
Abstract
Protein and DNA-advanced glycation end-products (DNA-AGEs) are toxic by-products of metabolism and are also assimilated by high temperature processed foods. AGEs may be generated rapidly or over long times stimulated by distinct triggering mechanisms, thereby accounting for their roles in multiple settings and disease states. Neurodegenerative diseases (NDDs) are associated with the misfolding and deposition of specific proteins, DNA adduct formation either intra or extra-cellularly in the nervous system. There is also evidence that brain tissue in patients with NDD is exposed to DNA oxidation and glycoxidation during the course of the disease. Although familial mutations play an important role in protein misfolding and aggregation, the majority of cases of NDD are sporadic, suggesting that other factors must contribute to the onset and progression of these disorders. High levels of refined and carbohydrate enriched diets, hyper caloric diets and sedentary lifestyles drive endogenous formation of AGEs via accumulation of highly reactive glycolysis intermediates and activation of the reductase pathway (polyol/aldose) producing high intracellular reducing sugars are the important modifiable environmental factors. Some of these modifications might affect proteins in detrimental ways and lead to their misfolding and accumulation. Reducing sugars play important roles in modifying proteins, forming AGEs in a non-enzymatic process named glycation. Several proteins linked to NDDs, such as amyloid β, tau, prions and transthyretin, were found to be glycated in patients, and this is thought to be associated with increased protein stability through the formation of crosslinks that stabilize protein aggregates causing NDDs like Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), familial amyloid polyneuropathy (FAP), and prion disease (PrD). Moreover, glycation may also be responsible, via the receptor for AGE (RAGE), for an increase in oxidative stress and inflammation through the formation of reactive oxygen species (ROS) and the induction of nuclear factor-κB (NF-κB). Here, we revised the role of protein and DNA-AGEs in the major NDDs and highlight the potential value of protein and DNA-AGEs glycation as a biomarker or target for therapeutic intervention. Additionally, the chapter covers several new therapeutic approaches that have been applied to treat these devastating disorders, including the use of various synthetic, natural and gold and silver conjugated nanoparticles (Au, Ag-NPs).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Agvanced glycation end products (AGE)
- Neurodegenerative diseases (NDDs)
- Environmental toxicity
- DNA glycation (DNA-AGE)
- Receptor for AGE (RAGE)
- Amyloid β
- Tau
- Prions
- Transthyretin
- Nanoparticles (Au, Ag-NPs)
1 Introduction
The incidence of neurodegenerative diseases (NDDs) increases with extended life expectancy observed over the last century has led to the emergence of a rope of age related disorders that pose novel challenges to current societies. Age-related degenerative diseases including Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis (ALS) and the prion diseases (PrD) are growing to epidemic proportions, are debilitating and so far deadly disorders that require rigorous research. Increased incidences of mild cognitive impairment due to various environmental factors toxicity in elderly populations are characterized by a slow and progressive loss of neuronal cells, and also intra or extracellular deposition of misfolded and aggregated proteins. Modifiable lifestyle factors cover a critical role in these diseases. The prevalence of rigorous cognitive impairment crucially depends on various factors which are influencing health like energy balance, micronutrient density of food, level of physical activity and exposure to tobacco smoke etc. Recent evidence validates that food production, processing and methods of cooking are life-threatening to health outcomes as well. Induced glycation and reactive oxygen species (ROS) formations are important mechanism by which lifestyle influences. Extracellular and intracellular deposition of amyloid β-peptide (Aβ) (amyloid plaques) and tau protein (neurofibrillary tangles) respectively are the key pathological hallmarks in Alzheimer’s disease (AD). Cytoplasmic proteinaceous inclusions, mainly composed of the protein α-synuclein (α-syn), named Lewy bodies (LBs), are the pathognomonic inclusions in Parkinson’s disease (PD) (Spillantini et al. 1997). Indeed, the frequency of neurodegenerative disorders, including AD and PD, has increased significantly by about four folds from 9.4 million in 1950 (total population 179.5 million) to 40.3 million in 2010 (total population 831.5 million) (Lekoubou et al. 2014). Production of advanced glycation end products (AGEs) has been regarded as a process of molecular and cellular ageing (Jacobs et al. 2014). There are several mutations are associated with the amyloidogenesis behavior of proteins such as Aβ, α -syn, LBs and transthyretin are very well known while the enormous number of cases of NDDs are intermittent (Haass et al. 1994; Kruger et al. 1998; Polymeropoulos et al. 1997; Zarranz et al. 2004; Saraiva 2001). Factors such as oxidative stress, protein cross-linking, the sequelae of RAGE signaling and changes of DNA integrity extrapolate this process from the molecular to the biological (cellular and tissue) level (Huttunen et al. 2002), leading to the thrashing of their normal function, to cell dysfunction and death. Various physiological consequences of DNA and protein glycation comprise the development of diabetes mellitus and their secondary complications like retinopathy, nephropathy and neuropathy. Glycation has key clinical relevance, since it may be used as a specific biomarker for several disorders and their secondary complications. AGEs may be used as markers of tissue damage and may predict long-term complications of diabetes mellitus like diabetes-neuropathy and cardiomyopathy (Akhter et al. 2016a, b). Non-invasive techniques like auto-fluorescence reader have been developed to assess the levels of AGE in the skin, which rapidly measures AGE accumulation (Shimasaki et al. 2011). In view of the emerging knowledge about the widespread occurrence of DNA and protein-AGEs in various tissues (Peppa et al. 2003) including the central nervous system (Vlassara et al. 1983) we wants to propose the following hypothesis based on a chapter of recent results and increasing evidence for a putative role of the various environmental factors induced DNA and protein-AGEs in NDDs. We are also proposing the probable approaches to stop the menace of reactions which may open novel avenues for therapeutic intervention.
2 Life Style Dependent Browning Reaction a Deadly Consequence with Sweets
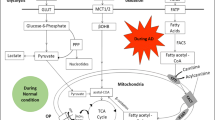
Despite the 100 or so years that have lapsed since French scientist Louis-Camille Maillard (1878–1936) (Maillard 1912), the first scientist to investigate the “browning reaction” in food and in the human body, who reported the story of glycation reaction in the year 1912 after whom the reaction is also known as the Maillard reaction (Maillard 1912). The non-enzymatic reaction starts with the addition of free carbonyl group of a reducing sugar to a nucleophilic free amino group of proteins, lipids and DNA macromolecule to form early, intermediate and advance glycation end-products (AGEs). Exogenous AGEs are acquired from tobacco, brown or high cooked food diet (exogenous sources). Reducing sugars in basic solutions and lipids by β-oxidation generate formyl (an aldehyde) and ketone groups. These aldehydes and ketones have a highly polarized carbonyl (C=O) group, the oxygen atom of which is highly electronegative and may react with nucleophiles in other biomolecules like protein, DNA and lipids. Under hyperglycemic condition (high glucose load), these biomolecules undergo glycation reaction leading to the formation of a complex series of AGEs. This, in turn, results in the altering their structural conformation or deprivation of the functions of the biological macromolecules.
Figure 1 schematically represents the promising pathway of reaction of biomacromolecules with reducing sugars to form protein advanced glycation end products (protein-AGEs) and DNA advanced glycation end products (DNA-AGEs), which is believed to be involved in the complications associated with several neuronal disorders through various pathways of glycation reaction (Aldini et al. 2013). There are also various pathways direct or indirect depends upon life style causing induced toxic effects of AGEs and ROS, involved to cause neuronal disorders as shown in Fig. 2.

Schematic representation of proposed pathway for macromolecules reacting with reduced sugars to form protein-AGEs and DNA-AGEs respectively

Schematic representation of direct or indirect modifiable environmental factors dependent various pathways induced toxic of glycation and their cause in neuronal apoptosis
Non-enzymatic glycation of proteins is a post-translational modification conventional process between sugars and proteins, occurring in all living systems. Protein glycation occurs through a complex series of very slow reactions in the body; it is quite dynamic in nature and starts with the formation of unstable Schiff base, which undergoes a series of reactions leading to the formation of heterogeneous AGEs molecules (Akhter et al. 2013a, b). Glucose and glucose-derived glyoxal, MG, glucosone and 3-deoxyglucosone (3-DG) dicarbonyls are the major precursors of AGEs. The levels of these precursors determine the formation of diverse types of AGEs including fructosyl-lysine (FL), carboxymethyl lysine (CML), carboxyethyl lysine (CEL) and pentosidine. Hyperglycemic condition promotes the formation of AGEs in vivo, thus, enhancing the overall accumulation. AGEs cause cell death at various levels, namely alteration of protein structure and function; protein aggregation, fibril formation and protease resistance (Wei et al. 2012), aberrant signaling through interaction with the RAGE and dysfunction of extracellular matrix. AGEs contribute drastically to the progression of diabetic secondary complications, including retinopathy, neuropathy, nephropathy, cardiomyopathy, accelerated aging and NDDs.
