
Abstract
Fungal diseases cause life-threatening illnesses such as meningitis and pneumonias, chronic asthma, other respiratory diseases, and recurrent diseases like oral and vaginal thrush. Invasive fungal infections are a consequence of underlying health problems often associated with immunosuppression [1]. Fungal infections often carry high mortality and successful patient management requires antifungal therapy. Yet, treatment options remain extremely limited due to limited classes of antifungal agents and by the emergence of prominent antifungal drug resistance. Currently registered antifungal drugs represented by polyenes and azoles, flucytosine, and echinocandins target the cell membrane, nucleic acid biosynthesis, and cell wall, respectively [2]. The latter and most recently approved class, the echinocandins, are now recommended as primary therapy for non-neutropenic patients with invasive candidiasis [3]. It is estimated that 60 % of candidemia patients now receive an echinocandin for treatment or prophylaxis [4]. As worldwide use of echinocandins broadens, clinical failures due to resistant organisms are a concern, especially among certain Candida species. The development of echinocandin resistance among most susceptible organisms like Candida albicans is an uncommon event. Yet, there is a disturbing trend of increased resistance among strains of Candida glabrata, which are frequently cross-resistant to azole drugs. Echinocandin resistance is acquired during therapy and its mechanism is firmly established to involve amino acid changes in “hot-spot” regions of the Fks subunits of the target glucan synthase. These changes significantly decrease the sensitivity of the enzyme to drug resulting in higher MIC values and reduced pharmacodynamic responses. Biological factors that promote selection of Fks-resistant strains involve complex cellular stress response pathways. The use of broth microdilution assays to assess susceptibility can be problematic with some drug- and species-related variability among clinical microbiology laboratories. Clinical factors promoting resistance include expanding use of echinocandins for therapy and prophylaxis, and localized reservoirs such as those in the gastrointestinal tract or intra-abdominal infections, which can seed emergence of resistant organisms. A basic understanding of the resistance mechanism, along with cellular and clinical factors promoting resistance, will promote better strategies to overcome and prevent echinocandin resistance.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Antifungal therapy
- Patient management
- Glucan synthase
- Candida albicans
- “Hot-spot” regions
- Broth microdilution assays
- Biofilms
- FKS resistance
1 Introduction
Fungal diseases cause life-threatening illnesses such as meningitis and pneumonias, chronic asthma, other respiratory diseases, and recurrent diseases like oral and vaginal thrush. Invasive fungal infections are a consequence of underlying health problems often associated with immunosuppression [1]. Fungal infections often carry high mortality and successful patient management requires antifungal therapy. Yet, treatment options remain extremely limited due to restricted classes of antifungal agents and by the emergence of prominent antifungal drug resistance. Currently registered antifungal drugs represented by polyenes and azoles, flucytosine, and echinocandins target the cell membrane, nucleic acid biosynthesis, and cell wall, respectively [2]. The latter and most recently approved class, the echinocandins, are now recommended as primary therapy for non-neutropenic patients with invasive candidiasis [3]. It is estimated that 60 % of candidemia patients now receive an echinocandin for treatment or prophylaxis [4]. As worldwide use of echinocandins broadens, clinical failures due to resistant organisms are a concern, especially among certain Candida species. The development of echinocandin resistance among most susceptible organisms like Candida albicans is an uncommon event. Yet, there is a disturbing trend of increased resistance among strains of Candida glabrata, which are frequently cross-resistant to azole drugs. Echinocandin resistance is acquired during therapy and its mechanism is firmly established to involve amino acid changes in “hot-spot” regions of the Fks subunits of the target glucan synthase. These changes significantly decrease the sensitivity of the enzyme to drug resulting in higher MIC values and reduced pharmacodynamic responses. Biological factors that promote selection of Fks-resistant strains involve complex cellular stress response pathways. The use of broth microdilution assays to assess susceptibility can be problematic with some drug- and species-related variability among clinical microbiology laboratories. Clinical factors promoting resistance include expanding use of echinocandins for therapy and prophylaxis, and localized reservoirs such as those in the gastrointestinal tract or intra-abdominal infections, which can seed emergence of resistant organisms. A basic understanding of the resistance mechanism, along with cellular and clinical factors promoting resistance, will promote better strategies to overcome and prevent echinocandin resistance.
2 Fungal Cell Walls and 1,3-β-d-Glucan
The cell wall of human fungal pathogens is essential for maintaining cell shape and rigidity. It consists primarily of an interwoven mesh of glucans, mannoproteins, and chitin. In yeasts like Candida albicans, branched fibrils of 1,3-β-d glucan form a network that acts as a scaffold for other macromolecules [5, 6]. Short 1-6-β-d-glucan chains establish bridges between linear 1,3-β-d glucan and cell wall proteins that coat the external surface of the cell wall. The majority of these proteins are heavily mannosylated through both O- and N-glycosidic linkages. Most cell wall proteins are covalently linked to the growing wall structure via 1-6-β-d-glucan. Chitin is found both below the network of 1,3-β-d glucan and as a linker between glucans. In other pathogenic fungi, including Aspergillus fumigatus and Cryptococcus neoformans, many of the same polysaccharides and mannoproteins are found in the cell wall, but the organization is somewhat different [7, 8] as polymers occur with other linkages between glucose units or sugars (e.g., galactomannan) [9]. When synthesis of a functional cell wall is reduced or eliminated, either by gene disruption or by inhibition with an antifungal inhibitor, cell growth is often adversely impacted leading to lysis and death.
3 Glucan Synthase
The fungal-specific enzyme 1,3-β-d glucan synthase (GS) is responsible for the biosynthesis of the central cell wall building block 1,3-β-d glucan. The enzyme is a membrane-associated complex that uses UDP-glucose to synthesize a 1,3-β-d glucan polysaccharide product 60 to 80 glucose residues in length. The enzyme has been extensively studied in S. cerevisiae [10], although it has also been studied in other yeasts and molds including Neurospora crassa, Aspergillus nidulans, and Aspergillus fumigatus; Schizosaccharomyces pombe; various Candida species; and Cryptococcus neoformans. GS is minimally a heterodimer involving a large integral membrane protein, encoded by FKS genes, that catalyzes the biosynthesis of 1,3-β-d-glucan and Rho, a regulatory GTP-binding protein. The FKS and RHO1 genes are conserved across numerous fungal genera. A high degree of homology among members of the FKS gene family aided cloning of paralogs from C. albicans [11, 12], C. neoformans [13], A. fumigatus [14], Neurospora crassa [15], P. carinii [16], and other fungi [10]. Conservation of FKS extends to the plant kingdom as well, where an FKS homolog is associated with synthesis of plant 1,3-β-d glucan (callose) in cotton and barley [17, 18]. Likewise, RHO1 genes have been identified and characterized in C. albicans [19], C. neoformans [20], and A. fumigatus [14]. Most yeast have three FKS genes, FKS1, FKS2, and FKS3. The FKS1 gene is essential in C. albicans [12, 13] and other Candida spp., while in C. glabrata, FKS1 and FKS2 are functionally redundant [21]. The FKS3 gene is expressed at a very low level relative to the other genes and its role is uncertain [22]. The GS enzyme complex has not been crystallized but it can be studied in an enriched form by a product entrapment technique [23, 24], which has allowed an evaluation of its kinetic properties [25].
4 Glucan Synthase Inhibitors and Echinocandins
There are three structural classes that define natural product inhibitors of 1,3-β-d glucan synthesis [10]. The first class are the lipopeptides including echinocandins, aerothricin lipopeptidolactones, and arborcandins. A second class comprises the glycolipid papulacandins, and a third class, the terpenoids, are represented by enfumafungin, ascosteroside, arundifungin, and ergokonin A. All GS inhibitor classes are noncompetitive with the biosynthetic substrate UDP-glucose. Cells exposed to GS inhibitors distort and lyse due to changes in cell wall glucans [26–28]. Of the three GS inhibitor classes, the echinocandins are best studied. The echinocandins are cyclic hexapeptides with an amide-linked fatty acyl side chain [29]. An early striking feature of this class was the potent activity of echinocandins in animal infection models due to C. albicans [30] and Pneumocystis jiroveci [31]. This led to medicinal chemistry efforts at Merck, Eli Lilly, and Fujisawa (Astellas) and the development of current semisynthetic echinocandins caspofungin, anidulafungin, and micafungin, respectively [32]. The US Food and Drug Administration has approved echinocandin drugs for the treatment of esophageal and invasive candidiasis, including candidemia, empirical therapy in febrile neutropenic patients, and prophylaxis in patients undergoing hematopoietic stem cell transplantation (HSCT) [33, 34]. The first in-class drug caspofungin was also approved for salvage therapy for patients with invasive aspergillosis refractory to conventional therapy [35]. Echinocandin drugs show in vitro fungicidal activity against susceptible Candida spp. [36, 37], although they are fungistatic against molds where they alter morphology, cell wall composition, and organization [38, 39]. The echinocandins are largely inactive against invasive Zygomycetes, Cryptococcus spp., or Fusarium spp. As echinocandin drugs have a distinct mechanism of action specific for glucan synthase, they are highly effective against yeasts with reduced susceptibility to azoles, such as C. glabrata and C. krusei [40–42]; they are also active against some Candida biofilms [43–46]. The echinocandins have an excellent therapeutic index with a low potential for renal or hepatic toxicity or serious drug-drug interactions [47, 48].
