
Abstract
Platelets are circulating blood cells classically known for their key roles in mediating hemostasis and vascular wall repair. Platelets also possess a dynamic repertoire of effector functions in the immune continuum that span from rapid innate immune responses to more delayed adaptive and acquired immune activities. Platelets express a wide array of structural and functional characteristics of host defense effector cells that augment host defenses to bacterial infections. Platelets display a diverse range of surface ligands and receptors that recognize and bind bacteria, including complement receptors, FcγRII, toll-like receptors (TLRs), and integrins conventionally described in the hemostatic response, such as αIIbβ3 and GPIb. Both direct and indirect binding of bacteria, bacterial toxins, and other agonists to platelets via fibrinogen, fibronectin, C1q, or von Willebrand factor (vWF) may result in platelet activation. Platelets may also internalize bacteria, although the function and fate of these host defense mechanisms remain incompletely understood. Once activated, platelets transform from quiescent discoid forms to amoeboid cells that chemotax to and target microbial pathogens or ligands displayed by tissues injured during infectious insults. Upon activation and subsequent degranulation, platelets secrete an array of multifunctional host defense and antimicrobial peptides that act as direct anti-infective agents and coordinate additional molecular and cellular host defenses. Platelets also release soluble immunomodulatory factors that play crucial roles in the formation of neutrophil extracellular traps (NETs), resulting in bacterial elimination but also enhancing thrombosis in disease situations. Therefore, platelets are increasingly recognized as key effector cells in immune and inflammatory responses to host infection. The multiplicity of events underscores the complexity of platelet–bacterial interaction and illustrates the emerging view that platelets are important sentinel and effector cells in host defenses against pathogens.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Platelet Aggregation
- Platelet Activation
- Disseminate Intravascular Coagulation
- Platelet Surface
- Bacterial Interaction
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Due to their high number in the circulation, platelets may be the first blood cell that encounters bacteria circulating systemically. As such, platelets have been suggested to be key sentinels residing within the vascular space. The amount and variety of functional immunoreceptors define these small cells as complex and unique blood “all-purpose” elements and weapons. Nevertheless, the recognition of platelets as fully functional immune cells is still controversial, primarily due to their lack of a nucleus and their structural “simplicity.” In the last decade, a multitude of previously unrecognized and dynamic functions have been discovered for platelets. These emerging functions are comprehensive and adaptive and span the immune continuum. Many of these newly recognized functions are essential for pathogen surveillance, host defenses and containment, and wound healing (Hamzeh-Cognasse et al. 2015; Vieira-de-Abreu et al. 2012; Weyrich et al. 2003; Weyrich and Zimmerman 2004). In this chapter, we will discuss the role of platelets as innate and adaptive immune cells and the relationship of immune activities of platelets to their hemostatic effector functions.
We will first focus on platelet–bacterial interactions, platelet-released immunomodulatory molecules, and platelet-mediated host defense systems. We will also discuss the intersection of bacterial infection and thrombosis, the interactions of platelets with other classic immune cells, and the necessary cross talk between platelets and neutrophils in neutrophil extracellular trap (NET) formation.
Platelet–Bacterial Interactions: One-on-One
When talking about platelets and their potential to interact with bacteria, there are several different possible mechanisms of interaction (Fig. 1). First, bacteria themselves bind host plasma proteins, which have a corresponding receptor on the platelet surface. Second is the direct binding or adhesion to platelet receptors or the platelet surface. Third is the interaction of secreted bacterial products, exotoxins, or bacterial degradation product with platelet surface receptors. Since platelets express a diverse array of surface receptors, these interactions are highly complex and may result in different activation patterns.

Human platelets directly interact with bacteria. Human platelets were incubated with bacteria for 2 h. (a) Platelets were incubated with S. aureus or (b) E. coli, fixed, spun down, and subsequently stained using Topro-3 for DNA-containing bacteria (yellow) and wheat germ agglutinin (WGA, magenta). White arrows indicate sites of platelet–bacterial interaction. Platelets were analyzed using transmission electron microscopy (TEM) after being incubated with S. aureus or (c) E. coli (d). White arrows indicate sites of platelet–bacterial interaction
Indirect Interactions
The Integrin αIIbβ3 (GPIIb–IIIa)
The platelet fibrinogen receptor αIIbβ3 is a key mediator of the hemostatic function of platelets. The engagement of the integrin by fibrinogen leads to platelet adhesion, aggregation, and activation via outside-in signaling events and is a pharmaceutical target in several disease scenarios (Bennett et al. 1999, 2009; Coller 1997). One family of bacteria that provides an example for direct fibrinogen and fibronectin binding to platelets is the genus Staphylococci. Binding of Staphylococci to tissues and the extracellular matrix is a critical step in establishing infection of a host organism. To achieve this, Staphylococci express surface receptors belonging to the microbial surface components recognizing adhesive matrix molecules (MSCRAMM) family (Josefsson et al. 1998). Some of the most common examples of MSCRAMM are listed in Table 1. While these various MSCRAMMs are highly similar proteins, they bind to fibrinogen via very heterogeneous binding sites. For example, the C-terminal region of the fibrinogen g-chain is the binding site for ClfA, Fbl, and FnbpA and B (McDevitt et al. 1994; Mitchell et al. 2004; Ni Eidhin et al. 1998). In contrast, ClfB binds to the C-terminal region of the fibrinogen a-chain, and SdrG on the b-chain (Flock et al. 1987; Brennan et al. 2009). Besides Staphylococci there are other bacteria, which can engage with fibrinogen. These include the two members of the Streptococcus family. Streptococcus pyogenes and Streptococcus mitis can bind fibrinogen via the M1 protein and lysine, respectively (Cox et al. 2011). While serving as an indirect receptor for bacterial adhesion to platelets, αIIbβ3 can also be directly bound by several bacterial surface proteins, which is discussed in more detail below.
Involvement of Glycoprotein Ibα (GPIbα)
GP1bα is an essential and integral part of the von Willebrand factor (vWF) binding complex on the platelet surface, which is comprised of GPIbα, GPIbβ, GPIX, and GPV (Bennett et al. 2009). The GPIb/IX/V complex is a key mediator of primary hemostasis. In addition, some bacteria are capable of binding to vWF, docking onto the platelet surface via engagement of the GPIb/IX/V complex. Similarly, protein A from S. aureus is capable of binding to vWF, promoting thrombus formation in vitro and under conditions of flow (Claes et al. 2014; O’Seaghdha et al. 2006; Thomer et al. 2013). Helicobacter pylori also directly binds to platelet vWF, although the ligand on H. pylori mediating this interaction has not yet been identified. These interactions ultimately cause platelet aggregation (Byrne et al. 2003). Besides the vWF-mediated binding of GPIb by bacteria, the glycoprotein can also be directly bound with some bacterial surface proteins, a mechanism discussed in more detail below.
Binding to Platelets via Complement Receptors
It is well known that the complement system can activate platelets by inducing the expression of procoagulant factors, such as prothrombinase complex, on the surface of the cells (Peerschke et al. 2010). Furthermore, both complement pathways—the conventional as well as the alternative pathway—are known to interact with bacteria via complement proteins bound to the bacterial surface (Gadjeva 2014). Upon activation, platelet surface expression of gC1q-R, the receptor for C1q, increases. This enables platelets to indirectly bind bacteria coated with C1q on their surface (Peerschke et al. 2003). Moreover, platelet activation also triggers the increased surface expression of CD62P, a potent binding partner for the complement protein C3b, leading to platelet-complement factor-coated bacteria interaction (Hamad et al. 2010). Complement-mediated platelet–bacterial interactions were also demonstrated for S. sanguinis and S. aureus, which induce complement-dependent platelet aggregation (Ford et al. 1996; Loughman et al. 2005).