When DNA reacts with a reducing sugar in vitro at a physiological temperature, the formation of DNA-AGEs is observed (Akhter et al. 2015; Shahab et al. 2014). DNA glycation significantly alter the structure of DNA macromolecule, which leads to depurination, strand breaks and mutations such as insertions, deletions and transposition (Ahmad et al. 2011a, b). Therefore, DNA-AGEs could contribute to the loss of genomic integrity, which occurs during aging and may contribute to the age-related complications like NDDs. Few studies on the stability and dynamics of DNA showed that glycation leads to partial unwinding and/or fragmentation of the DNA double helix (Mustafa et al. 2012). The mutagenic potential of the predominant DNA-glycation adduct carboxy ethyl deoxygunaosine (CEdG) was investigated by Wuenschell et al. (2010) and exhibited that CEdG within the template DNA and the corresponding triphosphate possess different syn/anti conformations during replication which influence base pairing preferences. Some reports have shown in the recent past that genotoxicity and immunogenicity are incurred in DNA and proteins by carcinogens and reactive oxygen species (ROS) as well (Ahmad et al. 2014; Shahab et al. 2012a, b, 2013). Moreover, most recently, it has also been investigated that glycation-induced oxidative stress leads to the modification of DNA macromolecule and results in alteration of its structure (Akhter et al. 2013a, b). The consequence of genotoxic effect is due to the structural perturbations in the glycated DNA macromolecules (Ahmad et al. 2011b). Induced levels of these DNA-AGEs have been implicated in the pathological complications of diabetes and NDDs.
3 Toxic Role of Various Glycated Protein (Protein-AGEs) in NDDs
In the world, millions of elder people suffer from NDDs and diagnosis and treatment of these diseases costs millions of dollars per year. Life style dependent alterations in glycosylated protein have critical role in various human NDDs states, such as AD, PD and Creutzfeldt-Jakob disease (CJD) (Saez-Valero et al. 2003; Silveyra et al. 2006). Although the glycoprotein’s structural elucidation is a challenge because of their inherent complexity and heterogeneity in biological systems, advances have been made to identify a few proteins where glycosylation appears to be important in the disease processes of AD and PD (Sihlbom et al. 2004). The role of a few key proteins involved in AD and PD pathogenesis are discussed below.
3.1 Toxic Effect of Protein-AGEs in AD
Among the NDDs, AD is the most common, touching ∼5% of people aged 65–75 and nearly 50% of people over the age of 85 (Grossman et al. 2006). The most common reason of NDDs is protein misfolding which leads to protein aggregation and accumulation. The characteristic features of this disease are progressive loss of memory, speech, ability to recognize people and objects followed by confusion, disorientation and disordered thinking. The dysfunction involves degeneration of neurons especially in hippocampus, amygdala, nucleus basalis and entorhinal cortex. Most AD cases are intermittent, whereas approximately 6% show genetic origin (Campion et al. 1999). The great majority of genetic cases are allied to the occurrence of the ε4 allele of the apolipoprotein E (APOEε4) and also to mutations in the amyloid β precursor protein (APP) (Rademakers and Rovelet-Lecrux 2009). It has already been identified that glycation of amyloid-β peptide, the constituent of the senile plaques in AD may contribute to its cross-linking. There are some strong indications that the glycation of amyloid-βeta (Aβ) peptide and tau protein (τ- protein) occurs in the early stages of AD, although it has been hypothesized that peptide free radicals generated by Aβ would cross-link peptides with sugars resulting in protein glycation (Mattson et al. 1995).
The diagnosis of AD can only be confirmed through the presence of characteristic pathological signs such as Aβ plaques and neurofibrillary tangles (NFTs) (Cummings et al. 1998). Some studies have suggested that over-expression of microtubule-associated τ-protein, hyper phosphorylation, and mutations is known to contribute to its aggregation and accumulation of Aβ in mitochondria is linked with the neuronal toxicity observed in this disorder (Andorfer et al. 2005; Sato et al. 2002; Wittmann et al. 2001; Caspersen et al. 2005; Du et al. 2008; Lustbader et al. 2004). However, AGEs are also formed from the reaction of free reactive carbonyl species with free lysine or arginine side chains of proteins (Munch et al. 1999), and are believed to play an important role in NFTs formation as well as in the development of β-amyloid plaques. Increased levels of AGEs in the Aβ plaques and NFTs play an important role in AD (Vitek et al. 1994; Smith et al. 1994). The plaque fractions of AD brains hold higher levels of AGEs than samples from age-matched controls (normal human brain). An immunohistochemically AGEs can be identified in both SPs and NFTs (Sasaki et al. 1998). Furthermore, neurons, microglial cells, and astrocytes in the normal human brain express RAGE (Li et al. 1998), while its expression by cortical neurons increases and becomes more widespread in AD (Yan et al. 1996). Since it was reported that RAGE might be the nerve cell receptor for Aβ protein, the role of RAGE in the pathogenesis of AD has attracted substantial attention (Yan et al. 1997).
Glycation of τ-proteins enhances the formation of paired helical filaments in AD and in vitro reduces its ability to bind microtubules (Ledesma et al. 1994). In addition, AGE modified τ-proteins are responsible for increased fibrillization of the protein. Aβ aggregation follows a nucleation-dependent polymerization mechanism, which is considerably accelerated by AGE-mediated crosslinking (Munch et al. 1999). The Aβ aggregation consists of two characteristic stages i.e. slow, reversible nucleus formation step, involving oligomer formation, followed by a rapid elongation phase. Once the oligomers reach a decisive size, a fast, linear elongation of the aggregates or fibril formation can occur via the addition of Aβ peptides to the eNDDs. The fibril creation step is an irreversible process. Finally, these fibrils grow and form amyloid plaques.
Methylglyoxal (MGO) (AGE precursor), is able to activate p38 MAP kinase, which is able to phosphorylate tau (Liu et al. 2003; Reynolds et al. 2000), a progression believed to occur in neurons in AD (Zhu et al. 2000). MGO glycated Aβ promotes the development of β-sheets, oligomers and protofibrils and also increases the size of the aggregates (Chen et al. 2006). Aβ is also recognized by RAGE, an important player in AD (Li et al. 1998). RAGE is expressed in astrocytes and monocytes across the blood-brain barrier, activating nuclear factor-κB (NF-κB) which consequently increasing reactive oxygen species (ROS) (Sato et al. 2006; Takeuchi et al. 2000; Yan et al. 1998). RAGE-mediated GSK-3 activation, leads to the toxic effect of AGEs, which induces tau protein hyperphosphorylation and consequently impairs synapse signaling and memory in rats (Li et al. 2012).
Surprisingly, levels of protein-AGEs in AD are three-fold higher than healthy subjects (Vitek et al. 1994). A paired helical filament of glycated A68 proteins, a known precursor of NFTs, suggests that it could be an early result (Takeda et al. 2000). Although protein-AGEs levels increase with age (37.5%), in AD the increase is much greater (72.6%) (Luth et al. 2005). ApoE also seems to be modified under glycation reaction, since in AD the staining patterns of AGEs and ApoE are similar (Dickson et al. 1996). Presence of these protein-AGEs in the cerebrospinal fluid (CSF) of AD patients, signifying that this may be explored as a biomarker for AD.
3.2 Role of Protein-AGEs Toxicity in PD
PD is the second most common progressive neurodegenerative disorder, is caused by a progressive degeneration of the dopaminergic neurons in the substantia nigra of the midbrain, which are involved in the control of voluntary movements (Izumi et al. 2011). The prevalence of PD is around 2% in people over age 60 (Burke 1999). Muscular rigidity, tremor, slow movement, loss of balance and coordination are the most common symptoms of the disease (Forno 1996). Dopaminergic cell loss, excessive accumulation of iron in midbrain neurons, formation of LBs containing α-syn aggregates, presence of extracellular melanin released from degenerating neurons, and increased oxidative stress in substantia nigra region of the brain, known to induce apoptosis, are the main pathological hallmarks of PD.