5 Antifungal Spectrum and Breakpoints
The CLSI and EUCAST have established standardized microbroth dilution susceptibility tests for Candida and echinocandins, which show uniformly potent activity against most Candida species including C. albicans, C. glabrata, Candida tropicalis, and Candida krusei [49, 50]. The C. parapsilosis complex (Candida parapsilosis sensu stricto, C. orthopsilosis, and C. metapsilosis) and C. guilliermondii are notable exceptions displaying higher echinocandin antifungal MIC values relative to other highly susceptible Candida species [51–56]. Intrinsic reduced susceptibility has an unclear clinical significance, as patients infected with these strains are successfully treated with echinocandin drugs [57], although clinical response may vary with patient population [58–60]. The effect of echinocandins on filamentous fungi in vitro is less prominent with molds like A. fumigatus and other Aspergillus spp., showing reduced growth and altered hyphae morphology [39]. The multidrug-resistant pathogen Aspergillus lentulus is largely unresponsive to echinocandin action [61]. For A. fumigatus, the echinocandin-induced change in cell wall morphology correlates with exposure of masked epitopes (e.g., 1,3-β-d glucan), which promote a robust immune response contributing to in vivo efficacy [62]. Echinocandins show similar in vitro behavior with black molds such as Alternaria spp., and hyalohyphomycetes such as Scedosporium apiospermum [63]. In contrast, Rhizopus oryzae and other zygomycetes are largely unaffected by caspofungin [64]. Micafungin is active against mycelial forms of Histoplasma capsulatum, Blastomyces dermatitidis, and Coccidioides immitis but it is less active against yeast-like forms [65]. Like Aspergillus species, dermatophytes Trichophyton rubrum and Microsporum canis show diminished growth and malformed hyphae in response to echinocandins [66]. Finally, the neurotropic pathogen Cryptococcus neoformans is unresponsive to echinocandins [67, 68]. However, in vitro susceptibility can be overcome by addition of the calcineurin inhibitor FK506 [69]. Epidemiologic cutoff values (ECVs) have been determined for echinocandins against the most clinically important yeasts and molds from numerous global surveillance studies verifying the potent behavior of these drugs [70, 71]. The CLSI and EUCAST have also established species- and drug-specific clinical breakpoints (CBP) for echinocandin drugs based on extensive pharmacokinetic, microbiological, enzyme kinetic, and clinical response data [72, 73] (Table 29.1). See section on “Standardized Testing for Resistance.”
6 Epidemiology of Echinocandin Resistance
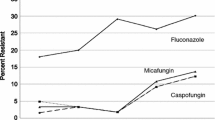
Candida species isolates resistant to echinocandin drugs were first reported in 2005 [74]. Their frequency remains relatively low at less than 2–3 % with C. albicans and most other Candida species [75–78]. Yet, consistent with the broader application of echinocandin therapy, high MIC clinical isolates associated with clinical failures are more commonly reported [22, 25, 79–89]. Despite these reports, echinocandin resistance among most Candida species has been largely unchanged in the past decade [90]. However, this is not the case for C. glabrata, where echinocandin resistance is rising and there is serious cause for concern since many isolates also display azole resistance [91–93], which greatly limits therapy. The SENTRY Antimicrobial Surveillance Program reported echinocandin resistance of 8.0–9.3 % among bloodstream isolates (BSI) of C. glabrata from 2006 to 2010 [92]. In a study of C. glabrata bloodstream isolates from Duke hospital spanning 10 years, echinocandin resistance of C. glabrata rose from 2 to 3 % in 2001–2006 to more than 13 % in 2009–2010 [91]. Resistance is not uniform, as a study involving 1380 isolates of C. glabrata collected between 2008 and 2013 from four US cities showed that 3.1–3.6 % of the isolates were resistant to the echinocandin drugs [93]. This is consistent with rates of 3.6 and 5.7 % from anidulafungin and caspofungin, respectively, obtained from regional data of Candida non-albicans strains at US medical centers over a 6-year period (2006–2011) [90]. Yet, echinocandin resistance among C. glabrata has also coincided with a nearly parallel rise in azole resistance resulting in multidrug-resistant strains (Fig. 29.1). In a recent study covering 1032 isolates, nearly all isolates containing an FKS mutation were resistant to at least one echinocandin and 36 % were also resistant to fluconazole [93]. The expanding use of echinocandin and azole prophylaxis in many healthcare centers has prompted an epidemiologic shift with C. glabrata emerging as the most dominant fungal bloodstream pathogen [94, 95]. The development of echinocandin resistance typically occurs after prolonged therapy (3–4 weeks or longer) [87]. Yet, it has been observed to emerge shortly after the start of therapy [88, 96]. Echinocandin resistance in molds is rarely encountered but it has been reported for A. fumigatus [97] and the inherently multidrug-resistant A. lentulus [61].

Rise in antifungal resistance of Candida glabrata to azole (fluconazole) and echinocandin (anidulafungin, caspofungin, and micafungin) drugs from 2001 to 2010. Adapted from Alexander et al. [91]
7 Mechanism of Acquired Resistance
Echinocandin resistance resulting in clinical failures due to high MIC isolates involves modification of the catalytic subunit of glucan synthase, which is encoded by genes FKS1 and/or FKS2. Echinocandin drugs are not substrates for multidrug transporters like azole drugs [42], and other cellular mechanisms conferring azole resistance do not affect echinocandin susceptibility. This has led to the recommendation of echinocandins as preferred therapy for infections involving azole-resistant strains of Candida. Echinocandin resistance is well characterized and known to be conferred by restricted mutations in two highly conserved “hot-spot” regions of the FKS genes [34] (Table 29.2). These fks mutations result in amino acid substitutions that induce elevated MIC values from 20- to 100-fold and reduced sensitivity of glucan synthase (IC50) to drug by 50- to 3000-fold [22, 25, 99]. These less susceptible fks mutant strains respond poorly to echinocandin drugs in pharmacodynamic models of infection [100–103], and the manifestation of characteristic fks mutations is associated with reduced clinical response [104–106]. The presence of an FKS mutation was found to be the only independent risk factor associated with echinocandin failure among C. glabrata isolates in a study of patients with invasive candidiasis [105]. The FKS resistance mechanism has been observed in many Candida species including C. albicans, C. glabrata, C. tropicalis, C. krusei, C. kefyr, and C. lusitaniae [96, 107, 108]. In all Candida species, except C. glabrata, mutations occur within two “hot-spot” regions of FKS1, encoding residues Phe641-Pro649 and Arg1361 (Table 29.2). In C. albicans, amino acid substitutions at Ser645 and Phe641 are the most abundant (Table 29.2). In C. glabrata, mutations occur in the homologous hot-spot regions of FKS1 and FKS2 [22, 99], although mutations are observed within FKS2 at twice the frequency of FKS1 [22, 34, 109]. Amino acid substitutions at Fks1 positions F625 and S629 and Fks2 positions F659 and S663 are most prominent inducing elevated MIC values (Table 29.2) [98]. In some cases, nonsense mutations and deletions are observed in FKS1 or FKS2 in C. glabrata [22, 98, 112]. Mutations in FKS1 or FKS2 can significantly alter the relative expression of their genes [21, 22], which can influence susceptibility. In C. glabrata, FKS2 expression is calcineurin dependent and downregulated by FK506 [111], and echinocandin resistance conferred by mutations in FKS2 are mitigated with FK506 [21]. A third hot-spot modification W695 (S. cerevisiae) was recently identified by in vitro selection [112], but it is not associated with clinical failures.
8 Biofilms
Biofilms also play a factor in resistance. They are one of the most important microbial communities encountered in nature, and they are well established to contribute to antifungal drug resistance [113]. It has been shown for echinocandin drugs that the extensive production of β-glucan within the extracellular glucan matrix helps sequester drugs by decreasing their concentration at the cell membrane surface [114]. Decreasing glucan productions, either by genetic or chemical means, increases the susceptibility to antifungal agents [115]. Genetic factors that regulate glucan formation promoting drug-sequestering biofilms include Rlm, Smi1, and glucan synthase Fks1 [115].
9 Acquired Resistance and Microbial Fitness
It is a well-established microbial paradigm that drug resistance often carries a fitness cost for microorganisms. The most prominent amino acid substitutions (e.g., Ser645 in C. albicans) in hot-spot regions of Fks subunits have been shown to decrease the catalytic efficiency for glucan biosynthesis [22, 25]. This reduced capacity for glucan production results in compensatory changes that alter cell wall morphology [116], which can reduce the fitness of such mutants. In C. albicans, reduced fitness has been observed for fks mutants in animal models [21, 22, 116]. The fks mutant strains compete weakly with their wild-type equivalents [116]. This reduced competition may account for the observation that resistance is with acquired during therapy and patient-patient transmission is not observed.
10 Cellular Stress and Drug Tolerance
The inhibition of glucan synthase following exposure of cells to an echinocandin drug induces significant cellular stress. In response, fungi activate a wide range of adaptive mechanisms that promote survival by helping protect against cell stress [117, 118]. These stress adaptation responses often result in drug-tolerant cells with elevated in vitro MIC values to echinocandins. Yet, they are not typically associated with clinical failures [119–121], as drug-exposed cells are less robust because glucan synthase is inhibited. Cell wall stress is sensed by receptors such Mtl2 and Wsc1, which induce stress tolerance involving cell wall integrity, protein kinace C (PKC), calcineurin-Crz1, and HOG [122, 123] interacting pathways. Hsp90 is an important protein that helps induce tolerance through its major client proteins calcineurin, along with its effector Crz1 [124–126]. Genetic or chemical impairment of Hsp90 function diminishes the ability of C. albicans and C. glabrata to develop tolerance in the presence of caspofungin [126, 127].
