
Abstract
Reads alignment is an essential step of next generation sequencing) data analyses. One challenging issue is represented by unmapped reads that are usually discarded and considered as not informative. Instead, it is important to fully understand the source of those reads, to assess the quality of the whole experiment. Moreover, it is of interest to get some insights on possible “contamination” from non-human sequences (e.g., viruses, bacteria, and fungi). Contamination may take place during the experimental procedures leading to sequencing, or be due to the presence of microorganisms infecting the sampled tissues. Here we propose a pipeline for the detection of viral, bacterial, and fungi contamination in human sequenced data. Similarities between input reads (query) and putative contaminating organism sequences (subject) are detected using a local alignment strategy (MegaBLAST). For each organism database DecontaMiner provides two main output files: one containing all the reads matching only a single organism; the second one containing the “ambiguous” matching reads. In both files, data is sorted by organism and classified by taxonomic group. Low quality, unaligned sequences, and those discarded by user criteria are also provided as output. Other information and summary statistics on the number of matched/filtered/discarded reads and organisms are generated. This pipeline has successfully detected foreign sequences in human Cancer RNA-seq data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The study of the human genome and its relationship with the environment is a crucial task in the context of modern biology.
The application of next generation sequencing technologies allows to characterize the genome-wide map of organisms. Genome investigation has been made possible by the construction of the reference genomes. Sequencing experiments produce a large amount of small sequences that have to be mapped to the reference. The alignment is probably the most challenging step of next generation sequencing (NGS) data analyses. It allows to obtain several information—such as read density, gene lists, and variant lists—crucial to the definition of the biological meaning underlying the data.
Typically the amount of reads that correctly map onto the human reference genome ranges between 70 and 90 % [1] leaving in some cases a consistent fraction of unmapped reads. Underestimating this portion may determine loss of precious information. Unmapped reads can be explained by errors during sequencing protocols, by the presence of repeat elements difficult to map, by novel transcripts that can be investigated by de novo assembly, and lastly, they can derive from non-human sequences. Indeed, microorganisms contamination can occur during samples processing or can be part of the normal or pathological tissues microbiome [2].
The interest in detecting microorganisms-derived sequences has grown up together with the spread of high-throughput approaches, allowing the extraction of information both about the quality of the experimental procedures and about the link between diseases and infections. The main appeal of these investigations is represented by the possibility to find new pathogen-disease associations. In literature there are many evidences which underline the importance of detecting contaminating organisms. Worth to note are the detection of polyomavirus in human Merkel cell carcinoma [3] and a novel Old World arenavirus in a cluster of patients with fatal transplant-associated disease [4]. Assembly of a novel bacterial draft genome starting from tissue specimens sequencing of cord colitis patients suggested an opportunistic pathogenic role for Bradyrhizobium enterica in humans [5].
Besides, environmental contaminations are routinely found in NGS datasets. Downstream contaminations or cross-contaminations can compromise the reliability of the whole experimental procedure. Strong et al. detected bacterial sequences, belonging to different taxa, in cell line data coming from different sequencing experiments and suggested the idea that a good portion of these bacterial reads did not derive from the specimens themselves but from downstream contamination. This suggestion has been supported by the detection of bacterial sequences in polyA RNA-seq [6]. Indeed, the polyA selection step should remove upstream contamination since bacteria are poorly polyadenylated. Moreover, to strengthen the hypothesis of downstream contamination occurrence, the authors analyzed RNA-sequencing data of five Epstein-Barr virus (EBV)-positive lymphoblastoid cell lines obtained in six different Illumina laboratories. Across these labs the level of bacterial reads per million human mapped reads (RPMHs) differed by as much as 30-fold, while the transcript levels of the EB virus were similar.
Furthermore, another study also confirmed this laboratory-peculiar contamination, showing that different sequencing centers had specific signatures of contaminating genomes as “time stamps” [7]. Unmapped ChIP-Seq reads from A. thaliana, Z. mays, H. sapiens, and D. melanogaster datasets were investigated and found contaminated by foreign sequences. Taxonomic classification of these reads allowed authors to define the contaminants and to calculate the relative abundance for each dataset [8].
