
Chapter Summary
The innate immune response to malaria has always attracted the interest of researchers trying to understand the basis for the high fevers observed in malaria patients during blood-stage infection and the lack of an apparent response to the liver-stage infection. Research targeting specific parts of the immune response has contributed to a basic understanding of the concepts that play a role in malaria-induced inflammation. Given the complexity of the immune response in general and to the parasite in particular, some findings have been contradictory. Here we summarize a large body of work including the host innate immune response to a Plasmodium liver and blood-stage infection, focusing on the different parasite- and host-derived molecules that trigger inflammation, the immune cell types involved, and the role of different cytokines in inflammation and pathology of malaria.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Malaria
- Innate immunity
- Plasmodium
- Liver stage
- Blood stage
- Dendritic cells
- Natural killer cells
- Natural killer T cells
- Macrophages
- Interleukins
- Interferon
- Toll-like receptors
1 Plasmodium Infection and Disease
Malaria infection starts when an infected Anopheles mosquito injects Plasmodium sporozoites into the skin of the vertebrate host. After traversing the dermis, the parasites enter the circulation and home to the liver, initiating the hepatic stage of infection. Within the liver, hepatocyte traversal precedes parasite invasion and replication in the host cell, ultim ately maturing into erythrocyte-infectious merozoites [1]. These are later released into the lumen of liver sinusoids [2] invading, developing, and multiplying inside erythrocytes, the blood stage of infection, associated with the establishment of disease and all its complications.
The blood stage of malaria is characterized by high cyclical fevers and elevated levels of inflammatory mediators in the circulation. Excessive and persistent inflammation during P. falciparum infections contributes to severe pathology and to the development of associated complications such as cerebral malaria and severe malarial anemia [3].
Acute malaria is always characterized by high fevers, but it is known that different species of Plasmodium need to reach higher parasitemias to induce inflammatory responses in the host. Although the mechanisms underlying this effect are not clear, it seems apparent that P. vivax is more effecti ve at inducing inflammation and therefore needs lower parasitemias to induce a high fever response compared to P. falciparum [4]. This difference has also been observed in the levels of TNF compared to parasitemia in patients, where P. vivax or P. ovale induce much higher levels of inflammation per infected erythrocyte compared to P. falciparum [5, 6], although this observation has been recently challenged [7, 8].
2 Parasite- and Host-Derived Inflammatory Molecules
The asymptomatic nature of the liver stage, the first step of Plasmodium infection in the mammalian host, has led to the long-lasting view that the parasite can establish and replicate within the host hepatocyte without being detected [1]. However, such notion has been challenged by the presence of inflammatory cell foci in the liver during exoerythrocytic parasite development [9–12], as well as by the mounting of a strong inflammat ory reaction aiming at controlling Plasmodium hepatic burden, as is the case of the rate-limiting enzyme of heme catabolism heme oxygenase 1 [9]. Taken together, these findings suggest that the host is able to sense Plasmodium hepatocyte infection and respond to it.
In contrast, the inflammatory nature of the blood stage of infection has long been recognized. As described by Golgi [13], the synchronized rupture of infected erythrocytes in the peripheral circulation is followed by a peak of fever in malaria patients. This observation led to the hypothesis that the high levels of inflammation in malaria during the blood stage of infection were caused by molecules released from infected erythrocytes during parasite egress, including merozoites and erythrocyte cellular contents. The search for these molecules for the past century has led to the identification of several pro-inflammatory molecules that are either derived directly from Plasmodium or generated as a result of infection from erythrocyte components.
Among the molecules generated by the parasite, glycosylphosphatidylinositol (GPI) anchors were identified early as inflammato ry mediators [14]. These glycolipid structures anchor parasite proteins to the plasma membrane in the merozoite [15] and activate toll-like receptors (TLR), preferentially TLR-2/TLR-6 and TLR-2/TLR-1 heterodimers, but also TLR-4 homodimers [16]. Although purified GPI anchors induce an inflammatory response in mice [14], in vitro this response is downregulated in the presence of other P. falciparum lipids [17], which may explain why in vitro inflammatory responses do not require TLR-2 or TLR-4 [18]. The association of TLR-4 [19], TLR-2, TLR-1, and TLR-6 [20, 21] polymorphisms with malaria severity may be compatible with a role for GPI anchors in malaria inflammation, but this association may also be caused by other Plasmodium-derived inflammatory mediators. The role of GPI in malaria-induced inflammation in patients remains unclear.
Another parasite-derived molecule with inflammatory effects is hemozoin , a crystal polymer of heme that Plasmodium generates after degradation of hemoglobin within infected erythrocytes. Hemozoin is generated in the parasite’s food vacuole and is released after erythrocyte rupture and merozoites egress. It is important to consider than the inflammatory propertie s of crystals, such as hemozoin, depend on the size, charge, and association with protein, lipids, or other elements of the crystal [22]. Different groups have found conflicting results regarding the inflammatory effects of hemozoin, which are probably due to the variations in the protocols used to obtain hemozoin that would result in crystals with different inflammatory characteristics. When synthetic hemozoin, or also hemozoin purified from infected erythrocytes and stripped of any binding molecules, was used as starting material, activation and binding to TLR-9 [23, 24] was observed. Inflammatory pathways dependent on nitric oxide and NF-kB [25], as well as activation of the NLRP3 inflammasome [26, 27], were also identified.
It is likely that hemozoin obtained from cultured parasites that naturally rupture and release their contents would more accurately resemble the characteristics of the hemozoin generated during disease. This kind of hemozoin was used in studies that found parasite and host components bound to it, including Plasmodium DNA [28] and host fibrinogen [29]. The bound materials DNA and fibrinogen, not the hemozoin per se, were found to mediate the inflammatory response observed in vitro through the activation of TLR-9 and TLR-4 or the integrin CD11b/CD18, respectively [28, 30]. Indee d, the inflammatory activity of hemozoin was lost after treatment to remove associated proteins or DNA, probably because the proteins provide a link to bind DNA to the hemozoin crystal [31]. These results indicate that hemozoin could act as a “carrier” for other molecules, increasing their inflammatory potential. Another effect of hemozoin, also caused by other crystals, is the destabilization of the phagosome [32], which results in the release of hemozoin and DNA to the cytosol of the phagocytic cell and in the activation of the AIM2 and NLRP3 inflammasomes, respectively [31]. Accordingly, synthetic hemozoin is being developed as a vaccine adjuvant for other diseases [33, 34].
The “carrier” effect was also proposed for Plasmodium histones, which are bound to parasite DNA and mediate inflammation in vitro [35]. Additionally, immune complexes formed by DNA and anti-DNA antibodies, which are found in high concentrations in the sera of malaria patients, were found to induce cytokine secretion from immune cells in vitro [36], suggesting that another Plasmodium DNA-carrier complex is also contributing to the inflammatory pathway. Plasmodium DNA can activate not only TLR-9, which recognizes CpG motifs, but also an alternative inflammatory pathway that recognizes AT-rich hairpin motifs, involves STING/TBK1/IRF3 signaling, and results in the production of type I interferon [37].
Interestingly, whole lysates of P. falciparum-infected erythrocytes injected into mice are more immunogenic in wt compared to TLR-9-deficient mice, independently of Plasmodium DNA [24]; however, infection of TLR-9-deficient mice with P. berghei did not show a reduction in inflammatory cytokine response [38, 39]. Stud ies in malaria patients have found an association of TLR-9 polymorphisms with malaria susceptibility and development of anemia [40, 41], but not with malaria severity [19, 40, 42]. The role of hemozoin and Plasmodium DNA in the inflammatory response in malaria patients is still not well defined.
Plasmodium RNA is probably also an inflammatory activator during the blood stage of infection, since TLR-7, which recognizes ssRNA, was found to be essential for cytokine production in mice [39]. Since TLR-7 and 9, which recognize ssRNA and dsDNA, respectively, are not found in the plasma membrane of immune cells, the “carrier” effect is thought to facilitate phagocytosis of the nucleic acids, allowing the contact with the endosomes in the phagolysosome. In the liver stage, however, it is established that Plasmodium dsRNA is sensed by the host hepatocyte by a mechanism involving the intracellular RIG-I-like receptor melanoma differentiation-associated protein 5 (MDA5 also known as IFIH1), its adaptor protein mitochondrial antiviral-signaling protein (MAVS ), and the transcription factors IRF3 and IRF7. Notwithstanding, the ensuing immune response is partially MDA5 independent, supporting the idea that several parallel mechanisms could contribute to Plasmodium recognition by the host during the liver stage of infection [43]. The question remains, however, on how Plasmodium spp. dsRNA becomes accessible to the host cytosolic receptors. Several hypotheses can be put forward including: (1) the active transport of this ligand across the parasitophorous vacuole, (2) passive release of dsRNA through vacuolar membrane pores, (3) leakage of dsRNA into the cytosol from nonviable parasites, and/or (4) vesicular transfer of parasite dsRNA from infected cells to liver-resident immune cells, such as Kupffer cells. Overall, we cannot exclude that several o f these mechanisms operate simultaneously to trigger the immune response against Plasmodium during the liver stage of infection.
Another pro-inflammatory mediator that could contribute to the inflammatory response to Plasmodium infection is precipitated uric acid. Plasmodium-infected erythrocytes import hypoxanthine, which is the precursor of uric acid, for the synthesis of nucleic acids required during parasite replication. After requirements for hypoxanthine decrease at the end of the replication cycle, hypoxanthine accumulates in the infected erythrocyte [44, 45]. Precipitates of uric acid are also observed in the cytoplasm of the Plasmodium parasitophorous vacuole [46]. Upon rupture of infected erythrocytes, precipitates of uric acid and soluble hypoxanthine, which would be degraded into uric acid in the tissues, are released and can become inflammatory [44–46]. Indeed, activation of the NLRP3 inflammasome by uric acid precipitates is well characterized [47], and activation of both NLRP3 and NLRP12 inflammasomes has been observed in infected mice and malaria patients [48], possibly caused by hemozoin and/or uric acid crystals. Uric acid was also found to mediate the activation of mast cells in a mouse model of malaria, leading to the regulation of a subset of dendritic cells , which then activate pathogenic CD8+ T cell responses directed against the parasite [49]. Interestingly, treatment of malaria patients with an inhibitor of xanthine oxidase, an enzyme that produces uric acid, results in a more rapid decrease of the inflammatory response [50], suggesting that uric acid may be involved in the inflammatory response in patients.