Chitin and glucans comprise the major structural components of the fungal cell wall and there is a prominent biosynthetic interdependence for both constituents [128]. Therefore, it is not surprising that echinocandin exposure results in compensatory increases in chitin synthesis to strengthen the cell wall and resistant drug action. Cell wall mutants with higher basal chitin contents are less susceptible to caspofungin [122, 123, 129, 130] and they confer reduced pharmacodynamics responses in animal model [131]. Paradoxical growth at very high drug levels has also been linked to prominent compensatory responses in chitin biosynthesis [132, 133]. Finally, defects in sphingolipid biosynthesis can differentially alter in drug-dependent fashion responses to echinocandin drugs. This mixed susceptibility phenotype is linked to interactions of the aliphatic tail of echinocandins and membrane sphingolipids [134, 135].
In general, tolerance pathways are insufficient to result in clinical drug failure. Yet, they are important for stabilizing cells in the presence of drug, and may account for stasis behavior of cells exposed to echinocandin drugs in animal model systems [102]. Even though these cells are not sufficiently resistant to induce therapeutic failures, they are poised to develop higher level resistance, as the drug-tolerant state allows cells sufficient time to overcome drug action by forming stable FKS mutations. It is not entirely clear how this ultimately occurs, although it may involve defects in DNA repair. Genome plasticity, observed widely in C. albicans and C. glabrata in response to azole drugs [136, 137], may also emerge as a factor for echinocandin drugs [138].
11 Mechanisms of Inherent Reduced Susceptibility
Candida parapsilosis complex (C. parapsilosis sensu stricto, Candida orthopsilosis, and Candida metapsilosis) and C. guilliermondii are intrinsically less susceptible in vitro to echinocandin drugs (MIC 0.5–8 μg/mL) relative to other highly susceptible Candida species [70, 95, 139], which prompted the CLSI to adopt higher breakpoints [73]. The clinical significance of this reduced susceptibility is unclear since patients can be successfully treated with echinocandins at standard dosages [54–56]; however, clinical efficacy may vary with patient population [58–60]. The underlying molecular mechanism appears to be naturally occurring polymorphisms in FKS hot-spot regions, which confer reduced sensitivity of glucan synthase to drug [140]. In C. parapsilosis complex, a highly conserved Pro660 is converted to alanine at the distal edge of hot-spot 1. Enzyme kinetic inhibition studies demonstrated that glucan synthase from the C. parapsilosis group were 10- to 50-fold less to echinocandin drugs than from enzymes obtained from highly susceptible species like C. albicans [140]. Furthermore, an engineered lab strain and clinical isolates of C. albicans and C. glabrata strains containing amino acid substitutions at this position display comparable decreases in target enzyme sensitivity and increased MIC values [140]. An additional I1359V polymorphism is observed in hot-spot 2 of C. orthopsilosis and S. cerevisiae, which confers higher MIC values. C. guilliermondii shows several additional amino acid polymorphisms in HS1 [140], although their relative contribution to overall insensitivity is unclear.
Cryptococcus neoformans is inherently resistant to echinocandin drugs even though 1,3 glucan synthase is essential and appears fully inhibited by echinocandin drugs in vitro [141]. It has been suggested that capsular melanin may help protect but capsule-deficient strains are also unresponsive to drug [142]. Finally, Aspergillus lentulus, a sibling species of A. fumigatus, is inherently resistant to a wide range of antifungal drugs including the echinocandins. The mechanism of this resistant is unclear but appears to be independent of FKS mutations [143].
12 Serum Effects on Drug Action
The echinocandin drugs are highly serum protein bound (>98 %), which reduces their relative in vitro efficacy causing a shift in MIC [144–146]. The magnitude of the shift depends on the specific drugs with anidulafungin and micafungin showing a larger relative shift than caspofungin. A consequence of this shift in efficacy is that serum alters the relative fungicidal properties of the drugs, often resulting in fungistatic behavior against certain Candida species [147, 148]. The serum effects are more pronounced with mutant strains carrying FKS mutations [149].
13 Standardized Testing for Resistance
The Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) have established comparable standards for broth microdilution (BMD) antifungal susceptibility testing of echinocandins against Candida species [53, 150, 151]. The objective for susceptibility testing is to establish an in vitro assessment that differentiates infecting strains as either susceptible or likely to respond to therapy or as resistant with an enhanced probability to fail therapy. In the case of echinocandin drugs, it is essential to capture high MIC strains containing FKS mutations. Initially, the CLSI used clinical and microbiological data to establish a preliminary common clinical breakpoint (CBP) for all three echinocandins against Candida spp. [120]. However, resistant strains with FKS mutations were often misclassified by this CBP [25, 152]. In response, the CLSI revised the CBP based on pharmacokinetic, microbiological, enzyme kinetic, and clinical data and established new species- and drug-specific breakpoints that better accounted for strains containing FKS mutations [73] (Table 29.3). However, the lower CBPs presented a clinical microbiology testing challenge, as BMD testing using either CLSI and EUCAST failed to promote consistent inter-laboratory test results without major errors (misclassifying wild-type strains as resistant or fks-containing mutants as susceptible) between laboratory groups [153–154]. Disturbingly, there were wide modal ranges encountered with C. glabrata and caspofungin [153–155]. Consistent MIC results were obtained for micafungin and anidulafungin, and it was suggested that they could serve as testing surrogates for the class to assess resistance [98, 156, 157]. EUCAST has now established species-specific clinical breakpoints for micafungin against C. albicans, C. glabrata, and C. parapsilosis [72], and they have established breakpoints for anidulafungin to accommodate use of these compounds in some clinical situations [72, 158]. EUCAST has not set caspofungin breakpoints and does not currently recommend caspofungin MIC testing for clinical decision making involving echinocandin drugs [72]. Epidemiological cutoff values (ECV or ECOFF), which define the upper limit of the wild-type MIC population in the absence of a known resistance mechanism [49, 159], have been defined for anidulafungin and micafungin against common Candida species (Table 29.3). The ECV does not replace the BP, but it provides additional information for clinical decision making when a BP is not available. Although the designation of NWT does not allow a clinician to determine whether a particular isolate will respond to a particular antifungal agent, it does allow for a more informed decision based on how wild-type organisms would likely respond to therapy.
Rather than seeking testing surrogates or special conditions for BMD to distinguish wild-type strains from resistant isolates containing an FKS hot-spot mutation, it has been suggested that direct molecular testing for resistance mutations may provide a reliable alternative [160]. Direct DNA sequencing or real-time probing with allele-specific molecular probes provides an easy and unequivocal assessment of the resistance potential. The presence of an FKS mutation is the most important independent risk factor in predicting echinocandin therapeutic responses among patients with invasive candidiasis [104, 105, 110], which is well supported by extensive pharmacodynamics, MIC, and biochemical data [161, 162]. One criticism of this approach is that molecular testing requires specific knowledge of known resistance mechanisms and an unknown mechanism would not be detected. Yet, this probability is sufficiently remote given the large body of current data. Molecular testing to directly identify mutant strains containing FKS mutations would eliminate the current controversy surrounding some susceptibility testing, which prevents an accurate determination of resistance.
14 Paradoxical Growth Effects
The “paradoxical effect” refers to the unusual behavior of echinocandin drugs in susceptibility testing assays to show strong growth inhibition at low and moderate levels of drugs and then loss of inhibition at supra high drug concentrations, well in excess of the MIC. First described by Stevens and colleagues, it is a commonly observed property of echinocandin drugs [163]. This behavior is largely conditional as paradoxical strains show normal susceptibility properties following culture. The mechanism responsible for paradoxical growth is unclear, but is unrelated to mutations in FKS [124, 164]. It is not due to antifungal degradation or instability. The drug-induced growth behavior is more consistent with adaptive stress responses, which can lead to reduced susceptibility. In one instance, a paradoxical C. albicans strain showed a 900 % increase in chitin content [133]. Consistent with changes in cell wall composition, remodeling is observed [165, 166]. The paradoxical effect is eliminated by serum, chitin synthase inhibitor nikkomycin Z, and calcineurin pathway inhibitors [167], and in C. albicans mutants that lack phosphatidylinositol-(4,5)-bisphosphate 5′-phosphatase [167]. Paradoxical behavior has been observed in a murine model of pulmonary aspergillosis [168] and in a patient with pulmonary aspergillosis [169]. Paradoxical growth in response to caspofungin in Candida species does not confer survival advantage in a Drosophila or moth model of candidiasis [165, 170]. The clinical significance of the paradoxical growth remains unclear, as the drug levels necessary to induce it exceed normal human dosing levels.