Several tools, based on different computational approaches, have been developed and used for the detection of pathogens in high-throughput sequencing data, especially in cancer samples. In particular, PathSeq [9] and CaPSID [10] are worth mentioning. Both are available as integrated open source softwares.
PathSeq applies a subtraction approach in which the reads are aligned on six different human genomes. After, it uses local aligners such as Mega BLAST and BLASTN [11] to re-align reads to microbial reference sequences and to two additional human sequence databases. PathSeq is implemented in a cloud-computing environment. However, the PathSeq pipeline can be computationally intensive, mostly due to the numerous subtraction steps. CaPSID overcame this limit using a single human reference genome with splice junctions. Although CaPSID might face the risk to fail the correct alignment, it provides a large reduction in elaboration time. Furthermore, PathSeq discards the ambiguous reads that map both to human and pathogen genomes, while CaPSID stores them in a database.
It should be noted that PathSeq also requires a commercial computing platform (i.e., Amazon Elastic Compute Cloud, EC2) to be used. CaPSID does not have this kind of restriction but it requires two files in bam format as input, obtained by the user with a separate alignment software. The user should take care of aligning the sequences both to human and to each pathogen (bacteria, viruses, and fungi) reference genome of interest, thus performing the most computationally intensive steps before CaPSID. Hence, the CaPSID pipeline is lighter and faster, and it can provide even gene annotations and a user-friendly web application that integrates a genome browser.
Another cloud-compatible bioinformatics pipeline aimed to pathogen discovery is SURPI (“Sequence-based Ultrarapid Pathogen Identification”) [12], which provides a very useful and complete tool for the analysis of complex metagenomic NGS data. However, its purpose is the detection of microorganisms from complex clinical metagenomic samples open to the environment, using the entire NCBI nt and/or NCBI nr protein databases in comprehensive mode. The algorithm is particularly sensitive but, as consequence, the pipeline is likely not appropriate for a rapid analysis of the unmapped reads.
As far as we know, all the pipelines mentioned above are designed to analyze data primarily aimed to the detection of pathogens in human samples. Due to this, some of them, such as PathSeq and SURPI, provide intensive pipeline including alignment to host genome, while CaPSID, in order to reduce the required time and computational efforts, works on BAM files provided by the user, containing the resulted alignments to the human and to all the pathogen reference sequences.
Here we propose DecontaMiner, a pipeline designed and developed to detect contaminating sequences in NGS data. Our main purpose is to understand the nature of those reads that fail to map to the reference genome, as well as to provide an automatic pipeline that allows the quality filtering and the processing of these sequences.
From the detected output it is straightforward to extract information about the eventual samples contamination and/or tissue infection. As in the above-mentioned papers [6–8] the experimental setup and the study of the detected microorganism species might suggest the possible contamination sources. In general, it is not possible to automatically discriminate between upstream and downstream contamination.
Concluding, it can be said that DecontaMiner lies in the middle between the complex, intensive pipelines of PathSeq and SURPI, and the post-alignment approach of CaPSID.
2 DecontaMiner Pipeline
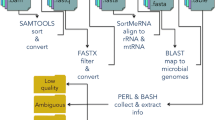
The DecontaMiner pipeline is a suite composed of several command-line tools wrapped together to identify, through digital subtraction, non-human nucleotide sequences generated by high-throughput sequencing of RNA or DNA samples. It is mainly written using Bash scripting and the Perl language. It requires in input the BAM files or the raw fastQ files containing the unmapped reads (i.e., all the reads discarded during the alignment on the human reference genome) if any. A schematic view of the pipeline is shown in Fig. 1.