Complement bacteria–platelet interactions provide a mechanism by which platelets may facilitate the destruction of bacteria by enhancing the complement cascade via surface-bound complement factors and exposed complement receptors. This process may also result in the potential destruction of platelets themselves by being targeted by the lytic activity of the complement system. The complement modulatory potential demonstrated by platelets highlights the immunologic function of this dynamic anucleate cell.
Expression and Functional Significance of the FcγRIIa Receptor
While the expression of the immune receptor FcγRIIa was previously thought to be reserved for classic immune cells, blood platelets also surface-express this receptor (Cassel et al. 1993), although only the Fcγ receptor has been identified in platelets to date. The general function of the immune receptor of the FcγRIIa class is to recognize and bind the Fc domain of immunoglobulin G (IgG). The Fcγ receptor demonstrates a higher affinity for complexed IgG and has a weaker affinity for monomeric IgG. Platelet FcγRIIa is also a key mediator of heparin-induced thrombocytopenia, where autoantibodies recognize the platelet factor 4 (PF4)–heparin complexes that bind to the platelet FcγRIIa (Greinacher 2015). In addition, bacteria recognized and bound by IgG can be attached to this platelet receptor and subsequently internalized (Worth et al. 2006). As platelets are the second most abundant circulating blood cell and express the FcγRIIa in a high copy number on their surface (Cox et al. 2011), platelets may be a significant, yet underappreciated mediator, of antibacterial responses during human infectious settings.
Direct Interactions
Role of Integrin αIIbβ3 (GPIIb–IIIa)
In addition to indirect platelet–bacterial interactions utilizing platelet surface receptors traditionally recognizing extracellular matrix, those receptors directly engage with bacterial surface proteins. The aforementioned S. epidermidis surface protein SdrG, in addition to binding fibrinogen, also directly targets platelet glycoprotein IIb–IIIa (Brennan et al. 2009). Furthermore, S. aureus uses one of its iron-regulated surface determinant (Isd) proteins (IsdB), which are usually used to ensure iron supply for the microbe via heme binding, to bind to integrin αIIbβ3 in the absence of plasma protein (Miajlovic et al. 2010). Specificity of this interaction was demonstrated using platelets treated with αIIbβ3 antibodies, a treatment resulting in complete inhibition of platelet–bacterial adhesion. Recently, S. gordonii was found to express a surface factor, termed platelet adherence protein A (PadA), which directly interacts with αIIbβ3 (Petersen et al. 2010).
Interaction with Glycoprotein Ibα (GPIbα)
GPIbα can also serve as a binding ligand by bacteria for direct engagement with human platelets. This interaction involves a family of highly glycosylated, serine-rich bacterial proteins. These proteins include serine-rich protein A (SrpA) from S. sanguinis (Plummer et al. 2005), glycosylated streptococcal protein B (GspB), and hemagglutinin salivary antigen (Hsa) from S. gordonii (Bensing et al. 2004). Moreover, the staphylococcal accessory regulator (Sar) P protein expressed by S. aureus also mediates direct adhesion of bacteria to platelets (Siboo et al. 2005).
Effects of Platelet–Bacteria Interactions on Aggregation
As discussed above, platelet–bacterial interactions include the engagement of platelet surface receptors traditionally viewed as primary mediators of hemostasis. As such, published evidence demonstrates that platelet aggregation is, at least, partially dependent on docking of bacteria with platelets. Once bacteria have bound to platelet surface αIIbβ3 via fibrinogen or fibronectin, platelet aggregation is initiated in a manner similar as seen with other fibrinogen-coated surfaces (Cox et al. 2011). The SrpA and GspB proteins of S. sanguinis and S. gordonii, respectively, bind platelets and are requisite for platelet aggregation. If these two proteins are genetically deleted, aggregation is eliminated (Cox et al. 2011). Bacteria-induced platelet aggregation can be shear dependent and shear independent. For example, S. pyogenes and S. aureus shear forces appear to be dispensable for the induction of platelet aggregation.
There remain aspects of platelet–bacterial interactions and resulting aggregation responses that are incompletely understood. For example, S. gordonii, which directly interacts with αIIbβ3 on platelets through its PadA protein, does not induce platelet aggregation (Petersen et al. 2010). It has been hypothesized that for some of these bacterial groups, binding of GPIb might result in rearrangements that bring other platelet surface receptors, such as FcγRIIa or αIIbβ3, together in closer proximity, thus promoting thrombus formation (Cox et al. 2011).
In addition to direct interactions leading to aggregation, other mechanisms are involved. Platelet activation induced by surface-bound S. sanguinis (via GPIb) results in the release of platelet dense granule content, namely, the vasoactive substances ATP and ADP, which can act via autocrine signaling to further amplify platelet activation. This mechanism can be enhanced by S. sanguinis through the hydrolyzation of ATP to ADP by the surface-expressed ecto-ATPases. The additionally produced ADPs will increase platelet activation using the P2Y signaling pathway (Cox et al. 2011). Several reports highlighted the need for FcγRIIa involvement on platelet aggregation after bacterial adhesion (Cox et al. 2011). Nevertheless, the observed aggregation seemed to be independent of IgG, and therefore FcγRIIa receptors may have alternative functions in addition to what has been traditionally described. In this context, two publications reported a functional interplay between FcγRIIa and GPIb during bacterial stimulation, which might be the first step in signal transduction (Sullam et al. 1998; Sun et al. 1999). Since GPIb binds to FcγRIIa using the same binding motif as when binding to actin, one might speculate that an alternative functional role of FcγRIIa is mediated by cytoskeletal remodeling (Sun et al. 1999). Similarly, Newman et al. showed that the Src residue from αIIbβ3 phosphorylates the immunoreceptor tyrosine-based activation motif (ITAM) residue of FcγRIIa and thus amplifies the platelet activation signals (Boylan et al. 2008).
In other aspects, bacteria-induced platelet aggregation may be distinct from conventional agonist (ADP, ATP, and thrombin)-induced aggregation. For example, in “binary” aggregation, there is a bacterial density below which platelet aggregation is not observed and above which platelet aggregation is maximal (Kerrigan and Cox 2010). Furthermore, in the presence of bacteria, the lag time before platelet aggregation begins is longer than that observed with hemostatic activation. For some bacteria the lag time might be as long as 20 min, although in some settings, the lag time is dependent on bacterial density (Kerrigan and Cox 2010). There could be several explanations for these lag time variations, including different times required for platelet to bind and interact with bacteria or induction of a weak receptor response.
Bacterial Toxins and Platelets
It is well recognized that bacteria secrete toxins into the host circulation or release endotoxins when lysed or destroyed in other fashion. Members of the Staphylococcus family express alpha-toxin, which binds to the lipid bilayer membrane of platelets to form a pore, followed by a flow of calcium, similar to that induced by calcium ionophore (Arvand et al. 1990). Alpha-toxin-induced platelet activation was described to induce RNA splicing and protein synthesis events in platelets as well as the formation of platelet–bacterial aggregates (Rondina et al. 2011; Schubert et al. 2011). Other pore-forming toxins engaging with the platelet surface have been described. These include streptolysin O from S. pyogenes (Bryant et al. 2005) and pneumolysin from Streptococcus pneumoniae (Johnson et al. 1981). Furthermore, Porphyromonas gingivalis secretes a family of cysteine proteases called gingipains. These secreted toxins recognize the platelet protease-activated receptor (PAR) 1, cleaving it in a similar fashion to the natural ligand thrombin, thereby making it functional (Fitzpatrick et al. 2009). Some bacterial toxins have known superantigenic effects. Among those toxins are the family of staphylococcal superantigen-like (SSL) toxins, released by Staphylococcus aureus and S. pyogenes. One of them, SSL5, can directly interact with GPIba as well as having a direct affinity for GPIV (Cox et al. 2011).