Microvesicles are shed by almost all cell types in response to different stimuli, such as activation or response to environmental stress [51]. Microvesicles derived from Plasmodium-infected erythrocytes, induced probably in response to oxidative stress during infection [52], can activate macrophages and trigger the secretion of inflammatory cytokines in vitro through the activation of TLR-4 [53, 54]. Endothelial microvesicles can also induce the proliferation of T cells [55]. In malaria patients, increa sed levels of circulating microvesicles are derived preferentially from uninfected erythrocytes, but also infected erythrocytes, lymphocytes, platelets, and endothelial cells [52, 56, 57]. Correlations of microvesicle levels with inflammatory markers or disease severity has been observed in malaria patients. Microvesicle levels correlate with TNF in cerebral malaria patients [58], erythrocyte [52] and endothelial [57] microvesicles correlate with the severity of P. falciparum malaria, and platelet microvesicles also correlate with fever in P. vivax infections [56]. It is still not clear whether microvesicles are causing inflammation and malaria severe pathology in patients or their formation is induced as a consequence of the high inflammation [59], which is characteristic of severe malaria.
Although Plasmodium converts heme derived from hemoglobin degradation into nontoxic hemozoin, which in itself can be inflammatory (see below), up to 40 % of the hemoglobin of an infected red blood cell can be released and oxidized. This leads to the formation of toxic heme in the circulation of infected individuals [60]. In vitro and in vivo experiments suggest that heme induces apoptosis of brain vascular endothelial cells, which affects the stability of the blood-brain barrier in experimental cerebral malaria [61, 62]. Heme-induced apoptosis of brain vascular endothelial cells or endothelial progenitor cells was shown to be mediated by the transcription factor STAT3 [61, 62], the tumor protein p73 [63], or TLR-4-induced CXCL10 [64].
Another source of inflammation in mala ria appears to be oxidative stress that is generated during infection. Malaria patients exhibit high levels of oxidative stress, as measured by lipid peroxidation, and at the same time lower anti-oxidative factors compared to healthy controls [65–68]. The source of increased reactive oxygen species (ROS ) leading to oxidative stress has been subject of speculation. While some reports suggest that ROS might be produced by the parasite [69–71], others indicate that the human host can be a potent source of ROS to combat Plasmodium, most notably through the oxidative burst of phagocytes [72, 73] and ROS-producing enzymes like xanthine oxidase [74]. This implicates that ROS production by the host might be an important inflammatory response to control parasitemia; however, elevated levels of oxidative stress also correlate with increased disease severity during the infection [75, 76], suggesting a role in pathology. It is not clear whether oxidative stress is a cause or consequence of inflammation during malaria. Given the severe nature of complications often leaving traces of oxidative damage like impaired memory after cerebral malaria, the use of antioxidants as adjunctive therapy has been discussed. Although of great potential benefit, antioxidants could increase parasite survival by interfering with the host inflammatory response or the action of antimalarial drugs (reviewed in [77]).
All the parasite and host components described above are able to induce inflammatory reactions in vitro and/or when injected in mice, but their relati ve importance in the inflammation observed in malaria patients remains unclear. Assessing their real contribution during disease has not been possible because of the difficulties in specifically inhibiting each of them in vivo; therefore, most of the available evidence comes from correlations of inflammatory parameters in the blood of malaria patients that cannot establish a causative relation. In vitro inhibition of DNA and/or uric acid is possible using P. falciparum lysates or merozoites treated with DNAse and/or uricase, which suggest an important role for these molecules in the activation of human dendritic cells [18, 46]. The role of hemozoin in the inflammatory response in vivo may now be tested in mice using P. berghei parasites that produce very low levels of hemozoin [78]. Inhibition of uric acid formation in vivo is possible by treatment of malaria patients with allopurinol, an inhibitor of the enzyme that produces uric acid. In this case, a more rapid decrease of the inflammatory response was observed in treated patients [50].
There is an unsolved paradox in the study of the innate immune response to malaria blood stage, where patients present all signs of an intense inflammatory response, including high fevers and circulating inflammatory cytokines, but innate immune cells in vitro respond weakly to the parasite when incubated together. When naïve peripheral blood mononuclear cells (PBMCs ), isolated dendritic cells, or macrophages are incubated with P. falciparum-infected red blood cells in vitro, the levels of inflammatory cytokines released, such as TNF, are undetectable or substantially lower than responses triggered by well-characterized activators (LPS, CpG, or β-hematin [79, 80]) or inflammasome activators (uric acid). Conversely, cells from infected individuals were much more responsive to parasite stimuli in the secretion of cytokines [81, 82] when compared to cells from healthy individuals or recovered patients. These results suggest that there still may be components of the innate immune response to blood-stage Plasmodium infection that have not been identified yet and play an important role d uring infection.
3 Cellular Responses During Malaria
3.1 Hepatocytes
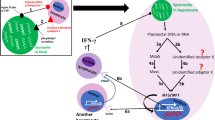
Within hepatocytes, Plasmodium parasites settle inside a parasitophorous vacuole where it undergoes a remarkable transformation differentiating into highly metabolically active merozoites. By the end of the liver stage, as the vacuole expands, a single parasite generates thousands of merozoites. During this proces s plasmodial dsRNA gains access to the host cell cytosol and activates the MDA5/MAVS/IRF3/7 cytosolic signaling pathway leading to the release of type I interferon (IFN) into the extracellular environment. The initial signal is consequently propagated in an autocrine and paracrine manner through the activation of the interferon alpha receptor (Ifnar) in neighboring hepatocytes via the production of interferon-stimulated genes (ISGs ). This type I IFN-mediated response enables the host to control hepatic parasite burden. However such control does not seem to be mediated directly by hepatocytes but rather relies on the recruitment of accessory immune cells [43, 83]. In fact, liver-stage infection results in the recruitment of natural killer T (NKT) cells in an IFNAR-dependent manner, that through an IFNγ-mediated mechanism are responsible for the control of Plasmodium liver infection [83]. Although IFNγ has been shown to directly kill liver-stage parasites [84, 85], it is possible that an IFNγ-independent killing mechanism within the hepatocyte could also take place.
3.2 Granulocytes
The levels of circulating neutrophils during malaria are significantly increased [86] and correlate with inflammation and severe disease [87, 88]. Adoptive transfer of neutrophils from infected rats provided partial protection against infection, suggesting that they play a role in protection against malaria [89]. However, the chemokine CXCL10 that is secreted by neutrophils during P. berghei infection in mice inhibits the control of blood-stage parasitemia and is required for the development of experimental cerebral malaria [90], although depletion of neutrophils did not prevent the development of this pathology [91]. Interestingly, activated neutrophils correlate with cerebral malaria vasculopathy, which presents with higher cytoadhesion levels of infected erythrocytes to endothelial cells in the brain [92], suggesting a role for neutrophils in P. falciparum malaria-induced pathology. In P. vivax infections expression of type I interferon in neutrophils was correlated with liver damage [93]; however, neutrophils present an atypical activation profile since phagocytic activity and superoxide production were increased but molecular markers of activation and secretion of cytokines are very low in response to stimulation [94].
Mast cells contribute to parasite clearance and TNF production in rodent malaria [95] and appear to have a role in promoting innate immune activation since they produce Flt3 ligand during malaria in mice, which, in turn, induces proliferation of a subpopulation of dendritic cells. Both Flt3 ligand and this subpopulation of dendritic cells are also elevated in malaria patients [49].
3.3 Monocytes and Macrophages
Macrophages are essential for clearance of infected erythrocytes as observed in mouse models of infection [96, 97]. Macrophages efficiently phagocytose infected erythrocytes, as early as ring stage [98], which are subsequently degraded in acidic phagosomes [99]. Non-opsonic phagocytosis is mediated by binding of infected erythrocytes to CD36 [100], while opsonic phagocytosis is mediated by complemen t receptor-1 [101, 102] and Fc-γ receptors [103].
3.4 Dendritic Cells
Dendritic cells (DCs ) are crucial for the initiation of the adaptive immune responses and regulate both innate and adaptive immunity to infections. DCs activate, or mature, in response to different pathogen signals, enabling their capacity as antigen-presenting cells that efficiently activate naïve T cells [104]. In vitro studies incubating DCs with Plasmodium showed efficient phagocytosis and phagosomal maturation of infected erythrocytes [99], but demonstrate that there is a dose-dependent inhibition of DC maturation [105] that takes place only at high concentrations of infected erythrocytes, where DCs do not upregulate co-stimulatory molecules [106]. Studies using isolated human DCs and Plasmodium lysates have found upregulation of classical co-stimulatory molecules in DCs [46, 79]. Also, plasmacytoid DCs are activated through TLR-9 by infected erythrocytes [79]. During early infection [107] and in asymptomatic patients [108], the level of expression of surface HLA-DR in circulating DCs maintains normal levels, but during the acute phase of disease, these levels are reduced [49, 109, 110]. The decreased levels of HLA-DR could affect antigen presentation and T cell activation during disease, although their role in mal aria immune response remains unclear.