Several genes like α-syn, Parkin, DJ-1, and Pink1 have already been identified to play an important role in the development of PD (Nuytemans et al. 2010; Martin et al. 2010, 2011) as well as various environmental factors are also lead to the onset of PD, including brief exposure to herbicides, pesticides, heavy metals, increased stress and brain injuries (Nuytemans et al. 2010; Martin et al. 2010, 2011; Hegde et al. 2010; Defebvre 2010). Glycation was first reported to be present in substantia nigra and locus coeruleus of periphery LB (Vitek et al. 1994). It is suggested to be involved in chemical crosslinking making the protein resistant to proteolytic degradation. Glycation of α-syn constitutes a factor that affects the aggregation of the protein into LBs in PD. α-syn has a total of 15 lysine residues which are all candidate sites for glycation, influences the nucleation of protein aggregates (Padmaraju et al. 2011) and induces α-syn oligomerization, thereby stabilizing oligomers. Glycated α-syn may interact with RAGE and activate the release of NF-κB. These signaling proteins trigger the signaling cascades in the brain and damage neuronal cells. By inducing the expression of RAGE proteins, NF-κB also regulates the expression of RAGE. Glycation is also reported to be found in cerebral cortex, amygdala, and substantia nigra of healthy subjects, but levels of glycation are higher in PD patients. The evidence of glycation was first reported in the substantia nigra and locus coeruleus of peripheral LB. The glycated α-syn is one of the important factors that affect the aggregation of the protein into LBs in PD (Guerrero et al. 2013). In comparison to age-matched control induced level of RAGE are expressed in AD patients (Dalfo et al. 2005), suggesting the key role of protein-AGEs in the disease (Fig. 3).

Presenting probable role of protein glycation in protein aggregation and misfolding. After synthesis, proteins may acquire different misfolded conformations and may form dimers, oligomers and, in later stages, amyloid fibrils, which may be sequestered into inclusion bodies (such as LBs for the case of α-syn in PD). Reducing sugars, increased in hyper-glycaemia conditions, may affect the biology of proteins in early or later stages of the aggregation process (glycating protein monomers, dimers, oligomers and amyloid fibrils). AGEs may lead to cell damage by interacting with RAGE, inducing oxidative stress and inflammatory cell responses causative response in neuronal toxicity
Acute diminish in the levels of cellular glutathione (GSH) in early stage is a fundamental characteristics of PD, results in a lower activity of the glyoxalase system. In catabolic pathway methylglyoxal is the important glycation agent in vivo (Thornalley 1998). The carbonyl content raise oxidative stress will then be responsible for an increase in AGEs concentrations might be responsible to the cell damage. Dopamine auto-oxidation and its deprivation by monoamine oxidase is likely to contribute to an induced level of oxidative stress. Prominently, an in vitro MGO and GO are able to induce oligomerization of α-syn (Lee et al. 2009). Study have been suggested that the membrane-binding ability of glycated α-syn was also decreased (Lee et al. 2009), thus, it is hypothesize that in a glycation-prone environment, more cytotoxic α-syn aggregates or oligomers are present in the cytoplasm, contributing to the development of PD.
3.3 Role of Protein-AGEs Toxicity in Other NDDs
3.3.1 Effects of AGEs in ALS
ALS, also known as Lou Gehrig’s disease, is a very common motor neuron disease affecting 4–6/100,000 people, and shares with other disorders of the ageing nervous system a polygenic, multifactorial origin (Choonara et al. 2009). ALS is polygenic and multifactorial in origin, characterized by the degeneration of motor neurons in the brain and spinal cord leading to progressive muscle weakness, atrophy, and spasticity (Véronique et al. 2010). It causes selective loss of upper and lower motor neurons of the brain and spinal cord. The causes of ALS are still unidentified and most of the cases are sporadic and inherited in nature (Gros-Louis et al. 2006). Several inherited cases of the disease are related to copper–zinc superoxide dismutase 1 (Cu, Zn-SOD1) (Gros-Louis et al. 2006; Pasinelli and Brown 2006). This protein catabolizes superoxide radicals, signifying that oxidative stress may participate an imperative role in the disease (Kikuchi et al. 2003). Glycation was first identified in both sporadic and inherited forms of ALS, in the spinal cord and brain of ALS patients. Firstly it was postulated that glycation could be implicated in the time-dependent cross-linking of neurofilament protein, subunits contain multiple Lys–Ser–Pro sequences. Glycation of these lysine residues impairs the self-assembly process; endorse cross-linking in the neurofilament protein, leads to ALS. Studies have revealed that AGE levels were higher in the presence of the Cu, Zn-SOD-1 mutation, while in control human and mouse subjects; AGE immunoreactivities were nearly absent (Shibata et al. 2002). The AGEs presence in astrocytes in the spinal cord demonstrates that carbonyl stress in SOD1 mutant subjects may stimulate the potentially deleterious effects of AGEs. Remarkably, levels of soluble RAGE (sRAGE), a C-terminal truncated isoform of RAGE, are significantly lower in the serum of ALS patients (Ilzecka 2008). sRAGE, deficient the transmembrane-anchoring domain, found to ameliorate the deleterious effects of RAGE by forming a complex with the ligand (Sakaguchi et al. 2003; Wendt et al. 2003). Therefore, one conspicuous role of sRAGE is to protect humans from ALS, indicates that low sRAGE levels may be a risk factor for ALS.
Arai et al. demonstrated that glycation, modifies Cu, Zn-SOD1, specifically at Lys3, Lys9, Lys39, Lys36m, Lys122 and Lys128 residues (Arai et al. 1987). Higher susceptibility of mutated Cu, Zn-SOD1 to glycation was observed at Lys122 and Lys128 (Arai et al. 1987; Takamiya et al. 2003). Recently, Kaufmann et al. demonstrated that the concentration of N-ε-(carboxymethyl) lysine (CML) significantly increases in serum and cerebrospinal fluid (CSF) of ALS patients, possibly representing a novel biomarker for diagnosis (Kaufmann et al. 2004). These results advocate that glycation might be accountable for the observed oxidative stress in ALS.
3.3.2 Effects of AGE Toxicity in FAP
Familial amyloid polyneuropathy (FAP) was first described in 1952 by Andrade, is a systemic amyloid disease, characterized by the extracellular deposition of fibrillar transthyretin (TTR), particularly in the peripheral nervous system (PNS) also affecting the autonomic nervous system. TTR is normally an innocuous protein, present in the plasma and CSF, responsible for the transport of thyroxin hormone (T4) and retinol in the blood (Kanai et al. 1968).
More than 80 TTR point mutations are linked with amyloidotic diseases. The most widely accepted disease model suggests that TTR tetramer instability is associated to point mutations. Nevertheless, this model fails to elucidate that the native TTR also forms amyloids in systemic senile amyloidosis, a geriatric disease and the age onset of disease varies by decades for patients bearing the similar mutation. Indeed, a few mutation carrying individuals are asymptomatic throughout their lives. Protein glycation, play an important role in disease development as methylglyoxal-derived AGE is found in FAP patients (da Costa et al. 2011). The glycated TTR (TTR-AGE) may contribute to cytotoxicity via a mechanism involving oxidative stress, or by interaction TTR-AGE with RAGE (Matsunaga et al. 2002; Sousa et al. 2001; Shorter and Lindquist 2005). Interaction between TTR-AGE fibrils and RAGE results in the translocation of NF-κB to the nucleus, where it induce tumor necrosis factor-α (TNFα) and interleukin-1β (IL-1β) (97). The activations of TNF-α and IL-1β were abrogated by an anti-RAGE antibody or sRAGE (Sousa et al. 2001). Thus, these studies suggest that glycation plays an important role in FAP. Thus, these data suggest that glycation plays an important role in FAP and blocking RAGE may constitute a good target for therapeutic intervention in FAP.