Lipopolysaccharide (LPS) is one of the most important endotoxins expressed on the surface of Gram-negative pathogens. It can be recognized by toll-like receptors (TLRs) expressed by platelets. Platelet surface-expressed TLR-4 is capable of interacting with LPS, leading to platelet activation, P-selectin surface exposure, pre-mRNA splicing events, induction NETs (see later paragraphs), and protein synthetic events and may also contribute to the thrombocytopenia that commonly occurs in sepsis (Akinosoglou and Alexopoulos 2014; Rondina et al. 2011; Semple et al. 2011).
Bacterial-Triggered Release of Platelet-Derived Immunomodulatory Factors
Numerous inflammatory and immune modulating factors are stored in platelet α- and dense granules (Table 2). These molecules are released during platelet activation, modifying host immune responses (Fong et al. 2011; Karshovska et al. 2013; Vieira-de-Abreu et al. 2012; Weyrich and Zimmerman 2004). We will focus in this chapter on several of these molecules and their key signaling events. CD40L (CD154) is a well-established immunoregulatory molecule released by platelets. Platelets were also identified as the major source of circulating soluble CD40L (sCD40L) (Andre et al. 2002). It is known that bacterial ligands interacting with platelet surface TLR-2 (Assinger et al. 2012; Assinger et al. 2011; Cognasse et al. 2007) and TLR-4 (Assinger et al. 2012; Berthet et al. 2012; Cognasse et al. 2007; Ward et al. 2005) induce the release of sCD40L. CD40L is first exposed on the platelet surface membrane in trimeric form (the biologically most active form) and is then cleaved by proteolytic activity, yielding a soluble biologically active fragment. Although not definitely established, there is some evidence suggesting that matrix metalloproteinase (MMP)-9 cleaves the trimeric form of CD40L. The fragment, sCD40L, is recognized by a receptor, CD40, on B lymphocytes, monocytes, and endothelial cells (Blumberg et al. 2009). It should also be pointed out that the soluble form of platelet CD40L may also have an autocrine effect due to the presence of CD40 on the platelet surface (Garraud et al. 2013). sCD40L can facilitate T and B cell interactions leading to antibody production or inducing class switching of B lymphocytes (Elzey et al. 2005). In addition, sCD40L released from platelet storage pools (granules) induces cytokines, chemokines, and lipid mediators through the activation of CD40+ cells (Blumberg et al. 2009). These cytokines potentiate and modulate neutrophil functions in defense against Gram-positive and Gram-negative bacteria (Miedzobrodzki et al. 2008). Thus, sCD40L is one archetypal molecule that platelets use to orchestrate host defense responses with lymphocytes, endothelial cells, and other host defense effector cells.
Another platelet cytokine being of importance is platelet factor 4 (PF4), a known platelet kinocidin (this term will be elucidated in more detail in the next paragraph). Numerous studies have shown that PF4 plasma levels increase dramatically in the setting of a microbial challenge in vivo. For example, PF4 plasma levels increase during bacterial septicemia (Lorenz and Brauer 1988) and streptococcal nephritis (Mezzano et al. 1992). Soluble PF4 can bind to bacteria through its positive charge, thus forming a new recognition site for IgG and effector immune cells (Krauel et al. 2011, 2012). This mechanism is also true for Gram-negative bacteria, since PF4 presents an affinity for bisphosphorylated lipid A of lipopolysaccharide (LPS) bacteria. The newly formed complex is taken up the phagocytic cells. Platelet PF4 might thus facilitate the clearance of certain bacteria. In addition to the lymphocyte modulatory function, an array of bioactive molecules, released from platelets activated by microbial challenge, are chemoattractants for monocytes and neutrophils. These include platelet-derived growth factor (PDGF), platelet-activating factor (PAF), regulated on activation normal T cell expressed and secreted (RANTES) protein, macrophage inflammatory protein (MIP-1 alpha), monocyte chemoattractant protein, and PF4 (Clark-Lewis et al. 1993; Deuel et al. 1981; Tzeng et al. 1985; Semple et al. 2011; Weyrich and Zimmerman 2004).
Platelets Are an Integral Part of the Antimicrobial Host Defense System
The Internalization of Bacteria
In the 1970s, Clawson and colleagues studied the interaction between platelets and bacteria and observed the internalization of S. aureus into some platelets (Clawson 1973; Clawson et al. 1975; Clawson and White 1980). These observations were confirmed by other investigators, especially for S. aureus (Youssefian et al. 2002). Youssefian et al. proposed that platelets have the ability to actively internalize or phagocytose microorganisms. This hypothesis was based on finding bacteria inside of vacuoles that were not connected to the open canalicular system (OCS) (Youssefian et al. 2002). Furthermore, it appears that bacterial internalization resembles morphologic evidence of platelet activation. In turn, platelets seem to gain enhanced capacity for bacterial internalization if been activated by bacteria or even conventional agonists (ADP or thrombin), indicating a common mechanism between activation and internalization. Interestingly, immunolabeling revealed that the engulfing vacuoles and the OCS were composed of distinct antigens. When S. aureus was engulfed by platelets, membranes expressed CD62P and αIIbβ3, but not GPIb, similar expression markers seen an activated platelet membrane. Moreover, endocytotic vacuoles containing S. aureus were demonstrated to fuse with α-granules, which contain platelet antimicrobicidal proteins. Because of its close proximity to the plasma membrane and the differential surface marker array, these vacuoles may be formed through invagination of the plasma membrane. Internalization of S. aureus has also been demonstrated in settings of ADP-induced platelet activation (Li et al. 2008). Under these activating conditions, P. gingivalis can also be internalized by platelets (Li et al. 2008). P. gingivalis, however, may also be internalized in the absence of activation, an observation not noted with S. aureus. While internalization of both bacteria results in them being found in vacuoles independent of the OCS, these findings suggest there may be unique mechanisms regulating the internalization of different bacterial species.
Platelet binding to bacteria using the FcγRII receptor could also initiate the internalization of IgG-bacteria complexes (Worth et al. 2006). In 2011, Antczak et al. demonstrated that platelets are capable of internalizing IgG-coated polystyrene beads (0.5–1.5 μm diameter) in a FcγRII-specific mechanism (Antczak et al. 2011). This endocytotic mechanism was inhibited by the actin-polymerization inhibitor cytochalasin D, demonstrating that actin cytoskeletal dynamics are an integral process of bacteria internalization. While these and other studies support the concept that platelets can internalize or endocytose bacteria, the final fate of these internalized microbes remains uncertain. Some have suggested that the absence of phagolysosomes in platelets precludes their ability to degrade or destroy bacteria (White 2006). Other lines of evidence suppose that the fusion of bacteria with platelet α-granules, which contain antimicrobicidal proteins, could result in pathogen destruction (Youssefian et al. 2002).
Platelets Release Antibacterial Effector Molecules Contributing to Host Defense
In addition to platelets’ ability to directly interact with and internalize bacteria, platelets also release antimicrobial proteins and peptides. In 1901, Gengou demonstrated that the bactericidal activity of a previously identified molecule within serum (Fodor 1887), β-lysin, was derived from cells involved in the clotting of blood in a mechanism that was independent of complement (Gengou 1901). This may have been the first study to identify a role for platelets in antimicrobial host defense responses.
