
Abstract
Bone morphogenetic proteins (BMPs) are originally identified with their ability to induce heterotopic ossification. Several decades of studies have demonstrated that BMPs have pleiotropic functions in numbers of tissues for many different aspects. This review focuses on the effects of BMP signaling on skeletogenesis and craniofacial development. We will summarize recent progresses on in vitro studies, animal models, and human genetics to uncover highly context-dependent functions of BMP signaling, including unexpected outcomes, and the mechanisms of how BMP signaling regulates bone mass. We will also summarize reported findings about BMP signaling-related genes identified as causes of human diseases in skeletal system such as chondrodysplasia, facial cleft, and craniosynostosis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Osteoblast
- Chondrocyte
- Osteocyte
- Osteoclast
- Mesenchyme
- Neural crest
- BMP signaling
- Wnt signaling
- Hedgehog
- FGF
- Facial process
- Cleft palate
- Cleft lip
- Craniosynostosis
- Chondrodysplasia
- Temporomandibular joint
1 Introduction
Bone morphogenetic proteins (BMPs) were discovered and named in 1965 by Marshall Urist, who initially identified the ability of a then unknown factor in the bone to induce ectopic bones in muscle [204]. In the past 50 years, the osteogenic function of BMPs has been extensively examined [188]. The US Food and Drug Administration (FDA) has approved BMP2 and BMP7 for clinical use in long bone open fractures, nonunion fractures, and spinal fusion. Therefore, the exogenous role of BMPs in the bone is well known in orthopedics. However, it is crucial to understand endogenous or physiological roles of BMPs during skeletogenesis and bone remodeling.
BMP signaling plays important roles in a variety of cell types in the skeleton including osteoblasts, chondrocytes, and osteoclasts. The osteogenic function of BMPs and BMP signaling has been further investigated over the last decade using gene-targeting technology in animals. This chapter focuses on the physiological roles of BMP signaling on bone formation, bone resorption, and bone mass control, specifically via its action on osteoblasts or chondrocytes by reviewing mouse genetic studies of skeletal development and bone remodeling. This chapter also focuses on roles of BMP during craniofacial development including formation of calvaria and mandible.
2 Embryonic Skeletogenesis
2.1 Developmental Stages of Ossification
One of the key components derived from the paraxial mesoderm is the bone. The skeleton which includes the bone is generated from three distinct lineages: (1) the somites which generate the axial skeleton, (2) the lateral plate mesoderm which generates the limb skeleton, and (3) the cranial neural crest which generates the branchial and craniofacial bones and cartilage. The skeleton in mammals is formed through two distinct processes during embryogenesis: intramembranous ossification and endochondral ossification [51, 110]. Both processes involve the transformation of a preexisting mesenchymal tissue into the bone tissue as they are called “bone formation” or “osteogenesis.” The intramembranous ossification is a direct conversion of mesenchymal tissue into the bone, which primarily occurs in flat bones including the skull, the mandible, and the clavicle. On the other hand, endochondral ossification, which occurs in long bones, is an indirect conversion of mesenchymal tissue into the bone; i.e., the mesenchymal tissue differentiates into cartilage and this cartilage is later replaced by the bone.
2.2 BMP and Osteogenesis
At cellular levels, the in vivo physiological process of “osteogenesis” or “bone formation” can be described as two distinct processes: (1) intramembranous ossification through osteoblastogenesis that is direct differentiation of mesenchymal cells into bone cells (i.e., osteoblasts) and (2) the endochondral ossification, which includes an initial chondrogenesis that is differentiation from mesenchymal cells into cartilage cells (i.e., chondrocytes) followed by the apoptosis of chondrocyte secondary differentiation from osteoblast precursor to osteoblasts via osteogenesis. Therefore, “osteogenesis” encompasses osteoblastogenesis and chondrogenesis. The key molecules of BMP pathway involved in osteogenesis are listed (Table 1). Note that BMPRIA is a potent receptor of BMP2 and BMP4 [66], as is ACVRI for BMP7 [136].
In mice, BMP2 is expressed in a variety of sites including the developing limb buds [134], mesenchymal derivatives of which undergo endochondral ossification. The osteogenic (i.e., anabolic) roles of BMPs have been extensively examined over 50 years, and human recombinant BMP2, BMP4, BMP6, and BMP7 proteins have been vigorously used for mammalian cells to induce their differentiation in culture. To induce chondrogenesis or osteogenesis, primary cells or pluripotent mesenchymal cell lines such as C3H10T1/C3H10T2 [43], C2C12 [99], ATDC5 [186], N1511 [90], MC3T3 [13], and ST2 [219] have been treated with BMPs. In these cells, BMPs directly activate Sox9 and Cbfa1, transcriptional master genes required for chondrogenesis and osteoblastogenesis, respectively [114, 160, 232], to secondarily induce expression of chondrogenic (i.e., aggrecan, ColII, ColIX, ColX, etc.) or osteogenic (i.e., ALP, osteocalcin, BSP, Col1, etc.) markers. Based on the accumulated evidence of anabolic actions of BMPs, BMP2 and BMP7 have been approved by the US FDA for clinical application [56, 62]. It is noted that average circulating serum levels of BMPs are around 300~600 pg/ml [102, 206] while a typical dosage range of BMPs in culture experiments is 0~300 ng/ml. Also expression levels of BMPs by primary osteoblasts and pluripotent mesenchymal cell lines are quite low demonstrating a significant discrepancy between levels of BMPs found in tissues and those used for pharmacological experiments.
In addition to BMP signaling, the impacts of Wnt signaling on skeletogenesis and bone formation have been investigated for a decade [16, 57, 63, 64, 109]. The relationship of BMP signal with Wnt signal in the skeletal system is of interest. In vitro experiments using pluripotent mesenchymal cell lines or primary osteoblasts to test the interaction between BMP and Wnt signaling in osteoblasts have yielded both synergistic and antagonistic results: C2C12 cells and primary osteoblasts induce Wnt3a expression and stabilize Wnt/β-catenin signaling upon BMP2 treatment [7, 33, 141]. Alternatively, C3H10T1/2 cells treated with Wnt3a induce BMP4 expression [215]. These facts suggest the presence of a positive autocrine loop between BMP and Wnt signaling pathways [33, 171]. In contrast, primary osteoblasts show increased Wnt canonical signaling when BMP signaling is inhibited upon treatment with Dorsomorphin, an inhibitor for BMP type I receptors [92]. Wnt3a treatment represses BMP2-dependent Id1 expression in C2C12 cells [152]. Similarly, treatment of cultured skull bone with a BMP antagonist Noggin increases Wnt canonical signaling [95]. Moreover, one study investigated intracellular cross talk between BMP and Wnt pathways using uncommitted bone marrow stromal cells [127]. Dishevelled homolog 1 (Dvl1) is a cytoplasmic protein known to act as a signaling molecule for Wnt pathway. This study found that BMP2 antagonizes Wnt3a-induced proliferation and Wnt/β-catenin activation through an interaction between Smad1 and Dvl1. Another intracellular interaction via Pten/Akt pathway has been reported in hair follicle stem/progenitor cells [234]; however, this pathway is less likely functional in osteoblasts [68]. Taken together, these facts suggest that both positive and negative feedback loops are present between the two signaling pathways, BMP and Wnt, in a context-dependent manner.
2.3 Functional Studies in Animal Models
As detailed in Chap. 4, the BMP family members are involved with early patterning of the mouse embryo. Conventional knockout mice for the key genes (i.e., BMP2, BMP4, and BMP7 and their receptors BMPRIA and ACVRI) are lethal, and, thus, it is not possible to investigate bone development and remodeling using these mouse models [49, 61, 132, 143, 146, 216, 233]. To avoid the embryonic lethality, a strategy of conditional knockout mice using a Cre-loxP system has been employed.
Both osteoblasts and chondrocytes are derived from mesenchymal cells and are responsible for the bone and cartilage, respectively. Recent animal studies have been designed to investigate the physiologic function of BMP signaling in these different cell types (mesenchymal cells, chondrocytes, and osteoblasts) independently (Table 2). Interestingly, BMP signaling both in chondrocytes and mesenchymal cells positively controls bone size and mass while negatively controls the same in osteoblasts. Accumulated evidence has revealed similarities between mesenchymal cells and chondrocytes and differences between these cells and osteoblasts regarding how BMP signaling affects their behavior (i.e., bone size).
2.3.1 BMP and Osteoblasts
An osteoblast-specific conditional deletion of Bmpr1a using the Og2-Cre mouse line, in which Cre recombination is restricted in differentiated osteoblasts under the osteocalcin promoter, was first reported in 2004 [145]. The co-Smad, Smad4, was also conditionally deleted in osteoblasts using another Og2-Cre mouse line [197]. Interestingly, these two studies demonstrated that the response of osteoblasts after loss of BMP signaling is age dependent; trabecular bone volume is lower in young mutant mice but higher in aged mutant mice. In addition, the activity of osteoclasts is reduced in aged osteoblast-specific Bmpr1a-deficient mice, which may have led to the complex skeletal phenotype [145, 197]. These facts suggest that BMP signaling in differentiated osteoblasts controls the balance between bone formation by osteoblasts and resorption by osteoclasts, thereby affecting the final outcome of the amount of bone mass in an age-dependent manner. Increased bone mass in Bmpr1a-deficient mice appeared to be challenging to the general concept of BMPs as osteogenic inducers.
Comprehensive functions of BMP signaling in skeletogenesis have been further investigated and led to a new paradigm that alternation of Wnt signal by BMP is the key modulator of skeletal development. The loss of function of BMP signaling via BMPRIA in osteoblasts upregulates Wnt canonical signaling during embryonic and postnatal bone development, suggesting a negative regulation of Wnt signaling by BMP [92, 95]. These studies show that the upregulation of Wnt signaling is at least in part mediated by suppression of Wnt inhibitors including Sost/sclerostin and Dkk1 because both Sost/sclerostin and Dkk1 are direct targets of BMP signaling (Fig. 1). In addition, Sost expression was severely downregulated in Bmpr1a-deficient bones as assessed by microarray analysis [92, 95]. Interestingly, both Smad-dependent and Smad-independent pathways appear to contribute to Dkk1 expression, whereas Sost/sclerostin requires only Smad-dependent signaling, suggesting differential regulation of these genes by BMP signaling via BMPRIA [92]. BMP and Wnt signaling regulate the development and remodeling of many tissues and interact synergistically or antagonistically in a context- and age-dependent manner in vivo [17, 77]. Lastly, the role of BMPR1A in osteocytes was recently investigated by conditional disruption of Bmpr1a using Dmp1-Cre mouse line from two independent groups [93, 124]. The resulting mutant mice demonstrated an increased bone mass concomitant with accelerated cell proliferation and SOST reduction [93, 124]. It is interesting that the increased bone phenotype was much stronger in the osteocyte-specific condition (i.e. Dmp1Cre:Bmpr1a mice) compared with osteoblast-specific condition (i.e. Col1Cre:Bmpr1a mice). In addition, similar to the Col1Cre:Bmpr1a mice, Wnt signal is activated while RANKL is suppressed in the Dmp1Cre:Bmpr1a mice [93]. This fact is very intriguing because recent reports show osteocytes as a primary source of RANKL production [153, 219] and therefore BMPR1A can be a key molecule in osteocytes by regulating RANKL production.