The dendritic cell cytokine response to Plasmodium in vitro was found to be low for common cytokines such as IL-12, IL-8, IL-6, IL-1β, IL-10, and TNF [80, 105], with the exception of IFN-α that is secreted by plasmacytoid DCs [79]. However, other authors found upregulation of inflammatory cytokines upon incubation with infected erythrocytes [111, 112].
When DCs were extracted from mal aria patients, they show an impaired capacity to mature, capture, and present antigens to T cells. They also undergo high levels of apoptosis probably as a result of increased IL-10 during infection [113, 114]. It appears that malaria, despite the high levels of inflammation, does not induce classical activation of DCs. However, studies in patients are limited to circulating DCs, and it is possible that effective, mature DCs are migrating into tissues and lymphatic organs and are not being detected in the studies.
During severe malaria, the numbers of BDCA3 DCs, which are a minor subset of myeloid DCs, are increased in peripheral blood [115] and correlate with high levels of Flt3L, a factor that induces expansion of DCs. Other DC populations, BDCA1 and plasmacytoid DCs (BDCA2), were not increased in malaria patients [49]. Conversely, in early uncomplicated human malaria infections, the frequency of BDCA3 DCs was not increased, but plasmacytoid DCs were [116], suggesting that the DC response may vary at different stages of infection.
3.5 NK Cells
In vitro studies show that incubatio n of PBMC with P. falciparum-infected erythrocytes results in the rapid activation of natural killer (NK) cells to secrete IFN-γ [117]. This activation requires the help of cytokines such as IL-2, IL-12, and IL-18 [117–119] from T cells but also contact-dependent signals from monocytes and dendritic cells [120]. In mice, NK cells are important for the control of parasitemia during early infection probably through the production of IFN-γ [121, 122]. Also in human malaria infections, restimulation of PBMCs show that NK cells contribute moderately to the production of IFN-γ [107].
3.6 γδ T Cells
γδ T cells are a minor population of T cells in the peripheral circulation that recognize self and non-self antigens without the restriction of MHC antigen presentation. γδ T cells expand during malaria to constitute a high percentage of circulating T cells in humans infected with P. falciparum [123, 124]. They get activated by parasite phosphoantigens produced by Plasmodium apicoplast [125] and release cytotoxic granules containing granulysin that are effective against merozoites [126]. γδ T cells proliferate and produce IFNγ and TNF after in vitro stimulation with P. falciparum-infected red blood cells which is dependent on IL-2 or autologous irradiated PBMC [127–131]. γδ T cells are the main producers of IFNγ in response to P. falciparum in vitro [127] and upon in vitro restimulation of PBMCs from humans infected with P. fa lciparum [107]. In mice, γδ T cells contribute to parasite clearance [132–134], probably due to the high production of IFNγ and their role in activation of dendritic cells [132].
In human malaria patients, γδ T cells are important contributors to inflammatory cytokines and have been associated with severe malaria [129]. When single experimental P. falciparum infections were analyzed, both γδ T cells and NK cells showed an enhanced IFNγ response upon restimulation with P. falciparum-infected red blood cells, even several weeks after the parasite clearance, indicating a memory-like activation [107, 135]. However, when patients with repeated exposure to malaria in endemic areas were analyzed, loss and dysfunction of γδ T cells was observed in the most exposed patients, which is associated with reduced symptoms and clinical tolerance upon reinfection [136]. This suggests that γδ T cells have roles in both clearance of the parasite as well as pathology.
4 Cytokine Responses in Malaria
The inflammatory response during acute malaria is often described as a “cytokine storm” to convey that there are high levels of a broad range of cytokines in the circulation. Earlier studies already correlated the levels o f IFN-α and IFN-γ with levels of parasite [137, 138] that were followed up by the confirmation that plasma levels of inflammatory cytokines—IFN-γ, TNF, IL1β, and IL-6—are elevated in patients with malaria and directly correlate with disease severity in P. falciparum and P. vivax infections [139–150]. Gene expression profiles also confirm high levels of inflammatory cytokines in peripheral blood mononuclear cells [148] and in tissues such as the brain of cerebral malaria patients [151, 152]. The use of mice models of malaria has allowed the continued evaluation of cytokine production during a self-resolving infection, where it was observed that there is an early production of pro-inflammatory cytokines that start decreasing before the parasitemia [153].
TNF is a pro-inflammatory cytokine produced early in mouse Plasmodium infection that is important in the clearance of parasites both in the liver and blood stages [154–157]. This protection is induced through the generation of nitric oxide [158]. Studies on malaria patients showed that TNF not only correlates with disease severity but was ten times as high in fatal cases of cerebral malaria [144]. Further studies confirmed the importance of TNF in malaria pathology showing that different alleles of the promoter region of TNF confer either decreased or increased susceptibility to cerebral malaria and severe anemia in populations of children in endemic areas [159–163]. High levels of TNF are also correlated with a rapid parasitological cure in patients supporting the hypothesis that inflammatory cytokines were effective and necessary for clearance of the parasite, but could also lead to severe forms of the disease [143]. Based on the evidences for the strong association of TNF with malaria severity, a clinical trial for the use of anti-TNF antibodies as adjunctive treatment for cerebral malaria patients was performed. However, no improvement in survival was found in the patients [164], a finding that revealed the complexity of the antimalarial immune response and our limited knowledge in the mechanisms underlying immune-mediated pathology.
Another inflammatory cytokine that has been implicated in parasite clearance and is highly increased during acute malaria is IFN-γ, with the particularity that increased levels of this cytokine during liver-stage infection were correlated with lack of blood-stage development in humans [165] and monkeys [166], suggesting that this cytokine may be important to inhibit progression of the disease, a finding that is specially relevant for vaccines targ eting the liver stage. Studies in mice have shown that IFN-γ is key in the elimination of Plasmodium, since infected mice given exogenous IFN-γ showed lower parasitemias and delayed mortality, while mice deficient in the IFN-γ gene or treated with anti-IFN-γ monoclonal antibodies had higher parasitemia and increased mortality [153, 155, 156, 167–175].
Mouse studies on IL-12 and IL-18 have established that these cytokines contribute to parasite clearance [176]. The increased protection granted by these cytokines is probably mediated by the positive effect on IFN-γ production [175, 177, 178]. However, high IFN-γ levels can be dangerous, since experimental Plasmodium infections also induce liver injury mediated by IL-12-dependent IFN-γ production [179, 180].
Regulatory, or anti-inflammatory, cytokines such as IL-10 are also highly increased during malaria and correlate with severe disease [149, 181, 182]; although in fatal severe cases, low IL-10 was observed in late stages as death approached [183]. Since regulatory cytokines are probably elevated as a response to the high levels of pro-inflammatory cytokines, it is considered more informative to study the ratio between both types of cytokine responses rather than the absolute levels of each specific cytokine. Different studies have confirmed that, in general, high pro-inflammatory versus regulatory cytokine ratios are indicative of severe disease, with specific examples such as high ratios of IL-6/IL-10 or IFN-γ/IL-10 being associated with severe P. falciparum malaria [139, 183]. However, the ratios of TGF-β1/IL-12 and IL-10/IL-12 were significantly higher in the severe malaria patients, suggesting that the generally considered pro-inflammatory cytokine IL-12 could have protective effects [184]. Further studies have shown that high IL-10/TNF ratios were found in children with uncomplicated malaria, while a low IL-10/TNF ratio is associated with malarial anemia in falciparum patients [184–187]. The role of IL-10 in malaria anemia is also supported by the finding that different IL-10 promoter haplotypes that result in low levels of IL-10 increase susceptibility to severe anemia in falciparum patients [188]. As observed before for inflammatory cytokines that contribute to parasite clearance but can promote malaria-associated pathologies, high levels of regulatory cytokines appear to be protective against severe malaria complications but induce a less effective clearance of P. falciparum parasites [189, 190].