3.3.3 Effects of AGE Toxicity in Prion Diseases
Prion diseases are a group of NDDs caused by prions, which are “proteinaceous infectious particles” or abnormally folded cellular prion protein (PrPC). These diseases affect a lot of different mammals in addition to humans—for instance, there is scrapie in sheep, mad cow disease in cows, and chronic wasting disease in deer (Knight and Will 2004). The human forms of prion disease are most often the names Creutzfeldt-Jakob disease (CJD), Gertsmann-Straussler-Scheinker syndrome (GSS), variably protease-sensitive prionopathy (VPSPr), kuru and fatal familial insomnia (FFI) (Prusiner 1998). Prion diseases are mostly age-related in nature that can be spontaneous, genetic, or infection-related. Spontaneously occurring prion diseases. All of these diseases are caused by just slightly different versions of the same protein. The ‘protein only’ hypothesis states that these diseases are caused by the conversion of a normally folded prion protein (PrPC) into an abnormal isoform, which is resistant to degradation by proteinase K (PrPres). However, the precise mechanism is still the subject of extensive discussion (Soto and Castilla 2004). Interestingly recent findings indicate that PrP (C) is involved in signal transduction (Didonna 2013). The presence of AGEs and RAGE in the occipital lobe of CJD patients in higher amounts than in control individuals were confirmed by Sasaki et al. (2002) which revealed a co-localization of PrP-positive granule AGEs and RAGE.
The authors hypothesized that glycation would advance over time and like Aβ, PrP would be degraded through the lysosomal pathway in a RAGE-mediated process. Choi et al. reported that this post-translational modification occurs in the N-terminus of the protein, since upon PK digestion. It cleaves approx 90 amino acid residues from the PrPres N-terminal; glycation was no longer detected (Choi et al. 2004). Although human PrP contains 21 lysine and arginine residues, glycation does not appear to be an arbitrary process. Only 23rd, 24th, 27th lysines and 37th arginine of PrPres can be glycated (Choi et al. 2004). Choi et al. reported glycation in several mouse models infected with 139A, ME7, 22L and 87V scrapie strains, in hamster adapted 263K and 139H scrapie strains, and in both sporadic and genetic CJD. Glycated PrP was detected by using western blot analysis, to form oligomeric species, where immunoreactivity was observed for monomeric and dimeric forms of PrP (Choi et al. 2004). So the role of AGEs in the formation of PrPSc remains unknown, but two hypotheses exist i.e. all AGE modification occurs at the time of PrPSc formation and glycation only occurs after the formation of PrPSc. Hence, glycation plays a key role in the pathogenesis of prion diseases, because glycation would provide enhanced protection for the PrPSc molecules against cellular degradation. Therefore, although the role of glycation in the formation of PrPres cannot be expelled, some discussions still need to be addressed to better understand the role of glycation in prion aggregation and disease progress.
4 Effects of Toxic DNA-AGE in NDDs
DNA bases are vulnerable to glycoxidative stress damage involving hydroxylation, protein carbonylation and nitration (Lovell and Markesbery 2007; Gabbita et al. 1998; Collins et al. 1996). It has been reported that under glycation reaction some free radicals i.e. superoxides and hydroxyl generates (Akhter et al. 2014) that caused oxidative damage to DNA molecules that might be caused the neuronal damaged having direct implication of NDDs. It has been observed in AD that brain ROS induces calcium influx, via glutamate receptors and triggers an excitotoxic response leading to cell death (Mattson and Chan 2003). DNA and RNA oxidation are marked by increased level of 8-hydroxy-2-deoxyguanosine (8OHdG) and 8-hydroxyguanosine (8OHD) (Nunomura et al. 1999, 2001; Lovell et al. 1999). Furthermore, these markers have been localized in Aβ plaques and NFTs (Mecocci et al. 1994). Increased levels of DNA strand breaks have been found in AD. They were first considered to be part of apoptosis, but it is now widely accepted that oxidative damage is responsible for DNA strand breaks and this is consistent with the increased free carbonyls in the nuclei of neurons and glia in AD. Among all dicarbonyl intermediates formed in the glycation reaction, MG is a potent and most reactive AGE precursor, forming adducts on deoxyguanosine (dG) residues. Furthermore, it has been shown that DNA can be glycated in vitro yielding N2-carboxyethyl-2′-deoxyguanosine (CEdG) as a major product. In vitro, nucleobases and ds-DNA react with sugars in a similar way as proteins (Lee and Cerami 1987; Knerr and Severin 1993; Singh et al. 2001). The exocyclic amino group of 2′-deoxyguanosine is particularly prone to glycation; leading to the formation of N2-carboxyethyl, N2-carboxymethyl, as well as cyclic dicarbonyl adducts (Ochs and Severin 1994). The CEdG is a stable reaction product, formed from a variety of glycating agents, such as glucose, ascorbic acid, glyceraldehyde, dihydroxyacetone (DHA) and MG (Frischmann et al. 2005). Apart from in vitro DNA glycation, under hyperglycemic condition the glycation of DNA (CEdG A and CEdG B) (Fig. 4) occurs in vivo and is considered to be a pathogenic factor for NDDs (Padmaraju et al. 2011) (Fig. 5).

A structurally representation of glycated DNA adducts i.e. CEdG A (a) and CEdG B (b) as DNA-AGEs

Probable mechanistic representation of glycation reaction between reducing sugars and adenine/guanine bases, and cause of neuronal toxicity
Oxygen free radicals as well as possibly signaling of AGEs to the nucleus through their specific receptor(s) (RAGE) may induce DNA strand scission (Kashimura et al. 1990; Morita and Kashimura 1990) e.g., by stimulation of nuclear factor NF-κB (Lander et al. 1997; Li and Schmidt 1997). DNA replication may become inhibited by intermediate products of the Maillard reaction such as 3-deoxyglucosone, which may lead to impairment of cell proliferation (Shinoda et al. 1990).
Non-enzymatic glycation with production of AGEs has been regarded as a process of molecular (Bunn 1981) and cellular (Li et al. 1995) ageing. Factors such as oxidant stress, molecular cross-linking, the sequelae of RAGE signaling and changes of DNA integrity extrapolate this process from the molecular to the biological (cellular and tissue) level (Huttunen et al. 1999, 2000, 2002). This pertains to ageing (Bjorksten 1977; Monnier et al. 1990) and AD (Heitner and Dickson 1997; Smith et al. 1994).
A few evidences suggests the role of DNA-AGEs in NDDs, yet a comprehensive, potential and prospective study is required to know the deleterious effects of DNA-AGEs in the assessment of neuronal diseases.
5 Anti-AGE System to Halt the Menace of Toxicity of Glycation
5.1 Natural and Synthetic Inhibitor
The deterrence of glycation reaction is one of the strategies to reduce DNA-AGEs/protein-AGEs. Both synthetic compounds and natural products have been evaluated as inhibitors against the formation of AGEs. The formation of ketoamine moieties or intermediate products can be inhibited by reacted biomolecule that react with the reducing sugars, thereby inhibiting the deleterious reaction. Anti AGE systems must be able to grasp with preexistent forms and their formation by scavenging or degrading antiglycation agents. Some protective enzymatic mechanisms are also involved in the degradation of glycation reaction. Glyoxal (GO) and methylglyoxal (MGO) are the most reactive species, able to damage various biomolecules like proteins, DNA and lipoproteins through the Maillard reaction, which can culminate in cell apoptosis (Akhter et al. 2016b; Maeta et al. 2005). Not only glyoxalase system but also some other oxide reductase and dehydrogenases like α-oxoaldehyde dehydrogenase, aldehyde dehydrogenase, aldose reductase, methylglyoxal reductase, and pyruvate dehydrogenase are able to convert MGO into its oxidized or reduced form (Kalapos 1999). Glyoxalase-I (GO-I) is evidently decreased in AD, as already stated by Kuhla et al. (2007), resulting in the accretion of α-dicarbonyls and subsequent deleterious effects. Some specific natural and synthetic inhibitors may also prevent glycation induced cross-linking of proteins and DNA adducts formation. The trapping of the reactive carbonyl intermediates formed in the first stage of Maillard reaction can be carried out by using guanidine compound such as aminoguanidine (AG). AG was the first AGE inhibitor proposed to scavenge the dicarbonyl compounds such as glyoxal, MG and 3-DG, via its fast reaction with α-dicarbonyl compounds, forms 3-amino-1,2,4-triazine derivatives (Thornalley et al. 2000). Tenilsetam is a known potent AGEs cross-linking inhibitor (Munch et al. 1994), is successfully used for the treatment of patients suffering from AD (Choonara et al. 2009). This is covalently attached to protein-AGEs and blocks the reactive sites for further polymerization reactions. Interestingly, both AG and Tenilsetam having anti neurodegenerative property as were found to protect against the neurotoxic effects of methylglyoxal (Webster et al. 2005). Another category of AGE inhibitors, “Amadorin” (the post-Amadori inhibitors), inhibits the conversion of Amadori intermediates to AGE (Khalifah et al. 1999). Pyridoxamine was the first