In 2010, Yeaman and colleagues proposed calling these antimicrobial molecules platelet microbicidal proteins (PmPs) (Yeaman 2010). These proteins are also known as thrombocidins. PmPs (predominantly PmP1 and PmP2) are released when platelets are activated by thrombin or bacteria. PmPs differ from classically described defensins by their molecular mass, their sequence, and the chains of lysine and arginine residues, which gives them a cationic charge. As with PARs (protease-activated receptors), PmPs must be cleaved by thrombin to become fully functional. The two PmP subunits then act in an autonomous, but complementary, manner, by alternating the permeability of the bacterial wall (Yeaman 2010). The ATP/ADP signaling pathway, utilizing the P2 receptors, primarily triggers the release of PmPs. The initial signal is further amplified by the release of platelet ADP, resulting in potentially autocrine activation of even neighboring platelets (Trier et al. 2008). Over the years, the PmP family was enlarged through the integration of kinocidins, which include those platelet-derived cytokines that have a direct bactericidal effect (Yang et al. 2003). This broad collection of antimicrobial peptides is released from human platelets following thrombin or bacterial stimulation. Similar to chemokines, kinocidins have been organized into classes, which are related to their structural immunobiology. The hallmark CXC motif is the defining element of α-kinocidins. This group includes PF4 (platelet factor 4), platelet basic protein (PBP), connective tissue-activating peptide (CTAP3), and neutrophil-activating peptide (NAP2). In contrast, RANTES is a member of the β-kinocidins, containing the typical CC-chemokine motif. Importantly, members of the α-kinocidin and the β-kinocidin subgroups have synergistic effects. For example, CTAP3 does not have an effect on the viability of E. coli. However, the presence of PF4 potentiates CTAP3 activity and thereby reduces the bacterial density significantly. This result is not obtained for PF4 alone (Tang et al. 2002). Using elaborate structural biochemical analyses, the 60–74 structural domain in PF4 was identified as being responsible for this synergistic activity (Yeaman et al. 2007). Kinocidins still also maintain their primary role, which is the chemoattraction of leukocytes. This mechanism fosters cooperation between platelet and leukocyte factors in bacterial clearance, thus integrating different mechanisms in innate immunity (Agerberth and Gudmundsson 2006; Yeaman et al. 2007).
Kraemer et al. demonstrated that S. aureus growth decreases in the presence of human platelets (Kraemer et al. 2011). This platelet bactericidal effect was attributed to the expression of human β-defensin-1 (hBD-1), an antimicrobial peptide primarily expressed by epithelial cells, but also detectable in human blood (Fang et al. 2003). hBD-1 is present in CD34+ hematopoietic progenitor cell-derived megakaryocytes and human platelets at both the messenger RNA (mRNA) and protein level and was localized to an extragranular cytoplasmic compartment. Moreover, treating platelets with S. aureus-derived α-toxin, a bacterial pore-forming product, resulted in hBD-1 secretion. Subsequent studies have found that other family members, including hBD-2 and hBD-3, are present in human platelets and possess potent microbicidal activity (Tohidnezhad et al. 2011, 2012).
Boilard and colleagues recently extended these findings, describing a new mechanism by which platelets modulate inflammatory responses by using bactericidal phospholipase (Boudreau et al. 2014). They demonstrate that activated platelets release respiratory-competent mitochondria inside microparticles or as free organelles. The authors further show that the platelet-derived mitochondria are a substrate for the secreted phospholipase A2 IIA (sPLA2-IIA), a phospholipase otherwise specific for bacteria. This substrate specificity may reflect the ancestral Proteobacteria origin of mitochondria. Upon mitochondrial membrane phospholipid hydrolysis by sPLA2-IIA, platelets released a number of lysophospholipids (lyso-PAF, lyso-PC, lysoPE) and free fatty acids. The release of these products resulted in damaged structural integrity and the generation of mitochondrial DNA (mtDNA), a potent pro-inflammatory molecule. Taken together, these and other observations suggest that platelets are involved in infectious immune responses both directly through the release of antimicrobial factors and indirectly through the release of cytokines and other mediators.
Role of Platelets in the Development and Pathophysiology of Bacterial Sepsis
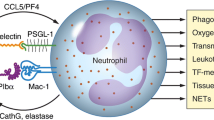
Sepsis is a common and often lethal infectious syndrome that is increasing in incidence. Dysregulated immune and hemostatic responses to pathogens, pathogen-associated molecular patterns, and microbial toxins are central to the pathogenesis of septic syndromes (Fig. 2) (Vincent and Abraham 2006).

Platelets interact with bacteria, recruit immune cells, and subsequently trigger inflammatory and thrombotic events. Activation of platelets by bacteria or microbial toxins can trigger platelet–bacterial aggregation or promote the interaction of activated platelets with monocytes, lymphocytes, and neutrophils. Interaction of platelets with leukocytes can also lead to NETosis and bacterial destruction. Platelet or platelet–bacteria/immune cell aggregate sequestration in microvascular beds can mediate increased local inflammatory signaling. In addition, the interaction with fibrinogen and the deposition of fibrin is a central pathophysiological process in sepsis. Furthermore, fibrin degradation products can modify platelet–cell interactions and contribute to the dysregulated hemostasis characteristic of sepsis
Coagulopathy Induced by Septic Triggers
Up to 30–50 % of patients with severe sepsis or septic shock develop disseminated intravascular coagulation (DIC). DIC, in conjunction with peripheral consumption and destruction of platelets, impaired thrombopoiesis, and sequestration of platelets in the microvasculature in platelet–fibrin deposits contributes to thrombocytopenia in sepsis. Moreover, severe thrombocytopenia or a delayed recovery in the platelet count is associated with adverse outcomes from sepsis. As a result of the inflammatory response in sepsis, neutrophils synthesize and release tissue factor, which may initiate or amplify activation of the coagulation cascade, ultimately activating circulating platelets. Activated platelets have increased surface expression of adhesion molecules, promoting interactions with and binding to the damaged endothelial surface and subendothelial matrix proteins (Warkentin et al. 2003). Dysregulated activation of coagulation cascade leads to the deposition of platelet–fibrin thrombi and platelet sequestration in microvessels (Warkentin et al. 2003). Moreover, the formation of homotypic platelet–platelet aggregates and heterotypic platelet–bacteria, platelet–monocyte, and platelet–neutrophil aggregates (in conjunction with excessive thrombin generation and conversion of fibrinogen to fibrin) may further contribute to micro- and macrovascular sequestration and thrombosis (Evans et al. 1969; Gawaz et al. 1995). Thus, platelets are key mediator and effector cells involved in the pathophysiology of sepsis.
The Inflammatory Face of Platelets during Sepsis
First evidence for the inflammatory potential of platelets in sepsis comes from studies demonstrating that the amount of circulating sCD40L is increased in patients with sepsis when compared to healthy control subjects, although levels of sCD40L were independent of sepsis severity (Chew et al. 2010; Inwald et al. 2006; Lorente et al. 2011; Luo et al. 2014; Rahman et al. 2013; Zhang et al. 2011). In murine models of sepsis, sCD40L is released from platelets (Rahman et al. 2009), and matrix metalloproteinase-9, which cleaves platelet-derived CD40L, is increased (Rahman et al. 2012). sCD40L is a key molecule for the recruitment of neutrophils, via macrophage-1 antigen (Mac-1) expression on neutrophils to sites of infection or injury. Mice deficient in CD40L cannot activate neutrophils and do not develop edema or neutrophil infiltration in the lungs in settings of sepsis induced by cecal ligation puncture (CLP) (Asaduzzaman et al. 2009; Rahman et al. 2009).
Another host synthesized pro-inflammatory mediator generated during sepsis is PAF (Yost et al. 2010). PAF binds to its receptor, platelet-activating factor receptor (PAFR), a G-protein coupled receptor expressed by immune cells and platelets, leading to platelet degranulation, the release of inflammatory factors, and initiation of the coagulation cascade. The PAFR-related signaling cascade is usually tightly regulated to prevent excessive thrombo-inflammatory response. Nevertheless, during sepsis, this regulatory balance is shifted, and excessive PAF signaling leads to the activation of neutrophils, monocytes, and platelets, as well as to the formation of heterotypic platelet aggregates (Yost et al. 2010). As discussed above, activation of circulating platelets by bacterial triggers also causes the expression of cell surface receptors and the release of molecules that may further amplify immune responses (Semple et al. 2011). P-selectin (CD62P), which is expressed on the surface of platelets, and its receptor, P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes, are additional molecules with key functions during inflammatory and immune responses (Diacovo et al. 1996; Katz et al. 2011; Russwurm et al. 2002; Semple et al. 2011). The functional cross talk between P-selectin and PSGL-1 in conjunction with the leukocyte integrin αMβ2 (Mac-1) helps orchestrating the molecular events necessary for leukocyte recruitment (Semple et al. 2011; Weyrich and Zimmerman 2004).
