Hamouly, Characterization of electron beam irradiated natural rubber/modified starch composites. J. Ind. Eng. Chem. 18, 1654–1661 (2013)
Z.X. Ooi, H. Ismail, A.A. Bakar, Optimisation of oil palm ash as reinforcement in natural rubber vulcanisation: a comparison between silica and carbon black fillers. Polym. Test. 32, 625–630 (2013)
R. Rajasekar, G.C. Nayak, A. Malas, S. Sahoo, C.K. Das, Effect of dual fillers on the properties of acrylonitrile butadiene rubber nanocomposites in presence of compatibilizer. Elastomery 15, 3–13 (2011)
T.V. Varghese, H.A. Kumar, S. Anitha, S. Ratheesh, R.S. Rajeev, R.V. Lakshmana Rao, Reinforcement of acrylonitrile butadiene rubber using pristine few layer graphene and its hybrid fillers. Carbon 61, 476–486 (2013)
S. Attharangsan, H. Ismail, M.A. Bakar, J. Ismail, Carbon black (CB)/rice husk powder (RHP) hybrid filler-filled natural rubber composites: effect of CB/RHP ratio on property of the composites. Polym.-Plast. Technol. Eng. 51(7), 655–662 (2012)
Y.P. Wu, G.H. Liang, L.Q. Zhang, Influence of starch on the properties of carbon black filled styrene—butadiene rubber composites. J. Appl. Polym. Sci. 114(4), 2254–2260 (2009)
M.-S. Kim, U.R. Cho, Manufacture and properties of PMMA grafted starch/carbon black/NBR composites. Polymer (Korea) 37, 764–769 (2013)
B. Pradhan, S.K. Srivastava, Layered double hydroxide/multiwalled carbon nanotube hybrids as reinforcing filler in silicone rubber. Compos. A Appl. Sci. Manuf. 56, 290–299 (2014)
B. Pradhan, S.K. Srivastava, Synergistic effect of three-dimensional multiwalled carbon nanotube–graphene nanofiller in enhancing the mechanical and thermal properties of high performance silicone rubber. Polym. Int. (2013)
Y. Zhang, Q. Zhang, Q. Liu, H. Cheng, R.L. Frost, Thermal stability of styrene butadiene rubber (SBR) composites filled with kaolinite/silica hybrid filler. J. Therm. Anal. Calorim. 115(2), 1013–1020 (2014)
J. Wang, H. Jia, L. Ding, L. Zhu, X. Dai, X. Fei, F. Li, X. Gong, Utilization of silane functionalized carbon nanotubes-silica hybrids as novel reinforcing fillers for solution styrene butadiene rubber. Polym. Compos. 34(5), 690–696 (2013)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Li, MC., Cho, U.R. (2017). Starch in Rubber Based Blends and Micro Composites. In: Visakh P. M. (eds) Rubber Based Bionanocomposites. Advanced Structured Materials, vol 56. Springer, Cham. https://doi.org/10.1007/978-3-319-48806-6_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-48806-6_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48804-2
Online ISBN: 978-3-319-48806-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)