Amadorin identified that showed a great potential for treatment of diseases by inhibiting AGE formation at different levels. It scavenges carbonyl products of reducing sugars and lipid degradation, sequestering catalytic metal ions, blocking oxidative degradation of glycation intermediates and trapping of ROS. Lalezari-Rahbar (LR) compounds are potent Cu2+ chelator, that is more efficiently than PM and which inhibit post-Amadori AGE formation in vitro (Rahbar and Figarola 2003). Benfotiamine; (a lipophilic derivative of vitamin B1), improves the activity of transketolase by reducing the accumulation of hexose and triose phosphates by shunting these intermediates to pentose phosphate pathway (Stracke et al. 2001). Animal model experimental studies have shown that bioactive compounds from therapeutic plant can reduce diabetic complications like neuropathy (Akhter et al. 2013a). The presence of specific enzymes like deglycases or amadoriases is also able to degrade glycated proteins, these include fructosamine 3-kinase enzyme that phosphorylates protein-bound fructosamines and spontaneously breaks down the Amadori rearrangement into inorganic phosphate, along with 3-deoxyglucosone and the amino compound (Akhter et al. 2014; Ahmad et al. 2013). Delpierre et al. observed this process in vivo, leading to deglycation of haemoglobin and protein-bound ribulosamines and psicosamines in erythrocytes (Collard et al. 2004). fructoselysine 6-kinase and Fructose lysine oxidase are also believed to play a role in deglycation of protein (Takahashi et al. 1997; Wiame et al. 2002). Several novel inhibitors like aldehyde, pyridoxal-phosphate, or the acetylating agent aspirin have recently been discovered, react with and thereby cap the free amino groups of proteins and prevent sugar attachment. Thiol-based antioxidants also act as a carbonyl scavenger like glutathione (GSH). Besides this, there are some other dicarbonyl scavengers including L-arginine that trap glyoxal, glycoaldehyde, and glucosones to form substituted triazines. These drugs are potent curative agents for treating neurodegenerative maladies. The ROS and free transition metal ions are known to play an important role in the formation of AGEs. Where, the glycoxidation reactions could be efficiently inhibited by redox metal chelators (Baynes 1991; Price et al. 2001; Nagai et al. 2012), including diethylene triamine penta acetic acid, phytate, and penicillamine. Hence, these redox-metal chelators could be used in the therapy of NDDs. Nucleation-dependent polymerization of Aβ, the major component of plaques in patients with AD, is significantly accelerated by AGEs in vitro. Drugs like metformin (Ruggiero-Lopez et al. 1999; Beisswenger and Ruggiero-Lopez 2003; Beisswenger et al. 1999) and buformin (Kiho et al. 2005) also showed the potential to protect proteins against in vitro glycation and cross-linking (Bonnefont-Rousselot 2001; Hipkiss 1998). Furthermore, isoferulc acid (IFA) suppressed the formation of β-cross-linked amyloid structures of BSA, is a promising anti-glycation agent, which can be used for the prevention of NDDs. There is one other significant approach to reduce accumulation of AGE formation is to reverse the process by AGE cross-link breakers such as N-phenacyl-4,5-dimethylthiazolium chloride (ALT-711/alagebrium) and N-phenacyl thiazolinium bromide. “RAGE blockers” comprise of agents that trap AGE with soluble RAGE (sRAGE), block RAGE and inhibit signal transduction mediated by AGE–RAGE interaction. Administration of recombinant sRAGE in animal models has shown to suppress neuronal dysfunction (Park et al. 1998). Although the synthetic compounds are powerful drugs that inhibit AGE formation or break cross-links, they can also have severe side effects. The importance of anti-AGEs is still divisive, since the resulting degraded compounds are able to react once more with amino groups of DNA, proteins and lipoproteins. However, the inhibition of AGEs and intermediate dicarbonyl compounds formation may correspond to a therapeutic approach also to be explored in various NDDs.
5.2 Anti-AGE Effect of Bio-conjugated Gold and Silver Nanoparticles (Au, Ag-NPs)
Anti-AGEs activity of gold nanoparticles (GNPs) was first reported in year 2009, inhibit the consequence of the glycation reaction (Singha et al. 2009). The prevention of glycation of α-crystalline was done by conjugation with GNPs. Formation of protein and using bio-conjugated GNPs prevents DNA-AGEs, even if a strong glycated agent such as fructose is used. In addition, the nano-conjugation approach can provide some important information on the structural distribution of any dynamic chaperone protein and DNA. Because GNPs are biocompatible, their reported antiglycation property may have therapeutic implications.
Furthermore apart from GNPs, silver nanoparticles (Ag-NPs) have also shown to efficiently diminish the menace of the glycation reaction. It has been determined the effects of Ag-NPs on AGEs-induced cell permeability. The AGE-bovine serum albumin (BSA) increased the dextran flux across a PREC monolayer and Ag-NPs blocked the solute flux induced by AGE-BSA. It was also demonstrated that Ag-NPs could inhibit the AGE-BSA-induced permeability via Src kinase pathway. There are reports where researchers have proved that GNPs alone (Seneviratne et al. 2012) and GNPs bio-conjugated with protein (Nowacek et al. 2009) can reduce the rate of non-enzymatic modification of proteins responsible for glycation. The role of GNPs in the targeted delivery of drugs is one of the most promising aspects. For the bio-conjugation of drug with GNPs a robust and stable protein, albumin was chosen as a capping agent over GNPs. The role of albumin as a drug carrier for last several years has emerged wonderfully because it does not only provide the stability to the drugs but also improves the half-life of drugs/active proteins/peptides. The conjugation was carried out either by simple physical adsorption of the drugs onto GNPs or via the use of alkanethiol linkers (Kratz 2008).
In fact, proteins glycated to different extents showed the formation of nanoparticles of different size and eventually their plasmon intensities were also different. This implied that glycated proteins cause some attenuation of the particle formation that led to only smaller nanoparticle formation. One possible example of such negative control on the particle size formation may arise from proximity of glycation-prone sites on which GNPs preferentially form on the protein surface. This as well as other structural studies with nanoparticle protein conjugates, prompted to explore the possibility of prevention of glycation (Neely et al. 2009). The origin of such resistance against glycation is intriguing. The amino acids containing the free amino group (lysine) are potent sites for glycation in addition to the N-terminal amino acid. Few antiglycating agents have been already reported in the literature, and apparently a nontoxic agent like gold showing antiglycating properties may itself have important clinical significance (Rahim et al. 2014).
In the light of the above explanations, this review is aiming to exploit its preventive effect on glycation by reducing the concentrations of the AG or other drugs showing toxicity at higher concentrations but has antiglycation effect. These drugs might be used for nano-conjugation using GNPs, thus reducing the toxic concentration to minimal. The present concern on bio-conjugation of AG with GNPs is the need of the hour. To enhance activity of the entire above novel drugs at reduced dozes, such as LR-74, LR-90 and others including AG in bio-conjugation with GNPs, might prove to be more accurate and specific novel inhibitor of DNA and protein glycation at reduced doses. Table 1 enlists the several bio-conjugated of GNPs with various proteins and their association with neurodegenerative disorders.
6 Concluding Interpretation and Future Prospects
Modifiable environmental factors producing toxicity of oxidized DNA and DNA-AGEs, accumulate in a wide variety of environments and has potential role in the cause of neurological disorders (Wuenschell et al. 2010). These overall studies have conclusively verified that AGEs are complex and heterogeneous in nature. Their mechanism of formation is only partially understood. AGEs play an important role in various NDDs, including AD, PD, ALS, FAP and PrD. In all of these, pathological amyloid glycation induces the formation of α-sheet structure in the amyloid-β- protein, α-synuclein, TTR, SOD1 protein, and prions and causes these NDD. More recently, it has been suggested that oligomeric species of glycated α-synuclein and prion are more toxic than fibrils alone. The query of whether glycation induce protein fibrilization remains unclear and is under intense dispute. Although it seems to induce Aβ fibrilization, α-syn and PrP oligomerization, a direct relationship between glycation and β-sheet formation in vivo is still mislaid. Since glycation is understood as a non-enzymatic process, the modifications are identified to occur at arbitrary. Moreover, only four of the 21 PrPres putative glycation sites are modified. These results suggest that glycation may modify specific targets and be understood as a posttranslational modification that alters protein function, similarly to phosphorylation or acetylation.