A proposed model of the relationship between the BMP signaling via BMPRIA and the canonical Wnt signaling in osteoblasts. Both Dkk1 and Sost/sclerostin are downstream targets of the BMP signaling. The BMP signaling upregulates Sost expression primarily through the Smad-dependent signaling while it upregulates Dkk1 expression through both the Smad and non-Smad signaling pathways (p38 MAPK). As DKK1 and SOST/sclerostin act as Wnt signaling inhibitors, BMP signaling in osteoblasts, in turn, inhibits osteogenesis and decreases bone mass. DKK1 and Sost/sclerostin play an important role in regulating bone mass and mechanical strength as downstream effectors of BMPR1A signaling in bone by taking balances between BMP signaling and Wnt signaling
Similarly, the loss of function of BMP signaling in osteoblasts via ACVR1, another type I receptor, results in increased bone mass [91]. In this mouse model, upregulation of Wnt canonical signaling is observed concomitant with reduction in Dkk1 and Sost expression during embryonic and postnatal bone development [91]. Because the resulting Acvr1 mutant mice show similar bone phenotypes to those found in Bmpr1a mutant mice, despite structural and functional similarities between two receptors, the other does not compensate loss of one receptor.
Sost/sclerostin was originally reported as a member of the BMP antagonist DAN family [111, 214]. Although DAN family members modulate both BMP and Wnt signaling in Xenopus [19, 79, 167], recent studies suggest a primary role of Sost/sclerostin in Wnt signaling in mouse and humans: Sost/sclerostin is not a BMP antagonist [207] but rather a Wnt inhibitor [208] that binds the Wnt co-receptors low-density lipoprotein receptor-related proteins 5 and 6 (LRP5 and LRP6) [123, 183]. It is known that both DKK1 and Sost/sclerostin inhibit Wnt/β-catenin signaling by binding to co-receptors. As both Dkk1 and Sost/sclerostin are secreted proteins expressed by osteoblasts, their role in regulating bone mass has been investigated using human and mouse genetic approaches. Although conventional knockouts of Dkk1 die in utero from defective head induction and limb formation [151], mice heterozygous for Dkk1 (Dkk1 +/− mice) exhibit a high bone mass (HBM) phenotype [150], while overexpression of Dkk1 in osteoblasts causes osteopenia [118]. In addition, increased DKK1 expression in bone marrow has also been associated with lytic bone lesions in patients with multiple myeloma [199].
Similar to Dkk1 +/− mice, conventional knockouts of Sost are viable and exhibit increased bone mass [122]. In humans, the loss of function and hypomorphic mutations in SOST cause sclerosteosis [9, 30] and van Buchem disease [10, 191], respectively, with a high bone mass (HBM) phenotype. These mutants share the HBM phenotypes with other gain of function of LRP5 mutation effects, due to defect in Dkk1-mediated regulation of LRP5 in humans [26, 125, 209] and overexpression of Lrp5 in mice [6]. In contrast, the loss of function of LRP5 leads to OPPG with low bone mass [59], which is similar to the bone phenotype of mice overexpressing Sost [214]. In addition, recent genome-wide SNP-based analyses identified a significant association between bone mineral density and the SOST gene locus [76, 194, 226]. Consistent with these observations, conditional knockouts of Bmpr1a, which show reductions in expressions of Dkk1 and Sost, show an HBM phenotype [92–95]. Furthermore, increased expression of Dkk1 and Sost in osteoblasts by constitutive activation of BMPRIA signaling is associated with a partial rescue of the bone phenotype of Bmpr1a-deficient mice [92]. These facts support the interpretation that Dkk1 and Sost/sclerostin act physiologically as inhibitors of Wnt canonical signaling and therefore as negative regulators of bone mass.
2.3.2 BMP and Chondrocytes
When BMP signaling was enhanced by overexpression of Bmp4 in chondrocytes using a chondrocyte-specific Cre mouse line, the mutant mice demonstrated an increase in bone mass [202]. By contrast, when the BMP signaling was attenuated by overexpression of Noggin, an antagonist for BMPs (BMP2, BMP4, BMP5, BMP6, and BMP7) [236], in chondrocytes, the mutant mice showed a decrease in bone mass [202]. Similarly, the loss of function of BMP signaling via BMPRIA in chondrocytes, which is a potent receptor for BMP2 and BMP4, demonstrated impairment of articular cartilage and growth plate cartilage, resulting in decreased bone size [86, 176, 227]. Mice deficient for Bmpr1a or Bmpr1b in chondrocytes can form intact cartilage during skeletal development, while double mutant embryos deficient for both Bmpr1a and Bmpr1b exhibit a severe defect in cartilage (i.e., chondrodysplasia) around embryonic day 12.5 (E12.5) to E16.5 [227]. These facts suggest a possible functional compensation mechanism between BMPR1A and BMPR1B in chondrocytes during early cartilage development in growth plates [227]. Mice deficient in Acvr1 in chondrocytes using a Col2-Cre-driven conditional deletion are viable but exhibit defects in the development of cranial and axial structures [174]. The mutant mice exhibit shortened cranial base, and cervical vertebrae are hypoplastic. Unlike compound mutant mice for Bmpr1a and Bmpr1b, compound mutant mice for Avcr1 and Bmpr1b can develop cartilage primordia and subsequent bones through endochondral ossification [174], suggesting that BMP signaling through ACVR1 plays a relatively minor role compared with other type 1 receptors during chondrogenesis.
Recent study using aggrecan CreERT2-Cre mice to conditionally disrupt Bmpr1a in chondrocytes demonstrated a severe reduction in bone length and bone mass in the mutant femur at the age of 1 month [86], indicating a more distinct role of BMPR1A in chondrocytes postnatally which is not redundant with other receptors. Note that cell proliferation assessed by BrdU incorporation was strikingly reduced in the mutant mice at 2 weeks of age, which may reduce the size of cartilaginous foundation during the process of endochondral bone formation, leading finally to reduced bone length and mass. Taken together, these facts strongly demonstrate that BMP signal in chondrocytes plays a positive and potent role in regulating bone mass.
2.3.3 BMP and Mesenchymal Cells
Similar to chondrocytes, BMP signaling in mesenchymal cells contributes to an increase in bone mass (Table 2). A mesenchymal cell-specific Cre mouse line, Prx1-Cre, is used for these studies since Cre is active in mesenchymal cells as early as E9.5 in this line [128]. The simultaneous disruption of Bmp2 and Bmp4 in mesenchymal cells resulted in impairment of osteogenesis with reduced bone size [11]. Disruption of Bmp2 in mesenchymal cells impaired the initiation of fracture healing, presumably due to a defect in endochondral bone formation after a bone fracture, in which chondrocytes derived from mesenchymal cells play an important role [201]. These facts demonstrate the necessity of BMP signaling in mesenchymal cells for proper bone mass during development and remodeling. Recently, the role of type 2 receptor, BMPRII, in the skeleton was investigated using the Prx1-Cre mouse line. The resulting mutant mice are expectedly normal probably due to the compensation mechanism by other type 2 receptors, ACVR2A and ACVR2B, suggesting BMPRII is not required for endochondral ossification in the limb [54]. The same group further investigated the mutant mice and found increased bone mass at 2 months after birth [130]. While BMP signal is unchanged, activin signal is impaired in mutant mice, leading to increased osteoblast activity. This study raises the possibility that type 2 receptor segregation and/or competition could be a generalized mechanism by which BMP and activin signaling interact.
2.3.4 BMP and Osteoclasts
A putative coupling theory in bone metabolism states that in general, bone anabolism is locally induced by bone catabolism [71]. Osteoblasts control bone resorption by expressing RANK ligand (RANKL) and its decoy receptor, osteoprotegerin (OPG) [112, 187]. BMPs induce osteoclastogenesis via the RANKL-OPG pathway in an osteoblast-dependent manner. Exogenous treatment of BMP2 in vitro induces osteoclastogenesis by upregulating RANKL while treatment with BMP antagonist Noggin blocks osteoclastogenesis [1, 80, 159, 163]. In vivo studies using genetically engineered mutant mice demonstrated similar results (Table 2). Gain of function of BMP signaling by Bmp4 overexpression in osteoblasts results in an increase of osteoclastogenesis and reduced bone mass [158]. In contrast, the loss of function of BMP signaling by disruption of Bmpr1a or Noggin overexpression results in reduction of osteoclastogenesis, leading to an increase of bone mass [145, 158] due to a decrease in the RANKL-OPG ratio [94, 95]. Taken together, these facts indicate that BMP signal has an indirect positive role in osteoclast function through osteoblast as a secondary effect. In a nonhuman primate bone defect model, treatment with BMP2 increases the size of the defect in association with increased osteoclast number and bone resorption, which is followed by bone formation [182].
In addition, it is also possible that BMPs directly control osteoclasts since Bmp2 and its receptor Bmpr1a both are expressed in osteoclasts [55, 98]. When BMP signaling through BMPR1A is conditionally ablated in osteoclasts using a cathepsin K promoter (CtsK) to drive Cre, bone mass increased in association with reduced osteoclast number in the bone as expected [157] (Table 2). Interestingly, both bone formation rate and osteoblast number assessed by bone histomorphometric analysis are greater in the mutant mice compared to their control littermates. This evidence suggests a possibility that BMPR1A signaling in osteoclasts negatively regulates osteoblast function though its downstream target genes within osteoclasts. Several recent reports have emerged revealing factors secreted by osteoclasts such as sphingosine-1-phosphate regulate osteogenesis [164]. It is an interesting future direction how BMP signaling involves osteoclast-mediated osteoblast differentiation.
2.3.5 BMP and Other Cell Types in Skeletal System
Angiogenesis is another necessary step in new bone formation in skeletal development as well as in bone remodeling after fracture [31, 97]. Both BMP2 and BMP7 are known to induce angiogenesis by associating with other growth factors such as VEGF (vascular endothelial growth factor), bFGF (basic fibroblast growth factor), and TGF-β1 [40]. Overexpression of BMP9 in muscle induces heterotopic bone formation similar to BMP2 [34, 166]. As BMP9 is abundantly expressed in endothelial cells that are a primary cell type for angiogenesis [38], it is possible that BMP signaling in endothelial cells synergizes anabolic bone formation. The mechanism and origin of precursor cells for heterotopic bone formation, which is pathologically observed in fibrodysplasia ossificans progressiva (FOP) patients, is under investigation [96, 129, 229]. Taken together, the fact that BMPs implanted subcutaneously induce ectopic bone and increase bone mass [204] is likely due to the primary effects of BMP signaling on cells that are positive regulators for bone mass, including mesenchymal cells, chondrocytes, and endothelial cells (Table 3).