15 Risk Factors for Resistance Emergence
The gastrointestinal (GI) tract is colonized with Candida species, often at very high burdens [171–178], which are in the form of a complex microbial biofilm [179]. Typically, drug penetration varies across the biofilm and drug concentrations in the glucan matrix are irregular [114]. This creates a drug exposure environment that can select for resistant variants, which may desorb from the biofilm and cause systemic infections. As biofilms are difficult to eradicate, they can form a resistance reservoir that seeds resistant infections. Similarly, intra-abdominal candidiasis occurs in 40 % or more of patients following repeated gastrointestinal surgery, GI perforation, or necrotizing pancreatitis [180]. The high burden of Candida in this protected space with poor drug penetration creates a strong selection for resistant variants. Prophylaxis is another potential source for resistance. Prior and repeated exposure to echinocandin drugs is a risk factor development of resistance. As the FKS resistance mechanism is a prominent risk factor for therapeutic failure [105], resistance emergence is directly linked prior to exposure [106, 181, 182]. Antifungal prophylaxis with an azole or echinocandin class drug is standard prevention in many clinical settings with immunosuppressed patients at high risk for development of invasive fungal infections. Echinocandin drugs have been used because they have favorable pharmacokinetics and safety profile, and they are active against azole-resistant yeasts and molds. Both micafungin and caspofungin have been successfully applied for this purpose in adults [183–186] and children [187]. Meta-analyses have confirmed that echinocandin prophylaxis reduces the incidence of invasive fungal infections greater than fluconazole or itraconazole [188, 189]. Micafungin is FDA approved for prophylaxis of Candida infections in patients undergoing hematopoietic SCT or expected to be neutropenic for at least 10 days [190] and the European Society of Clinical Microbiology and Infectious Diseases guidelines also recommend micafungin for prophylaxis against Candida infections in allogeneic HSCT adult and pediatric patients, as well as in pediatric patients with acute myeloid and recurrent leukemia [191]. A consequence of the expanding use of echinocandins for prophylaxis is that patient drug exposure is on the rise, which has implication for inducing higher rates of echinocandin drug resistance, especially among resistance-prone organisms like C. glabrata. Even more concerning is the high prevalence of multidrug-resistant C. glabrata isolates cross-resistant to both azole- and echinocandin-class drugs [91, 192–196]. The coevolution of azole and echinocandin multidrug resistance among C. glabrata is an alarming trend [91]. Breakthrough infections involving C. albicans are also reported in patients following transplantation who received micafungin prophylaxis [197]. It is not surprising that broadening patient exposure to echinocandin drugs would promote development of resistance. Echinocandin prophylaxis may continue to fuel an increase in the frequency of isolates that are resistant to multiple classes of antifungal drugs. Furthermore, prior antifungal exposure, especially with fluconazole, leads to genomic instability, which increases azole resistance [138] and may potentially predispose for enhanced mutations leading to FKS-mediated drug resistance.
16 Conclusions
Echinocandin resistance among Candida species is low but significant, especially among C. glabrata where high-frequency resistance is often associated with azole resistance resulting in multidrug-resistant strains. Characteristic mutations in hot-spot regions of FKS genes encoding glucan synthase remain the most significant factor responsible for resistant isolates that are refractory to therapy. However, in response to echinocandin action, cellular stress response pathways induce drug-adapted persister states, which can ultimately facilitate development of stable FKS-resistant genotypes. Host factors that promote resistance include biofilm formation within the gastrointestinal tract and intra-abdominal candidiasis. The widespread use of echinocandin prophylaxis needs to be monitored for its effects on promoting enhanced drug exposure and resistance emergence. Effective antibiotic stewardship is required, especially in certain settings where resistance is prominent. Finally, new drug- and species-specific breakpoints have resulted in testing challenges, which may require drug surrogates for the class, but it may be more prudent to transition to sequence-based evaluation of FKS genotypes as the new gold standard for resistance assessment for all echinocandin drugs.
References
Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, et al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4:165rv113.
Odds FC, Brown AJ, Gow NA. Antifungal agents: mechanisms of action. Trends Microbiol. 2003;11:272–9.
Pappas PG, Kauffman CA, Andes D, Benjamin Jr DK, Calandra TF, et al. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:503–35.
Cleveland AA, Farley MM, Harrison LH, Stein B, Hollick R, et al. Changes in incidence and antifungal drug resistance in candidemia: results from population-based laboratory surveillance in Atlanta and Baltimore, 2008–2011. Clin Infect Dis. 2012;55:1352–61.
Klis FM, Mol P, Hellingwerf K, Brul S. Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2002;26:239–56.
Klis FM, de Groot P, Hellingwerf K. Molecular organization of the cell wall of Candida albicans. Med Mycol. 2001;39 Suppl 1:1–8.
Bernard M, Latge JP. Aspergillus fumigatus cell wall: composition and biosynthesis. Med Mycol. 2001;39:9–17.
Reese AJ, Doering TL. Cell wall alpha-1,3-glucan is required to anchor the Cryptococcus neoformans capsule. Mol Microbiol. 2003;50:1401–9.
Fukazawa Y, Kagaya K, Shinoda T. Cell wall polysaccharides of pathogenic yeasts. Curr Top Med Mycol. 1995;6:189–219.
Douglas CM. Fungal beta(1,3)-D-glucan synthesis. Med Mycol. 2001;39:55–66.
Douglas CM, D’Ippolito JA, Shei GJ, Meinz M, Onishi J, et al. Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-beta-D-glucan synthase inhibitors. Antimicrob Agents Chemother. 1997;41:2471–9.
Mio T, Adachi-Shimizu M, Tachibana Y, Tabuchi H, Inoue SB, et al. Cloning of the Candida albicans homolog of Saccharomyces cerevisiae GSC1/FKS1 and its involvement in beta-1,3-glucan synthesis. J Bacteriol. 1997;179:4096–105.
Thompson JR, Douglas CM, Li W, Jue CK, Pramanik B, et al. A glucan synthase FKS1 homolog in cryptococcus neoformans is single copy and encodes an essential function. J Bacteriol. 1999;181:444–53.
Beauvais A, Bruneau JM, Mol PC, Buitrago MJ, Legrand R, et al. Glucan synthase complex of Aspergillus fumigatus. J Bacteriol. 2001;183:2273–9.
Schimoler-O’Rourke R, Renault S, Mo W, Selitrennikoff CP. Neurospora crassa FKS protein binds to the (1,3)beta-glucan synthase substrate, UDP-glucose. Curr Microbiol. 2003;46:408–12.
Kottom TJ, Limper AH. Cell wall assembly by Pneumocystis carinii. Evidence for a unique gsc-1 subunit mediating beta-1,3-glucan deposition. J Biol Chem. 2000;275:40628–34.
Li J, Burton RA, Harvey AJ, Hrmova M, Wardak AZ, et al. Biochemical evidence linking a putative callose synthase gene with (1 → 3)-beta-D-glucan biosynthesis in barley. Plant Mol Biol. 2003;53:213–25.
Cui X, Shin H, Song C, Laosinchai W, Amano Y, et al. A putative plant homolog of the yeast beta-1,3-glucan synthase subunit FKS1 from cotton (Gossypium hirsutum L.) fibers. Planta. 2001;213:223–30.
Kondoh O, Tachibana Y, Ohya Y, Arisawa M, Watanabe T. Cloning of the RHO1 gene from Candida albicans and its regulation of beta-1,3-glucan synthesis. J Bacteriol. 1997;179:7734–41.
Tanaka K, Nambu H, Katoh Y, Kai M, Hidaka Y. Molecular cloning of homologs of RAS and RHO1 genes from Cryptococcus neoformans. Yeast. 1999;15:1133–9.
Katiyar SK, Alastruey-Izquierdo A, Healey KR, Johnson ME, Perlin DS, et al. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob Agents Chemother. 2012;56:6304–9.
Garcia-Effron G, Lee S, Park S, Cleary JD, Perlin DS. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-D-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob Agents Chemother. 2009;53:3690–9.
Kang MS, Elango N, Mattia E, Au-Young J, Robbins PW, et al. Isolation of chitin synthetase from Saccharomyces cerevisiae. Purification of an enzyme by entrapment in the reaction product. J Biol Chem. 1984;259:14966–72.
Awald P, Zugel M, Monks C, Frost D, Selitrennikoff CP. Purification of 1,3-b-glucan synthase from Neurospora crassa by product entrapment. Exper Mycol. 1993;17:130–41.
Garcia-Effron G, Park S, Perlin DS. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrob Agents Chemother. 2009;53:112–22.
Mizoguchi J, Saito T, Mizuno K, Hayano K. On the mode of action of a new antifungal antibiotic, aculeacin A: inhibition of cell wall synthesis in Saccharomyces cerevisiae. J Antibiot (Tokyo). 1977;30:308–13.
Baguley BC, Rommele G, Gruner J, Wehrli W. Papulacandin B: an inhibitor of glucan synthesis in yeast spheroplasts. Eur J Biochem. 1979;97:345–51.
Onishi J, Meinz M, Thompson J, Curotto J, Dreikorn S, et al. Discovery of novel antifungal (1,3)-beta-D-glucan synthase inhibitors. Antimicrob Agents Chemother. 2000;44:368–77.
Hammond M. Chemical and structure activity studies on the echinocandin lipopeptides. In: Rippon J, Fromtling R, editors. Cutaneous antifungal agents. New York: Marcel Dekker; 1993. p. 395–420.
Bartizal K, Abruzzo G, Trainor C, Krupa D, Nollstadt K, et al. In vitro antifungal activities and in vivo efficacies of 1,3-b-D-glucan synthesis inhibitors L-671,329, L-646,991, tetrahydroechinocandin B, and L-687,781, a papulacandin. Antimicrob Agents Chemother. 1992;36:1648–57.
Schmatz DM, Powles M, McFadden DC, Pittarelli LA, Liberator PA, et al. Treatment and prevention of Pneumocystis carinii pneumonia and further elucidation of the P. carinii life cycle with 1,3-b-glucan synthesis inhibitor L-671,329. J Protozool. 1991;38:151S–3.
Wiederhold NP, Lewis RE. The echinocandin antifungals: an overview of the pharmacology, spectrum and clinical efficacy. Expert Opin Investig Drugs. 2003;12:1313–33.
Turner MS, Drew RH, Perfect JR. Emerging echinocandins for treatment of invasive fungal infections. Expert Opin Emerg Drugs. 2006;11:231–50.
Perlin DS. Current perspectives on echinocandin class drugs. Future Microbiol. 2011;6:441–57.
Walsh TJ, Anaissie EJ, Denning DW, Herbrecht R, Kontoyiannis DP, et al. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:327–60.