Abbreviations
- DCs:
-
Dendritic cells
- GPI:
-
Glycosylphosphatidylinositol
- IFN:
-
Interferon
- Ifnar:
-
Interferon alpha receptor
- ISG:
-
Interferon-stimulated gene
- MAVS:
-
Mitochondrial antiviral-signaling protein
- MDA5:
-
Melanoma differentiation-associated protein 5
- NK:
-
Natural killer cells
- NKT:
-
Natural killer T cells
- PBMC:
-
Peripheral blood mononuclear cells
- ROS:
-
Reactive oxygen species
- TLR:
-
Toll-like receptors
References
Prudencio M, Rodriguez A, Mota MM (2006) The silent path to thousands of merozoites: the Plasmodium liver stage. Nat Rev Microbiol 4(11):849–856. doi:10.1038/nrmicro1529
Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, Krueger A, Pollok JM, Menard R, Heussler VT (2006) Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 313(5791):1287–1290. doi:10.1126/science.1129720
Mackintosh CL, Beeson JG, Marsh K (2004) Clinical features and pathogenesis of severe malaria. Trends Parasitol 20(12):597–603
Kwiatkowski D (1995) Malarial toxins and the regulation of parasite density. Parasitol Today 11(6):206–212
Hemmer CJ, Holst FG, Kern P, Chiwakata CB, Dietrich M, Reisinger EC (2006) Stronger host response per parasitized erythrocyte in Plasmodium vivax or ovale than in Plasmodium falciparum malaria. Trop Med Int Health 11(6):817–823. doi:10.1111/j.1365-3156.2006.01635.x
Yeo TW, Lampah DA, Tjitra E, Piera K, Gitawati R, Kenangalem E, Price RN, Anstey NM (2010) Greater endothelial activation, Weibel-Palade body release and host inflammatory response to Plasmodium vivax, compared with Plasmodium falciparum: a prospective study in Papua, Indonesia. J Infect Dis 202(1):109–112. doi:10.1086/653211
Goncalves RM, Scopel KK, Bastos MS, Ferreira MU (2012) Cytokine balance in human malaria: does Plasmodium vivax elicit more inflammatory responses than Plasmodium falciparum? PLoS One 7(9), e44394. doi:10.1371/journal.pone.0044394
Branch O, Casapia WM, Gamboa DV, Hernandez JN, Alava FF, Roncal N, Alvarez E, Perez EJ, Gotuzzo E (2005) Clustered local transmission and asymptomatic Plasmodium falciparum and Plasmodium vivax malaria infections in a recently emerged, hypoendemic Peruvian Amazon community. Malar J 4:27
Epiphanio S, Mikolajczak SA, Goncalves LA, Pamplona A, Portugal S, Albuquerque S, Goldberg M, Rebelo S, Anderson DG, Akinc A, Vornlocher HP, Kappe SH, Soares MP, Mota MM (2008) Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine Plasmodium liver infection. Cell Host Microbe 3(5):331–338. doi:10.1016/j.chom.2008.04.003
Khan ZM, Ng C, Vanderberg JP (1992) Early hepatic stages of Plasmodium berghei: release of circumsporozoite protein and host cellular inflammatory response. Infect Immun 60(1):264–270
Leiriao P, Mota MM, Rodriguez A (2005) Apoptotic Plasmodium-infected hepatocytes provide antigens to liver dendritic cells. J Infect Dis 191(10):1576–1581. doi:10.1086/429635
van de Sand C, Horstmann S, Schmidt A, Sturm A, Bolte S, Krueger A, Lutgehetmann M, Pollok JM, Libert C, Heussler VT (2005) The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol Microbiol 58(3):731–742. doi:10.1111/j.1365-2958.2005.04888.x
Golgi C (1893) Sulle febbri malariche estivo-autunnali di Roma. Gazzetta Medica di Pavia 2:2:481–493; 505–520; 553–559
Schofield L, Hackett F (1993) Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J Exp Med 177(1):145–153
Bautista JM, Marin-Garcia P, Diez A, Azcarate IG, Puyet A (2014) Malaria proteomics: insights into the parasite-host interactions in the pathogenic space. J Proteome 97:107–125. doi:10.1016/j.jprot.2013.10.011
Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC (2005) Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem 280(9):8606–8616. doi:10.1074/jbc.M413541200
Debierre-Grockiego F, Schofield L, Azzouz N, Schmidt J, Santos de Macedo C, Ferguson MA, Schwarz RT (2006) Fatty acids from Plasmodium falciparum down-regulate the toxic activity of malaria glycosylphosphatidylinositols. Infect Immun 74(10):5487–5496. doi:10.1128/IAI.01934-05
Wu X, Gowda NM, Kumar S, Gowda DC (2010) Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol 184(8):4338–4348. doi:10.4049/jimmunol.0903824
Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh NR, Otchwemah RN, Dietz E, Ehrhardt S, Schroder NW, Bienzle U, Schumann RR (2006) Toll-like receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. Proc Natl Acad Sci U S A 103(1):177–182. doi:10.1073/pnas.0506803102
Panigrahi S, Kar A, Tripathy S, Mohapatra MK, Dhangadamajhi G (2016) Genetic predisposition of variants in TLR2 and its co-receptors to severe malaria in Odisha, India. Immunol Res 64(1):291–302. doi:10.1007/s12026-015-8749-7
Manning L, Cutts J, Stanisic DI, Laman M, Carmagnac A, Allen S, O’Donnell A, Karunajeewa H, Rosanas-Urgell A, Siba P, Davis TM, Michon P, Schofield L, Rockett K, Kwiatkowski D, Mueller I (2016) A Toll-like receptor-1 variant and its characteristic cellular phenotype is associated with severe malaria in Papua New Guinean children. Genes Immun 17(1):52–59. doi:10.1038/gene.2015.50
Franklin BS, Mangan MS, Latz E (2016) Crystal formation in inflammation. Annu Rev Immunol 34:173–202. doi:10.1146/annurev-immunol-041015-055539
Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S (2005) Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med 201(1):19–25
Coban C, Igari Y, Yagi M, Reimer T, Koyama S, Aoshi T, Ohata K, Tsukui T, Takeshita F, Sakurai K, Ikegami T, Nakagawa A, Horii T, Nunez G, Ishii KJ, Akira S (2010) Immunogenicity of whole-parasite vaccines against Plasmodium falciparum involves malarial hemozoin and host TLR9. Cell Host Microbe 7(1):50–61
Jaramillo M, Gowda DC, Radzioch D, Olivier M (2003) Hemozoin increases IFN-gamma-inducible macrophage nitric oxide generation through extracellular signal-regulated kinase- and NF-kappa B-dependent pathways. J Immunol 171(8):4243–4253
Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, Sutterwala FS, Bohle DS, Descoteaux A, Flavell RA, Olivier M (2009) Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog 5(8), e1000559. doi:10.1371/journal.ppat.1000559
Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G, Tschopp J (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4(8), e6510. doi:10.1371/journal.pone.0006510
Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT (2007) Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A 104(6):1919–1924
Barrera V, Skorokhod OA, Baci D, Gremo G, Arese P, Schwarzer E (2011) Host fibrinogen stably bound to hemozoin rapidly activates monocytes via TLR-4 and CD11b/CD18-integrin: a new paradigm of hemozoin action. Blood 117(21):5674–5682. doi:10.1182/blood-2010-10-312413
Pichyangkul S, Saengkrai P, Webster HK (1994) Plasmodium falciparum pigment induces monocytes to release high levels of tumor necrosis factor-alpha and interleukin-1 beta. Am J Trop Med Hyg 51(4):430–435
Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, Fitzgerald KA (2014) Dual engagement of the NLRP3 and AIM2 inflammasomes by Plasmodium-derived hemozoin and DNA during malaria. Cell Rep 6(1):196–210. doi:10.1016/j.celrep.2013.12.014
Tyberghein A, Deroost K, Schwarzer E, Arese P, Van den Steen PE (2014) Immunopathological effects of malaria pigment or hemozoin and other crystals. Biofactors 40(1):59–78. doi:10.1002/biof.1119
Onishi M, Kitano M, Taniguchi K, Homma T, Kobayashi M, Sato A, Coban C, Ishii KJ (2014) Hemozoin is a potent adjuvant for hemagglutinin split vaccine without pyrogenicity in ferrets. Vaccine 32(25):3004–3009. doi:10.1016/j.vaccine.2014.03.072
Uraki R, Das SC, Hatta M, Kiso M, Iwatsuki-Horimoto K, Ozawa M, Coban C, Ishii KJ, Kawaoka Y (2014) Hemozoin as a novel adjuvant for inactivated whole virion influenza vaccine. Vaccine 32(41):5295–5300. doi:10.1016/j.vaccine.2014.07.079
Gowda NM, Wu X, Gowda DC (2011) The nucleosome (histone-DNA complex) is the TLR9-specific immunostimulatory component of Plasmodium falciparum that activates DCs. PLoS One 6(6), e20398. doi:10.1371/journal.pone.0020398
Hirako IC, Gallego-Marin C, Ataide MA, Andrade WA, Gravina H, Rocha BC, de Oliveira RB, Pereira DB, Vinetz J, Diamond B, Ram S, Golenbock DT, Gazzinelli RT (2015) DNA-containing immunocomplexes promote inflammasome assembly and release of pyrogenic cytokines by CD14+ CD16+ CD64high CD32low Inflammatory Monocytes from Malaria Patients. MBio 6(6):e01605–e01615. doi:10.1128/mBio.01605-15
Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, Chan J, Bartholomeu DC, Lauw F, Hall JP, Barber GN, Gazzinelli RT, Fitzgerald KA, Golenbock DT (2011) Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35(2):194–207. doi:10.1016/j.immuni.2011.05.016
Lepenies B, Cramer JP, Burchard GD, Wagner H, Kirschning CJ, Jacobs T (2008) Induction of experimental cerebral malaria is independent of TLR2/4/9. Med Microbiol Immunol 197(1):39–44. doi:10.1007/s00430-007-0057-y