The current application of BMP therapy via systemic and local treatment can affect multiple cell types simultaneously in bone tissue including mesenchymal cells, chondrocytes, osteocytes, osteoblasts, osteoclasts, and endothelial cells, because typically a BMP2-soaked collagen sponge is applied around bone defects in orthopedic surgeries and the BMP2 diffuses to other tissues around the bone. Thus, it is important to evaluate the effects of BMPs on more than just osteoblasts. In addition to these cell types, we recently investigated the effects of high-dose BMP2 on periosteum and found that high concentration of BMP2 can reduce cell proliferation and increase apoptosis via DKK1 and SOST by inhibiting Wnt activity in human primary periosteal cells [102]. Interestingly, a lower concentration of BMP2 (i.e., 50–200 ng/ml) shows a trend of decreased caspase activity which is opposite to the effect of higher concentrations of BMP2 (500–2000 ng/ml) that shows an increased caspase activity, suggesting a “biphasic nature” of BMP2 depending on its concentration. Note that BMP2 belongs to the TGF-beta superfamily and TGF-beta also has biphasic effects in a concentration-dependent manner with distinct molecular mechanisms [218]. This study is clinically significant because BMP2 is generally applied around the periosteum in orthopedic surgeries for fracture repair and spinal fusion and, therefore, it is important to delineate the effects of the BMP2 concentration on human periosteum-derived cells. In addition, the BMP2 concentration of clinical applications is extremely high (i.e., 1.5 mg/ml [InFUSE Bone Graft/LT-CAGE Lumbar Tapered Fusion Device. Summary of safety and effective data premarket approval application P000058, 2002, US Food and Drug Administration, Sliver Spring, MD, http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cftopic/pma/pma.cfm?num=P000058]), compared with the BMP2 concentration of cell basis studies (i.e., 0~300 ng/ml) as described before. It is possible that the negative role of BMP2 on cell proliferation leads to a reduction in bone mass because the cell proliferation is an initial phase prior to the cell differentiation phase that is required for new bone formation (Table 3).
The potential effects of BMP signal on mesenchymal cells, chondrocytes, and osteoblasts have been discussed. It is possible that chondrocytes or mesenchymal cells increase bone mass by responding to BMPs while osteoblasts or osteocytes reduce net bone mass (Fig. 2). This possibility supports a physiological role of BMPs in endogenous bone formation and remodeling, while the current view that BMPs enhances bone formation reflects a pharmacological role. Apparently, BMP signal has a different function depending on each context (i.e., endogenous vs. exogenous, low dose vs. high dose, chondrocyte vs. osteoblast).

Possible effects of BMP signal induced by BMPs on mesenchymal cells, chondrocytes, osteoblasts, and osteocytes. Based on the recent progresses shown in Tables 2 and 3, it is possible that BMP signaling in chondrocytes or mesenchymal cells can function to increase cell proliferation, bone size, mass, density, and mechanical strength while BMP signaling in osteoblasts or osteocytes may have opposite outcomes through regulating balance between bone formation and resorption
2.4 BMP and Bone-Related Diseases
Studies of human mutations also suggest the importance of BMP signaling for skeletogenesis and bone-related diseases such as chondrodysplasia and fibrodysplasia ossificans progressiva [185, 198]. Mutations in genes involving BMP signaling associated with skeletal abnormalities in humans are summarized in Table 4 [5, 8–10, 30, 37, 41, 42, 115, 116, 168, 191, 225]. While the association of each molecule with its skeletal abnormality is known (Table 4), precise molecular mechanisms including tissue source and cell type responsible for the pathogenesis are still under investigation.
3 Craniofacial Development
3.1 Head Induction
Soon after implantation and before gastrulation, one group of cells formed at the distal tip of the visceral endoderm moves along one direction to form the anterior visceral endoderm (AVE). The AVE acts as a signaling center to instruct underneath epiblast (embryonic ectoderm) to form the future head [103, 193]. Nodal signaling plays a critical role for migration of the AVE [44, 222]. BMP signaling mediated by BMPR1A is critical to orient migration of the AVE [147, 221]. Similarly, BMPR1A signaling in epiblast regulates functions in the AVE for head induction [39]. The loss of Bmp4 and Bmp2 affect normal head formation; however, usage of different receptors in this context is not fully understood [32, 216, 233]. These facts suggest that BMP signaling is critical for induction of the head structure around the gastrulation stage and causes of some of craniofacial abnormalities may be traced back to such early stages.
3.2 Facial Development and Abnormalities
Fetuses (by the end of 5 weeks for humans and at 10.5 days of mice) develop the frontonasal prominence (FNP) [154] (Fig. 3). Neural crest cells (NCCs) formed at the dorsal ectodermal midline in vertebrate embryos migrate laterally and ventrally on all axial levels [24]. Cranial neural crest cells (CNCCs) migrate into the FNP and branchial arches and differentiate to most of the facial tissues. The FNP further splits into four processes, a pair of the medial nasal process and a pair of the lateral nasal processes [195, 200] (Fig. 3). The maxillary and mandibular processes are derived from first branchial arch. The face is formed by fusion of these primordial structures, namely, four processes developed from the FNP and the paired maxillary and mandibular processes. Fusion of the two medial nasal processes at the midline provides the continuity of the nose, the middle upper lip, and the primary palate. Fusion of the medial nasal and maxillary prominences provides the continuity of the upper lip and jaw.

Facial development and fusion of facial processes. During early facial development, pairs of the medial nasal processes and the lateral nasal processes are developed from the frontonasal prominences. Pairs of the maxillary process and the mandibular processes are developed from the first and second pharyngeal arches, respectively. Failure of the fusion of these processes causes facial clefts as detailed in the text. BMPs and related molecules play a critical role in the fusion process
3.2.1 BMP and Cleft Lip
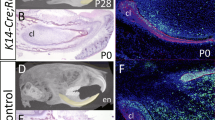
Failure of fusions of any processes will develop facial cleft. For examples, failure of fusion between the medial nasal and the maxillary processes results in uni- or bilateral cleft lip and that between the lateral nasal and the maxillary processes results in oblique facial cleft. The fusion of these processes is critical for formation of the lip and the alveolar ridge in the primary palate. Following closure of the primary palate, closure of the secondary palate takes place by elevation of the palatal shelves. In some cases, these facial clefts occur alone (cleft lip without cleft palate), while other cases, these clefts accompany cleft palate (cleft lip with cleft palate) [45]. Studies in human genetics and animal models reveal several genes involved in development of cleft lips such as mutations in MSX1, tumor protein 63 (TP63), interferon regulatory factor 6 (IRF6), and fibroblast growth factor receptor 1 (FGFR1) [27, 28, 45]. Since MSX1 is one of the established downstream targets of BMP signaling, involvement of BMP signaling during fusion process for lip formation has been speculated. Disruption of Bmpr1a in a dental epithelial-specific manner using Nestin-Cre results in bilateral cleft lip in association with increased apoptosis in the medial nasal processes [126] (Fig. 3).
3.2.2 BMP, Facial Cleft, and Midline Structure
Craniofacial syndromes that include median facial cleft are believed to be caused by dysplasia of the frontonasal prominence [181]. When the fusion between left and right medial nasal processes fails, that likely results in midface clefting [23]. In humans, it is reported that mutations in aristaless-related homeobox transcription factor 3 and 4 (Alx3 and Alx4) are identified in frontonasal dysplasia patients (FND OMIM ID, 136760; FND2 OMIM ID, 613451) [22, 203, 205]. FND is characterized by hypertelorism, severely depressed nasal bridge and ridge, and bifid nasal tip. In the mouse, similar phenotypes are seen in Alx3/Alx4 or Alx1/Alx4 compound mutant mice [23, 169]. A significant increase of apoptosis is detected in the outgrowing frontonasal prominence at E10, which is proposed to be the underlying cause of the subsequent nasal cleft [23]. Potential involvement of BMP signaling in FND is poorly understood. However, it is reported that a gain-of-function mutation in Msx2 causes midface clefting [217]. Neural crest-specific expression of caBmpr1a results in short nasal septum due to increased cell death [67] (Fig. 3). The amount of Hedgehog signaling is known to be strongly associated with alterations in midline facial structures [29, 231]. Since BMP signaling and Hedgehog signaling regulate each other in highly context-dependent manner, it is possible to speculate that BMP signaling may also play a critical role in midline development and failure of precise control of signaling activity may result in medial facial cleft and FND.
There are several evidences indicating that increased BMP signaling leads to a reduction or loss of the midline structure. Noggin mutant mice develop a microform of holoprosencephaly (HPE) [113]. The fact that compound mutations of Noggin and Chordin results in variable forms of HPE [104] suggests that levels of BMP signaling are associated with severity of HPE. It is reported that BMP ligands interact with NODAL, another TGF-beta superfamily ligand, and it is possible that increased availability of BMP ligands because of the loss of their binding antagonists (Noggin and Chordin) secondarily influences NODAL signaling activity that plays a critical role in head formation soon after gastrulation [223, 224]. Alternatively, but not exclusively, it is also possible to speculate that increased BMP signaling activity may suppress Hedgehog activity. In tooth development, BMP signaling has been shown to negatively regulate Hedgehog signaling activity [117]. It is reported that disruption of Shh in mice results in holoprosencephaly and cyclopia [35] and mutations in SHH in human are associated with holoprosencephaly [20, 175, 190]. Thus, there exists a possibility of cross talk with increased BMP signaling activity suppressing Hedgehog signaling leading to midline hypoplasia.
3.2.3 BMP and Cleft Palate
During palatogenesis, first a pair of palatal shelves is formed downward around 7 weeks of gestation in humans and E11.5 in mice with interposition of the tongue. Fetal growth allows downward movement of the tongue to reorient palatal shelves to medial direction around 8 weeks in human and E13–14 in mice. These shelves grow, come closer, and then fuse to separate the oral and nasal cavity by 9 weeks in human and E15.5 in mice [45, 107] (Fig. 4). Thus, failure of fetal growth, movement of tongue reorientation of the palatal shelves, and/or growth of the palatal shelves may result in cleft palate. The final step of palatogenesis is dissolution of the medial edge epithelium (MEE) likely due to the cell death of this population. Persistence of the MEE results in submucosal cleft, i.e., the soft tissue has fused, while underlying palatal bone and muscle layer remain unfused.