Barchiesi F, Spreghini E, Tomassetti S, Arzeni D, Giannini D, et al. Comparison of the fungicidal activities of caspofungin and amphotericin B against Candida glabrata. Antimicrob Agents Chemother. 2005;49:4989–92.
Ernst EJ, Klepser ME, Ernst ME, Messer SA, Pfaller MA. In vitro pharmacodynamic properties of MK-0991 determined by time-kill methods. Diagn Microbiol Infect Dis. 1999;33:75–80.
Bowman JC, Abruzzo GK, Flattery AM, Gill CJ, Hickey EJ, et al. Efficacy of caspofungin against Aspergillus flavus, Aspergillus terreus, and Aspergillus nidulans. Antimicrob Agents Chemother. 2006;50:4202–5.
Bowman JC, Hicks PS, Kurtz MB, Rosen H, Schmatz DM, et al. The antifungal echinocandin caspofungin acetate kills growing cells of Aspergillus fumigatus in vitro. Antimicrob Agents Chemother. 2002;46:3001–12.
Pfaller MA, Messer SA, Boyken L, Rice C, Tendolkar S, et al. Caspofungin activity against clinical isolates of fluconazole-resistant Candida. J Clin Microbiol. 2003;41:5729–31.
Bachmann SP, Patterson TF, Lopez-Ribot JL. In vitro activity of caspofungin (MK-0991) against Candida albicans clinical isolates displaying different mechanisms of azole resistance. J Clin Microbiol. 2002;40:2228–30.
Niimi K, Maki K, Ikeda F, Holmes AR, Lamping E, et al. Overexpression of Candida albicans CDR1, CDR2, or MDR1 does not produce significant changes in echinocandin susceptibility. Antimicrob Agents Chemother. 2006;50:1148–55.
Bachmann SP, Ramage G, VandeWalle K, Patterson TF, Wickes BL, et al. Antifungal combinations against Candida albicans biofilms in vitro. Antimicrob Agents Chemother. 2003;47:3657–9.
Ferreira JA, Carr JH, Starling CE, de Resende MA, Donlan RM. Biofilm formation and effect of caspofungin on biofilm structure of Candida species bloodstream isolates. Antimicrob Agents Chemother. 2009;53:4377–84.
Kuhn DM, George T, Chandra J, Mukherjee PK, Ghannoum MA. Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob Agents Chemother. 2002;46:1773–80.
Simitsopoulou M, Peshkova P, Tasina E, Katragkou A, Kyrpitzi D, et al. Species-specific and drug-specific differences in susceptibility of Candida biofilms to echinocandins: characterization of less common bloodstream isolates. Antimicrob Agents Chemother. 2013;57:2562–70.
Chen SC, Slavin MA, Sorrell TC. Echinocandin antifungal drugs in fungal infections: a comparison. Drugs. 2011;71:11–41.
Kofla G, Ruhnke M. Pharmacology and metabolism of anidulafungin, caspofungin and micafungin in the treatment of invasive candidosis: review of the literature. Eur J Med Res. 2011;16:159–66.
Pfaller MA, Espinel-Ingroff A, Bustamante B, Canton E, Diekema DJ, et al. Multicenter study of anidulafungin and micafungin MIC distributions and epidemiological cutoff values for eight Candida species and the CLSI M27-A3 broth microdilution method. Antimicrob Agents Chemother. 2014;58:916–22.
Pfaller MA, Messer SA, Woosley LN, Jones RN, Castanheira M. Echinocandin and triazole antifungal susceptibility profiles for clinical opportunistic yeast and mold isolates collected from 2010 to 2011: application of new CLSI clinical breakpoints and epidemiological cutoff values for characterization of geographic and temporal trends of antifungal resistance. J Clin Microbiol. 2013;51:2571–81.
Garcia-Effron G, Canton E, Peman J, Dilger A, Roma E, et al. Epidemiology and echinocandin susceptibility of Candida parapsilosis sensu lato species isolated from bloodstream infections at a Spanish university hospital. J Antimicrob Chemother. 2012;67:2739–48.
Spreghini E, Orlando F, Tavanti A, Senesi S, Giannini D, et al. In vitro and in vivo effects of echinocandins against Candida parapsilosis sensu stricto, Candida orthopsilosis and Candida metapsilosis. J Antimicrob Chemother. 2012;67:2195–202.
Pfaller MA, Castanheira M, Diekema DJ, Messer SA, Moet GJ, et al. Comparison of European Committee on Antimicrobial Susceptibility Testing (EUCAST) and Etest methods with the CLSI broth microdilution method for echinocandin susceptibility testing of Candida species. J Clin Microbiol. 2010;48:1592–9.
Mora-Duarte J, Betts R, Rotstein C, Colombo AL, Thompson-Moya L, et al. Comparison of caspofungin and amphotericin B for invasive candidiasis. N Engl J Med. 2002;347:2020–9.
Kale-Pradhan PB, Morgan G, Wilhelm SM, Johnson LB. Comparative efficacy of echinocandins and nonechinocandins for the treatment of Candida parapsilosis infections: a meta-analysis. Pharmacotherapy. 2010;30:1207–13.
Colombo AL, Perfect J, DiNubile M, Bartizal K, Motyl M, et al. Global distribution and outcomes for Candida species causing invasive candidiasis: results from an international randomized double-blind study of caspofungin versus amphotericin B for the treatment of invasive candidiasis. Eur J Clin Microbiol Infect Dis. 2003;22:470–4.
Fernandez-Ruiz M, Aguado JM, Almirante B, Lora-Pablos D, Padilla B, et al. Initial use of echinocandins does not negatively influence outcome in Candida parapsilosis bloodstream infection: a propensity score analysis. Clin Infect Dis. 2014;58:1413–21.
Ghannoum MA, Chen A, Buhari M, Chandra J, Mukherjee PK, et al. Differential in vitro activity of anidulafungin, caspofungin and micafungin against Candida parapsilosis isolates recovered from a burn unit. Clin Microbiol Infect. 2009;15:274–9.
Kabbara N, Lacroix C, Peffault de Latour R, Socie G, Ghannoum M, et al. Breakthrough C. parapsilosis and C. guilliermondii blood stream infections in allogeneic hematopoietic stem cell transplant recipients receiving long-term caspofungin therapy. Haematologica. 2008;93:639–40.
Forrest GN, Weekes E, Johnson JK. Increasing incidence of Candida parapsilosis candidemia with caspofungin usage. J Infect. 2008;56:126–9.
Balajee SA, Gribskov JL, Hanley E, Nickle D, Marr KA. Aspergillus lentulus sp. nov., a new sibling species of A. fumigatus. Eukaryot Cell. 2005;4:625–32.
Hohl TM, Feldmesser M, Perlin DS, Pamer EG. Caspofungin modulates inflammatory responses to Aspergillus fumigatus through stage-specific effects on fungal beta-glucan exposure. J Infect Dis. 2008;198:176–85.
Pfaller MA, Marco F, Messer SA, Jones RN. In vitro activity of two echinocandin derivatives, LY303366 and MK-0991 (L-743,792), against clinical isolates of Aspergillus, Fusarium, Rhizopus, and other filamentous fungi. Diagn Microbiol Infect Dis. 1998;30:251–5.
Espinel-Ingroff A. Comparison of In vitro activities of the new triazole SCH56592 and the echinocandins MK-0991 (L-743,872) and LY303366 against opportunistic filamentous and dimorphic fungi and yeasts. J Clin Microbiol. 1998;36:2950–6.
Nakai T, Uno J, Ikeda F, Tawara S, Nishimura K, et al. In vitro antifungal activity of Micafungin (FK463) against dimorphic fungi: comparison of yeast-like and mycelial forms. Antimicrob Agents Chemother. 2003;47:1376–81.
Motyl M, Nielsen Kahn J, Giacobbe R. In vitro susceptibiity of dermatophytes to CANCIDAS (caspofungin acetate); Chicago: ASM; 2003. p. Abstract M-1210.
Abruzzo GK, Flattery AM, Gill CJ, Kong L, Smith JG, et al. Evaluation of the echinocandin antifungal MK-0991 (L-743,872): efficacies in mouse models of disseminated aspergillosis, candidiasis, and cryptococcosis. Antimicrob Agents Chemother. 1997;41:2333–8.
Bartizal K, Gill CJ, Abruzzo GK, Flattery AM, Kong L, et al. In vitro preclinical evaluation studies with the echinocandin antifungal MK-0991 (L-743,872). Antimicrob Agents Chemother. 1997;41:2326–32.
Del Poeta M, Cruz MC, Cardenas ME, Perfect JR, Heitman J. Synergistic antifungal activities of bafilomycin A(1), fluconazole, and the pneumocandin MK-0991/caspofungin acetate (L-743,873) with calcineurin inhibitors FK506 and L-685,818 against Cryptococcus neoformans. Antimicrob Agents Chemother. 2000;44:739–46.
Pfaller MA, Boyken L, Hollis RJ, Kroeger J, Messer SA, et al. Wild-type MIC distributions and epidemiological cutoff values for the echinocandins and Candida spp. J Clin Microbiol. 2010;48:52–6.
Pfaller MA, Boyken L, Hollis RJ, Kroeger J, Messer SA, et al. Wild-type minimum effective concentration distributions and epidemiologic cutoff values for caspofungin and Aspergillus spp. as determined by Clinical and Laboratory Standards Institute broth microdilution methods. Diagn Microbiol Infect Dis. 2010;67:56–60.