A number of AGE-inhibitors have been discovered that inhibit the toxicity of glycation pathway. These inhibitors are either synthetic or natural. One novel approach is bio-conjugated nanoparticles as an antiglycating agent i.e. Au-NPs and Ag-NPs that might be effective at reduced doses for the treatment of NDD. The inhibitors cap the amino groups of DNA and proteins, scavenge free carbonyls or dicarbonyls, block Amadori adducts, break existing cross-links, chelate metal ions, and possess anti-amyloidogenic, anti-oxidative, and anti-inflammatory activities. Thus, they can inhibit glycation reactions. The AGE–RAGE damaging axis is now considered to be a promising drug target. Additionally, there are defense enzymes and protein present in the body, such as glyoxylase systems I and II, fructose-3-kinase, aldose reductase, and carnosine. These enzymes and protein protect the neuronal cell from glycation and carbonyl stress. The formation of toxic oligomeric species could be controlled by blocking conformational changes in monomeric species of these pathological proteins using theses novel inhibitors. More efficient drugs could be designed to be more hydrophobic so that it can easily cross the lipid-bilayer membrane of the brain and prevent efficiently NDD. Using combination therapies, novel drugs could be designed that simultaneously target multiple pathways and may obviously be more efficient than those drugs that modify a single pathway and thereby decrease the risk of side effects.
Eventually, based on anti neurological disorders characteristics of natural products, it is hypothesized that these therapeutic compounds might exhibit antiglycating properties as well with no side effects. AGEs inhibition using nanoparticles as drug delivery system (Kim et al. 2012; Seneviratne et al. 2012); therefore, it would be interesting to see the inhibition of AGEs using various bio-conjugated inhibitors (IFA, AG and Tenilsetam) with GNPs. In the light of above explanations, in general, this study holds strong future prospects with the development of new, more effective and more specific inhibitors. The inhibitors will be developed based on structure–activity liaison. This study will also help to identify the new targets for AGEs. Since the above explanations confirm that the NDDs such as Alzheimer’s and Parkinson’s diseases are associated with toxicity of glycation intermediate (dicarbonyl compounds and keto-amine moieties) and their end products (AGEs), which direct or indirect depends on various environmental factors too; therefore, targeting a site and changes in life styles could result in prevention or protection against these drowning and dreaded diseases.
References
Ahmad MI, Ahmad S, Moinuddin (2011a) Preferential recognition of methyl glyoxal-modified calf thymus DNA by circulating antibodies in cancer patients. Int J Biochem Biophys 48:290–296
Ahmad S, Dixit K, Moinuddin et al (2011b) Genotoxicity and immunogenicity of DNA-advanced glycation end products formed by methylglyoxal and lysine in presence of Cu2+. Biochem Biophys Res Commun 407:568–574
Ahmad S, Akhter F, Moinuddin et al (2013) Studies on glycation of human low density lipoprotein: a functional insight into physico chemical analysis. Int J Biol Macromol 62:167– 171
Ahmad S, Khan MS, Khan MS et al (2014) Glycoxidation of biological macromolecules: a critical approach to halt the menace of glycation. Glycobiology 24:979–990
Akhter F, Hashim A, Khana MS et al (2013a) Antioxidant, a-amylase inhibitory and oxidative DNA damage protective property of Boerhaavia diffusa (Linn.) root. S Afr J Biol 88:265–272
Akhter F, Khan MS, Shahab U et al (2013b) Bio-physical characterization of ribose induced glycation: a mechanistic study on DNA perturbations. Int J Biol Macromol 58:206–210
Akhter F, Khan MS, Singh S, Ahmad S (2014) An immunohistochemical analysis to validate the rationale behind the enhanced immunogenicity of D-ribosylated low density lipo-protein. PLoS ONE 9:0113144
Akhter F, Khan MS, Ahmad S (2015) Acquired immunogenicity of calf thymus DNA and LDL modified by D-ribose: a comparative study. Int J Biol Macromol 72:1222–1227
Akhter F, Khan MS, Alatar AA et al (2016a) Antigenic role of the adaptive immune response to d-ribose glycated LDL in diabetes, atherosclerosis and diabetes atherosclerotic patients. Life Sci 151:139–146
Akhter F, Khan MS, Faisal M, Ahmad S (2016b) Detection of circulating auto-antibodies against ribosylated-LDL in diabetes patients. J Clin Lab Anal. doi:10.1002/jcla.22039
Aldini G, Vistoli G, Stefek M et al (2013) Molecular strategies to prevent, inhibit and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radical Res 47:93–137
Andorfer C, Acker CM, Kress Y et al (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25:5446–5454
Arai K, Maguchi S, Fujii S et al (1987) Glycation and inactivation of human Cu–Zn-superoxide dismutase. Identification of the in vitro glycated sites. J Biol Chem 262:16969–16972
Baynes JW (1991) Role of oxidative stress in development of complication in diabetes. Diabetes 40:405–412
Beisswenger P, Ruggiero-Lopez D (2003) Metformin inhibition of glycation processes. Diabetes Metab 29:6S95–6S103
Beisswenger PJ, Howell SK, Touchette AD et al (1999) Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 48:198–202
Bjorksten J (1977) Some therapeutic implications of the crosslinkage theory of aging. Adv Exp Med Biol 86B:579–602
Bonnefont-Rousselot D (2001) Antioxidant and anti-AGE therapeutics: evaluation and perspectives. J Soc Biol 195:391–398
Bunn AF (1981) Non-enzymatic glycosylation of protein: a form of molecular aging. Schweiz Med Wschr 111:1503–1507
Burke RE (1999) α-Synuclein and Parkinson’s disease. Brain Res Bull 50:465–466
Campion D, Dumanchin C, Hannequin D et al (1999) Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65:664–670
Caspersen C, Wang N, Yao J et al (2005) Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 19:2040–2041
Chen K, Maley J, Yu PH (2006) Potential inplications of endogenous aldehydes in β-amyloid misfolding, oligomerization and fibrillogenesis. J Neurochem 99:1413–1424
Choi YG, Kim JI, Jeon YC et al (2004) Nonenzymatic glycation at the N-terminus of pathogenic prion protein in transmissible spongiform encephalopathies. J Biol Chem 279:30402–30409
Choonara YE, Pillay V, du Toit LC et al (2009) Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int J Mol Sci 10:2510–2557
Collard F, Wiame E, Bergans N et al (2004) Fructosamine 3-kinase-related protein and deglycation in human erythrocytes. Biochem J 382:137–143
Collins AR, Dusinská M, Gedik CM, Stetina R (1996) Oxidative damage to DNA: do we have a reliable biomarker? Environ Health Perspect 104:465–469
Cummings JL, Vinters HV, Cole GM, Khachaturian ZS (1998) Alzheimer’s disease: etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 51(S2–17): discussion, S65–17
da Costa G, Gomes RA, Guerreiro A et al (2011) Beyond genetic factors in familial amyloidotic polyneuropathy: protein glycation and the loss of fibrinogen’s chaperone activity. PLoS ONE 6:e24850
Dalfo E, Portero-Otin M, Ayala V et al (2005) Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol 64:816–830
Defebvre L (2010) Parkinson’s disease: role of genetic and environment factors. Involvement in everyday clinical practice. Rev Neurol (Paris) 166:764–769
Dickson DW, Sinicropi S, Yen SH et al (1996) Glycation and microglial reaction in lesions of Alzheimer’s disease. Neurobiol Aging 17:733–743
Didonna A (2013) Prion protein and its role in signal transduction. Cell Mol Biol Lett 18:209–230
Du H, Guo L, Fang F et al (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14:1097–1105
Forno LS (1996) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55:259–272
Frischmann M, Bidmon C, Angerer J, Pischetsrieder M (2005) Identification of DNA adducts of methylglyoxal. Chem Res Toxicol 18:1586–1592
Gabbita SP, Lovell MA, Markesbery WR (1998) Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem 71:2034–2040
Gros-Louis F, Gaspar C, Rouleau GA (2006) Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:956–972
Grossman H, Bergmann C, Parker S (2006) Dementia: a brief review. Mt Sinai J Med 73:985–992
Guerrero E, Vasudevaraju P, Hegde ML et al (2013) Recent advances in α-synuclein functions, advanced glycation, and toxicity: implications for Parkinson’s disease. Mol Neurobiol 47:525–536