Baccarella A, Fontana MF, Chen EC, Kim CC (2013) Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun 81(12):4431–4442. doi:10.1128/IAI.00923-13
Esposito S, Molteni CG, Zampiero A, Baggi E, Lavizzari A, Semino M, Daleno C, Groppo M, Scala A, Terranova L, Miozzo M, Pelucchi C, Principi N (2012) Role of polymorphisms of toll-like receptor (TLR) 4, TLR9, toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) and FCGR2A genes in malaria susceptibility and severity in Burundian children. Malar J 11:196. doi:10.1186/1475-2875-11-196
Munde EO, Okeyo WA, Anyona SB, Raballah E, Konah S, Okumu W, Ogonda L, Vulule J, Ouma C (2012) Polymorphisms in the Fc gamma receptor IIIA and Toll-like receptor 9 are associated with protection against severe malarial anemia and changes in circulating gamma interferon levels. Infect Immun 80(12):4435–4443. doi:10.1128/IAI.00945-12
Campino S, Forton J, Auburn S, Fry A, Diakite M, Richardson A, Hull J, Jallow M, Sisay-Joof F, Pinder M, Molyneux ME, Taylor TE, Rockett K, Clark TG, Kwiatkowski DP (2009) TLR9 polymorphisms in African populations: no association with severe malaria, but evidence of cis-variants acting on gene expression. Malar J 8:44. doi:10.1186/1475-2875-8-44
Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, Konert M, Hanson KK, Carret C, Lassnig C, Muller M, Kalinke U, Saeed M, Chora AF, Golenbock DT, Strobl B, Prudencio M, Coelho LP, Kappe SH, Superti-Furga G, Pichlmair A, Vigario AM, Rice CM, Fitzgerald KA, Barchet W, Mota MM (2014) Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med 20(1):47–53. doi:10.1038/nm.3424
Orengo JM, Evans JE, Bettiol E, Leliwa-Sytek A, Day K, Rodriguez A (2008) Plasmodium-induced inflammation by uric acid. PLoS Pathog 4(3), e1000013
Orengo JM, Leliwa-Sytek A, Evans JE, Evans B, van de Hoef D, Nyako M, Day K, Rodriguez A (2009) Uric acid is a mediator of the Plasmodium falciparum-induced inflammatory response. PLoS One 4(4), e5194
van de Hoef DL, Coppens I, Holowka T, Ben Mamoun C, Branch O, Rodriguez A (2013) Plasmodium falciparum-derived uric acid precipitates induce maturation of dendritic cells. PLoS One 8(2), e55584. doi:10.1371/journal.pone.0055584
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440(7081):237–241
Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, Elian S, Martins FS, Pereira D, Reed G, Fitzgerald KA, Golenbock DT, Gazzinelli RT (2014) Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog 10(1), e1003885. doi:10.1371/journal.ppat.1003885
Guermonprez P, Helft J, Claser C, Deroubaix S, Karanje H, Gazumyan A, Darasse-Jeze G, Telerman SB, Breton G, Schreiber HA, Frias-Staheli N, Billerbeck E, Dorner M, Rice CM, Ploss A, Klein F, Swiecki M, Colonna M, Kamphorst AO, Meredith M, Niec R, Takacs C, Mikhail F, Hari A, Bosque D, Eisenreich T, Merad M, Shi Y, Ginhoux F, Renia L, Urban BC, Nussenzweig MC (2013) Inflammatory Flt3l is essential to mobilize dendritic cells and for T cell responses during Plasmodium infection. Nat Med 19(6):730–738. doi:10.1038/nm.3197
Sarma PS, Mandal AK, Khamis HJ (1998) Allopurinol as an additive to quinine in the treatment of acute complicated falciparum malaria. Am J Trop Med Hyg 58(4):454–457
Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, Shah P, Wisler J, Eubank TD, Tridandapani S, Paulaitis ME, Piper MG, Marsh CB (2013) Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 121(6):984–995. doi:10.1182/blood-2011-08-374793
Nantakomol D, Dondorp AM, Krudsood S, Udomsangpetch R, Pattanapanyasat K, Combes V, Grau GE, White NJ, Viriyavejakul P, Day NP, Chotivanich K (2011) Circulating red cell-derived microparticles in human malaria. J Infect Dis 203(5):700–706. doi:10.1093/infdis/jiq104
Couper KN, Barnes T, Hafalla JC, Combes V, Ryffel B, Secher T, Grau GE, Riley EM, de Souza JB (2010) Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog 6(1), e1000744. doi:10.1371/journal.ppat.1000744
Mantel PY, Hoang AN, Goldowitz I, Potashnikova D, Hamza B, Vorobjev I, Ghiran I, Toner M, Irimia D, Ivanov AR, Barteneva N, Marti M (2013) Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe 13(5):521–534. doi:10.1016/j.chom.2013.04.009
Wheway J, Latham SL, Combes V, Grau GE (2014) Endothelial microparticles interact with and support the proliferation of T cells. J Immunol 193(7):3378–3387. doi:10.4049/jimmunol.1303431
Campos FM, Franklin BS, Teixeira-Carvalho A, Filho AL, de Paula SC, Fontes CJ, Brito CF, Carvalho LH (2010) Augmented plasma microparticles during acute Plasmodium vivax infection. Malar J 9:327. doi:10.1186/1475-2875-9-327
Combes V, Taylor TE, Juhan-Vague I, Mege JL, Mwenechanya J, Tembo M, Grau GE, Molyneux ME (2004) Circulating endothelial microparticles in malawian children with severe falciparum malaria complicated with coma. JAMA 291(21):2542–2544. doi:10.1001/jama.291.21.2542-b
Sahu U, Sahoo PK, Kar SK, Mohapatra BN, Ranjit M (2013) Association of TNF level with production of circulating cellular microparticles during clinical manifestation of human cerebral malaria. Hum Immunol 74(6):713–721. doi:10.1016/j.humimm.2013.02.006
McGinn CM, MacDonnell BF, Shan CX, Wallace R, Cummins PM, Murphy RP (2016) Microparticles: a pivotal nexus in vascular homeostasis and disease. Curr Clin Pharmacol 11(1):28–42
Francis SE, Sullivan DJ Jr, Goldberg DE (1997) Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51:97–123. doi:10.1146/annurev.micro.51.1.97
Liu M, Amodu AS, Pitts S, Patrickson J, Hibbert JM, Battle M, Ofori-Acquah SF, Stiles JK (2012) Heme mediated STAT3 activation in severe malaria. PLoS One 7(3), e34280. doi:10.1371/journal.pone.0034280
Liu M, Wilson NO, Hibbert JM, Stiles JK (2013) STAT3 regulates MMP3 in heme-induced endothelial cell apoptosis. PLoS One 8(8), e71366. doi:10.1371/journal.pone.0071366
Liu M, Dickinson-Copeland C, Hassana S, Stiles JK (2016) Plasmodium-infected erythrocytes (pRBC) induce endothelial cell apoptosis via a heme-mediated signaling pathway. Drug Des Devel Ther 10:1009–1018. doi:10.2147/DDDT.S96863
Dickinson-Copeland CM, Wilson NO, Liu M, Driss A, Salifu H, Adjei AA, Wilson M, Gyan B, Oduro D, Badu K, Botchway F, Anderson W, Bond V, Bacanamwo M, Singh S, Stiles JK (2015) Heme-mediated induction of CXCL10 and depletion of CD34+ progenitor cells is toll-like receptor 4 dependent. PLoS One 10(11), e0142328. doi:10.1371/journal.pone.0142328
Metzger A, Mukasa G, Shankar AH, Ndeezi G, Melikian G, Semba RD (2001) Antioxidant status and acute malaria in children in Kampala, Uganda. Am J Trop Med Hyg 65:115–119
Narsaria N, Mohanty C, Das BK, Mishra SP, Prasad R (2012) Oxidative stress in children with severe malaria. J Trop Pediatr 58:147–150. doi:10.1093/tropej/fmr043
Sohail M, Kaul A, Raziuddin M, Adak T (2007) Decreased glutathione-S-transferase activity: diagnostic and protective role in vivax malaria. Clin Biochem 40:377–382. doi:10.1016/j.clinbiochem.2007.01.005
Yazar S, Kilic E, Saraymen R, Ozbilge H (2004) Serum malondialdehyde levels in patients infected with Plasmodium vivax. West Indian Med J 53:147–149
Atamna H, Ginsburg H (1993) Origin of reactive oxygen species in erythrocytes infected with Plasmodium falciparum. Mol Biochem Parasitol 61:231–241
Dondorp AM, Omodeo-Sale F, Chotivanich K, Taramelli D, White NJ (2003) Oxidative stress and rheology in severe malaria. Redox Rep 8(5):292–294. doi:10.1179/135100003225002934
Huber SM, Uhlemann A-C, Gamper NL, Duranton C, Kremsner PG, Lang F (2002) Plasmodium falciparum activates endogenous Cl– channels of human erythrocytes by membrane oxidation. EMBO J 21:22–30. doi:10.1093/emboj/21.1.22
Jaramillo M, Godbout M, Olivier M (2005) Hemozoin induces macrophage chemokine expression through oxidative stress-dependent and -independent mechanisms. J Immunol 174:475–484
Potter SM, Mitchell AJ, Cowden WB, Sanni LA, Dinauer M, de Haan JB, Hunt NH (2005) Phagocyte-derived reactive oxygen species do not influence the progression of murine blood-stage malaria infections. Infect Immun 73:4941–4947. doi:10.1128/IAI.73.8.4941-4947.2005
Iwalokun BA, Bamiro SB, Ogunledun A (2006) Levels and interactions of plasma xanthine oxidase, catalase and liver function parameters in Nigerian children with Plasmodium falciparum infection. APMIS 114(12):842–850
Das BS, Patnaik JK, Mohanty S, Mishra SK, Mohanty D, Satpathy SK, Bose TK (1993) Plasma antioxidants and lipid peroxidation products in falciparum malaria. Am J Trop Med Hyg 49(6):720–725
Das BS, Thurnham DI, Patnaik JK, Das DB, Satpathy R, Bose TK (1990) Increased plasma lipid peroxidation in riboflavin-deficient, malaria-infected children. Am J Clin Nutr 51(5):859–863
Isah MB, Ibrahim MA (2014) The role of antioxidants treatment on the pathogenesis of malarial infections: a review. Parasitol Res 113(3):801–809. doi:10.1007/s00436-014-3804-1