Arendrup MC, Cuenca-Estrella M, Lass-Florl C, Hope WW. Breakpoints for antifungal agents: an update from EUCAST focussing on echinocandins against Candida spp. and triazoles against Aspergillus spp. Drug Resist Updat. 2013;16:81–95.
Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, et al. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat. 2011;14:164–76.
Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother. 2005;49:3264–73.
Pfaller MA, Messer SA, Moet GJ, Jones RN, Castanheira M. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008–2009). Int J Antimicrob Agents. 2011;38:65–9.
Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M. Geographic variations in species distribution and echinocandin and azole antifungal resistance rates among Candida bloodstream infection isolates: report from the SENTRY Antimicrobial Surveillance Program (2008 to 2009). J Clin Microbiol. 2011;49:396–9.
Castanheira M, Woosley LN, Diekema DJ, Messer SA, Jones RN, et al. Low prevalence of fks1 hot spot 1 mutations in a worldwide collection of Candida strains. Antimicrob Agents Chemother. 2010;54:2655–9.
Castanheira M, Messer SA, Jones RN, Farrell DJ, Pfaller MA. Activity of echinocandins and triazoles against a contemporary (2012) worldwide collection of yeast and moulds collected from invasive infections. Int J Antimicrob Agents. 2014;44:320–6.
Cleary JD, Garcia-Effron G, Chapman SW, Perlin DS. Reduced Candida glabrata susceptibility secondary to an FKS1 mutation developed during candidemia treatment. Antimicrob Agents Chemother. 2008;52:2263–5.
Garcia-Effron G, Chua DJ, Tomada JR, DiPersio J, Perlin DS, et al. Novel FKS mutations associated with echinocandin resistance in Candida species. Antimicrob Agents Chemother. 2010;54:2225–7.
Kahn JN, Garcia-Effron G, Hsu MJ, Park S, Marr KA, et al. Acquired echinocandin resistance in a Candida krusei isolate due to modification of glucan synthase. Antimicrob Agents Chemother. 2007;51:1876–8.
Laverdiere M, Lalonde RG, Baril JG, Sheppard DC, Park S, et al. Progressive loss of echinocandin activity following prolonged use for treatment of Candida albicans oesophagitis. J Antimicrob Chemother. 2006;57:705–8.
Miller CD, Lomaestro BW, Park S, Perlin DS. Progressive esophagitis caused by Candida albicans with reduced susceptibility to caspofungin. Pharmacotherapy. 2006;26:877–80.
Garcia-Effron G, Kontoyiannis DP, Lewis RE, Perlin DS. Caspofungin-resistant Candida tropicalis strains causing breakthrough fungemia in patients at high risk for hematologic malignancies. Antimicrob Agents Chemother. 2008;52:4181–3.
Wiederhold NP, Grabinski JL, Garcia-Effron G, Perlin DS, Lee SA. Pyrosequencing to detect mutations in FKS1 that confer reduced echinocandin susceptibility in Candida albicans. Antimicrob Agents Chemother. 2008;52:4145–8.
Pfeiffer CD, Garcia-Effron G, Zaas AK, Perfect JR, Perlin DS, et al. Breakthrough invasive candidiasis in patients on micafungin. J Clin Microbiol. 2010;48:2373–80.
Thompson III GR, Wiederhold NP, Vallor AC, Villareal NC, Lewis II JS, et al. Development of caspofungin resistance following prolonged therapy for invasive candidiasis secondary to Candida glabrata infection. Antimicrob Agents Chemother. 2008;52:3783–5.
Lewis II JS, Wiederhold NP, Wickes BL, Patterson TF, Jorgensen JH. Rapid emergence of echinocandin resistance in Candida glabrata resulting in clinical and microbiologic failure. Antimicrob Agents Chemother. 2013;57(9):4559–61.
Dannaoui E, Desnos-Ollivier M, Garcia-Hermoso D, Grenouillet F, Cassaing S, et al. Candida spp. with acquired echinocandin resistance, France, 2004–2010. Emerg Infect Dis. 2012;18:86–90.
Pfaller MA, Jones RN, Castanheira M. Regional data analysis of Candida non-albicans strains collected in United States medical sites over a 6-year period, 2006–2011. Mycoses. 2014;57:602–11.
Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, et al. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis. 2013;56:1724–32.
Pfaller MA, Castanheira M, Lockhart SR, Ahlquist AM, Messer SA, et al. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J Clin Microbiol. 2012;50:1199–203.
Pham CD, Iqbal N, Bolden CB, Kuykendall RJ, Harrison LH, et al. Role of FKS Mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob Agents Chemother. 2014;58:4690–6.
Lortholary O, Desnos-Ollivier M, Sitbon K, Fontanet A, Bretagne S, et al. Recent exposure to caspofungin or fluconazole influences the epidemiology of candidemia: a prospective multicenter study involving 2,441 patients. Antimicrob Agents Chemother. 2011;55:532–8.
Tortorano AM, Prigitano A, Lazzarini C, Passera M, Deiana ML, et al. A 1-year prospective survey of candidemia in Italy and changing epidemiology over one decade. Infection. 2013;41:655–62.
Fekkar A, Meyer I, Brossas JY, Dannaoui E, Palous M, et al. Rapid emergence of echinocandin resistance during Candida kefyr fungemia treatment with caspofungin. Antimicrob Agents Chemother. 2013;57:2380–2.
Arendrup MC, Perkhofer S, Howard SJ, Garcia-Effron G, Vishukumar A, et al. Establishing in vitro-in vivo correlations for Aspergillus fumigatus: the challenge of azoles versus echinocandins. Antimicrob Agents Chemother. 2008;52:3504–11.
Arendrup MC, Perlin DS. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis. 2014;27:484–92.
Katiyar S, Pfaller M, Edlind T. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob Agents Chemother. 2006;50:2892–4.
Arendrup MC, Perlin DS, Jensen RH, Howard SJ, Goodwin J, et al. Differential in vivo activity of Anidulafungin, Caspofungin and Micafungin against C. glabrata with and without FKS resistance mutations. Antimicrob Agents Chemother. 2012;56(5):2435–42.
Howard SJ, Lestner JM, Sharp A, Gregson L, Goodwin J, et al. Pharmacokinetics and pharmacodynamics of posaconazole for invasive pulmonary aspergillosis: clinical implications for antifungal therapy. J Infect Dis. 2011;203:1324–32.
Slater JL, Howard SJ, Sharp A, Goodwin J, Gregson LM, et al. Disseminated Candidiasis caused by Candida albicans with amino acid substitutions in Fks1 at position Ser645 cannot be successfully treated with micafungin. Antimicrob Agents Chemother. 2011;55:3075–83.
Wiederhold NP, Najvar LK, Bocanegra RA, Kirkpatrick WR, Patterson TF. Caspofungin dose escalation for invasive candidiasis due to resistant Candida albicans. Antimicrob Agents Chemother. 2011;55:3254–60.
Lackner M, Tscherner M, Schaller M, Kuchler K, Mair C, et al. Position and numbers of FKS mutations in C. albicans selectively influence in vitro and in vivo susceptibility to echinocandin treatment. Antimicrob Agents Chemother. 2014;58(7):3626–35.
Shields RK, Nguyen MH, Press EG, Kwa AL, Cheng S, et al. The presence of an FKS mutation rather than MIC is an independent risk factor for failure of echinocandin therapy among patients with invasive candidiasis due to Candida glabrata. Antimicrob Agents Chemother. 2012;56:4862–9.
Beyda ND, John J, Kilic A, Alam MJ, Lasco TM, et al. FKS mutant Candida glabrata: risk factors and outcomes in patients with candidemia. Clin Infect Dis. 2014;59:819–25.
Jensen RH, Johansen HK, Arendrup MC. Stepwise development of a homozygous S80P substitution in Fks1p, conferring echinocandin resistance in Candida tropicalis. Antimicrob Agents Chemother. 2013;57:614–7.
Pasquale T, Tomada JR, Ghannoun M, Dipersio J, Bonilla H. Emergence of Candida tropicalis resistant to caspofungin. J Antimicrob Chemother. 2008;61:219.
Perlin DS. Echinocandin-resistant Candida: molecular methods and phenotypes. Curr Fungal Infect Rep. 2011;5(3):113–9.
Castanheira M, Woosley LN, Messer SA, Diekema DJ, Jones RN, et al. Frequency of fks mutations among Candida glabrata isolates from a 10-year global collection of bloodstream infection isolates. Antimicrob Agents Chemother. 2014;58:577–80.
Eng WK, Faucette L, McLaughlin MM, Cafferkey R, Koltin Y, et al. The yeast FKS1 gene encodes a novel membrane protein, mutations in which confer FK506 and cyclosporin A hypersensitivity and calcineurin-dependent growth. Gene. 1994;151:61–71.
Johnson ME, Katiyar SK, Edlind TD. A new Fks hotspot for acquired echinocandin resistance in yeast, and its contribution to intrinsic resistance of Scedosporium species. Antimicrob Agents Chemother. 2011;55(8):3774–81.
d’Enfert C. Biofilms and their role in the resistance of pathogenic Candida to antifungal agents. Curr Drug Targets. 2006;7:465–70.
Mitchell KF, Taff HT, Cuevas MA, Reinicke EL, Sanchez H, et al. Role of matrix beta-1,3 glucan in antifungal resistance of non-albicans Candida biofilms. Antimicrob Agents Chemother. 2013;57:1918–20.
Desai JV, Bruno VM, Ganguly S, Stamper RJ, Mitchell KF, et al. Regulatory role of glycerol in Candida albicans biofilm formation. MBio. 2013;4:e00637-12.