The pipelineA scheme of the DecontaMiner pipeline. On the right, in blue are the input files, and in red the tools used to process the data. In the central part, as a flux, the processing steps are described. On the left, the parameters that can be set for each step are indicated in green. Several tab-delimited files and one matrix are the pipeline outputs. All the discarded reads are also provided, as well as all the different file formats generated (fastQ, FASTA, etc.). The matrix, containing all the samples, can be easily used to create a bar plot
All the files that have to be submitted to DecontaMiner can be collected in the same directory, and its path given as input. The entire pipeline can be subdivided into three main phases.
The first phase involves the filtering and file format conversion steps, needed to remove low quality reads and to obtain reads in fasta-format files, ready to be aligned to the genome databases. More in detail, DecontaMiner wraps in its pipeline two of the most used toolkits, Samtools [13] and Bedtools [14] used for the format conversions, and FastX [15] for the quality filtering. The filtering is mainly based on two parameters set by the user, namely the Phred quality threshold and the minimum percentage of bases within that threshold.
DecontaMiner works both on paired- and single-end experiments, a parameter that must be specified by the user. The conversion steps allow to sort the reads and switch from bam to fastq and then to fasta formats.
Once terminated the conversion phase, the mapping module can start. In the case of RNA-seq data, it is crucial to remove the ribosomal RNA (rRNA). Indeed, rRNA represents up to 90 % of the total RNA. Although the wet lab procedures provide an rRNA removal step, often this procedure is not totally satisfactory, due to high number of rRNA copies. We downloaded the fasta sequences of human ribosomal RNA (28S, 18S, 5S, 5.8S and mitochondrial 12S, 16S) from NCBI website. The rRNA alignment is performed using the SortmeRNA tool [16], which is a software designed to this aim. All the reads that do not map to the human rRNA will undergo mapping to bacteria, viruses, and fungi genome databases (NCBI nt) using the MegaBLAST [17] algorithm.
The rRNA alignment reliability is evaluated using the E-value score. This threshold can be either set by the user or left at the default SortMeRna value. The user can specify also the alignment length and number of allowed mismatches/gaps when aligning to contaminating genomes.
The BLAST outputs, in table format, are then submitted to the third and last phase, that involves the collection and extraction of information from the local alignments.
This module, mainly composed of Perl scripts, is executed accordingly to some user-specified parameters specifying the filtering and collecting options. In particular, the filtering is based on the threshold number of total reads successfully mapped and on the minimum threshold of reads mapped to a single organism. Instead, the collecting options involve the choice of organizing the results according either to genus or to species names.
DecontaMiner stores the output reads into three main files: unaligned, ambiguous, and aligned. The “unaligned” file contains the reads that do not satisfy the filtering parameters (i.e., length of alignment, number of allowed gaps, and mismatches). The ambiguous reads are those that map to different Genera or, in case of paired-end reads, those having mates mapping to different genera. Ambiguous reads mapping to more than one Genus might derive from ortholog sequences. Since Reads matching all the filtering criteria are stored into the “aligned” file.
The results are available in a tabular format, one for each sample, containing the names of the detected organisms and the relative reads count. Furthermore, DecontaMiner generates a matrix that can be easily used to create a barplot or other types of diagrams in which all the data are collected together.
Lastly, the summary statistics about the number of matched/filtered/discarded reads and organisms are generated and stored into tabular textual files.
3 Case Studies
3.1 Cancer Datasets
In order to assess the usefulness of the DecontaMiner pipeline and its efficiency in detecting non-human sequences in NGS data, we used two publicly available datasets downloaded from the GEO portal (GSE68086 and GSE69240).
The first study, from which the dataset GSE68086 was generated, concerns the total RNA-sequencing experiments of blood platelet samples from patients with six different malignant tumors (non-small cell lung cancer, colorectal cancer, pancreatic cancer, glioblastoma, breast cancer, and hepato-biliary carcinomas) and from healthy donors [18]. The experiment was performed with single-end 100 bp reads.