Platelet Microparticles Fuel the Inflammatory Fire in Sepsis
Increased levels of circulating microparticles are also commonly detected in sepsis patients (Ogura et al. 2001). These membrane-enclosed vesicles can arise from different cell types, including granulocytes, monocytes, endothelial cells, and platelets (George 2008). Here, we will focus on platelet-derived microparticles in sepsis. Platelet microparticles (PMPs) are small phospholipid vesicles between 100–1000 nm in diameter that are released after budding from the platelet plasma membrane. As a result, PMP express the same antigens as their parent cells, i.e., αIIbβ3, GPIb, CD31, CD61, and CD62P (Vasina et al. 2010). Platelet-derived microparticles comprise the majority (e.g., approximately 70–90 %) of the pool of circulating microparticles (Burnier et al. 2009). In sepsis, microparticles can be produced via the stimulation of platelet TLR-4 by bacterial LPS (Brown and McIntyre 2011; Hashimoto et al. 2009; Kappelmayer et al. 2013), but also in response to the Shiga toxin (Ge et al. 2012). Formation of platelet microparticles results in an asymmetrical distribution of membrane phospholipids (Flaumenhaft et al. 2010; Italiano et al. 2010). Therefore, PMPs express highly procoagulant phosphatidylserines (PS) on their surface, although microparticles derived from platelets may be less procoagulant than monocyte-derived microparticles (Owens and Mackman 2011). Furthermore, in recent studies it was shown that two distinct subpopulations of microparticles exist: (1) PS-expressing and (2) PS-nonexpressing MPs. As surface phosphatidylserine-positive microparticles participate in the coagulation process, these data suggest that PS-nonexpressing MPs might have roles separate from the maintenance of primary hemostasis (Arraud et al. 2014; Cloutier et al. 2013).
Platelet microparticles are also capable of delivering immunomodulatory factors, such as IL-1β (Boilard et al. 2010), as well as interacting with other cells. For example, Provost et al. recently demonstrated that human platelet-derived microparticles contained functional Argonaute 2 (Ago2)–miR-223 complexes that were capable of regulating the expression of two endogenous genes in endothelial cells (Laffont et al. 2013). These findings support the concept that microparticles may act as intercellular carriers of functional cell components, thereby influencing the function of other circulating cell types. Boilard et al. even further refined the concept of microparticles being transport vehicles. These investigators demonstrated that platelet-derived microparticles were internalized by activated neutrophils in the endomembrane system via 12(S)-HETE, a hydroxyeicosatetraenoic acid produced through the activity of sPLA2-IIA, which is present in inflammatory fluids (Duchez et al. 2015). Platelet microparticles can also be internalized by macrophages, leading to the differential expression of numerous miRNAs and mRNAs, including transcripts encoding for chemokine (C–C motif) ligand 4 (CCL4/MIP-1β), colony-stimulating factor 1 (CSF1), and TNF (Laffont et al. 2015). Additionally, platelet microparticle uptake by macrophages led to an increase in their phagocytic capacity (Laffont et al. 2015). These recently discovered functions of platelets and platelet-derived microparticles highlight once more the versatility of this anucleate cell and its key roles in inflammatory and immune disorders.
Platelets Induce NETosis in Sepsis
NET formation in response to bacterial invasion was first demonstrated by Brinkmann and colleagues in 2004 (Brinkmann et al. 2004). NETs consist of expulsed nuclear DNA that forms lattices, containing histones (H1, H2A, H2B, and H4), granule enzymes (elastase, myeloperoxidase), and antimicrobial proteins released by the PMN in parallel with extrusion of nuclear material (Brinkmann et al. 2004). NETs can be generated under both flow and static conditions, although there is some evidence that flow conditions enhance bacterial trapping (analogous to the human circulatory system) (Clark et al. 2007). NETs are long lattices that can be as large as 25 nm in diameter (Clark et al. 2007). These DNA/enzyme complexes accomplish both capture and killing of extracellular bacteria. In addition, antimicrobial activities can be blunted or subverted by endonucleases and DNases expressed by some microorganisms (Beiter et al. 2006; Buchanan et al. 2006). NETs are also implicated in vascular injury associated with neutrophil–platelet interactions in human sepsis (Clark et al. 2007). For example, Ma and Kubes demonstrated that a bacterial environment or stimulation using LPS resulted in the in vivo formation of NETs which trapped bacteria, reducing their growth, and blunted the severity of sepsis (Ma and Kubes 2008). Ma and colleagues further demonstrated that NETs were able to trap both Gram-negative and Gram-positive bacteria.
NETosis is an active mechanism occurring within 5–10 min after neutrophil stimulation and being distinct from apoptosis and necrosis (Brinkmann et al. 2004). There is evidence that reactive oxygen species (ROS) generation is a key event in NET formation (Fuchs et al. 2007; Yost et al. 2009). However, ROS generation and NET formation can be dissociated under some conditions, suggesting that the pathways and mechanisms regulating NET formation are more complex than previously appreciated (Chow et al. 2010; Marcos et al. 2010). Yost et al. found that the mammalian target of rapamycin (mTOR) regulates NET formation through induction of hypoxia-inducible factor 1α (HIF-1α). Citrullination of histones, release of mtDNA, and platelet hBD-1 also mediate key aspects of NET formation (Kraemer et al. 2011; Yousefi et al. 2009). In addition, platelet-derived microparticles containing mitochondria as well as mitochondria released from platelets into the circulation have been linked to NET formation. The hydrolysis of the mitochondrial membrane by sPLA2-IIA also leads to the formation of inflammatory mediators inducing robust NET formation in an in vitro assay (Boudreau et al. 2014).
Nevertheless, while NETs may be key host defense mechanisms in response to bacterial invasion, NET formation may also have adverse effects on the microvascular circulation by promoting the formation of microthrombi, thereby also potentially preventing immune cells from reaching the bacteria. Emerging evidence implicates NET formation as injurious in many inflammatory and thrombotic diseases, including sepsis (Xu et al. 2009), coronary artery disease (Borissoff et al. 2013), microvascular thrombosis (Xu et al. 2009), and deep vein thrombosis (Fuchs et al. 2010). Recent work by Wagner and colleagues demonstrates that thrombin-stimulated platelets induce NET formation via P-selectin/PSGL-1 interactions (Etulain et al. 2015). These intriguing findings suggest that the P-selectin/PSGL-1 axis may be a potential therapeutic target for settings where dysregulated NET formation is considered injurious (Etulain et al. 2015).
Conclusion
Established and emerging evidence supports the role of platelets as dynamic sentinels and effector cells during bacterial infections. These previously unrecognized functions broaden the view of platelets as having complementary roles in both hemostasis and immune pathways. Furthermore, as platelets orchestrate key aspects of immune and inflammatory responses, platelets are being studied as a potential therapeutic target in sepsis.
Take Home Messages
-
Platelets are key sentinels and effector cells involved in the pathophysiology of sepsis.
-
Platelet integrins mediate platelet–bacterial interactions.
-
FcgRIIa on platelets coordinates antibacterial host responses.
-
The final fate of internalized bacteria remains unclear and warrants further investigations.