Lin JW, Spaccapelo R, Schwarzer E, Sajid M, Annoura T, Deroost K, Ravelli RB, Aime E, Capuccini B, Mommaas-Kienhuis AM, O’Toole T, Prins F, Franke-Fayard BM, Ramesar J, Chevalley-Maurel S, Kroeze H, Koster AJ, Tanke HJ, Crisanti A, Langhorne J, Arese P, Van den Steen PE, Janse CJ, Khan SM (2015) Replication of Plasmodium in reticulocytes can occur without hemozoin formation, resulting in chloroquine resistance. J Exp Med 212(6):893–903. doi:10.1084/jem.20141731
Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, Heppner DG, Stewart VA, Hasegawa H, Looareesuwan S, Shanks GD, Miller RS (2004) Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol 172(8):4926–4933
Giusti P, Urban BC, Frascaroli G, Albrecht L, Tinti A, Troye-Blomberg M, Varani S (2011) Plasmodium falciparum-infected erythrocytes and beta-hematin induce partial maturation of human dendritic cells and increase their migratory ability in response to lymphoid chemokines. Infect Immun 79(7):2727–2736. doi:10.1128/IAI.00649-10
Walther M, Woodruff J, Edele F, Jeffries D, Tongren JE, King E, Andrews L, Bejon P, Gilbert SC, De Souza JB, Sinden R, Hill AV, Riley EM (2006) Innate immune responses to human malaria: heterogeneous cytokine responses to blood-stage Plasmodium falciparum correlate with parasitological and clinical outcomes. J Immunol 177(8):5736–5745
Franklin BS, Parroche P, Ataide MA, Lauw F, Ropert C, de Oliveira RB, Pereira D, Tada MS, Nogueira P, da Silva LH, Bjorkbacka H, Golenbock DT, Gazzinelli RT (2009) Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci U S A 106(14):5789–5794. doi:10.1073/pnas.0809742106
Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH (2014) Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep 7(2):436–447. doi:10.1016/j.celrep.2014.03.018
Schofield L, Ferreira A, Altszuler R, Nussenzweig V, Nussenzweig RS (1987) Interferon-gamma inhibits the intrahepatocytic development of malaria parasites in vitro. J Immunol 139(6):2020–2025
Vergara U, Ferreira A, Schellekens H, Nussenzweig V (1987) Mechanism of escape of exoerythrocytic forms (EEF) of malaria parasites from the inhibitory effects of interferon-gamma. J Immunol 138(12):4447–4449
Abdalla SH (1988) Peripheral blood and bone marrow leucocytes in Gambian children with malaria: numerical changes and evaluation of phagocytosis. Ann Trop Paediatr 8(4):250–258
Mahanta A, Kar SK, Kakati S, Baruah S (2015) Heightened inflammation in severe malaria is associated with decreased IL-10 expression levels and neutrophils. Innate Immun 21(5):546–552. doi:10.1177/1753425914561277
Tangteerawatana P, Krudsood S, Kanchanakhan N, Troye-Blomberg M, Khusmith S (2014) Low monocyte to neutrophil ratio in peripheral blood associated with disease complication in primary Plasmodium falciparum infection. Southeast Asian J Trop Med Public Health 45(3):517–530
Pierrot C, Adam E, Hot D, Lafitte S, Capron M, George JD, Khalife J (2007) Contribution of T cells and neutrophils in protection of young susceptible rats from fatal experimental malaria. J Immunol 178(3):1713–1722
Ioannidis LJ, Nie CQ, Ly A, Ryg-Cornejo V, Chiu CY, Hansen DS (2016) Monocyte- and neutrophil-derived CXCL10 impairs efficient control of blood-stage malaria infection and promotes severe disease. J Immunol 196(3):1227–1238. doi:10.4049/jimmunol.1501562
Schumak B, Klocke K, Kuepper JM, Biswas A, Djie-Maletz A, Limmer A, van Rooijen N, Mack M, Hoerauf A, Dunay IR (2015) Specific depletion of Ly6Chi inflammatory monocytes prevents immunopathology in experimental cerebral malaria. PLoS One 10(4), e0124080. doi:10.1371/journal.pone.0124080
Feintuch CM, Saidi A, Seydel K, Chen G, Goldman-Yassen A, Mita-Mendoza NK, Kim RS, Frenette PS, Taylor T, Daily JP (2015) Activated neutrophils are associated with pediatric cerebral malaria vasculopathy in Malawian children. MBio 7(1):e01300–e01315. doi:10.1128/mBio.01300-15
Rocha BC, Marques PE, Leoratti FM, Junqueira C, Pereira DB, Antonelli LR, Menezes GB, Golenbock DT, Gazzinelli RT (2015) Type I interferon transcriptional signature in neutrophils and low-density granulocytes are associated with tissue damage in malaria. Cell Rep 13(12):2829–2841. doi:10.1016/j.celrep.2015.11.055
Leoratti FM, Trevelin SC, Cunha FQ, Rocha BC, Costa PA, Gravina HD, Tada MS, Pereira DB, Golenbock DT, Antonelli LR, Gazzinelli RT (2012) Neutrophil paralysis in Plasmodium vivax malaria. PLoS Negl Trop Dis 6(6), e1710. doi:10.1371/journal.pntd.0001710
Furuta T, Kikuchi T, Iwakura Y, Watanabe N (2006) Protective roles of mast cells and mast cell-derived TNF in murine malaria. J Immunol 177(5):3294–3302
Stevenson MM, Ghadirian E, Phillips NC, Rae D, Podoba JE (1989) Role of mononuclear phagocytes in elimination of Plasmodium chabaudi AS infection. Parasite Immunol 11(5):529–544
Couper KN, Blount DG, Hafalla JC, van Rooijen N, de Souza JB, Riley EM (2007) Macrophage-mediated but gamma interferon-independent innate immune responses control the primary wave of Plasmodium yoelii parasitemia. Infect Immun 75(12):5806–5818. doi:10.1128/IAI.01005-07
Ayi K, Patel SN, Serghides L, Smith TG, Kain KC (2005) Nonopsonic phagocytosis of erythrocytes infected with ring-stage Plasmodium falciparum. Infect Immun 73(4):2559–2563. doi:10.1128/IAI.73.4.2559-2563.2005
Bettiol E, Van de Hoef DL, Carapau D, Rodriguez A (2010) Efficient phagosomal maturation and degradation of Plasmodium-infected erythrocytes by dendritic cells and macrophages. Parasite Immunol 32(6):389–398. doi:10.1111/j.1365-3024.2010.01198.x
Patel SN, Serghides L, Smith TG, Febbraio M, Silverstein RL, Kurtz TW, Pravenec M, Kain KC (2004) CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis 189(2):204–213. doi:10.1086/380764
Su Z, Fortin A, Gros P, Stevenson MM (2002) Opsonin-independent phagocytosis: an effector mechanism against acute blood-stage Plasmodium chabaudi AS infection. J Infect Dis 186(9):1321–1329. doi:10.1086/344576
Fernandez-Arias C, Lopez JP, Hernandez-Perez JN, Bautista-Ojeda MD, Branch O, Rodriguez A (2013) Malaria inhibits surface expression of complement receptor 1 in monocytes/macrophages, causing decreased immune complex internalization. J Immunol 190(7):3363–3372. doi:10.4049/jimmunol.1103812
Yoneto T, Waki S, Takai T, Tagawa Y, Iwakura Y, Mizuguchi J, Nariuchi H, Yoshimoto T (2001) A critical role of Fc receptor-mediated antibody-dependent phagocytosis in the host resistance to blood-stage Plasmodium berghei XAT infection. J Immunol 166(10):6236–6241
Steinman RM (2012) Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30:1–22. doi:10.1146/annurev-immunol-100311-102839
Elliott SR, Spurck TP, Dodin JM, Maier AG, Voss TS, Yosaatmadja F, Payne PD, McFadden GI, Cowman AF, Rogerson SJ, Schofield L, Brown GV (2007) Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect Immun 75(7):3621–3632. doi:10.1128/IAI.00095-07
Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM, Roberts DJ (1999) Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature 400(6739):73–77. doi:10.1038/21900
Teirlinck AC, McCall MB, Roestenberg M, Scholzen A, Woestenenk R, de Mast Q, van der Ven AJ, Hermsen CC, Luty AJ, Sauerwein RW (2011) Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLoS Pathog 7(12), e1002389. doi:10.1371/journal.ppat.1002389
Kho S, Marfurt J, Noviyanti R, Kusuma A, Piera KA, Burdam FH, Kenangalem E, Lampah DA, Engwerda CR, Poespoprodjo JR, Price RN, Anstey NM, Minigo G, Woodberry T (2015) Preserved dendritic cell HLA-DR expression and reduced regulatory T cell activation in asymptomatic Plasmodium falciparum and P. vivax infection. Infect Immun 83(8):3224–3232. doi:10.1128/IAI.00226-15
Loughland JR, Minigo G, Burel J, Tipping PE, Piera KA, Amante FH, Engwerda CR, Good MF, Doolan DL, Anstey NM, McCarthy JS, Woodberry T (2016) Profoundly reduced CD1c+ myeloid dendritic cell HLA-DR and CD86 expression and increased TNF production in experimental human blood-stage malaria infection. Infect Immun 84(5):1403–1412. doi:10.1128/IAI.01522-15
Urban BC, Mwangi T, Ross A, Kinyanjui S, Mosobo M, Kai O, Lowe B, Marsh K, Roberts DJ (2001) Peripheral blood dendritic cells in children with acute Plasmodium falciparum malaria. Blood 98(9):2859–2861
Mukherjee P, Chauhan VS (2008) Plasmodium falciparum-free merozoites and infected RBCs distinctly affect soluble CD40 ligand-mediated maturation of immature monocyte-derived dendritic cells. J Leukoc Biol 84(1):244–254. doi:10.1189/jlb.0807565
Gowda NM, Wu X, Kumar S, Febbraio M, Gowda DC (2013) CD36 contributes to malaria parasite-induced pro-inflammatory cytokine production and NK and T cell activation by dendritic cells. PLoS One 8(10), e77604. doi:10.1371/journal.pone.0077604
Woodberry T, Minigo G, Piera KA, Amante FH, Pinzon-Charry A, Good MF, Lopez JA, Engwerda CR, McCarthy JS, Anstey NM (2012) Low-level Plasmodium falciparum blood-stage infection causes dendritic cell apoptosis and dysfunction in healthy volunteers. J Infect Dis 206(3):333–340. doi:10.1093/infdis/jis366