Development of the palatal shelf and formation of the secondary palate. During mid-gestation, a pair of palatal shelves are formed from the mandibular processes and grow downwards. Along with the growth of the mandibular, the position of the tongue lowers allowing the shelves elevate. The elevated shelves further grow to reach each other and then fuse together to form the secondary palate. The primary palate is formed anterior to the secondary palate as a derivative of the medial nasal process and the frontonasal prominence
Mice homozygous for Tgfß3 null mutation develop cleft palate demonstrating for the first time that TGFß superfamily signaling plays a critical role in palatogenesis [89]. Tissue-specific inactivation of Tgfbr1, a type 1 receptor for TGFß, using Wnt1-Cre or K14-Cre also results in cleft palate [46, 47]. Involvement of BMP signaling in palatogenesis was initially suggested in a retinoic acid-induced cleft palate model [74, 131], where pathogenesis coincided with downregulation of BMP ligands such as Bmp2, Bmp3, Bmp4, Bmp5, and Bmp7. Msx1-null mice also develop cleft palate [180]. Msx1 is expressed in mesenchymal tissues in anterior palatal shelves, and the loss of Msx1 results in downregulation of Bmp4 [4, 235]. Detailed analyses suggest that in the palatal shelves, BMP4 induces Shh expression that in turn induces Bmp2 expression that positively regulates cell proliferation [4].
Neural crest-specific disruption of Acvr1, one of the type 1 receptors for BMPs, results in cleft palate along with multiple craniofacial defects including a hypomorphic mandible [48]. Neural crest-specific disruption of Bmpr1a results in mid-gestation lethality due to cardiac malfunctions [156, 192]. When the said cardiac malfunction is compensated by administration of isoproterenol, a beta-adrenergic agonist, the mutant embryos can survive until term and develop reduced projection of facial structures [148] and cleft palate [119]. In addition to the cleft lip mentioned above, deletion of Bmpr1a using Nestin-Cre resulted in cleft palate [126]. However, deletion of Bmpr1a in a neural crest-specific manner using Wnt1-Cre resulted in anterior clefting only [119], suggesting that BMP signaling mediated by ACVR1 and BMPR1A positively regulates proliferation of the cells in the anterior palatal shelve mesenchyme.
A BMP antagonist Noggin is highly expressed in the palatal shelf epithelium [138]. Disruption of Noggin results in cleft palate [70] suggesting that increased BMP signaling activity also affect normal palatogenesis. In the anterior regions of the secondary palate, the loss of Noggin results in upregulation of Bmp2 expression leading to an increase of cell proliferation. In the posterior regions of the secondary palate, in contrast, the loss of Noggin induces ectopic expression of Tgfß3 that coincides to ectopic fusion of palatal shelves to epithelia of the oral cavity and tongue [70]. Expression of a constitutively active form of Bmpr1a in the oral epithelium also leads to the similar phenotype [70]. Taken together, these facts suggest that suppression of BMP signaling is critical to prevent premature or ectopic fusions of palatal shelves to maintain structural integrity within the oral cavity. In contrast, expression of a constitutively active form of Acvr1 in the oral epithelium using K14-Cre results in submucosal cleft in association with a reduced cell death in the MEE [155]. These results might suggest that BMP signaling mediated by different receptors plays distinct roles during palatogenesis. Further investigation is required to address this exciting hypothesis.
3.3 Calvarial Vault and Cranial Base
Mammalian craniofacial skeleton consists of a little more than 20 bones. Bones comprising the cranial vault are generated through intramembranous ossification. In contrast, bones in cranial base are generated through endochondral ossification. The majority of cranial bones and cartilage residing in the anterior part of the head are derived from cranial neural crest cells (CNCCs), whereas the posterior part of elements is from paraxial mesoderm [137, 144, 177, 189, 212]. BMP signaling components are highly expressed in the migrating cranial neural crest cells and later in the cranial cartilage and bone [135]. These reports suggest that BMP signaling regulates skeletal development by organizing neural crest cell proliferation and cell death [36]. Both CNCC-derived and paraxial mesoderm-derived osteoprogenitor cells undergo intramembranous ossification to generate corresponding skull elements. Interestingly, osteoblasts from neural crest-derived bones show a higher level of activation of FGF signaling pathways compared with osteoblasts from paraxial mesoderm-derived bones [121, 170]. Osteoblasts from neural crest-derived bones also show lower apoptotic response when stimulated by TGFß signaling [120]. Regenerative ability of skull defects in the frontal bone is higher than that in parietal bones [18]. Taken together, these results suggest that neural crest-derived bones are more proliferative and less apoptotic than paraxial-derived bones due to enhanced signaling of FGF, BMP, and Wnt signaling pathways with a reduction in the TGF-beta pathway [184].
Sutures are a fibrous connective tissue found between bones in the cranial vault and cranial base. Sutures are critical growth sites in the skull. Mesenchymal cells proliferate and differentiate into osteoblasts that deposit collagen fibers and minerals to the bony plates to increase their size. Genetic studies in mice demonstrate that nasal and metopic sutures, which connect nasal bones and frontal bones, are of neural crest origin [83]. Coronal sutures are of mesodermal origin and are formed between the neural crest-derived frontal bones and the mesoderm-derived parietal bones. The sagittal suture is formed between the two mesoderm-derived parietal bones and is of neural crest origin. Since sutures are critical for growth of the skull, premature fusion of sutures results in cessation of skull growth at the site of fusion causing a pathological condition called craniosynostosis resulting in increased intracranial pressure and skull deformity [144, 149, 173].
3.3.1 BMP and Skull Formation
BMP signaling alters the homeobox Msx genes which are important for normal skull development [15, 21, 140]. A conventional gain-of-function mutation in Msx2 results in skeletal defects such as mandibular hypoplasia and aplasia of interparietal bone [217]. BMP signaling plays crucial roles in regulation of cranial suture morphogenesis [101]. BMP signaling components such as Bmp2, Bmp4, Msx1, and Msx2 are expressed in sagittal suture during its development [101]. Local application of BMP4 protein into mouse calvarial explants induces expression of Msx genes and obliteration of the mid-sutural space [101], which is probably through a BMP-responsive element located proximal to the Mxs2 promoter [12]. Both Msx1 and Msx2 mutant mice develop persistent calvarial foramina [78, 179, 180]. Compound heterozygous mutant mice for Msx1 and Msx2 lack formation of the frontal and parietal bones [78]. These results suggest that BMP signaling plays a critical role through expression of Msx1 and Msx2 on osteoblast differentiation for normal skull vault formation.
3.3.2 BMP and Suture Formation
Fibroblast growth factor (FGF) family is known to play critical role during facial development and cranial vault formation [65, 142, 144]. Gain-of-function mutations in FGF signaling are known to cause some types of craniosynostosis [162, 213]. For example, two missense mutations (S252W and P253R) have been found in the IgII-LgIII linker region of FGFR2 and are associated with Apert syndrome [149, 162]. Gain-of-function mutations in MSX2 also result in Boston-type craniosynostosis in human (OMIM ID: 604757) by inducing premature fusion in cranial sutures [82]. Noggin is present in postnatal sutures, and its expression is under negative regulation of FGF signaling. Fgf gain-of-function mutations in syndromic forms of craniosynostosis might inappropriately reduce Noggin expression such that the suture loses its patency [211]. Direct involvement of BMP signaling in skull deformity and craniosynostosis was recently demonstrated. Enhanced BMP signaling through constitutively active form of Bmpr1a (caBmpr1a) in neural crest cells results in craniosynostosis through premature fusion of the anterior frontal suture in mice [95, 105]. Increased BMP signaling in neural crest cells also leads to craniofacial skeletal defects. Constitutive activation of Bmpr1a in neural crest cell linage using P0-Cre or Wnt1-Cre leads to increased level of cell death in skeletal primordia. These mutant mice exhibited bone and cartilage defects of nasomaxillary complex such as nasal bone and nasal septum [60, 67, 105].
In contrasting to neural crest-specific augmentation of BMP signaling activity, osteoblast-specific augmentation of BMPR1A signaling does not cause overt skull deformity [105]. Increased apoptosis is found in the skull vault in this animal model, and the skull deformity is rescued by prevention of cell death by inhibition of p53 together suggesting that augmented BMP signaling increases p53-dependent cell death resulting in depletion of osteogenic progenitor cells leading to premature suture fusion [67, 105]. This is an interesting finding since it is believed that premature fusion of cranial sutures is a result of increased bone formation within the cranial suture [52].
In the craniosynostosis mouse model caused by neural crest-specific augmentation of BMPR1A signaling, it is shown that only a small increase in BMP signaling (50 %) is enough to result in skull deformity and craniosynostosis [105]. It is reasonable to speculate that several folds of changes in BMP signaling may result in early embryonic lethality [53, 75]. Recent genome-wide association studies find single-nucleotide polymorphisms (SNPs) located in proximity to BMP-related genes that are associated with skull morphology such as sagittal suture craniosynostosis [88]. Direct connection between mutations in BMP-related genes and human skull deformity has not been demonstrated; however, gain-of-function mutation in MSX2, a known downstream target gene of BMP signaling, results in Boston-type craniosynostosis as mentioned earlier [82]. Suture mesenchymal cells isolated from craniosynostosis patients show mutations in glypican-1 and glypican-3 (GPC1 and GPC3) that negatively regulate BMP signaling [50]. It is possible to speculate that SNIPs found in proximity of BMP2 may alter enhancer activity to increase BMP signaling in the sagittal suture [88]. Taken together, these circumstantial evidences imply that some human cases may be caused by augmented BMP signaling in suture mesenchymal cells.
3.3.3 BMP and Cranial Base
Unlike calvarial vault, bones in the calvarial base (ethmoid, presphenoid, and basisphenoid) are formed through endochondral ossification. Very little is known about involvement of BMP signaling in the calvarial base. Cartilage structures called synchondrosis connect between the bones in the skull base. The ethmoid bone and the basisphenoid bone are articulated to the frontal and the basioccipital bones, respectively, through synchondrosis. Bmp2, 3, 4, 5, and 6 are expressed in cranial base with a temporally dynamic manner [100]. Development of growth plates in synchondrosis are tightly regulated by SHH and FGF signaling like the ones in long bones [139, 228]. Expression of inhibitor of differentiation 2 (Id2) is regulated in part by BMP-Smad signaling. The mutant mice for Id2 are born without overt abnormalities; however, they show a narrower hypertrophic zone in the synchondrosis postnatally [178].
3.4 Mandibular Development and Temporomandibular Joint Formation
The mandible that forms the lower jaw is unique among bones in the body because it is formed through both intramembranous ossification and endochondral ossification [161] (Fig. 5). The body (or base) of the mandible and the ramus undergo intramembranous ossification; however, processes from these bones such as condyle, coronoid, and symphysis undergo endochondral ossification. The body of the mandible forms along with the Meckel’s cartilage; however, cells in the Meckel’s cartilage do not contribute the body. The body and the ramus form separately then fused together [45]. Cartilage is formed between 10 and 14 weeks in the human fetus at the head of condyle, coronoid, and symphysis. These cartilages are called as secondary cartilage since the cartilage primordia for endochondral ossification are formed at 5 weeks. The endochondral bone growth driven by the condylar cartilage is the most significant contributor to mandibular growth. The condylar process articulates to the temporal bone to form the temporomandibular joint (TMJ) [72, 73].