Ben-Ami R, Garcia-Effron G, Lewis RE, Gamarra S, Leventakos K, et al. The fitness and virulence cost of fks1 mutations causing echinocandin-resistance in Candida albicans. J Infect Dis. 2011;204(4):626–35.
Perlin DS. Resistance to echinocandin-class antifungal drugs. Drug Resist Updat. 2007;10:121–30.
Walker LA, Gow NA, Munro CA. Fungal echinocandin resistance. Fungal Genet Biol. 2010;47:117–26.
Kartsonis N, Killar J, Mixson L, Hoe CM, Sable C, et al. Caspofungin susceptibility testing of isolates from patients with esophageal candidiasis or invasive candidiasis: relationship of MIC to treatment outcome. Antimicrob Agents Chemother. 2005;49:3616–23.
Pfaller MA, Diekema DJ, Ostrosky-Zeichner L, Rex JH, Alexander BD, et al. Correlation of MIC with outcome for Candida species tested against caspofungin, anidulafungin, and micafungin: analysis and proposal for interpretive MIC breakpoints. J Clin Microbiol. 2008;46:2620–9.
Pfaller MA, Diekema DJ, Boyken L, Messer SA, Tendolkar S, et al. Effectiveness of anidulafungin in eradicating Candida species in invasive candidiasis. Antimicrob Agents Chemother. 2005;49:4795–7.
Munro CA, Selvaggini S, de Bruijn I, Walker L, Lenardon MD, et al. The PKC, HOG and Ca2+ signalling pathways coordinately regulate chitin synthesis in Candida albicans. Mol Microbiol. 2007;63:1399–413.
Walker LA, Munro CA, de Bruijn I, Lenardon MD, McKinnon A, et al. Stimulation of chitin synthesis rescues Candida albicans from echinocandins. PLoS Pathog. 2008;4, e1000040.
Cowen LE. Hsp90 orchestrates stress response signaling governing fungal drug resistance. PLoS Pathog. 2009;5, e1000471.
LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, et al. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010;6, e1001069.
Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, et al. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 2009;5, e1000532.
Singh-Babak SD, Babak T, Diezmann S, Hill JA, Xie JL, et al. Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Pathog. 2012;8, e1002718.
Firon A, Lesage G, Bussey H. Integrative studies put cell wall synthesis on the yeast functional map. Curr Opin Microbiol. 2004;7:617–23.
Gow NA, Netea MG, Munro CA, Ferwerda G, Bates S, et al. Immune recognition of Candida albicans beta-glucan by dectin-1. J Infect Dis. 2007;196:1565–71.
Plaine A, Walker L, Da Costa G, Mora-Montes HM, McKinnon A, et al. Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal Genet Biol. 2008;45:1404–14.
Lee KK, Maccallum DM, Jacobsen MD, Walker LA, Odds FC, et al. Elevated cell wall chitin in Candida albicans confers echinocandin resistance in vivo. Antimicrob Agents Chemother. 2012;56:208–17.
Stevens DA, Ichinomiya M, Koshi Y, Horiuchi H. Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for beta-1,6-glucan synthesis inhibition by caspofungin. Antimicrob Agents Chemother. 2006;50:3160–1.
Clemons KV, Espiritu M, Parmar R, Stevens DA. Assessment of the paradoxical effect of caspofungin in therapy of candidiasis. Antimicrob Agents Chemother. 2006;50:1293–7.
Healey KR, Katiyar SK, Raj S, Edlind TD. CRS-MIS in Candida glabrata: sphingolipids modulate echinocandin-Fks interaction. Mol Microbiol. 2012;86:303–13.
Healey KR, Katiyar SK, Castanheira M, Pfaller MA, Edlind TD. Candida glabrata mutants demonstrating paradoxical reduced caspofungin susceptibility but increased micafungin susceptibility. Antimicrob Agents Chemother. 2011;55:3947–9.
Coste A, Selmecki A, Forche A, Diogo D, Bougnoux ME, et al. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell. 2007;6:1889–904.
Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313:367–70.
Shin JH, Chae MJ, Song JW, Jung SI, Cho D, et al. Changes in karyotype and azole susceptibility of sequential bloodstream isolates from patients with Candida glabrata candidemia. J Clin Microbiol. 2007;45:2385–91.
Pfaller MA, Boyken L, Hollis RJ, Kroeger J, Messer SA, et al. In vitro susceptibility of invasive isolates of Candida spp. to anidulafungin, caspofungin, and micafungin: six years of global surveillance. J Clin Microbiol. 2008;46:150–6.
Garcia-Effron G, Katiyar SK, Park S, Edlind TD, Perlin DS. A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob Agents Chemother. 2008;52:2305–12.
Maligie MA, Selitrennikoff CP. Cryptococcus neoformans resistance to echinocandins: (1,3)beta-glucan synthase activity is sensitive to echinocandins. Antimicrob Agents Chemother. 2005;49:2851–6.
van Duin D, Casadevall A, Nosanchuk JD. Melanization of Cryptococcus neoformans and Histoplasma capsulatum reduces their susceptibilities to amphotericin B and caspofungin. Antimicrob Agents Chemother. 2002;46:3394–400.
Staab JF, Kahn JN, Marr KA. Differential Aspergillus lentulus echinocandin susceptibilities are Fksp independent. Antimicrob Agents Chemother. 2010;54:4992–8.
Odabasi Z, Paetznick V, Rex JH, Ostrosky-Zeichner L. Effects of serum on in vitro susceptibility testing of echinocandins. Antimicrob Agents Chemother. 2007;51:4214–6.
Paderu P, Garcia-Effron G, Balashov S, Delmas G, Park S, et al. Serum differentially alters the antifungal properties of echinocandin drugs. Antimicrob Agents Chemother. 2007;51:2253–6.
Wiederhold NP, Najvar LK, Bocanegra R, Molina D, Olivo M, et al. In vivo efficacy of anidulafungin and caspofungin against Candida glabrata and association with in vitro potency in the presence of sera. Antimicrob Agents Chemother. 2007;51:1616–20.
Foldi R, Szilagyi J, Kardos G, Berenyi R, Kovacs R, et al. Effect of 50% human serum on the killing activity of micafungin against eight Candida species using time-kill methodology. Diagn Microbiol Infect Dis. 2012;73:338–42.
Kovacs R, Gesztelyi R, Berenyi R, Doman M, Kardos G, et al. Killing rates exerted by caspofungin in 50 % serum and its correlation with in vivo efficacy in a neutropenic murine model against Candida krusei and Candida inconspicua. J Med Microbiol. 2014;63:186–94.
Arendrup MC, Rodriguez-Tudela JL, Park S, Garcia-Effron G, Delmas G, et al. Echinocandin susceptibility testing of Candida spp. Using EUCAST EDef 7.1 and CLSI M27-A3 standard procedures: analysis of the influence of bovine serum albumin supplementation, storage time, and drug lots. Antimicrob Agents Chemother. 2011;55:1580–7.
Arendrup MC, Garcia-Effron G, Lass-Florl C, Lopez AG, Rodriguez-Tudela JL, et al. Echinocandin susceptibility testing of Candida species: comparison of EUCAST EDef 7.1, CLSI M27-A3, Etest, disk diffusion, and agar dilution methods with RPMI and isosensitest media. Antimicrob Agents Chemother. 2010;54:426–39.
Pfaller MA, Castanheira M, Messer SA, Rhomberg PR, Jones RN. Comparison of EUCAST and CLSI broth microdilution methods for the susceptibility testing of 10 systemically active antifungal agents when tested against Candida spp. Diagn Microbiol Infect Dis. 2014;79:198–204.
Andes D, Diekema DJ, Pfaller MA, Bohrmuller J, Marchillo K, et al. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob Agents Chemother. 2010;54:2497–506.
Eschenauer GA, Nguyen MH, Shoham S, Vazquez JA, Morris AJ, et al. Real-world experience with echinocandin MICs against Candida species in a multicenter study of hospitals that routinely perform susceptibility testing of bloodstream isolates. Antimicrob Agents Chemother. 2014;58:1897–906.
Espinel-Ingroff A, Arendrup MC, Pfaller MA, Bonfietti LX, Bustamante B, et al. Interlaboratory variability of Caspofungin MICs for Candida spp. Using CLSI and EUCAST methods: should the clinical laboratory be testing this agent? Antimicrob Agents Chemother. 2013;57:5836–42.
Ben-Ami R, Hilerowicz Y, Novikov A, Giladi M. The impact of new epidemiological cutoff values on Candida glabrata resistance rates and concordance between testing methods. Diagn Microbiol Infect Dis. 2014;79:209–13.
Pfaller MA, Messer SA, Diekema DJ, Jones RN, Castanheira M. Use of micafungin as a surrogate marker to predict susceptibility and resistance to caspofungin among 3,764 clinical isolates of Candida by use of CLSI methods and interpretive criteria. J Clin Microbiol. 2014;52:108–14.
Pfaller MA, Diekema DJ, Jones RN, Castanheira M. Use of anidulafungin as a surrogate marker to predict susceptibility and resistance to caspofungin among 4,290 clinical isolates of Candida by using CLSI methods and interpretive criteria. J Clin Microbiol. 2014;52:3223–9.
Arendrup MC, Rodriguez-Tudela JL, Lass-Florl C, Cuenca-Estrella M, Donnelly JP, et al. EUCAST technical note on anidulafungin. Clin Microbiol Infect. 2011;17:E18–20.
Espinel-Ingroff A, Pfaller MA, Bustamante B, Canton E, Fothergill A, et al. Multilaboratory study of epidemiological cutoff values for detection of resistance in eight Candida species to fluconazole, posaconazole, and voriconazole. Antimicrob Agents Chemother. 2014;58:2006–12.