Pinzon-Charry A, Woodberry T, Kienzle V, McPhun V, Minigo G, Lampah DA, Kenangalem E, Engwerda C, Lopez JA, Anstey NM, Good MF (2013) Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J Exp Med 210(8):1635–1646. doi:10.1084/jem.20121972
Urban BC, Cordery D, Shafi MJ, Bull PC, Newbold CI, Williams TN, Marsh K (2006) The frequency of BDCA3-positive dendritic cells is increased in the peripheral circulation of Kenyan children with severe malaria. Infect Immun 74(12):6700–6706. doi:10.1128/IAI.00861-06
Teirlinck AC, Roestenberg M, Bijker EM, Hoffman SL, Sauerwein RW, Scholzen A (2015) Plasmodium falciparum infection of human volunteers activates monocytes and CD16+ dendritic cells and induces upregulation of CD16 and CD1c expression. Infect Immun 83(9):3732–3739. doi:10.1128/IAI.00473-15
Artavanis-Tsakonas K, Eleme K, McQueen KL, Cheng NW, Parham P, Davis DM, Riley EM (2003) Activation of a subset of human NK cells upon contact with Plasmodium falciparum-infected erythrocytes. J Immunol 171(10):5396–5405
Horowitz A, Newman KC, Evans JH, Korbel DS, Davis DM, Riley EM (2010) Cross-talk between T cells and NK cells generates rapid effector responses to Plasmodium falciparum-infected erythrocytes. J Immunol 184(11):6043–6052. doi:10.4049/jimmunol.1000106
Stegmann KA, De Souza JB, Riley EM (2015) IL-18-induced expression of high-affinity IL-2R on murine NK cells is essential for NK-cell IFN-gamma production during murine Plasmodium yoelii infection. Eur J Immunol 45(12):3431–3440. doi:10.1002/eji.201546018
Newman KC, Korbel DS, Hafalla JC, Riley EM (2006) Cross-talk with myeloid accessory cells regulates human natural killer cell interferon-gamma responses to malaria. PLoS Pathog 2(12), e118. doi:10.1371/journal.ppat.0020118
Kitaguchi T, Nagoya M, Amano T, Suzuki M, Minami M (1996) Analysis of roles of natural killer cells in defense against Plasmodium chabaudi in mice. Parasitol Res 82(4):352–357
Mohan K, Moulin P, Stevenson MM (1997) Natural killer cell cytokine production, not cytotoxicity, contributes to resistance against blood-stage Plasmodium chabaudi AS infection. J Immunol 159(10):4990–4998
Roussilhon C, Agrapart M, Ballet JJ, Bensussan A (1990) T lymphocytes bearing the gamma delta T cell receptor in patients with acute Plasmodium falciparum malaria. J Infect Dis 162(1):283–285
Ho M, Webster HK, Tongtawe P, Pattanapanyasat K, Weidanz WP (1990) Increased gamma delta T cells in acute Plasmodium falciparum malaria. Immunol Lett 25(1-3):139–141
Behr C, Poupot R, Peyrat MA, Poquet Y, Constant P, Dubois P, Bonneville M, Fournie JJ (1996) Plasmodium falciparum stimuli for human gammadelta T cells are related to phosphorylated antigens of mycobacteria. Infect Immun 64(8):2892–2896
Costa G, Loizon S, Guenot M, Mocan I, Halary F, de Saint-Basile G, Pitard V, Dechanet-Merville J, Moreau JF, Troye-Blomberg M, Mercereau-Puijalon O, Behr C (2011) Control of Plasmodium falciparum erythrocytic cycle: γδ T cells target the red blood cell-invasive merozoites. Blood 118(26):6952–6962. doi:10.1182/blood-2011-08-376111
D’Ombrain MC, Hansen DS, Simpson KM, Schofield L (2007) gammadelta-T cells expressing NK receptors predominate over NK cells and conventional T cells in the innate IFN-gamma response to Plasmodium falciparum malaria. Eur J Immunol 37(7):1864–1873. doi:10.1002/eji.200636889
Goodier MR, Lundqvist C, Hammarstrom ML, Troye-Blomberg M, Langhorne J (1995) Cytokine profiles for human V gamma 9+ T cells stimulated by Plasmodium falciparum. Parasite Immunol 17(8):413–423
Stanisic DI, Cutts J, Eriksson E, Fowkes FJ, Rosanas-Urgell A, Siba P, Laman M, Davis TM, Manning L, Mueller I, Schofield L (2014) gammadelta T cells and CD14+ monocytes are predominant cellular sources of cytokines and chemokines associated with severe malaria. J Infect Dis 210(2):295–305. doi:10.1093/infdis/jiu083
Jones SM, Goodier MR, Langhorne J (1996) The response of gamma delta T cells to Plasmodium falciparum is dependent on activated CD4+ T cells and the recognition of MHC class I molecules. Immunology 89(3):405–412
Waterfall M, Black A, Riley E (1998) Gammadelta+ T cells preferentially respond to live rather than killed malaria parasites. Infect Immun 66(5):2393–2398
Inoue S, Niikura M, Takeo S, Mineo S, Kawakami Y, Uchida A, Kamiya S, Kobayashi F (2012) Enhancement of dendritic cell activation via CD40 ligand-expressing gammadelta T cells is responsible for protective immunity to Plasmodium parasites. Proc Natl Acad Sci U S A 109(30):12129–12134. doi:10.1073/pnas.1204480109
Kobayashi F, Niikura M, Waki S, Matsui T, Fujino T, Tsuruhara T, Kamiya S (2007) Plasmodium berghei XAT: contribution of gammadelta T cells to host defense against infection with blood-stage nonlethal malaria parasite. Exp Parasitol 117(4):368–375. doi:10.1016/j.exppara.2007.05.002
Weidanz WP, LaFleur G, Brown A, Burns JM Jr, Gramaglia I, van der Heyde HC (2010) Gammadelta T cells but not NK cells are essential for cell-mediated immunity against Plasmodium chabaudi malaria. Infect Immun 78(10):4331–4340. doi:10.1128/IAI.00539-10
Obiero JM, Shekalaghe S, Hermsen CC, Mpina M, Bijker EM, Roestenberg M, Teelen K, Billingsley PF, Sim BK, James ER, Daubenberger CA, Hoffman SL, Abdulla S, Sauerwein RW, Scholzen A (2015) Impact of malaria preexposure on antiparasite cellular and humoral immune responses after controlled human malaria infection. Infect Immun 83(5):2185–2196. doi:10.1128/IAI.03069-14
Jagannathan P, Kim CC, Greenhouse B, Nankya F, Bowen K, Eccles-James I, Muhindo MK, Arinaitwe E, Tappero JW, Kamya MR, Dorsey G, Feeney ME (2014) Loss and dysfunction of Vdelta2+ gammadelta T cells are associated with clinical tolerance to malaria. Sci Transl Med 6(251):251ra117. doi:10.1126/scitranslmed.3009793
Druilhe P, Rhodes-Feuillette A, Canivet M, Gentilini M, Periês J (1982) Circulating interferon in patients with Plasmodium falciparum, P. ovale and P. vivax malaria. Trans R Soc Trop Med Hyg 76:422–423
Ojo-Amaize EA, Salimonu LS, Williams AI, Akinwolere OA, Shabo R, Alm GV, Wigzell H (1981) Positive correlation between degree of parasitemia, interferon titers, and natural killer cell activity in Plasmodium falciparum-infected children. J Immunol 127:2296–2300
Andrade BB, Reis-Filho A, Souza-Neto SM, Clarêncio J, Camargo LMA, Barral A, Barral-Netto M (2010) Severe Plasmodium vivax malaria exhibits marked inflammatory imbalance. Malar J 9:13. doi:10.1186/1475-2875-9-13
Angulo I, Fresno M (2002) Cytokines in the pathogenesis of and protection against malaria. Clin Vaccine Immunol 9:1145–1152. doi:10.1128/CDLI.9.6.1145-1152.2002
Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, Lambert PH (1989) Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med 320:1586–1591. doi:10.1056/NEJM198906153202404
Kern P, Hemmer CJ, Van Damme J, Gruss HJ, Dietrich M (1989) Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am J Med 87:139–143
Kremsner PG, Winkler S, Brandts C, Wildling E, Jenne L, Graninger W, Prada J, Bienzle U, Juillard P, Grau GE (1995) Prediction of accelerated cure in Plasmodium falciparum malaria by the elevated capacity of tumor necrosis factor production. Am J Trop Med Hyg 53:532–538
Kwiatkowski D, Sambou I, Twumasi P, Greenwood BM, Hill AVS, Manogue KR, Cerami A, Castracane J, Brewster DR (1990) TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet 336:1201–1204. doi:10.1016/0140-6736(90)92827-5
Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, Kone A, Harley R, Plowe CV, Doumbo OK, Sztein MB (2004) Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun 72:5630–5637. doi:10.1128/IAI.72.10.5630-5637.2004
Mordmüller BG, Metzger WG, Juillard P, Brinkman BM, Verweij CL, Grau GE, Kremsner PG (1997) Tumor necrosis factor in Plasmodium falciparum malaria: high plasma level is associated with fever, but high production capacity is associated with rapid fever clearance. Eur Cytokine Netw 8:29–35
Mshana RN, Boulandi J, Mshana NM, Mayombo J, Mendome G (1991) Cytokines in the pathogenesis of malaria: levels of IL-I beta, IL-4, IL-6, TNF-alpha and IFN-gamma in plasma of healthy individuals and malaria patients in a holoendemic area. J Clin Lab Immunol 34:131–139
Ockenhouse CF, Hu W-c, Kester KE, Cummings JF, Stewart A, Heppner DG, Jedlicka AE, Scott AL, Wolfe ND, Vahey M, Burke DS (2006) Common and divergent immune response signaling pathways discovered in peripheral blood mononuclear cell gene expression patterns in presymptomatic and clinically apparent malaria. Infect Immun 74:5561–5573. doi:10.1128/IAI.00408-06
Peyron F, Burdin N, Ringwald P, Vuillez JP, Rousset F, Banchereau J (1994) High levels of circulating IL-10 in human malaria. Clin Exp Immunol 95:300–303
Ringwald P, Peyron F, Vuillez JP, Touze JE, Le Bras J, Deloron P (1991) Levels of cytokines in plasma during Plasmodium falciparum malaria attacks. J Clin Microbiol 29:2076–2078