Mandibular development. A pair of mandibular condensation occurs along with Meckel’s cartilage that forms the body of mandibular. Another pair of condensation forms posteriorly to give rise ramus of mandibular that eventually fused with the body of the mandibular. Secondary cartilage is developed at the tip of the condylar process and participates formation of the temporomandibular joint
3.4.1 BMP and Mandible
Bmp2 and Bmp7 are expressed at early stages of the developing Meckel’s cartilage, while Noggin expression persists and is continuous [210]. Noggin-deficient mice that result in increased pSmad1/pSmad5/pSmad9 develop a significantly thicker Meckel’s cartilage that is later ossified instead of degenerating [210]. In contrast, the growth of Meckel’s cartilage is reduced in Bmp7-deficient mice [108]. This animal model develops small mandible (micrognathia) leading to cleft palate since the palatal shelves can fuse when whole upper jaws are cultured in vitro [108, 237]. Similar skeletal defects are observed when Tak1, a downstream component critical for non-Smad signaling pathway, is disrupted in a neural crest-specific manner and the cleft palate phenotype is rescued when the whole upper jaws are cultured [230]. These suggest that compromised BMP signaling during mandibular development may be one of the causes of the Pierre Robin syndrome [196].
Neural crest-specific disruption of both Bmp2 and Bmp4 using Wnt1-Cre results in mandibular and cranial bone defects in mice [25]. Subsequent analyses demonstrate that BMP signaling is required for self-renewal of cranial neural crest cells, and thus the loss of BMP signaling results in micrognathia and enlarged frontal fontanelle phenotype [25]. Similar skeletal phenotypes are reported in neural crest-specific mutant mice for Acvr1 [48]. In contrast, overexpression of Bmp4 in neural crest cells leads to syngnathia, a rare human bony birth defect manifested by a bony connection between maxilla and mandible [69].
3.4.2 BMP and the Temporomandibular Joint
The temporomandibular joint (TMJ) forms between the condyle process and the temporal bone in the calvarial vault and plays a critical role in jaw movement during chewing and articulating sound while speaking. The secondary cartilage found in the TMJ is different from primary cartilages by the fact that cells in the prechondroblastic layer produce type 1 collagens rather than type 2 collagens [73]. Cells in the prechondroblastic layer are dual potent, i.e., they can differentiate into either cartilage or bone depending on their mechanical environment [58, 133]. Direct transformation of chondrocytes in condylar cartilage into osteoblasts is recently demonstrated in vivo using a lineage tracing technique [87]. Genes affecting growth and differentiation of primary cartilages such as Sox9, Shh, and Pthrp play important roles in normal TMJ development [72, 84]. Neural crest-specific disruption of Bmpr1a results in malformation of TMJ including failure of articular disc separation from a hypoplastic condyle [60]. Similarly, cartilage-specific removal of Bmpr1a also develops chondrodysplastic phenotypes in TMJ, and mandibular condyle growth is significantly compromised [85]. In the global Bmp7 mutant mice, the secondary cartilage does not form at the anterior end of the mandible (symphysis) [106]. In this animal model, condylar cartilage however seems to be developed suggesting that requirement of BMP signaling activity in the secondary cartilage may be different depending on anatomical sites.
4 Perspective and Conclusions
The current review elucidates how BMP signal has multifaceted functions in different cell types, ages, and anatomical sites of bones. Knowledge gained from studies on genetically altered animal models and human genetics demonstrates that functions of BMP signaling are highly context dependent and that alterations of BMP signaling in one tissue type secondarily affect behavior of other tissues. It is noteworthy that levels of BMP2 or BMP7 clinically used for fracture healing are very high compared with endogenous levels of BMPs. The functions of BMPs that we have learned from clinical applications may be better applied to understand pathogenesis of genetically induced and trauma-induced heterotopic ossifications [2, 3, 165, 185]. It is now an established concept that both bone mass and bone quality such as collagen cross-linking and mineral crystallinity are important factors contributing to biomechanical properties of bones [14, 81]. How BMP signaling influences bone quality in addition to bone mass in a physiological condition is an interesting future direction.
References
Abe E, Yamamoto M, Taguchi Y, Lecka-Czernik B, O'Brien CA, Economides AN, Stahl N, Jilka RL, Manolagas SC (2000) Essential requirement of BMPs-2/4 for both osteoblast and osteoclast formation in murine bone marrow cultures from adult mice: antagonism by noggin. J Bone Miner Res 15:663–673
Agarwal S, Loder S, Brownley C, Cholok D, Mangiavini L, Li J, Breuler C, Sung HH, Li S, Ranganathan K et al (2016) Inhibition of Hif1alpha prevents both trauma-induced and genetic heterotopic ossification. Proc Natl Acad Sci U S A 113(3):E338–E347
Agarwal S, Loder SJ, Brownley C, Eboda O, Peterson JR, Hayano S, Wu B, Zhao B, Kaartinen V, Wong VC et al (2015) BMP signaling mediated by constitutively active Activin type 1 receptor (ACVR1) results in ectopic bone formation localized to distal extremity joints. Dev Biol 400:202–209
Alappat S, Zhang ZY, Chen YP (2003) Msx homeobox gene family and craniofacial development. Cell Res 13:429–442
Asai-Coakwell M, French CR, Ye M, Garcha K, Bigot K, Perera AG, Staehling-Hampton K, Mema SC, Chanda B, Mushegian A et al (2009) Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Hum Mol Genet 18:1110–1121
Babij P, Zhao W, Small C, Kharode Y, Yaworsky PJ, Bouxsein ML, Reddy PS, Bodine PV, Robinson JA, Bhat B et al (2003) High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res 18:960–974
Bain G, Muller T, Wang X, Papkoff J (2003) Activated beta-catenin induces osteoblast differentiation of C3H10T1/2 cells and participates in BMP2 mediated signal transduction. Biochem Biophys Res Commun 301:84–91
Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Martin A, Williams S, Lindley V et al (2008) Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet 82:304–319
Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P et al (2001) Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10:537–543
Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ et al (2002) Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39:91–97
Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ (2006) Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet 2:e216
Bandyopadhyay A, Yadav PS, Prashar P (2013) BMP signaling in development and diseases: a pharmacological perspective. Biochem Pharmacol 85:857–864
Banerjee C, Javed A, Choi JY, Green J, Rosen V, van Wijnen AJ, Stein JL, Lian JB, Stein GS (2001) Differential regulation of the two principal Runx2/Cbfa1 n-terminal isoforms in response to bone morphogenetic protein-2 during development of the osteoblast phenotype. Endocrinology 142:4026–4039
Banse X (2002) When density fails to predict bone strength. Acta Orthop Scand Suppl 73:1–57
Barlow AJ, Francis-West PH (1997) Ectopic application of recombinant BMP-2 and BMP-4 can change patterning of developing chick facial primordia. Development 124:391–398
Baron R, Rawadi G, Roman-Roman S (2006) Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol 76:103–127
Barrow JR, Thomas KR, Boussadia-Zahui O, Moore R, Kemler R, Capecchi MR, McMahon AP (2003) Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev 17:394–409
Behr B, Panetta NJ, Longaker MT, Quarto N (2010) Different endogenous threshold levels of Fibroblast Growth Factor-ligands determine the healing potential of frontal and parietal bones. Bone 47:281–294
Bell E, Munoz-Sanjuan I, Altmann CR, Vonica A, Brivanlou AH (2003) Cell fate specification and competence by Coco, a maternal BMP, TGFbeta and Wnt inhibitor. Development 130:1381–1389
Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV et al (1996) Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet 14:353–356
Bennett JH, Hunt P, Thorogood P (1995) Bone morphogenetic protein-2 and −4 expression during murine orofacial development. Arch Oral Biol 40:847–854
Bertola DR, Rodrigues MG, Quaio CR, Kim CA, Passos-Bueno MR (2013) Vertical transmission of a frontonasal phenotype caused by a novel ALX4 mutation. Am J Med Genet A 161A:600–604
Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F (2001) Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development 128:3975–3986
Bhatt S, Diaz R, Trainor PA (2013) Signals and switches in Mammalian neural crest cell differentiation. Cold Spring Harb Perspect Biol 5(2)
Bonilla-Claudio M, Wang J, Bai Y, Klysik E, Selever J, Martin JF (2012) Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development 139:709–719
Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP (2002) High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 346:1513–1521
Brown JM, Robertson KE, Wedden SE, Tickle C (1997) Alterations in Msx 1 and Msx 2 expression correlate with inhibition of outgrowth of chick facial primordia induced by retinoic acid. Anat Embryol (Berl) 195:203–207
Brown JM, Wedden SE, Millburn GH, Robson LG, Hill RE, Davidson DR, Tickle C (1993) Experimental analysis of the control of expression of the homeobox-gene Msx-1 in the developing limb and face. Development 119:41–48
Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA (2010) A primary cilia-dependent etiology for midline facial disorders. Hum Mol Genet 19:1577–1592
Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y et al (2001) Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 68:577–589
Carano RA, Filvaroff EH (2003) Angiogenesis and bone repair. Drug Discov Today 8:980–989
Castranio T, Mishina Y (2009) Bmp2 is required for cephalic neural tube closure in the mouse. Dev Dyn 238:110–122
Chen Y, Whetstone HC, Youn A, Nadesan P, Chow EC, Lin AC, Alman BA (2007) Beta-catenin signaling pathway is crucial for bone morphogenetic protein 2 to induce new bone formation. J Biol Chem 282:526–533
Cheng H, Jiang W, Phillips FM, Haydon RC, Peng Y, Zhou L, Luu HH, An N, Breyer B, Vanichakarn P et al (2003) Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J Bone Joint Surg Am 85-A:1544–1552
Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383:407–413
Creuzet S, Schuler B, Couly G, Le Douarin NM (2004) Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proc Natl Acad Sci U S A 101:4843–4847
Dathe K, Kjaer KW, Brehm A, Meinecke P, Nurnberg P, Neto JC, Brunoni D, Tommerup N, Ott CE, Klopocki E et al (2009) Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet 84:483–492
David L, Feige JJ, Bailly S (2009) Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev 20:203–212
Davis S, Miura S, Hill C, Mishina Y, Klingensmith J (2004) BMP receptor IA is required in the mammalian embryo for endodermal morphogenesis and ectodermal patterning. Dev Biol 270:47–63