The second one, GSE69240, derives from the expression profiling by high-throughput sequencing of High-Grade Ductal Carcinoma In Situ (DCIS) [19]. The dataset contains 25 pure HG-DCIS and 10 normal breast organoids samples. The reads are paired-end 76 nucleotides long. This second dataset was used for testing our pipeline on polyA RNA-seq data.
3.2 Pre-processing
The Sequence Read Archive (SRA) file of each sample was downloaded and converted to fastq format using the SRAToolkit [20]. The sequencing reads were cleaned by eventual poor quality ends by Trimmomatic [21]. The quality assessment of the trimmed reads was performed with FastQC [22]. The fast splice junction mapper TopHat [23] was chosen to align the fastq files to the reference genome (assembly hg19) guided by UCSC gene annotation. The sequence features in mapped data were checked by SamStat [24]. The unmapped bam files provided by TopHat were the input to our pipeline.
The parameter setting used for analyzing the two datasets is listed in Table 1.
3.3 Results
The analysis of the overall read mapping rate showed a high variability among the samples of the GSE68086 dataset, with a range of 5–40 % of unmapped reads.
In the case of the GSE69240 dataset, instead, we observed a good mapping rate in all the samples, with a percentage of unmapped reads below 10 %. The mapping statistics of the two datasets immediately suggested a different probability to detect non-human sequences.
In order to test the reliability of our pipeline we submitted to the analysis also the samples with a small amount of unmapped sequences.
As we expected, we did not find any significant match to contaminating genomes for the samples of the GSE69240 dataset. We also re-analyzed the data, lowering the stringency of the parameters in terms of allowed mismatches and gaps (2 for each), with the same negative outcome.
This result completely agrees with the type of experimental procedure used. As mentioned before, an efficient polyA RNA-seq process and a set of samples not contaminated by the environment should guarantee reads free of contamination. Hence, this result supports the reliability of the pipeline in terms of false positives detection.
Instead, in the GSE68086 dataset DecontaMiner detected several matches to bacterial reference sequences. In particular, we focused on those samples having more than 10 % of human-unaligned reads. A modest amount of reads matched to fungal genomes, whereas many reads aligned to bacteriophages specific for the identified bacteria (namely Enterobacteria phage and Propionibacterium phage). This last finding further confirmed the accuracy of the bacteria identification. As an example, the number of reads in two samples before and after the filtering and rRNA alignment processes are shown in Table 2. Sample A and Sample B had low mapping rates on the reference genome 69 and 63.4 %, respectively. However, the reason for such a high number of unmapped reads is completely different. Most of the alignment failure of the Sample B is due to the presence of low quality reads, that are approximately 22 % of the total raw reads, and only 0.0004 % reads matched correctly to bacteria, according to our setting. Instead, only 7.5 % of the sample A are low quality reads and almost 10 % significantly matched to bacteria.
As shown by the barplots generated by a Matlab in-house script, Figs. 2 and 3, both healthy and tumor samples contain non-human sequences. For each sample, we plotted only the organisms having number of matched reads greater than 20 % of the total. All the species that do not fit this criterion are reported as “Others.”

Healthy controls barplot. For each healthy sample a bar reports the detected contaminating organism (colors) and percentage of unmapped reads assigned to each of them

Tumor samples barplot
Propionibacterium acnes and Escherichia coli species were detected in almost all tumor samples and healthy donors, suggesting the possibility of a downstream contamination of the samples or some kind of machine artifacts. P. acnes is a gram-positive bacterium that forms part of the normal flora of the skin [25] and it is usually considered a contaminant of blood cultures [26]. E. coli is a gram-negative bacterium, host of the normal intestinal flora, but also one of the most common responsible of a wide variety of hospital and community-onset infections, affecting patients with normal immune systems as well as those immunodepressed [27].
One of the healthy samples, as well as one of the hepato-biliary carcinoma group, did not have a significant number of reads matching to any bacterial species, according to our thresholds.