Brown H, Turner G, Rogerson S, Tembo M, Mwenechanya J, Molyneux M, Taylor T (1999) Cytokine expression in the brain in human cerebral malaria. J Infect Dis 180:1742–1746
Maneerat Y, Pongponratn E, Viriyavejakul P, Punpoowong B, Looareesuwan S, Udomsangpetch R (1999) Cytokines associated with pathology in the brain tissue of fatal malaria. Southeast Asian J Trop Med Public Health 30:643–649
De Souza JB, Williamson KH, Otani T, Playfair JH (1997) Early gamma interferon responses in lethal and nonlethal murine blood-stage malaria. Infect Immun 65:1593–1598
Bate CA, Taverne J, Playfair JH (1988) Malarial parasites induce TNF production by macrophages. Immunology 64:227–231
Clark IA, Hunt NH, Butcher GA, Cowden WB (1987) Inhibition of murine malaria (Plasmodium chabaudi) in vivo by recombinant interferon-gamma or tumor necrosis factor, and its enhancement by butylated hydroxyanisole. J Immunol 139:3493–3496
Shear HL, Srinivasan R, Nolan T, Ng C (1989) Role of IFN-gamma in lethal and nonlethal malaria in susceptible and resistant murine hosts. J Immunol 143:2038–2044
Taverne J, Tavernier J, Fiers W, Playfair JH (1987) Recombinant tumour necrosis factor inhibits malaria parasites in vivo but not in vitro. Clin Exp Immunol 67:1–4
Rockett KA, Awburn MM, Aggarwal BB, Cowden WB, Clark IA (1992) In vivo induction of nitrite and nitrate by tumor necrosis factor, lymphotoxin, and interleukin-1: possible roles in malaria. Infect Immun 60:3725–3730
Knight JC, Udalova I, Hill AV, Greenwood BM, Peshu N, Marsh K, Kwiatkowski D (1999) A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat Genet 22:145–150. doi:10.1038/9649
McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D (1994) Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 371:508–510. doi:10.1038/371508a0
McGuire W, Knight JC, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D (1999) Severe malarial anemia and cerebral malaria are associated with different tumor necrosis factor promoter alleles. J Infect Dis 179:287–290. doi:10.1086/314533
Ribeiro BP, Cassiano GC, de Souza RM, Cysne DN, Grisotto MAG, de Azevedo Dos Santos APS, Marinho CRF, Machado RLD, Nascimento FRF (2016) Polymorphisms in Plasmodium vivax circumsporozoite protein (CSP) influence parasite burden and cytokine balance in a pre-amazon endemic area from Brazil. PLoS Negl Trop Dis 10, e0004479. doi:10.1371/journal.pntd.0004479
Wattavidanage J, Carter R, Perera KL, Munasingha A, Bandara S, McGuinness D, Wickramasinghe AR, Alles HK, Mendis KN, Premawansa S (1999) TNFα*2 marks high risk of severe disease during Plasmodium falciparum malaria and other infections in Sri Lankans. Clin Exp Immunol 115:350–355
van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, Memming H, Frenkel J, Enwere G, Bennett S, Kwiatkowski D, Greenwood B (1996) The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis 174:1091–1097. doi:10.1093/infdis/174.5.1091
Deloron P, Chougnet C, Lepers JP, Tallet S, Coulanges P (1991) Protective value of elevated levels of gamma interferon in serum against exoerythrocytic stages of Plasmodium falciparum. J Clin Microbiol 29:1757–1760
Hoffman SL, Crutcher JM, Puri SK, Ansari AA, Villinger F, Franke ED, Singh PP, Finkelman F, Gately MK, Dutta GP, Sedegah M (1997) Sterile protection of monkeys against malaria after administration of interleukin-12. Nat Med 3:80–83
Bienzle U, Fritsch KG, Hoth G, Rozdzinski E, Köhler K, Kalinowski M, Kremsner P, Rosenkaimer F, Feldmeier H (1988) Inhibition of Plasmodium vinckei-malaria in mice by recombinant murine interferon-gamma. Acta Trop 45:289–290
Favre N, Ryffel B, Bordmann G, Rudin W (1997) The course of Plasmodium chabaudi chabaudi infections in interferon-gamma receptor deficient mice. Parasite Immunol 19:375–383
Huang KY, Schultz WW, Gordon FB (1968) Interferon induced by Plasmodium berghei. Science 162:123–124
Jahiel RI, Nussenzweig RS, Vilcek J, Vanderberg J (1969) Protective effect of interferon inducers on Plasmodium berghei malaria. Am J Trop Med Hyg 18:823–835
Kobayashi F, Ishida H, Matsui T, Tsuji M (2000) Effects of in vivo administration of anti-IL-10 or anti-IFN-gamma monoclonal antibody on the host defense mechanism against Plasmodium yoelii yoelii infection. J Vet Med Sci 62:583–587
Meding SJ, Cheng SC, Simon-Haarhaus B, Langhorne J (1990) Role of gamma interferon during infection with Plasmodium chabaudi chabaudi. Infect Immun 58:3671–3678
Schultz WW, Huang KY, Gordon FB (1968) Role of interferon in experimental mouse malaria. Nature 220:709–710
Stevenson MM, Tam MF, Nowotarski M (1990) Role of interferon-gamma and tumor necrosis factor in host resistance to Plasmodium chabaudi AS. Immunol Lett 25:115–121
Su Z, Stevenson MM (2000) Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect Immun 68:4399–4406
Sam H, Stevenson MM (1999) In vivo IL-12 production and IL-12 receptors beta1 and beta2 mRNA expression in the spleen are differentially up-regulated in resistant B6 and susceptible A/J mice during early blood-stage Plasmodium chabaudi AS malaria. J Immunol 162:1582–1589
Singh RP, S-i K, Rao P, Okamura H, Mukherjee A, Chauhan VS (2002) The role of IL-18 in blood-stage immunity against murine malaria Plasmodium yoelii 265 and Plasmodium berghei ANKA. J Immunol 168:4674–4681
Stevenson MM, Tam MF, Wolf SF, Sher A (1995) IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J Immunol 155:2545–2556
Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O, Takeda K, Okumura K, Van Kaer L, Okamura H, Akira S, Nakanishi K (2001) Plasmodium berghei infection in mice induces liver injury by an IL-12- and toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol 167:5928–5934
Yoshimoto T, Takahama Y, Wang CR, Yoneto T, Waki S, Nariuchi H (1998) A pathogenic role of IL-12 in blood-stage murine malaria lethal strain Plasmodium berghei NK65 infection. J Immunol 160:5500–5505
Ho M, Sexton MM, Tongtawe P, Looareesuwan S, Suntharasamai P, Webster HK (1995) Interleukin-10 inhibits tumor necrosis factor production but not antigen-specific lymphoproliferation in acute Plasmodium falciparum malaria. J Infect Dis 172:838–844
Luty AJ, Perkins DJ, Lell B, Schmidt-Ott R, Lehman LG, Luckner D, Greve B, Matousek P, Herbich K, Schmid D, Weinberg JB, Kremsner PG (2000) Low interleukin-12 activity in severe Plasmodium falciparum malaria. Infect Immun 68:3909–3915
Day NP, Hien TT, Schollaardt T, Loc PP, Chuong LV, Chau TT, Mai NT, Phu NH, Sinh DX, White NJ, Ho M (1999) The prognostic and pathophysiologic role of pro- and antiinflammatory cytokines in severe malaria. J Infect Dis 180:1288–1297. doi:10.1086/315016
Perkins DJ, Weinberg JB, Kremsner PG (2000) Reduced interleukin-12 and transforming growth factor-beta1 in severe childhood malaria: relationship of cytokine balance with disease severity. J Infect Dis 182:988–992. doi:10.1086/315762
Kurtzhals JA, Adabayeri V, Goka BQ, Akanmori BD, Oliver-Commey JO, Nkrumah FK, Behr C, Hviid L (1998) Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet 351:1768–1772. doi:10.1016/S0140-6736(97)09439-7
May J, Lell B, Luty AJ, Meyer CG, Kremsner PG (2000) Plasma interleukin-10: tumor necrosis factor (TNF)-alpha ratio is associated with TNF promoter variants and predicts malarial complications. J Infect Dis 182:1570–1573. doi:10.1086/315857
Othoro C, Lal AA, Nahlen B, Koech D, Orago AS, Udhayakumar V (1999) A low interleukin-10 tumor necrosis factor-alpha ratio is associated with malaria anemia in children residing in a holoendemic malaria region in western Kenya. J Infect Dis 179:279–282. doi:10.1086/314548
Ouma C, Davenport GC, Were T, Otieno MF, Hittner JB, Vulule JM, Martinson J, Ong’echa JM, Ferrell RE, Perkins DJ (2008) Haplotypes of IL-10 promoter variants are associated with susceptibility to severe malarial anemia and functional changes in IL-10 production. Hum Genet 124:515–524. doi:10.1007/s00439-008-0578-5
Hugosson E, Montgomery SM, Premji Z, Troye-Blomberg M, Björkman A (2004) Higher IL-10 levels are associated with less effective clearance of Plasmodium falciparum parasites. Parasite Immunol 26:111–117. doi:10.1111/j.0141-9838.2004.00678.x
Zeyrek FY, Kurcer MA, Zeyrek D, Simsek Z (2006) Parasite density and serum cytokine levels in Plasmodium vivax malaria in Turkey. Parasite Immunol 28:201–207. doi:10.1111/j.1365-3024.2006.00822.x
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Götz, A., Ty, M., Chora, A.F., Zuzarte-Luís, V., Mota, M.M., Rodriguez, A. (2017). Innate Immunity to Malaria. In: Mota, M., Rodriguez, A. (eds) Malaria. Springer, Cham. https://doi.org/10.1007/978-3-319-45210-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-45210-4_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45208-1
Online ISBN: 978-3-319-45210-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)