Perlin DS. Antifungal drug resistance: do molecular methods provide a way forward? Curr Opin Infect Dis. 2009;22:568–73.
Pfaller MA, Andes D, Arendrup MC, Diekema DJ, Espinel-Ingroff A, et al. Clinical breakpoints for voriconazole and Candida spp. revisited: review of microbiologic, molecular, pharmacodynamic, and clinical data as they pertain to the development of species-specific interpretive criteria. Diagn Microbiol Infect Dis. 2011;70:330–43.
Perlin DS. Echinocandin resistance, susceptibility testing and prophylaxis: implications for patient management. Drugs. 2014;74:1573–85.
Stevens DA, Espiritu M, Parmar R. Paradoxical effect of caspofungin: reduced activity against Candida albicans at high drug concentrations. Antimicrob Agents Chemother. 2004;48:3407–11.
Stevens DA, White TC, Perlin DS, Selitrennikoff CP. Studies of the paradoxical effect of caspofungin at high drug concentrations. Diagn Microbiol Infect Dis. 2005;51:173–8.
Rueda C, Cuenca-Estrella M, Zaragoza O. Paradoxical growth of Candida albicans in the presence of caspofungin is associated with multiple cell wall rearrangements and decreased virulence. Antimicrob Agents Chemother. 2014;58:1071–83.
Bizerra FC, Melo AS, Katchburian E, Freymuller E, Straus AH, et al. Changes in cell wall synthesis and ultrastructure during paradoxical growth effect of caspofungin on four different Candida species. Antimicrob Agents Chemother. 2011;55:302–10.
Shields RK, Nguyen MH, Du C, Press E, Cheng S, et al. Paradoxical effect of caspofungin against Candida bloodstream isolates is mediated by multiple pathways but eliminated in human serum. Antimicrob Agents Chemother. 2011;55:2641–7.
Wiederhold NP, Kontoyiannis DP, Chi J, Prince RA, Tam VH, et al. Pharmacodynamics of caspofungin in a murine model of invasive pulmonary aspergillosis: evidence of concentration-dependent activity. J Infect Dis. 2004;190:1464–71.
Klont RR, Mennink-Kersten MA, Ruegebrink D, Rijs AJ, Blijlevens NM, et al. Paradoxical increase in circulating Aspergillus antigen during treatment with caspofungin in a patient with pulmonary aspergillosis. Clin Infect Dis. 2006;43:e23–5.
Zanette RA, Kontoyiannis DP. Paradoxical effect to caspofungin in Candida species does not confer survival advantage in a Drosophila model of candidiasis. Virulence. 2013;4:497–8.
Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–63.
Koh AY, Kohler JR, Coggshall KT, Van Rooijen N, Pier GB. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog. 2008;4, e35.
Kennedy MJ, Volz PA, Edwards CA, Yancey RJ. Mechanisms of association of Candida albicans with intestinal mucosa. J Med Microbiol. 1987;24:333–41.
Magill SS, Swoboda SM, Johnson EA, Merz WG, Pelz RK, et al. The association between anatomic site of Candida colonization, invasive candidiasis, and mortality in critically ill surgical patients. Diagn Microbiol Infect Dis. 2006;55:293–301.
Magill SS, Swoboda SM, Shields CE, Colantuoni EA, Fothergill AW, et al. The epidemiology of Candida colonization and invasive candidiasis in a surgical intensive care unit where fluconazole prophylaxis is utilized: follow-up to a randomized clinical trial. Ann Surg. 2009;249:657–65.
Miranda LN, van der Heijden IM, Costa SF, Sousa AP, Sienra RA, et al. Candida colonisation as a source for candidaemia. J Hosp Infect. 2009;72:9–16.
Voss A, Hollis RJ, Pfaller MA, Wenzel RP, Doebbeling BN. Investigation of the sequence of colonization and candidemia in nonneutropenic patients. J Clin Microbiol. 1994;32:975–80.
Richet HM, Andremont A, Tancrede C, Pico JL, Jarvis WR. Risk factors for candidemia in patients with acute lymphocytic leukemia. Rev Infect Dis. 1991;13:211–5.
Harriott MM, Noverr MC. Importance of Candida-bacterial polymicrobial biofilms in disease. Trends Microbiol. 2011;19:557–63.
Cheng S, Clancy C, Hartman D, Hao B, Nguyen M. Candida glabrata intra-abdominal candidiasis is characterized by persistence within the peritoneal cavity and abscesses. Infect Immun. 2014;82(7):3015–22.
Fekkar A, Dannaoui E, Meyer I, Imbert S, Brossas JY, et al. Emergence of echinocandin-resistant Candida spp. in a hospital setting: a consequence of 10 years of increasing use of antifungal therapy? Eur J Clin Microbiol Infect Dis. 2014;33(9):1489–96.
Blanchard E, Lortholary O, Boukris-Sitbon K, Desnos-Ollivier M, Dromer F, et al. Prior caspofungin exposure in patients with hematological malignancies is a risk factor for subsequent fungemia due to decreased susceptibility in Candida spp.: a case-control study in Paris, France. Antimicrob Agents Chemother. 2011;55:5358–61.
de la Torre P, Reboli AC. Micafungin: an evidence-based review of its place in therapy. Core Evid. 2014;9:27–39.
van Burik JA, Ratanatharathorn V, Stepan DE, Miller CB, Lipton JH, et al. Micafungin versus fluconazole for prophylaxis against invasive fungal infections during neutropenia in patients undergoing hematopoietic stem cell transplantation. Clin Infect Dis. 2004;39:1407–16.
Chou LS, Lewis RE, Ippoliti C, Champlin RE, Kontoyiannis DP. Caspofungin as primary antifungal prophylaxis in stem cell transplant recipients. Pharmacotherapy. 2007;27:1644–50.
Mattiuzzi GN, Alvarado G, Giles FJ, Ostrosky-Zeichner L, Cortes J, et al. Open-label, randomized comparison of itraconazole versus caspofungin for prophylaxis in patients with hematologic malignancies. Antimicrob Agents Chemother. 2006;50:143–7.
Doring M, Hartmann U, Erbacher A, Lang P, Handgretinger R, et al. Caspofungin as antifungal prophylaxis in pediatric patients undergoing allogeneic hematopoietic stem cell transplantation: a retrospective analysis. BMC Infect Dis. 2012;12:151.
Xu SX, Shen JL, Tang XF, Feng B. Newer antifungal agents for fungal infection prevention during hematopoietic cell transplantation: a meta-analysis. Transplant Proc. 2013;45:407–14.
Ziakas PD, Kourbeti IS, Mylonakis E. Systemic antifungal prophylaxis after hematopoietic stem cell transplantation: a meta-analysis. Clin Ther. 2014;36(2):292–306.e1.
Scott LJ. Micafungin: a review of its use in the prophylaxis and treatment of invasive Candida infections. Drugs. 2012;72:2141–65.
Hope WW, Castagnola E, Groll AH, Roilides E, Akova M, et al. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect. 2012;18 Suppl 7:38–52.
Ostrosky-Zeichner L. Candida glabrata and FKS mutations: witnessing the emergence of the true multidrug-resistant Candida. Clin Infect Dis. 2013;56:1733–4.
Zimbeck AJ, Iqbal N, Ahlquist AM, Farley MM, Harrison LH, et al. FKS mutations and elevated echinocandin MIC values among Candida glabrata isolates from U.S. population-based surveillance. Antimicrob Agents Chemother. 2010;54:5042–7.
Pfaller MA, Castanheira M, Messer SA, Moet GJ, Jones RN. Echinocandin and triazole antifungal susceptibility profiles for Candida spp., Cryptococcus neoformans, and Aspergillus fumigatus: application of new CLSI clinical breakpoints and epidemiologic cutoff values to characterize resistance in the SENTRY Antimicrobial Surveillance Program (2009). Diagn Microbiol Infect Dis. 2011;69:45–50.
Chapeland-Leclerc F, Hennequin C, Papon N, Noel T, Girard A, et al. Acquisition of flucytosine, azole, and caspofungin resistance in Candida glabrata bloodstream isolates serially obtained from a hematopoietic stem cell transplant recipient. Antimicrob Agents Chemother. 2010;54:1360–2.
Bizerra FC, Jimenez-Ortigosa C, Souza AC, Breda GL, Queiroz-Telles F, et al. Breakthrough candidemia due to multidrug-resistant Candida glabrata during prophylaxis with a low dose of micafungin. Antimicrob Agents Chemother. 2014;58:2438–40.
Ruggero MA, Topal JE. Development of echinocandin-resistant Candida albicans candidemia following brief prophylactic exposure to micafungin therapy. Transpl Infect Dis. 2014;16(3):469–72.
Acknowledgments
David S. Perlin is supported by grants from National Institutes of Health (AI069397 and AI109025) and Astellas Pharma.
Disclosures
Dr. Perlin serves on scientific advisory boards for Merck, Astellas, Amplyx, Cidara,and Synexis and he receives grant support from Astellas, Cidara and Amplyx. He is an inventor in US patent 8,753,819 entitled “Assays for Resistance to Echinocandin-Class Drugs.”
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Perlin, D.S. (2017). Echinocandin Resistance. In: Mayers, D., Sobel, J., Ouellette, M., Kaye, K., Marchaim, D. (eds) Antimicrobial Drug Resistance. Springer, Cham. https://doi.org/10.1007/978-3-319-46718-4_29
Download citation
DOI: https://doi.org/10.1007/978-3-319-46718-4_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46716-0
Online ISBN: 978-3-319-46718-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)