Haass C, Hung AY, Selkoe DJ, Teplow DB (1994) Mutations associated with a locus for familial Alzheimer’s disease result in alternative processing of amyloid β-protein precursor. J Biol Chem 269:17741–17748
Hegde ML, Vasudevaraju P, Rao KJ (2010) DNA induced folding/fibrillation of alpha-synuclein: new insights in Parkinson’s disease. Front Biosci 15:418–436
Heitner J, Dickson D (1997) Diabetic do not have increased Alzheimer-type pathology compared with age-matched control subjects. A retrospective postmortem immunocytochemical and histofluorescent study. Neurology 49:1306–1311
Hipkiss AR (1998) Carnosine, a protective, anti-ageing peptide? Int J Biochem Cell Biol 30:863–868
Huttunen HJ, Fages C, Rauvala H (1999) Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-jB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 274:19919–19924
Huttunen HJ, Kuja-Panula J, Sorci G et al (2000) Coregulation of neurite outgrowth and cell survival by amphoterin and S100 protein through receptor for advanced glycation end products (RAGE) activation. J Biol Chem 275:40096–40105
Huttunen HJ, Kuja-Panula J, Rauvala H (2002) Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J Biol Chem 277:38635–38646
Ilzecka J (2008) Serum-soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand
Izumi Y, Kume T, Akaike A (2011) Regulation of dopaminergic neuronal death by endogenous dopamine and proteasome activity. Yakugaku Zasshi 131:21–27
Jacobs COK, Haucke E, Santos AN et al (2014) Role of advanced glycation end products in cellular signaling. Redox Biol 2:411–429
Kalapos MP (1999) Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett 110:145–175
Kanai M, Raz A, Goodman DS (1968) Retinol-binding protein: the transport protein for vitamin A in human plasma. J Clin Invest 47:2025–2044
Kashimura N, Morita J, Nishikawa S, Kumazawa Z (1990) Autooxidation and DNA cleavage reaction of glycated proteins. In: Finot PA, Aeschbacher HU, Hurrell RF, Liardon R (eds) The Maillard reaction in food processing, human nutrition and physiology. Birkhäuser, Basel, pp 449–454
Kaufmann E, Boehm BO, Sussmuth SD et al (2004) The advanced glycation endproduct N-ε-(carboxymethyl)lysine level is elevated in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurosci Lett 371:226–229
Khalifah RG, Baynes JW, Hudson BG (1999) Amadorins: novel post-Amadori inhibitors of advanced glycation reactions. Biochem Biophys Res Commun 57:251–258
Kiho T, Kato M, Usui S, Hirano K (2005) Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin Chim Acta 358:139–145
Kikuchi S, Shinpo K, Takeuchi M (2003) Glycation—a sweet tempter for neuronal death. Brain Res Rev 41:306–323
Kim JH, Hong CO, Koo YC et al (2012) Anti-glycation effect of gold nanoparticles on collagen. Biol Pharm Bull 35:260–264
Knerr T, Severin T (1993) Reaction of glucose with guanosine. Tetrahedron Lett 34:7389–7390
Knight RS, Will RG (2004) Prion diseases. J Neurol Neurosurg Psychiat 75:36–42
Kratz F (2008) Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release 132:171–183
Kruger R, Kuhn W, Muller T et al (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Kuhla B, Boeck K, Schmidt A et al (2007) Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol Aging 28:29–41
Lander HM, Tauras JM, Ogiste JS et al (1997) Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem 272:17810–17814
Ledesma MD, Bonay P, Colaco C, Avila J (1994) Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem 269:21614–21619
Lee A, Cerami A (1987) The formation of reactive intermediate(s) of glucose 6-phosphate and lysine capable of rapidly reacting with DNA. Mutat Res 179:151–158
Lee D, Park CW, Paik SR, Choi KY (2009) The modification of α-ynuclein by dicarbonyl compounds inhibits its fibril-forming process. Biochim Biophys Acta 1794:421–430
Lekoubou A, Echouffo-Tcheugui JB, Kengne AP (2014) Epidemiology of neurodegenerative diseases in sub-Saharan Africa: a systematic review. BMC Public Health 14:653
Li J, Schmidt AM (1997) Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 272:16498–16506
Li JJ, Surini M, Catsicas S, Kawashima E, Bouras C (1995) Age-dependent accumulation of advanced glycosylation end products in human neurons. Neurobiol Aging 16:69–76
Li JJ, Dickson D, Hof PR, Vlassara H (1998) Receptors for advanced glycosylation endproducts in human brain: role in brain homeostasis. Mol Med 4:46–60
Li XH, Lv BL, Xie JZ et al (2012) AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging 33:400–410
Liu BF, Miyata S, Hirota Y et al (2003) Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int 63:947–957
Lovell MA, Markesbery WR (2007) Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 35:7497–7504
Lovell MA, Gabbita SP, Markesbery WR (1999) Increased DNA oxidation and decreassed levels of repair products in Alzheimer’s disease ventricular CSF. J Neurochem 72:771–776
Lustbader JW, Cirilli M, Lin C et al (2004) ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304:448–452
Luth HJ, Ogunlade V, Kuhla B et al (2005) Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb Cortex 15:211–220
Maeta K, Mori K, Takatsume Y et al (2005) Diagnosis of cell death induced by methylglyoxal, a metabolite derived from glycolysis, in Saccharomyces cerevisiae. FEMS Microbiol Lett 243:87–92
Maillard LC (1912) Action des acides amines sur les sucres; formation de me`lanoides par voie méthodique. C R Acad Sci 154:66–68
Martin I, Dawson VL, Dawson TM (2010) The impact of genetic research on our understanding of Parkinson’s disease. Prog Brain Res 183:21–41
Martin I, Dawson VL, Dawson TM (2011) Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet 12:301–325
Matsunaga N, Anan I, Forsgren S et al (2002) Advanced glycation end products (AGE) and the receptor for AGE are present in gastrointestinal tract of familial amyloidotic polyneuropathy patients but do not induce NF-κB activation. Acta Neuropathol 104:441–447
Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34:385–397
Mattson MP, Carney JW, Butterfield DA (1995) A tombstone in Alzheimer’s? Nature 373:481
Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36:747–751
Monnier VM, Sell DR, Miyata S, Nagaraj RH (1990) The Maillard reaction as a basis for a theory of aging. In: Finot PA, Aeschbacher HU, Hurrell RF, Liardon R (eds) The Maillard reaction in food processing, human nutrition and physiology. Birkhäuser, Basel, pp 393–414
Morita J, Kashimura N (1990) Involvement of monosaccharide autooxidation in DNA glycation under physiological conditions. In: Finot PA, Aeschbacher HU, Hurrell RF, Liardon R (eds) The Maillard reaction in food processing, human nutrition and physiology. Birkhäuser, Basel, pp 505–510
Munch G, Taneli Y, Schraven E et al (1994) The cognition-enhancing drug tenilsetam is an inhibitor of protein crosslinking by advanced glycosylation. J Neural Transm Park Dis Dement Sect 8:193–208
Munch G, Schicktanz D, Behme A et al (1999) Amino acid specificity of glycation and protein-AGE crosslinking reactivities determined with a dipeptide SPOT library. Nat Biotechnol 17:1006–1010
Mustafa I, Ahmad S, Dixit K et al (2012) Glycated human DNA is a preferred antigen for anti-DNA antibodies in diabetic patients. Diabetes Res Clin Pract 95:98–104
Nagai R, Murray DB, Metz TO, Baynes JW (2012) Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 61:549–559
Neely A, Perry C, Varisli B et al (2009) Ultrasensitive and highly selective detection of Alzheimer’s disease biomarker using two-photon Rayleigh scattering properties of gold nanoparticle. ACS Nano 3:2834–2840
Nowacek A, Kosloski LM, Gendelman HE (2009) Neurodegenerative disorders and nanoformulated drug development. Nanomedicine (Lond) 4:541–555
Nunomura A, Perry G, Pappolla MA (1999) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19:1959–1964
Nunomura A, Perry G, Aliev G et al (2001) Oxidative damage is the earliest event in Alzheimer’s disease. J Neuropathol Exp Neurol 60:759–767
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Gene tic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780
Ochs S, Severin T (1994) Reaction of 2′-deoxyguanosine with glyceraldehyde. Liebigs Ann Chem 5:851–853
Padmaraju V, Bhaskar JJ, Prasada Rao UJ et al (2011) Role of advanced glycation on aggregation and DNA binding properties of alpha-synuclein. J Alzheimers Dis 24:211–221
Park L, Raman KG, Lee KJ et al (1998) Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 4:1025–1031
Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723