Deckers MM, van Bezooijen RL, van der Horst G, Hoogendam J, van Der Bent C, Papapoulos SE, Lowik CW (2002) Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology 143:1545–1553
Degenkolbe E, Konig J, Zimmer J, Walther M, Reissner C, Nickel J, Ploger F, Raspopovic J, Sharpe J, Dathe K et al (2014) A GDF5 point mutation strikes twice--causing BDA1 and SYNS2. PLoS Genet 9:e1003846
Demirhan O, Turkmen S, Schwabe GC, Soyupak S, Akgul E, Tastemir D, Karahan D, Mundlos S, Lehmann K (2005) A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. J Med Genet 42:314–317
Denker AE, Haas AR, Nicoll SB, Tuan RS (1999) Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: I. Stimulation by bone morphogenetic protein-2 in high-density micromass cultures. Differentiation 64:67–76
Ding J, Yang L, Yan YT, Chen A, Desai N, Wynshaw-Boris A, Shen MM (1998) Cripto is required for correct orientation of the anterior-posterior axis in the mouse embryo. Nature 395:702–707
Dixon MJ, Marazita ML, Beaty TH, Murray JC (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet 12:167–178
Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V (2006) Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev Biol 296:298–314
Dudas M, Nagy A, Laping NJ, Moustakas A, Kaartinen V (2004) Tgf-beta3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev Biol 266:96–108
Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V (2004) Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev 121:173–182
Dudley AT, Lyons KM, Robertson EJ (1995) A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev 9:2795–2807
Dwivedi PP, Grose RH, Filmus J, Hii CS, Xian CJ, Anderson PJ, Powell BC (2013) Regulation of bone morphogenetic protein signalling and cranial osteogenesis by Gpc1 and Gpc3. Bone 55:367–376
Franz-Odendaal TA (2011) Induction and patterning of intramembranous bone. Front Biosci (Landmark Ed) 16:2734–2746
Fromigue O, Modrowski D, Marie PJ (2005) Apoptosis in membranous bone formation: role of fibroblast growth factor and bone morphogenetic protein signaling. Crit Rev Eukaryot Gene Expr 15:75–92
Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, Yamada M, Mishina Y (2006) Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 44:159–167
Gamer LW, Tsuji K, Cox K, Capelo LP, Lowery J, Beppu H, Rosen V (2011) BMPR-II is dispensable for formation of the limb skeleton. Genesis 49:719–724
Garimella R, Tague SE, Zhang J, Belibi F, Nahar N, Sun BH, Insogna K, Wang J, Anderson HC (2008) Expression and synthesis of bone morphogenetic proteins by osteoclasts: a possible path to anabolic bone remodeling. J Histochem Cytochem 56:569–577
Garrison KR, Donell S, Ryder J, Shemilt I, Mugford M, Harvey I, Song F (2007) Clinical effectiveness and cost-effectiveness of bone morphogenetic proteins in the non-healing of fractures and spinal fusion: a systematic review. Health Technol Assess 11:1–168
Glass DA 2nd, Karsenty G (2006) Molecular bases of the regulation of bone remodeling by the canonical Wnt signaling pathway. Curr Top Dev Biol 73:43–84
Glineburg RW, Laskin DM, Blaustein DI (1982) The effects of immobilization on the primate temporomandibular joint: a histologic and histochemical study. J Oral Maxillofac Surg 40:3–8
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D et al (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107:513–523
Gu S, Wu W, Liu C, Yang L, Sun C, Ye W, Li X, Chen J, Long F, Chen Y (2014) BMPRIA mediated signaling is essential for temporomandibular joint development in mice. PLoS One 9:e101000
Gu Z, Reynolds EM, Song J, Lei H, Feijen A, Yu L, He W, MacLaughlin DT, van den Eijnden-van Raaij J, Donahoe PK et al (1999) The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development 126:2551–2561
Gupta MC, Khan SN (2005) Application of bone morphogenetic proteins in spinal fusion. Cytokine Growth Factor Rev 16:347–355
Harada S, Rodan GA (2003) Control of osteoblast function and regulation of bone mass. Nature 423:349–355
Hartmann C (2006) A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol 16:151–158
Hatch NE (2010) FGF signaling in craniofacial biological control and pathological craniofacial development. Crit Rev Eukaryot Gene Expr 20:295–311
Hatta T, Konishi H, Katoh E, Natsume T, Ueno N, Kobayashi Y, Yamazaki T (2000) Identification of the ligand-binding site of the BMP type IA receptor for BMP-4. Biopolymers 55:399–406
Hayano S, Komatsu Y, Pan H, Mishina Y (2015) Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development 142:1357–1367
Hays E, Schmidt J, Chandar N (2009) Beta-catenin is not activated by downregulation of PTEN in osteoblasts. In Vitro Cell Dev Biol Anim 45:361–370
He F, Hu X, Xiong W, Li L, Lin L, Shen B, Yang L, Gu S, Zhang Y, Chen Y (2014) Directed Bmp4 expression in neural crest cells generates a genetic model for the rare human bony syngnathia birth defect. Dev Biol 391:170–181
He F, Xiong W, Wang Y, Matsui M, Yu X, Chai Y, Klingensmith J, Chen Y (2010) Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev Biol 347:109–121
Henriksen K, Neutzsky-Wulff AV, Bonewald LF, Karsdal MA (2009) Local communication on and within bone controls bone remodeling. Bone 44:1026–1033
Hinton RJ (2014) Genes that regulate morphogenesis and growth of the temporomandibular joint: a review. Dev Dyn 243:864–874
Hinton RJ, Jing J, Feng JQ (2015) Genetic influences on temporomandibular joint development and growth. Curr Top Dev Biol 115:85–109
Ho CT, Lau TY, Jin Y, Lu HB, Liong E, Leung KM, Tipoe GL (2004) Overexpression of iNOS and down-regulation of BMPs-2, 4 and 7 in retinoic acid induced cleft palate formation. Histol Histopathol 19:95–104
Hu Q, Ueno N, Behringer RR (2004) Restriction of BMP4 activity domains in the developing neural tube of the mouse embryo. EMBO Rep 5:734–739
Huang QY, Li GH, Kung AW (2009) The −9247 T/C polymorphism in the SOST upstream regulatory region that potentially affects C/EBPalpha and FOXA1 binding is associated with osteoporosis. Bone 45:289–294
Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W (2001) beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 105:533–545
Ishii M, Merrill AE, Chan YS, Gitelman I, Rice DP, Sucov HM, Maxson RE Jr (2003) Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development 130:6131–6142
Itasaki N, Jones CM, Mercurio S, Rowe A, Domingos PM, Smith JC, Krumlauf R (2003) Wise, a context-dependent activator and inhibitor of Wnt signalling. Development 130:4295–4305
Itoh K, Udagawa N, Katagiri T, Iemura S, Ueno N, Yasuda H, Higashio K, Quinn JM, Gillespie MT, Martin TJ et al (2001) Bone morphogenetic protein 2 stimulates osteoclast differentiation and survival supported by receptor activator of nuclear factor-kappaB ligand. Endocrinology 142:3656–3662
Iura A, McNerny EG, Zhang Y, Kamiya N, Tantillo M, Lynch M, Kohn DH, Mishina Y (2015) Mechanical Loading Synergistically Increases Trabecular Bone Volume and Improves Mechanical Properties in the Mouse when BMP Signaling Is Specifically Ablated in Osteoblasts. PLoS One 10:e0141345
Jabs EW, Muller U, Li X, Ma L, Luo W, Haworth IS, Klisak I, Sparkes R, Warman ML, Mulliken JB et al (1993) A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 75:443–450
Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM (2002) Tissue origins and interactions in the mammalian skull vault. Dev Biol 241:106–116
Jing J, Hinton RJ, Feng JQ (2015) Bmpr1a signaling in cartilage development and endochondral bone formation. Vitam Horm 99:273–291
Jing J, Hinton RJ, Mishina Y, Liu Y, Zhou X, Feng JQ (2014) Critical role of Bmpr1a in mandibular condyle growth. Connect Tissue Res 55(Suppl 1):73–78
Jing J, Ren Y, Zong Z, Liu C, Kamiya N, Mishina Y, Liu Y, Zhou X, Feng JQ (2013) BMP receptor 1 A determines the cell fate of the postnatal growth plate. Int J Biol Sci 9:895–906
Jing Y, Zhou X, Han X, Jing J, von der Mark K, Wang J, de Crombrugghe B, Hinton RJ, Feng JQ (2015) Chondrocytes directly transform into bone cells in mandibular condyle growth. J Dent Res 94:1668–1675
Justice CM, Yagnik G, Kim Y, Peter I, Jabs EW, Erazo M, Ye X, Ainehsazan E, Shi L, Cunningham ML et al (2012) A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet 44:1360–1364
Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J (1995) Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 11:415–421
Kamiya N, Jikko A, Kimata K, Damsky C, Shimizu K, Watanabe H (2002) Establishment of a novel chondrocytic cell line N1511 derived from p53-null mice. J Bone Miner Res 17:1832–1842
Kamiya N, Kaartinen VM, Mishina Y (2011) Loss-of-function of ACVR1 in osteoblasts increases bone mass and activates canonical Wnt signaling through suppression of Wnt inhibitors SOST and DKK1. Biochem Biophys Res Commun 414:326–330
Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM, Mishina Y (2010) Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J Bone Miner Res 25:200–210
Kamiya N, Shuxian L, Yamaguchi R, Phipps M, Aruwajoye O, Adapala NS, Yuan H, Kim HK, Feng JQ (2016 ) Targeted disruption of BMP signaling through type IA receptor (BMPR1A) in osteocyte suppresses SOST and RANKL, leading to dramatic increase in bone mass, bone mineral density and mechanical strength. Bone 91:53–63. doi: 10.1016/j.bone.2016.07.002. Epub 2016 Jul 8. PMID: 27402532
Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y (2008) Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res 23:2007–2017
Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y (2008) BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 135:3801–3811
Kan L, Hu M, Gomes WA, Kessler JA (2004) Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol 165:1107–1115
Kanczler JM, Oreffo RO (2008) Osteogenesis and angiogenesis: the potential for engineering bone. Eur Cell Mater 15:100–114
Kaneko H, Arakawa T, Mano H, Kaneda T, Ogasawara A, Nakagawa M, Toyama Y, Yabe Y, Kumegawa M, Hakeda Y (2000) Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)-2 and expression of BMP receptors in mature osteoclasts. Bone 27:479–486
Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawa-Sehara A, Suda T (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol 127:1755–1766
Kettunen P, Nie X, Kvinnsland IH, Luukko K (2006) Histological development and dynamic expression of Bmp2-6 mRNAs in the embryonic and postnatal mouse cranial base. Anat Rec A Discov Mol Cell Evol Biol 288:1250–1258
Kim HJ, Rice DP, Kettunen PJ, Thesleff I (1998) FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 125:1241–1251
Kim HK, Oxendine I, Kamiya N (2013) High-concentration of BMP2 reduces cell proliferation and increases apoptosis via DKK1 and SOST in human primary periosteal cells. Bone 54:141–150
Kishigami S, Mishina Y (2005) BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev 16:265–278
Klingensmith J, Matsui M, Yang YP, Anderson RM (2010) Roles of bone morphogenetic protein signaling and its antagonism in holoprosencephaly. Am J Med Genet C Semin Med Genet 154C:43–51