-
Platelets are involved in infectious immune responses both directly through the release of antimicrobial factors and indirectly through the release of cytokines and other mediators.
-
The P-selectin/PSGL-1 axis may be a potential therapeutic target for settings of injurious NET formation.
References
Agerberth B, Gudmundsson GH (2006) Host antimicrobial defence peptides in human disease. Curr Top Microbiol Immunol 306:67–90
Akinosoglou K, Alexopoulos D (2014) Use of antiplatelet agents in sepsis: a glimpse into the future. Thromb Res 133(2):131–138
Andre P, Nannizzi-Alaimo L, Prasad SK, Phillips DR (2002) Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circulation 106(8):896–899
Antczak AJ, Vieth JA, Singh N, Worth RG (2011) Internalization of IgG-coated targets results in activation and secretion of soluble CD40 ligand and RANTES by human platelets. Clin Vaccine Immunol 18(2):210–216
Arraud N, Linares R, Tan S, Gounou C, Pasquet JM, Mornet S, Brisson AR (2014) Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost 12(5):614–627
Arvand M, Bhakdi S, Dahlback B, Preissner KT (1990) Staphylococcus aureus alpha-toxin attack on human platelets promotes assembly of the prothrombinase complex. J Biol Chem 265(24):14377–14381
Asaduzzaman M, Lavasani S, Rahman M, Zhang S, Braun OO, Jeppsson B, Thorlacius H (2009) Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care Med 37(4):1389–1396
Assinger A, Laky M, Schabbauer G, Hirschl AM, Buchberger E, Binder BR, Volf I (2011) Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J Thromb Haemost 9(4):799–809
Assinger A, Laky M, Badrnya S, Esfandeyari A, Volf I (2012) Periodontopathogens induce expression of CD40L on human platelets via TLR2 and TLR4. Thromb Res 130(3):e73–78
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B (2006) An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 16(4):401–407
Bennett JS, Zigmond S, Vilaire G, Cunningham ME, Bednar B (1999) The platelet cytoskeleton regulates the affinity of the integrin alpha(IIb)beta(3) for fibrinogen. J Biol Chem 274(36):25301–25307
Bennett JS, Berger BW, Billings PC (2009) The structure and function of platelet integrins. J Thromb Haemost 7(Suppl 1):200–205
Bensing BA, Lopez JA, Sullam PM (2004) The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun 72(11):6528–6537
Berthet J, Damien P, Hamzeh-Cognasse H, Arthaud CA, Eyraud MA, Zeni F, Pozzetto B, McNicol A, Garraud O, Cognasse F (2012) Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin Immunol 145(3):189–200
Blumberg N, Spinelli SL, Francis CW, Taubman MB, Phipps RP (2009) The platelet as an immune cell-CD40 ligand and transfusion immunomodulation. Immunol Res 45(2–3):251–260
Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O‘Donnell E, Farndale RW, Ware J, Lee DM (2010) Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327(5965): 580–583
Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Ten Cate H, Hofstra L, Crijns HJ, Wagner DD, Kietselaer BL (2013) Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol 33(8):2032–2040
Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Pare A, Rousseau M, Naika GS, Levesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, Boilard E (2014) Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124(14):2173–2183
Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ (2008) Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood 112(7):2780–2786
Brennan MP, Loughman A, Devocelle M, Arasu S, Chubb AJ, Foster TJ, Cox D (2009) Elucidating the role of Staphylococcus epidermidis serine-aspartate repeat protein G in platelet activation. J Thromb Haemost 7(8):1364–1372
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535
Brown GT, McIntyre TM (2011) Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J Immunol 186(9):5489–5496
Bryant AE, Bayer CR, Chen RY, Guth PH, Wallace RJ, Stevens DL (2005) Vascular dysfunction and ischemic destruction of tissue in Streptococcus pyogenes infection: the role of streptolysin O-induced platelet/neutrophil complexes. J Infect Dis 192(6):1014–1022
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16(4):396–400
Burnier L, Fontana P, Kwak BR, Angelillo-Scherrer A (2009) Cell-derived microparticles in haemostasis and vascular medicine. Thromb Haemost 101(3):439–451
Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, Cox DM (2003) Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 124(7):1846–1854
Cassel DL, Keller MA, Surrey S, Schwartz E, Schreiber AD, Rappaport EF, McKenzie SE (1993) Differential expression of Fc gamma RIIA, Fc gamma RIIB and Fc gamma RIIC in hematopoietic cells: analysis of transcripts. Mol Immunol 30(5):451–460
Chew M, Rahman M, Ihrman L, Erson A, Zhang S, Thorlacius H (2010) Soluble CD40L (CD154) is increased in patients with shock. Inflamm Res 59(11):979–982., Official Journal of the European Histamine Research Society [et al]
Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, Gallo RL, Monestier M, Wang Y, Glass CK, Nizet V (2010) Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 8(5):445–454
Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, Missiakas D, Schneewind O, Hoylaerts MF, Heying R, Verhamme P (2014) Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood 124(10):1669–1676
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13(4):463–469
Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M (1993) Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci U S A 90(8):3574–3577
Clawson CC (1973) Platelet interaction with bacteria. 3. Ultrastructure. Am J Pathol 70(3):449–471
Clawson CC, White JG (1980) Platelet interaction with bacteria. V. Ultrastructure of congenital afibrinogenemic platelets. Am J Pathol 98(1):197–211
Clawson CC, Rao GH, White JG (1975) Platelet interaction with bacteria. IV. Stimulation of the release reaction. Am J Pathol 81(2):411–420
Cloutier N, Tan S, Boudreau LH, Cramb C, Subbaiah R, Lahey L, Albert A, Shnayder R, Gobezie R, Nigrovic PA, Farndale RW, Robinson WH, Brisson A, Lee DM, Boilard E (2013) The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes. EMBO Mol Med 5(2):235–249
Cognasse F, Lafarge S, Chavarin P, Acquart S, Garraud O (2007) Lipopolysaccharide induces sCD40L release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med 33(2):382–384
Coller BS (1997) Platelet GPIIb/IIIa antagonists: the first anti-integrin receptor therapeutics. J Clin Invest 99(7):1467–1471
Cox D, Kerrigan SW, Watson SP (2011) Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost 9(6):1097–1107
Deuel TF, Senior RM, Chang D, Griffin GL, Heinrikson RL, Kaiser ET (1981) Platelet factor 4 is chemotactic for neutrophils and monocytes. Proc Natl Acad Sci U S A 78(7):4584–4587
Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA (1996) Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood 88(1):146–157
Duchez AC, Boudreau LH, Bollinger J, Belleannee C, Cloutier N, Laffont B, Mendoza-Villarroel RE, Levesque T, Rollet-Labelle E, Rousseau M, Allaeys I, Tremblay JJ, Poubelle PE, Lambeau G, Pouliot M, Provost P, Soulet D, Gelb MH, Boilard E (2015) Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc Natl Acad Sci U S A 112(27):E3564–3573
Elzey BD, Grant JF, Sinn HW, Nieswandt B, Waldschmidt TJ, Ratliff TL (2005) Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukoc Biol 78(1):80–84
Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD (2015) P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126(2):242–246
Evans G, Lewis AF, Mustard JF (1969) The role of platelet aggregation in the development of endotoxin shock. Br J Surg 56(8):624