Peppa M, Uribarri J, Vlassara H (2003) Glucose, advanced glycation end products, and diabetes complications: what is new and what works. Clin Diab 21:186–187
Polymeropoulos MH, Lavedan C, Leroy E et al (1997) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Price DL, Rhett PM, Thorpe SR, Baynes JW (2001) Chelating activity of advanced glycation end product (AGE) inhibitors. J Biol Chem 276:48967–48972
Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383
Rademakers R, Rovelet-Lecrux A (2009) Recent insights into the molecular genetics of dementia. Trends Neurosci 32:451–461
Rahbar S, Figarola JL (2003) Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys 419:63–79
Rahim M, Iram S, Khan MS et al (2014) Glycation-assisted synthesized gold nanoparticles inhibit growth of bone cancer cells. Colloids Surf B Biointerfaces 117:473–479
Reynolds CH, Betts JC, Blackstock WP (2000) Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3β. J Neurochem 74:1587–1595
Ruggiero-Lopez D, Lecomte M, Moinet G et al (1999) Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol 58:1765–1773
Saez-Valero J, Fodero LR, Sjogren M et al (2003) Glycosylation of acetylcholinesterase and butyrylcholinesterase changes as a function of the duration of Alzheimer’s disease. J Neurosci Res 72:520–526
Sakaguchi T, Yan SF, Yan SD et al (2003) Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 111:959–972
Saraiva MJ (2001) Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat 17:493–503
Sasaki N, Fukatsu R, Tsuzuki K et al (1998) Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am J Pathol 153:1149–1155
Sasaki N, Takeuchi M, Chowei H et al (2002) Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt-Jakob disease with prion plaques. Neurosci Lett 326:117–120
Sato S, Tatebayashi Y, Akagi T et al (2002) Aberrant tau phosphorylation by glycogen synthase kinase-3β and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem 277:42060–42065
Sato T, Shimogaito N, Wu X et al (2006) Toxic advanced glycation end products (TAGE) theory in Alzheimer’s disease. Am J Alzheimers Dis Other Demen 21:197–208
Seneviratne C, Narayanan R, Liu W, Dain JA (2012) The in vitro inhibition effect of 2 nm gold nanoparticles on non-enzymatic glycation of human serum albumin. Biochem Biophys Res Commun 422:447–454
Shahab U, Ahmad A, Moinuddin et al (2012a) Hydroxyl radical modification of collagen type II increases its arthritogenicity and immunogenicity. PLoS ONE 7:e31199
Shahab U, Moinuddin, Ahmad S et al (2012b) Acquired immunogenicity of human DNA damaged by N-hydroxy- N-acetyl-4-aminobiphenyl. IUBMB Life 64:340–345
Shahab U, Moinuddin, Ahmad S et al (2013) Genotoxic effect of N-hydroxy-4-acetylaminobiphenyl on human DNA: implications in bladder cancer. PLoS ONE 8:e53205
Shahab U, Tabrez S, Khan MS et al (2014) Immunogenicity of DNA-advanced glycation end product fashioned through glyoxal and arginine in the presence of Fe3+: its potential role in prompt recognition of diabetes mellitus auto-antibodies. Chemico Biol Int 219:229–240
Shibata N, Hirano A, Hedley-Whyte ET et al (2002) Selective formation of certain advanced glycation end products in spinal cord astrocytes of humans and mice with superoxide dismutase-1 mutation. Acta Neuropathol 104:171–178
Shimasaki S, Kubota M, Yoshitomi M et al (2011) Nω-(carboxymethyl)arginine accumulates in glycated collagen and klotho-deficient mouse skin. Anti-Aging Med 8:82–87
Shinoda T, Chuyen NV, Hayase F, Kato H (1990) Physiological activity and metabolism of 3-deoxyglucosone. In: Finot PA, Aeschbacher HU, Hurrell RF, Liardon R (eds) The Maillard reaction in food processing, human nutrition and physiology. Birkhäuser, Basel, pp 309–14
Shorter J, Lindquist S (2005) Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 6:435–450
Sihlbom C, Davidsson P, Emmett MR et al (2004) Glycoproteomics of cerebrospinal fluid in neurodegenerative disease. Int J Mass Spectrom 234:145–152
Silveyra MX, Cuadrado-Corrales N, Marcos A et al (2006) Altered glycosylation of acetylcholinesterase in Creutzfeldt-Jakob disease. J Neurochem 96:97–104
Singh R, Barden A, Mori T, Beilin L (2001) Advanced glycation end-products: a review. Diabetologia 44:129–146
Singha S, Bhattacharya J, Datta H, Dasgupta AK (2009) Anti-glycation activity of gold nanoparticles. Nanomed Nanotech Biol Med 5:21–29
Smith MA, Taneda S, Richey PL et al (1994) Advanced Maillard reaction products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA 91:5710–5714
Soto C, Castilla J (2004) The controversial protein-only hypothesis of prion propagation. Nat Med 10:63–67
Sousa MM, Du Yan S, Fernandes R et al (2001) Familial amyloid polyneuropathy, receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurosci 21:7576–7586
Spillantini MG, Schmidt ML, Lee VM et al (1997) α-Synuclein in Lewy bodies. Nature 388:839–840
Stracke H, Hammes HP, Werkmann D et al (2001) Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats. Exp Clin Endocrinol Diabetes 109:330–336
Takahashi M, Pischetsrieder M, Monnier VM (1997) Molecular cloning and expression of amadoriase isoenzyme (fructosyl amine: oxygen oxidoreductase, EC 1.5.3) from Aspergillus fumigatus. J Biol Chem 272:12505–12507
Takamiya R, Takahashi M, Myint T et al (2003) Glycation proceeds faster in mutated Cu, Znsuperoxide dismutases related to familial amyotrophic lateral sclerosis. FASEB J 17:938–940
Takeda A, Smith MA, Avila J et al (2000) In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J Neurochem 75:1234–1241
Takeuchi M, Bucala R, Suzuki T et al (2000) Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J Neuropathol Exp Neurol 59:1094–1105
Thornalley PJ (1998) Glutathione-dependent detoxification of α-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interact 111–112:137–151
Thornalley PJ, Yurek-George A, Argirov OK (2000) Kinetics and mechanism of the reaction of aminoguanidine with the α-oxoaldehydes glyoxal, methylglyoxal, and 3-deoxyglucosone under physiological conditions. Biochem Pharmacol 60:55–65
Véronique VB, Hussein D, Patrick AD (2010) TDP-43, protein aggregation, and amyotrophic lateral sclerosis. US Neurology 5:35–38
Vitek MP, Bhattacharya K, Glendening JM et al (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 91:4766–4770
Vlassara H, Brownlee M, Cerami A (1983) Excessive nonenzymatic glycosylation of peripheral and central nervous system myelin components in diabetic rats. Diabetes 32:670–674
Webster J, Urban C, Berbaum K et al (2005) The carbonyl scavengers aminoguanidine and tenilsetam protect against the neurotoxic effects of methylglyoxal. Neurotox Res 7:95–101
Wei Y, Han CS, Zhou J et al (2012) D-ribose in glycation and protein aggregation. Biochim Biophys Acta 1820:488–894
Wendt TM, Tanji N, Guo J et al (2003) RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 162:1123–1137
Wiame E, Delpierre G, Collard F, Van Schaftingen E (2002) Identification of a pathway for the utilization of the Amadori product fructoselysine in Escherichia coli. J Biol Chem 277:42523–42529
Wittmann CW, Wszolek MF, Shulman JM et al (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714
Wuenschell GE, Tamae D, Cercillieux A et al (2010) Mutagenic potential of DNA glycation: Miscoding by (R)- and (S)-N2-(1-carboxyethyl)-2′-deoxyguanosine. Biochemistry 49:1814–1821
Yan SD, Chen X, Fu J et al (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Yan SD, Stern D, Schmidt AM (1997) What’s the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest 27:179–181
Yan SD, Stern D, Kane MD et al (1998) RAGE-Aβ interactions in the pathophysiology of Alzheimer’s disease. Restor Neurol Neurosci 12:167–173
Zarranz JJ, Alegre J, Gomez-Esteban JC et al (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173
Zhu X, Rottkamp CA, Boux H et al (2000) Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol 59:880–888
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Akhter, F., Akhter, A., Ahmad, S. (2017). Toxicity of Protein and DNA-AGEs in Neurodegenerative Diseases (NDDs) with Decisive Approaches to Stop the Deadly Consequences. In: Kesari, K. (eds) Perspectives in Environmental Toxicology. Environmental Science and Engineering(). Springer, Cham. https://doi.org/10.1007/978-3-319-46248-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-46248-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46247-9
Online ISBN: 978-3-319-46248-6
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)