The tumor samples barplot shows the presence of some bacterial species that are absent in control samples, or present with a very low reads number. Among them is worth to note the bacterium Acinetobacter baumannii. The percentage of reads aligned to A. baumannii is particularly evident in hepato-biliary carcinoma, although its presence seems to be independent of cancer type.
The genus Acinetobacter, as currently defined, comprises gram-negative, strictly aerobic, nonfermenting, nonfastidious, nonmotile, catalase-positive, and oxidase-negative bacteria [28]. A. baumannii normally inhabits human skin, mucous membranes, and soil [29]. Acinetobacter baumannii, in particular, has become one of the major causes of nosocomial infections during the past two decades [28, 30–32] and its correlation with outcomes of cancer patients is a clinical issue under study [33, 34].
4 Conclusions
The DecontaMiner pipeline was designed and developed to investigate the presence of contaminating sequences in NGS data. It has a dual utility, both as a filtering tool to remove foreign reads from the raw sequencing file, usually in fastq format, and as a detection tool to identify contaminating sequences among the unmapped reads, provided as a bam file. In order to test our pipeline we used two different RNA-seq datasets. The lack of matches to microorganisms in case of the polyA-RNA (GSE69240) demonstrates that the risk of incurring into false positive results is very low. The reliability of our pipeline is further proved on the total RNA (GSE68086) dataset analysis. Indeed, we found some kind of background contamination in almost all the samples. The most present organisms are P. acnes and E. coli and, in addition, some tumor samples significatively matched to A. baumannii, that it is a well-known nosocomial pathogen, even probably associated with outcomes of cancer diseases. It is important to underline that DecontaMiner can suggest the presence of contaminating sequences, but this results must be confirmed by an experimental validation. As an added value, the output fasta files and BLAST tables can be easily uploaded to MEGAN5 [35], a metagenome analyzer, which allows to obtain more detailed information about the taxonomy profile of the samples in several graphical modes. We are currently working to provide DecontaMiner as a Bash shell command-line tool, usable on a common laptop as well as in a distributed computing environment. We are also planning to put together the pipeline here developed and the Transcriptator tool [36] developed in our lab to provide an integrated environment for the analysis of omics data.
References
Conesa, A., et al.: A survey of best practices for RNA-seq data analysis. Genome Biol. 17 (1), 1–19 (2016)
Laurence, M., Hatzis, C., Brash, D.E.: Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS One 9 (5), e97876 (2014)
Feng, H., et al.: Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319(5866), 1096–1100 (2008)
Palacios, G., et al.: A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 358 (10), 991–998 (2008)
Bhatt, A.S., et al.: Sequence-based discovery of Bradyrhizobium enterica in cord colitis syndrome. N. Engl. J. Med. 369 (6), 517–528 (2013)
Strong, M.J., et al.: Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog. 10 (11), e1004437 (2014)
Tae, H., et al.: Large scale comparison of non-human sequences in human sequencing data. Genomics 104 (6), 453–458 (2014)
Ouma, W.Z., et al.: Important biological information uncovered in previously unaligned reads from chromatin immunoprecipitation experiments (ChIP-Seq). Sci. Rep. 5, 8635–8635 (2015)
Kostic, A.D., et al.: PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 29 (5), 393–396 (2011)
Borozan, I., et al.: CaPSID: a bioinformatics platform for computational pathogen sequence identification in human genomes and transcriptomes. BMC Bioinf. 13 (1), 206 (2012)
Altschul, S.F., et al.: Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410 (1990)
Naccache, S.N., et al.: A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 24 (7), 1180–1192 (2014)
Li, H., et al.: The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079 (2009)
Quinlan, A.R., Hall, I.M.: BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 (6), 841–842 (2010)
Gordon, A., Hannon, G.J.: FastX Toolkit (2010) http://hannonlab.cshl.edu/fastx_toolkit/index
Kopylova, E., Noé, L., Touzet, H.: SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28 (24), 3211–3217 (2012)
Zhang, Z., Schwartz, S., Wagner, L., Miller, W.: A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214 (2000)
Best, M.G., et al.: RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 28 (5), 666–676 (2015)
Abba, M.C., et al.: A molecular portrait of high-grade ductal carcinoma in situ. Cancer Res. 75 (18), 3980–3990 (2015)
Leinonen, R., Sugawara, H., Shumway, M.: The sequence read archive. Nucleic Acids Res. 39, D19–D21 (2010).