Komatsu Y, Yu PB, Kamiya N, Pan H, Fukuda T, Scott GJ, Ray MK, Yamamura K, Mishina Y (2013) Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J Bone Miner Res 28:1422–1433
Kouskoura T, El Fersioui Y, Angelini M, Graf D, Katsaros C, Chiquet M (2016) Dislocated tongue muscle attachment and cleft palate formation. J Dent Res 95(4):453–459
Kouskoura T, Fragou N, Alexiou M, John N, Sommer L, Graf D, Katsaros C, Mitsiadis TA (2011) The genetic basis of craniofacial and dental abnormalities. Schweiz Monatsschr Zahnmed 121:636–646
Kouskoura T, Kozlova A, Alexiou M, Blumer S, Zouvelou V, Katsaros C, Chiquet M, Mitsiadis TA, Graf D (2013) The etiology of cleft palate formation in BMP7-deficient mice. PLoS One 8:e59463
Krishnan V, Bryant HU, Macdougald OA (2006) Regulation of bone mass by Wnt signaling. J Clin Invest 116:1202–1209
Kronenberg HM (2003) Developmental regulation of the growth plate. Nature 423:332–336
Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N (2003) Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem 278:24113–24117
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S et al (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176
Lana-Elola E, Tylzanowski P, Takatalo M, Alakurtti K, Veistinen L, Mitsiadis TA, Graf D, Rice R, Luyten FP, Rice DP (2011) Noggin null allele mice exhibit a microform of holoprosencephaly. Hum Mol Genet 20:4005–4015
Lee MH, Javed A, Kim HJ, Shin HI, Gutierrez S, Choi JY, Rosen V, Stein JL, van Wijnen AJ, Stein GS et al (1999) Transient upregulation of CBFA1 in response to bone morphogenetic protein-2 and transforming growth factor beta1 in C2C12 myogenic cells coincides with suppression of the myogenic phenotype but is not sufficient for osteoblast differentiation. J Cell Biochem 73:114–125
Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, Kjaergaard S, Mahoney MJ, Morlot S, Reissner C et al (2007) A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet 81:388–396
Lehmann K, Seemann P, Stricker S, Sammar M, Meyer B, Suring K, Majewski F, Tinschert S, Grzeschik KH, Muller D et al (2003) Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci U S A 100:12277–12282
Li J, Feng J, Liu Y, Ho TV, Grimes W, Ho HA, Park S, Wang S, Chai Y (2015) BMP-SHH signaling network controls epithelial stem cell fate via regulation of its niche in the developing tooth. Dev Cell 33:125–135
Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W et al (2006) Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 39:754–766
Li L, Lin M, Wang Y, Cserjesi P, Chen Z, Chen Y (2011) BmprIa is required in mesenchymal tissue and has limited redundant function with BmprIb in tooth and palate development. Dev Biol 349:451–461
Li S, Meyer NP, Quarto N, Longaker MT (2013) Integration of multiple signaling regulates through apoptosis the differential osteogenic potential of neural crest-derived and mesoderm-derived Osteoblasts. PLoS One 8:e58610
Li S, Quarto N, Longaker MT (2010) Activation of FGF signaling mediates proliferative and osteogenic differences between neural crest derived frontal and mesoderm parietal derived bone. PLoS One 5:e14033
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, Kurahara C, Gao Y, Cao J, Gong J et al (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23:860–869
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 280:19883–19887
Lim J, Shi Y, Karner CM, Lee SY, Lee WC, He G, Long F (2016) Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in the mouse. Development 143(2):339–347
Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B et al (2002) A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 70:11–19
Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF (2005) Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132:1453–1461
Liu Z, Tang Y, Qiu T, Cao X, Clemens TL (2006) A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J Biol Chem 281:17156–17163
Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ (2002) Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33:77–80
Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS (2009) Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 91:652–663
Lowery JW, Intini G, Gamer L, Lotinun S, Salazar VS, Ote S, Cox K, Baron R, Rosen V (2015) Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J Cell Sci 128:1308–1315
Lu H, Jin Y, Tipoe GL (2000) Alteration in the expression of bone morphogenetic protein-2,3,4,5 mRNA during pathogenesis of cleft palate in BALB/c mice. Arch Oral Biol 45:133–140
Luo G, Hofmann C, Bronckers AL, Sohocki M, Bradley A, Karsenty G (1995) BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev 9:2808–2820
Lydiatt DD, Davis LF (1985) The effects of immobilization on the rabbit temporomandibular joint. J Oral Maxillofac Surg 43:188–193
Lyons KM, Pelton RW, Hogan BL (1990) Organogenesis and pattern formation in the mouse: RNA distribution patterns suggest a role for bone morphogenetic protein-2 A (BMP-2 A). Development 109:833–844
Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM (2003) Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development 130:6361–6374
Macias-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL (1998) Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J Biol Chem 273:25628–25636
Manzanares MC, Goret-Nicaise M, Dhem A (1988) Metopic sutural closure in the human skull. J Anat 161:203–215
Matsui M, Klingensmith J (2014) Multiple tissue-specific requirements for the BMP antagonist Noggin in development of the mammalian craniofacial skeleton. Dev Biol 392:168–181
Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bukulmez H, Balmes G, Krejci P, Mekikian PB, Otani K, Yamaura I et al (2009) FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet 18:227–240
Maxson R, Ishii M (2008) The Bmp pathway in skull vault development. Front Oral Biol 12:197–208
Mbalaviele G, Sheikh S, Stains JP, Salazar VS, Cheng SL, Chen D, Civitelli R (2005) Beta-catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J Cell Biochem 94:403–418
Miraoui H, Ringe J, Haupl T, Marie PJ (2010) Increased EFG- and PDGFalpha-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum Mol Genet 19:1678–1689
Mishina Y, Crombie R, Bradley A, Behringer RR (1999) Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Dev Biol 213:314–326
Mishina Y, Snider TN (2014) Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp Cell Res 325:138–147
Mishina Y, Starbuck MW, Gentile MA, Fukuda T, Kasparcova V, Seedor JG, Hanks MC, Amling M, Pinero GJ, Harada S et al (2004) Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem 279:27560–27566
Mishina Y, Suzuki A, Ueno N, Behringer RR (1995) Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev 9:3027–3037
Miura S, Singh AP, Mishina Y (2010) Bmpr1a is required for proper migration of the AVE through regulation of Dkk1 expression in the pre-streak mouse embryo. Dev Biol 341:246–254
Morikawa Y, Zehir A, Maska E, Deng C, Schneider MD, Mishina Y, Cserjesi P (2009) BMP signaling regulates sympathetic nervous system development through Smad4-dependent and -independent pathways. Development 136:3575–3584
Morriss-Kay GM, Wilkie AO (2005) Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat 207:637–653
Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P et al (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21:934–945
Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, Chen L, Tsukui T, Gomer L, Dorward DW, Glinka A, Grinberg A, Huang SP et al (2001) Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell 1:423–434
Nakashima A, Katagiri T, Tamura M (2005) Cross-talk between Wnt and bone morphogenetic protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J Biol Chem 280:37660–37668
Nakashima T1, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17(10):1231–4. doi: 10.1038/nm.2452
Neskey D, Eloy JA, Casiano RR (2009) Nasal, septal, and turbinate anatomy and embryology. Otolaryngol Clin North Am 42(193–205):vii
Noda K, Mishina Y, Komatsu Y (2016) Constitutively active mutation of ACVR1 in oral epithelium causes submucous cleft palate in mice. Dev Biol 415(2):306–313
Nomura-Kitabayashi A, Phoon CK, Kishigami S, Rosenthal J, Yamauchi Y, Abe K, Yamamura K, Samtani R, Lo CW, Mishina Y (2009) Outflow tract cushions perform a critical valve-like function in the early embryonic heart requiring BMPRIA-mediated signaling in cardiac neural crest. Am J Physiol Heart Circ Physiol 297:H1617–H1628
Okamoto M, Murai J, Imai Y, Ikegami D, Kamiya N, Kato S, Mishina Y, Yoshikawa H, Tsumaki N (2011) Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J Bone Miner Res 26:2511–2522
Okamoto M, Murai J, Yoshikawa H, Tsumaki N (2006) Bone morphogenetic proteins in bone stimulate osteoclasts and osteoblasts during bone development. J Bone Miner Res 21:1022–1033
Otsuka E, Notoya M, Hagiwara H (2003) Treatment of myoblastic C2C12 cells with BMP-2 stimulates vitamin D-induced formation of osteoclasts. Calcif Tissue Int 73:72–77
Pan Q, Yu Y, Chen Q, Li C, Wu H, Wan Y, Ma J, Sun F (2008) Sox9, a key transcription factor of bone morphogenetic protein-2-induced chondrogenesis, is activated through BMP pathway and a CCAAT box in the proximal promoter. J Cell Physiol 217:228–241
Parada C, Chai Y (2015) Mandible and tongue development. Curr Top Dev Biol 115:31–58
Passos-Bueno MR, Serti Eacute AE, Jehee FS, Fanganiello R, Yeh E (2008) Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol 12:107–143
Paul S, Lee JC, Yeh LC (2009) A comparative study on BMP-induced osteoclastogenesis and osteoblastogenesis in primary cultures of adult rat bone marrow cells. Growth Factors 27:121–131
Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ (2008) Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A 105:20764–20769
Peterson JR, De La Rosa S, Eboda O, Cilwa KE, Agarwal S, Buchman SR, Cederna PS, Xi C, Morris MD, Herndon DN et al (2014) Treatment of heterotopic ossification through remote ATP hydrolysis. Sci Transl Med 6:255ra132
Phillips FM, Bolt PM, He TC, Haydon RC (2005) Gene therapy for spinal fusion. Spine J 5:250S–258S
Piccolo S, Agius E, Leyns L, Bhattacharyya S, Grunz H, Bouwmeester T, De Robertis EM (1999) The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature 397:707–710
Polinkovsky A, Robin NH, Thomas JT, Irons M, Lynn A, Goodman FR, Reardon W, Kant SG, Brunner HG, van der Burgt I et al (1997) Mutations in CDMP1 cause autosomal dominant brachydactyly type C. Nat Genet 17:18–19
Qu S, Tucker SC, Zhao Q, deCrombrugghe B, Wisdom R (1999) Physical and genetic interactions between Alx4 and Cart1. Development 126:359–369
Quarto N, Wan DC, Kwan MD, Panetta NJ, Li S, Longaker MT (2010) Origin matters: differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones. J Bone Miner Res 25:1680–1694
Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S (2003) BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J Bone Miner Res 18:1842–1853
Retting KN, Song B, Yoon BS, Lyons KM (2009) BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136:1093–1104
Rice DP, Rice R, Thesleff I (2003) Molecular mechanisms in calvarial bone and suture development, and their relation to craniosynostosis. Eur J Orthod 25:139–148