Fang XM, Shu Q, Chen QX, Book M, Sahl HG, Hoeft A, Stuber F (2003) Differential expression of alpha- and beta-defensins in human peripheral blood. Eur J Clin Invest 33(1):82–87
Fitzpatrick RE, Wijeyewickrema LC, Pike RN (2009) The gingipains: scissors and glue of the periodontal pathogen, Porphyromonas gingivalis. Future Microbiol 4(4):471–487
Flaumenhaft R, Mairuhu AT, Italiano JE (2010) Platelet- and megakaryocyte-derived microparticles. Semin Thromb Hemost 36(8):881–887
Flock JI, Froman G, Jonsson K, Guss B, Signas C, Nilsson B, Raucci G, Hook M, Wadstrom T, Lindberg M (1987) Cloning and expression of the gene for a fibronectin-binding protein from Staphylococcus aureus. EMBO J 6(8):2351–2357
Fodor J (1887) Die Faehigkeit des Blutes Bakterien zu vernichten. Dtsch Med Wochenschr 13:745–747
Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, Speicher KD, Blair IA, Speicher DW, Grosser T, Brass LF (2011) Deciphering the human platelet sheddome. Blood 117(1):e15–26
Ford I, Douglas CW, Heath J, Rees C, Preston FE (1996) Evidence for the involvement of complement proteins in platelet aggregation by Streptococcus sanguis NCTC 7863. Br J Haematol 94(4):729–739
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176(2):231–241
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107(36):15880–15885
Gadjeva M (2014) The complement system. Overview. Methods Mol Biol 1100:1–9
Garraud O, Hamzeh-Cognasse H, Pozzetto B, Cavaillon JM, Cognasse F (2013) Bench-to-bedside review: Platelets and active immune functions - new clues for immunopathology? Crit Care 17(4):236
Gawaz M, Fateh-Moghadam S, Pilz G, Gurland HJ, Werdan K (1995) Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Invest 25(11):843–851
Ge S, Hertel B, Emden SH, Beneke J, Menne J, Haller H, von Vietinghoff S (2012) Microparticle generation and leucocyte death in Shiga toxin-mediated HUS. Nephrol Dial Transplant 27(7):2768–2775. Official publication of the European Dialysis and Transplant Association—European Renal Association
Gengou O (1901) De l‘origine de l‘axenine de serums normaux. Ann Inst Pasteur 15:232–245
George FD (2008) Microparticles in vascular diseases. Thromb Res 122(Suppl 1):S55–59
Greinacher A (2015) Clinical Practice. Heparin-Induced Thrombocytopenia. N Engl J Med 373(3):252–261
Hamad OA, Nilsson PH, Wouters D, Lambris JD, Ekdahl KN, Nilsson B (2010) Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J Immunol 184(5):2686–2692
Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O (2015) Platelets and infections—complex interactions with bacteria. Front Immunol 6:82
Hashimoto K, Jayachandran M, Owen WG, Miller VM (2009) Aggregation and microparticle production through toll-like receptor 4 activation in platelets from recently menopausal women. J Cardiovasc Pharmacol 54(1):57–62
Inwald DP, Faust SN, Lister P, Peters MJ, Levin M, Heyderman R, Klein NJ (2006) Platelet and soluble CD40L in meningococcal sepsis. Intensive Care Med 32(9):1432–1437
Italiano JE Jr, Mairuhu AT, Flaumenhaft R (2010) Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol 17(6):578–584
Johnson MK, Boese-Marrazzo D, Pierce WA Jr (1981) Effects of pneumolysin on human polymorphonuclear leukocytes and platelets. Infect Immun 34(1):171–176
Josefsson E, McCrea KW, Ni Eidhin D, O'Connell D, Cox J, Hook M, Foster TJ (1998) Three new members of the serine-aspartate repeat protein multigene family of Staphylococcus aureus. Microbiology 144(Pt 12):3387–3395
Kappelmayer J, Beke Debreceni I, Vida A, Antal-Szalmas P, Clemetson KJ, Nagy B Jr (2013) Distinct effects of Re- and S-forms of LPS on modulating platelet activation. J Thromb Haemost 11(4):775–778
Karshovska E, Weber C, von Hundelshausen P (2013) Platelet chemokines in health and disease. Thromb Haemost 110(5):894–902
Katz JN, Kolappa KP, Becker RC (2011) Beyond thrombosis: the versatile platelet in critical illness. Chest 139(3):658–668
Kerrigan SW, Cox D (2010) Platelet-bacterial interactions. Cell Mol Life Sci 67(4):513–523
Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WH, Lindemann S, Seizer P, Yost CC, Zimmerman GA, Weyrich AS (2011) Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog 7(11):e1002355
Krauel K, Potschke C, Weber C, Kessler W, Furll B, Ittermann T, Maier S, Hammerschmidt S, Broker BM, Greinacher A (2011) Platelet factor 4 binds to bacteria, [corrected] inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood 117(4):1370–1378
Krauel K, Weber C, Brandt S, Zahringer U, Mamat U, Greinacher A, Hammerschmidt S (2012) Platelet factor 4 binding to lipid A of Gram-negative bacteria exposes PF4/heparin-like epitopes. Blood 120(16):3345–3352
Laffont B, Corduan A, Ple H, Duchez AC, Cloutier N, Boilard E, Provost P (2013) Activated platelets can deliver mRNA regulatory Ago2*microRNA complexes to endothelial cells via microparticles. Blood 122(2):253–261
Laffont B, Corduan A, Rousseau M, Duchez AC, Lee CH, Boilard E, Provost P (2015) Platelet microparticles reprogram macrophage gene expression and function. Thromb Haemost 115(1). doi:10.1160/TH15-05-0389
Li X, Iwai T, Nakamura H, Inoue Y, Chen Y, Umeda M, Suzuki H (2008) An ultrastructural study of Porphyromonas gingivalis-induced platelet aggregation. Thromb Res 122(6):810–819
Lorente L, Martin MM, Varo N, Borreguero-Leon JM, Sole-Violan J, Blanquer J, Labarta L, Diaz C, Jimenez A, Pastor E, Belmonte F, Orbe J, Rodriguez JA, Gomez-Melini E, Ferrer-Aguero JM, Ferreres J, Lliminana MC, Paramo JA (2011) Association between serum soluble CD40 ligand levels and mortality in patients with severe sepsis. Crit Care 15(2):R97
Lorenz R, Brauer M (1988) Platelet factor 4 (PF4) in septicaemia. Infection 16(5):273–276
Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, Foster TJ (2005) Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 57(3):804–818
Luo L, Zhang S, Wang Y, Rahman M, Syk I, Zhang E, Thorlacius H (2014) Proinflammatory role of neutrophil extracellular traps in abdominal sepsis. Am J Physiol Lung Cell Mol Physiol 307(7):L586–596
Ma AC, Kubes P (2008) Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J Thromb Haemost 6(3):415–420
Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, Wiedenbauer EM, Krautgartner WD, Stoiber W, Belohradsky BH, Rieber N, Kormann M, Koller B, Roscher A, Roos D, Griese M, Eickelberg O, Doring G, Mall MA, Hartl D (2010) CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med 16(9):1018–1023
McDevitt D, Francois P, Vaudaux P, Foster TJ (1994) Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol Microbiol 11(2):237–248
Mezzano S, Burgos ME, Ardiles L, Olavarria F, Concha M, Caorsi I, Aranda E, Mezzano D (1992) Glomerular localization of platelet factor 4 in streptococcal nephritis. Nephron 61(1):58–63
Miajlovic H, Zapotoczna M, Geoghegan JA, Kerrigan SW, Speziale P, Foster TJ (2010) Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology 156(Pt 3):920–928
Miedzobrodzki J, Panz T, Plonka PM, Zajac K, Dracz J, Pytel K, Mateuszuk L, Chlopicki S (2008) Platelets augment respiratory burst in neutrophils activated by selected species of gram-positive or gram-negative bacteria. Folia histochemica et cytobiologica/Polish Academy of Sciences, Polish Histochemical and Cytochemical Society 46(3):383–388
Mitchell J, Tristan A, Foster TJ (2004) Characterization of the fibrinogen-binding surface protein Fbl of Staphylococcus lugdunensis. Microbiology 150(Pt 11):3831–3841
Ni Eidhin D, Perkins S, Francois P, Vaudaux P, Hook M, Foster TJ (1998) Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol 30(2):245–257