Bolger, A.M., Lohse, M., Usadel, B.: Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Andrews, S.: FastQC: a quality control tool for high throughput sequence data. Reference Source (2010)
Trapnell, C., Pachter, L., Salzberg, S.L.: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25 (9), 1105–1111 (2009)
Lassmann, T., Hayashizaki, Y., Daub, C.O.: SAMStat: monitoring biases in next generation sequencing data. Bioinformatics 27 (1), 130–131 (2011)
Perry, A., Lambert, P.: Propionibacterium acnes: infection beyond the skin. Expert Rev. Anti-Infect. Ther. 9 (12), 1149–1156 (2011)
Park, H.J., et al.: Clinical significance of Propionibacterium acnes recovered from blood cultures: analysis of 524 episodes. J. Clin. Microbiol. 49 (4), 1598–1601 (2011)
Pitout, J.D.D.: Extraintestinal pathogenic Escherichia coli: an update on antimicrobial resistance, laboratory diagnosis and treatment. Expert Rev. Anti-Infect. Ther. 10 (10), 1165–1176 (2012)
Peleg, A.Y., Seifert, H., Paterson, D.L.: Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21 (3), 538–582 (2008)
Manchanda, V., Sanchaita, S., Singh, N.P.: Multidrug resistant acinetobacter. J. Global Infect. Dis. 2 (3), 291 (2010)
Fukuta, Y., et al.: Risk factors for acquisition of multidrug-resistant Acinetobacter baumannii among cancer patients. Am. J. Infect. Control 41 (12), 1249–1252 (2013)
Al-Hassan, L., El Mehallawy, H., Amyes, S.G.B.: Diversity in Acinetobacter baumannii isolates from paediatric cancer patients in Egypt. Clin. Microbiol. Infect. 19 (11), 1082–1088 (2013)
Dijkshoorn, L., Nemec, A., Seifert, H.: An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 5 (12), 939–951 (2007)
Ñamendys-Silva, S.A., et al.: Outcomes of critically ill cancer patients with Acinetobacter baumannii infection. World J. Crit. Care Med. 4 (3), 258 (2015)
Nazer, L.H., et al.: Characteristics and Outcomes of Acinetobacter baumannii Infections in Critically Ill Patients with cancer: a matched case-control study. Microb. Drug Resist. 21(5), 556–561 (2015)
Huson, D.H., Weber, N.: Microbial community analysis using MEGAN. Methods Enzymol. 531, 465–485 (2012)
Tripathi, K.P., Evangelista, D., Zuccaro, A., Guarracino, M.R.: Transcriptator: an automated computational pipeline to annotate assembled reads and identify non coding RNA. PLoS One 10 (11), e0140268 (2015)
Acknowledgements
Ilaria Granata was supported by MIUR PON02-00619. Mara Sangiovanni was supported by Interomics Italian Flagship Project. Mario R. Guarracino work has been conducted at National Research University Higher School of Economics (HSE) and has been supported by the RSF grant n. 14-41-00039.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Granata, I., Sangiovanni, M., Guarracino, M. (2016). DecontaMiner: A Pipeline for the Detection and Analysis of Contaminating Sequences in Human NGS Sequencing Data. In: Rogato, A., Zazzu, V., Guarracino, M. (eds) Dynamics of Mathematical Models in Biology . Springer, Cham. https://doi.org/10.1007/978-3-319-45723-9_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-45723-9_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45722-2
Online ISBN: 978-3-319-45723-9
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)