Rigueur D, Brugger S, Anbarchian T, Kim JK, Lee YJ, Lyons K (2015) The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J Bone Miner Res 30(4):733–741
Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M (1996) Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 14:357–360
Rountree RB, Schoor M, Chen H, Marks ME, Harley V, Mishina Y, Kingsley DM (2004) BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol 2:e355
Sahar DE, Longaker MT, Quarto N (2005) Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev Biol 280:344–361
Sakata-Goto T, Takahashi K, Kiso H, Huang B, Tsukamoto H, Takemoto M, Hayashi T, Sugai M, Nakamura T, Yokota Y et al (2012) Id2 controls chondrogenesis acting downstream of BMP signaling during maxillary morphogenesis. Bone 50:69–78
Satokata I, Ma L, Ohshima H, Bei M, Woo I, Nishizawa K, Maeda T, Takano Y, Uchiyama M, Heaney S et al (2000) Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat Genet 24:391–395
Satokata I, Maas R (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6:348–356
Sedano HO, Cohen MM Jr, Jirasek J, Gorlin RJ (1970) Frontonasal dysplasia. J Pediatr 76:906–913
Seeherman HJ, Li XJ, Bouxsein ML, Wozney JM (2010) rhBMP-2 induces transient bone resorption followed by bone formation in a nonhuman primate core-defect model. J Bone Joint Surg Am 92:411–426
Semenov M, Tamai K, He X (2005) SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 280:26770–26775
Senarath-Yapa K, Li S, Meyer NP, Longaker MT, Quarto N (2013) Integration of multiple signaling pathways determines differences in the osteogenic potential and tissue regeneration of neural crest-derived and mesoderm-derived calvarial bones. Int J Mol Sci 14:5978–5997
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Shukunami C, Ohta Y, Sakuda M, Hiraki Y (1998) Sequential progression of the differentiation program by bone morphogenetic protein-2 in chondrogenic cell line ATDC5. Exp Cell Res 241:1–11
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Simpson AH, Mills L, Noble B (2006) The role of growth factors and related agents in accelerating fracture healing. J Bone Joint Surg Br 88:701–705
Snider TN, Mishina Y (2015) Cranial neural crest cell contribution to craniofacial formation, pathology, and future directions in tissue engineering. Birth Defects Res C Embryo Today 102:324–332
Solomon BD, Bear KA, Wyllie A, Keaton AA, Dubourg C, David V, Mercier S, Odent S, Hehr U, Paulussen A et al (2012) Genotypic and phenotypic analysis of 396 individuals with mutations in Sonic Hedgehog. J Med Genet 49:473–479
Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P et al (2002) A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 110:144–152
Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J (2004) BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development 131:2205–2218
Stower MJ, Srinivas S (2014) Heading forwards: anterior visceral endoderm migration in patterning the mouse embryo. Philos Trans R Soc Lond B Biol Sci 369:20130546
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Snorradottir S, Center JR et al (2009) New sequence variants associated with bone mineral density. Nat Genet 41:15–17
Sulik KK, Johnston MC (1982) Embryonic origin of holoprosencephaly: interrelationship of the developing brain and face. Scan Electron Microsc (Pt 1):309–322
Tan TY, Kilpatrick N, Farlie PG (2013) Developmental and genetic perspectives on Pierre Robin sequence. Am J Med Genet C Semin Med Genet 163C:295–305
Tan X, Weng T, Zhang J, Wang J, Li W, Wan H, Lan Y, Cheng X, Hou N, Liu H et al (2007) Smad4 is required for maintaining normal murine postnatal bone homeostasis. J Cell Sci 120:2162–2170
Thomas JT, Lin K, Nandedkar M, Camargo M, Cervenka J, Luyten FP (1996) A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet 12:315–317
Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD Jr (2003) The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 349:2483–2494
Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR (1999) Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev 13:3136–3148
Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V (2006) BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet 38:1424–1429
Tsumaki N, Nakase T, Miyaji T, Kakiuchi M, Kimura T, Ochi T, Yoshikawa H (2002) Bone morphogenetic protein signals are required for cartilage formation and differently regulate joint development during skeletogenesis. J Bone Miner Res 17:898–906
Twigg SR, Versnel SL, Nurnberg G, Lees MM, Bhat M, Hammond P, Hennekam RC, Hoogeboom AJ, Hurst JA, Johnson D et al (2009) Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet 84:698–705
Urist MR (1965) Bone: formation by autoinduction. Science 150:893–899
Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N et al (2010) Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet 86:789–796
van Baardewijk LJ, van der Ende J, Lissenberg-Thunnissen S, Romijn LM, Hawinkels LJ, Sier CF, Schipper IB (2013) Circulating bone morphogenetic protein levels and delayed fracture healing. Int Orthop 37:523–527
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199:805–814
van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, Quax PH, Vrieling H, Papapoulos SE, ten Dijke P et al (2007) Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res 22:19–28
Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y et al (2003) Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet 72:763–771
Wang Y, Zheng Y, Chen D, Chen Y (2013) Enhanced BMP signaling prevents degeneration and leads to endochondral ossification of Meckel's cartilage in mice. Dev Biol 381:301–311
Warren SM, Brunet LJ, Harland RM, Economides AN, Longaker MT (2003) The BMP antagonist noggin regulates cranial suture fusion. Nature 422:625–629
Warren SM, Greenwald JA, Spector JA, Bouletreau P, Mehrara BJ, Longaker MT (2001) New developments in cranial suture research. Plast Reconstr Surg 107:523–540
Wilkie AO, Morriss-Kay GM (2001) Genetics of craniofacial development and malformation. Nat Rev Genet 2:458–468
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K et al (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22:6267–6276
Winkler DG, Sutherland MS, Ojala E, Turcott E, Geoghegan JC, Shpektor D, Skonier JE, Yu C, Latham JA (2005) Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem 280:2498–2502
Winnier G, Blessing M, Labosky PA, Hogan BL (1995) Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev 9:2105–2116
Winograd J, Reilly MP, Roe R, Lutz J, Laughner E, Xu X, Hu L, Asakura T, vander Kolk C, Strandberg JD et al (1997) Perinatal lethality and multiple craniofacial malformations in MSX2 transgenic mice. Hum Mol Genet 6:369–379
Wu DT, Bitzer M, Ju W, Mundel P, Bottinger EP (2005) TGF-beta concentration specifies differential signaling profiles of growth arrest/differentiation and apoptosis in podocytes. J Am Soc Nephrol 16:3211–3221
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17(10):1235–41. doi: 10.1038/nm.2448. PMID: 21909103, PMCID: PMC3192296
Yamaguchi A, Ishizuya T, Kintou N, Wada Y, Katagiri T, Wozney JM, Rosen V, Yoshiki S (1996) Effects of BMP-2, BMP-4, and BMP-6 on osteoblastic differentiation of bone marrow-derived stromal cell lines, ST2 and MC3T3-G2/PA6. Biochem Biophys Res Commun 220:366–371
Yamamoto M, Beppu H, Takaoka K, Meno C, Li E, Miyazono K, Hamada H (2009) Antagonism between Smad1 and Smad2 signaling determines the site of distal visceral endoderm formation in the mouse embryo. J Cell Biol 184:323–334
Yamamoto M, Saijoh Y, Perea-Gomez A, Shawlot W, Behringer RR, Ang SL, Hamada H, Meno C (2004) Nodal antagonists regulate formation of the anteroposterior axis of the mouse embryo. Nature 428:387–392
Yang YP, Anderson RM, Klingensmith J (2010) BMP antagonism protects Nodal signaling in the gastrula to promote the tissue interactions underlying mammalian forebrain and craniofacial patterning. Hum Mol Genet 19:3030–3042
Yang YP, Klingensmith J (2006) Roles of organizer factors and BMP antagonism in mammalian forebrain establishment. Dev Biol 296:458–475
Ye M, Berry-Wynne KM, Asai-Coakwell M, Sundaresan P, Footz T, French CR, Abitbol M, Fleisch VC, Corbett N, Allison WT et al (2010) Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum Mol Genet 19:287–298
Yerges LM, Klei L, Cauley JA, Roeder K, Kammerer CM, Moffett SP, Ensrud KE, Nestlerode CS, Marshall LM, Hoffman AR et al (2009) High-density association study of 383 candidate genes for volumetric BMD at the femoral neck and lumbar spine among older men. J Bone Miner Res 24:2039–2049
Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM (2005) Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A 102:5062–5067
Young B, Minugh-Purvis N, Shimo T, St-Jacques B, Iwamoto M, Enomoto-Iwamoto M, Koyama E, Pacifici M (2006) Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Dev Biol 299:272–282
Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C et al (2008) BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med 14:1363–1369
Yumoto K, Thomas PS, Lane J, Matsuzaki K, Inagaki M, Ninomiya-Tsuji J, Scott GJ, Ray MK, Ishii M, Maxson R et al (2013) TGF-beta-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J Biol Chem 288:13467–13480
Zaghloul NA, Brugmann SA (2011) The emerging face of primary cilia. Genesis 49:231–246
Zehentner BK, Dony C, Burtscher H (1999) The transcription factor Sox9 is involved in BMP-2 signaling. J Bone Miner Res 14:1734–1741
Zhang H, Bradley A (1996) Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 122:2977–2986
Zhang J, He XC, Tong WG, Johnson T, Wiedemann LM, Mishina Y, Feng JQ, Li L (2006) BMP signaling inhibits hair follicle anagen induction by restricting epithelial stem/progenitor cell activation and expansion. Stem Cells 24(12):2826–2839
Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y (2002) Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129:4135–4146
Zimmerman LB, De Jesus-Escobar JM, Harland RM (1996) The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell 86:599–606
Zouvelou V, Luder HU, Mitsiadis TA, Graf D (2009) Deletion of BMP7 affects the development of bones, teeth, and other ectodermal appendages of the orofacial complex. J Exp Zool B Mol Dev Evol 312B:361–374
Acknowledgment
We thank Dr. Sudha Rajderkar for critical reading and Yoshiko Mishina for her artwork. We are sorry for not including all critical references due to the space limitation. Y.M. is supported by the National Institutes of Health (R01DE020843) and the Department of Defense (W81XWH-11-2-0073).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Mishina, Y., Kamiya, N. (2017). Embryonic Skeletogenesis and Craniofacial Development. In: Vukicevic, S., Sampath, K. (eds) Bone Morphogenetic Proteins: Systems Biology Regulators. Progress in Inflammation Research. Springer, Cham. https://doi.org/10.1007/978-3-319-47507-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-47507-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47505-9
Online ISBN: 978-3-319-47507-3
eBook Packages: MedicineMedicine (R0)