Ogura H, Kawasaki T, Tanaka H, Koh T, Tanaka R, Ozeki Y, Hosotsubo H, Kuwagata Y, Shimazu T, Sugimoto H (2001) Activated platelets enhance microparticle formation and platelet-leukocyte interaction in severe trauma and sepsis. J Trauma 50(5):801–809
O’Seaghdha M, van Schooten CJ, Kerrigan SW, Emsley J, Silverman GJ, Cox D, Lenting PJ, Foster TJ (2006) Staphylococcus aureus protein A binding to von Willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J 273(21):4831–4841
Owens AP 3rd, Mackman N (2011) Microparticles in hemostasis and thrombosis. Circ Res 108(10):1284–1297
Peerschke EI, Murphy TK, Ghebrehiwet B (2003) Activation-dependent surface expression of gC1qR/p33 on human blood platelets. Thromb Haemost 89(2):331–339
Peerschke EI, Yin W, Ghebrehiwet B (2010) Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol 47(13):2170–2175
Petersen HJ, Keane C, Jenkinson HF, Vickerman MM, Jesionowski A, Waterhouse JC, Cox D, Kerrigan SW (2010) Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect Immun 78(1):413–422
Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW (2005) A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol 129(1):101–109
Rahman M, Zhang S, Chew M, Ersson A, Jeppsson B, Thorlacius H (2009) Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann Surg 250(5):783–790
Rahman M, Roller J, Zhang S, Syk I, Menger MD, Jeppsson B, Thorlacius H (2012) Metalloproteinases regulate CD40L shedding from platelets and pulmonary recruitment of neutrophils in abdominal sepsis. Inflamm Res 61(6):571–579. Official journal of the European Histamine Research Society [et al]
Rahman M, Zhang S, Chew M, Syk I, Jeppsson B, Thorlacius H (2013) Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J Thromb Haemost 11(7):1385–1398
Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, Grissom CK, Weyrich AS, Zimmerman GA (2011) The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost 9(4):748–758
Russwurm S, Vickers J, Meier-Hellmann A, Spangenberg P, Bredle D, Reinhart K, Losche W (2002) Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock 17(4):263–268
Schubert S, Schwertz H, Weyrich AS, Franks ZG, Lindemann S, Otto M, Behr H, Loppnow H, Schlitt A, Russ M, Presek P, Werdan K, Buerke M (2011) Staphylococcus aureus alpha-toxin triggers the synthesis of B-cell lymphoma 3 by human platelets. Toxins 3(2):120–133
Semple JW, Italiano JE Jr, Freedman J (2011) Platelets and the immune continuum. Nat Rev Immunol 11(4):264–274
Siboo IR, Chambers HF, Sullam PM (2005) Role of SraP, a Serine-Rich Surface Protein of Staphylococcus aureus, in binding to human platelets. Infect Immun 73(4):2273–2280
Sullam PM, Hyun WC, Szollosi J, Dong J, Foss WM, Lopez JA (1998) Physical proximity and functional interplay of the glycoprotein Ib-IX-V complex and the Fc receptor FcgammaRIIA on the platelet plasma membrane. J Biol Chem 273(9):5331–5336
Sun B, Li J, Kambayashi J (1999) Interaction between GPIbalpha and FcgammaIIA receptor in human platelets. Biochem Biophys Res Commun 266(1):24–27
Tang YQ, Yeaman MR, Selsted ME (2002) Antimicrobial peptides from human platelets. Infect Immun 70(12):6524–6533
Thomer L, Schneewind O, Missiakas D (2013) Multiple ligands of von Willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J Biol Chem 288(39):28283–28292
Tohidnezhad M, Varoga D, Podschun R, Wruck CJ, Seekamp A, Brandenburg LO, Pufe T, Lippross S (2011) Thrombocytes are effectors of the innate immune system releasing human beta defensin-3. Injury 42(7):682–686
Tohidnezhad M, Varoga D, Wruck CJ, Podschun R, Sachweh BH, Bornemann J, Bovi M, Sonmez TT, Slowik A, Houben A, Seekamp A, Brandenburg LO, Pufe T, Lippross S (2012) Platelets display potent antimicrobial activity and release human beta-defensin 2. Platelets 23(3):217–223
Trier DA, Gank KD, Kupferwasser D, Yount NY, French WJ, Michelson AD, Kupferwasser LI, Xiong YQ, Bayer AS, Yeaman MR (2008) Platelet antistaphylococcal responses occur through P2X1 and P2Y12 receptor-induced activation and kinocidin release. Infect Immun 76(12):5706–5713
Tzeng DY, Deuel TF, Huang JS, Baehner RL (1985) Platelet-derived growth factor promotes human peripheral monocyte activation. Blood 66(1):179–183
Vasina E, Heemskerk JW, Weber C, Koenen RR (2010) Platelets and platelet-derived microparticles in vascular inflammatory disease. Inflamm Allergy Drug Targets 9(5):346–354
Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA (2012) Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol 34(1):5–30
Vincent JL, Abraham E (2006) The last 100 years of sepsis. Am J Respir Crit Care Med 173(3):256–263
Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I (2005) Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost 94(4):831–838
Warkentin TE, Aird WC, Rand JH (2003) Platelet-endothelial interactions: sepsis, HIT, and antiphospholipid syndrome. Hematology (Am Soc Hematol Educ Program): 497–519
Weyrich AS, Zimmerman GA (2004) Platelets: signaling cells in the immune continuum. Trends Immunol 25(9):489–495
Weyrich AS, Lindemann S, Zimmerman GA (2003) The evolving role of platelets in inflammation. J Thromb Haemost 1(9):1897–1905
White JG (2006) Why human platelets fail to kill bacteria. Platelets 17(3):191–200
Worth RG, Chien CD, Chien P, Reilly MP, McKenzie SE, Schreiber AD (2006) Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Exp Hematol 34(11):1490–1495
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT (2009) Extracellular histones are major mediators of death in sepsis. Nat Med 15(11):1318–1321
Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, Oppenheim JJ (2003) Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol 74(3):448–455
Yeaman MR (2010) Platelets in defense against bacterial pathogens. Cell Mol Life Sci 67(4):525–544
Yeaman MR, Yount NY, Waring AJ, Gank KD, Kupferwasser D, Wiese R, Bayer AS, Welch WH (2007) Modular determinants of antimicrobial activity in platelet factor-4 family kinocidins. Biochim Biophys Acta 1768(3):609–619
Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, Chandler NB, Rodesch CK, Albertine KH, Petti CA, Weyrich AS, Zimmerman GA (2009) Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood. doi: blood-2008-07-171629
Yost CC, Weyrich AS, Zimmerman GA (2010) The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie 92(6):692–697
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16(11):1438–1444
Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM (2002) Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 99(11):4021–4029
Zhang S, Rahman M, Zhang S, Qi Z, Thorlacius H (2011) Simvastatin antagonizes CD40L secretion, CXC chemokine formation, and pulmonary infiltration of neutrophils in abdominal sepsis. J Leukoc Biol 89(5):735–742
Acknowledgments
We thank Ms. Diana Lim for her excellent figure preparation. This work was supported by the NIH (HL112311 and HL126547 to M.T.R.), the NIA (AG048022 to M.T.R.), and the George E. Wahlen Department of Veterans Affairs Geriatric Research Education and Clinical Center (GRECC).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Rondina, M.T., Garraud, O., Schwertz, H. (2017). Platelets and Bacterial Infections. In: Gresele, P., Kleiman, N., Lopez, J., Page, C. (eds) Platelets in Thrombotic and Non-Thrombotic Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-47462-5_71
Download citation
DOI: https://doi.org/10.1007/978-3-319-47462-5_71
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47460-1
Online ISBN: 978-3-319-47462-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)