
Abstract
Formation of an oocyte involves a specialized cell division termed meiosis. In meiotic prophase I (the initial stage of meiosis), chromosomes undergo elaborate events to ensure the proper segregation of their chromosomes into gametes. These events include processes leading to the formation of a crossover that, along with sister chromatid cohesion, forms the physical link between homologous chromosomes. Crossovers are formed as an outcome of recombination. This process initiates with programmed double-strand breaks that are repaired through the use of homologous chromosomes as a repair template. The accurate repair to form crossovers takes place in the context of the synaptonemal complex, a protein complex that links homologous chromosomes in meiotic prophase I. To allow proper execution of meiotic prophase I events, signaling processes connect different steps in recombination and synapsis. The events occurring in meiotic prophase I are a prerequisite for proper chromosome segregation in the meiotic divisions. When these processes go awry, chromosomes missegregate. These meiotic errors are thought to increase with aging and may contribute to the increase in aneuploidy observed in advanced maternal age female oocytes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
Oocyte formation involves a process termed meiosis. Meiosis is not unique to oocytes; it occurs in spermatocytes as well as other type of gametes (e.g., spores). Meiosis includes a reductional division, which is not found in mitotic cells. In meiosis, cells replicate their DNA once, followed by two subsequent rounds of division: a reductional division—meiosis I (MI)—and then an equational division—meiosis II (MII). When diploid cells undergo meiosis to form gametes such as sperm or spores (in yeast), it results in four identical haploid cells (e.g., Tamaki 1965). In oogenesis, a polar body is extruded in both meiotic divisions, and only one meiotic product will become the oocyte (Rodman 1971). Since the polar bodies do not divide further, meiosis in oogenesis creates three meiotic products, two of which are not fertilized and their DNA is not transmitted to the next generation.
Meiotic prophase I includes unique chromosomal processes that are not found in mitotic prophase (Fig. 5.1). These include programmed DNA damage in the form of DNA double-strand breaks (DSBs) that occur multiple times in different positions on each chromosome (Kolodkin et al. 1986). In meiosis, homologous chromosomes identify each other and come in close proximity (pairing), a process largely unique to meiotic cells. The tight association between homologous chromosomes is mediated by a meiotic-specific protein structure known as the synaptonemal complex through a process termed synapsis (Fawcett 1956; Moses 1956, 1958). These unique meiotic processes are targeted toward the formation of crossovers between each pair of homologous chromosomes. Completion of crossover formation is a prerequisite for successful meiotic divisions; without crossovers, chromosome segregation in MI is random, leading most frequently to inviable progeny.

Key meiotic events from chromosomal perspective. (A) In premeiotic S phase, each homolog is replicated once and resulting sister chromatids are held together by sister chromatid cohesions (green). Two replicated homologs are shown in blue and orange. (B) Pairing begins concurrently with axial elements (yellow) binding to the chromosomes. DNA double-stranded breaks are also made at this time (red stars), starting the process of homologous recombination. (C) Pairing interactions are stabilized when central and lateral components of the synaptonemal complex (red zipper-like structure) are assembled along the chromosomes. (D) Homologous recombination results in crossovers between homologs (one crossover shown here). (E) SC disassembles (some residual SC protein localization to chromosome is not shown here) and centromeres (black circles) of homologs are pulled to opposite directions (gray arrows). (F) In MI (transition from E to F), homologs separate, yet sister chromatids are still held together by cohesion that is maintained at the centromeric regions. (G) In meiosis II (transition from F to G), sister chromatids separate when the cohesions are removed from chromosomes. Meiosis results in four distinct haploid daughter gametes that are allelically different than the parent organism
Meiotic prophase is divided into five ordered stages: leptotene, zygotene, pachytene, diplotene, and diakinesis (Zickler and Kleckner 1999). DSB formation and the initiation of pairing interactions occur at the entrance to prophase I, whereas complete synapsis and stabilizing pairing interactions are observed in the pachytene stage in which DSBs are repaired to form crossovers and noncrossovers (gene conversions without crossover). In diplotene, the synaptonemal complex disassembles, and in diakinesis, chromosomes are condensed and chiasmata are typically observed. A chiasma is the physical/visual representation of a crossover event holding together two homologous chromosomes to form a bivalent.
Here, we will review the processes that chromosomes undergo to form DSBs and repair them. We will focus on the process of homologous recombination as well as on key structural features unique to meiosis that also contribute to this repair. The progression of meiosis requires communication between different steps in the meiotic program, which is inter- and intra-connected by signaling events. Most importantly, these chromosomal events are responsible for setting the stage for chromosome disjunction, and we will discuss the current view about how perturbation in meiotic prophase I events may contribute to chromosome nondisjunction.
5.2 Homologous Recombination
5.2.1 DSB Formation
One key feature of meiosis is that the cell forms programmed DSBs in order to initiate homologous recombination (Fig. 5.2). Recombination maintains genetic diversity among species by breaking linkage disequilibria and further facilitates the efficient removal of deleterious mutations (Hill and Robertson 2007). The process of meiotic recombination must lead to repair of the genome in an error-free pathway, as error-prone pathways risk introducing chromosomal aberrations. Meiosis harbors a complex mechanism of DNA repair through recombination that bears resemblance to somatic DNA damage repair of DSBs.

Homologous recombination during meiotic prophase I. (A) Two homologous chromosomes have been replicated and are about to undergo meiosis (orange and blue lines). For simplicity, sister chromatids are not shown here, and two lines with similar colors represent the two strands of DNA. (B) The topoisomerase-like protein Spo11 (red balloon) and the MRX (blue triangle) complex localize to specific sites on the DNA, and Spo11 creates a double-strand break in the DNA. MRX then removes Spo11 by cleaving roughly 35 bp upstream of the DSB. This results in an oligonucleotide with Spo11 attached at one end being removed from the break site. (C) The nucleases Exo1 and Dna2 create longer ssDNA. This long-term resection may be executed by different protein complexes in different organisms; some may not require Exo–Dna2 for wild-type resection. Once ssDNA is formed, Ku proteins can no longer bind the cut site, thus inhibiting a mutagenic repair pathway known as NHEJ. (D) RPA, followed by Dmc1/Rad51 (here shown as a single green circle, though composing distinct complexes in vivo), coats the ssDNA and protects it from degradation. (E) Dmc1/Rad51 also conducts a homology search along the homolog. Once homology is found, polymerase extends the ssDNA to close the gap made by the initial exonucleases. (F) The crossover is resolved as either a “noncrossover” which results in gene conversion (dark blue line in the yellow homolog) when the newly synthesized DNA is removed from the invaded D-loop and is ligated to its original (yellow) homolog. Polymerase can then use the DNA of the invading strand (yellow) as a template for the top missing DNA (purple). (G) The “crossover” pathway. Proteins called resolvases cut the DNA (two pairs of red arrows), which, when religated, forms crossovers, DNA molecules that contain sequences originating from two different homologs
Chromosomal pairing and DSB repair are interdependent in some organisms, yet separable in others. In mice (Mus musculus), homolog pairing at chromosomal ends initiates before DSBs are formed (Boateng et al. 2013). While not initially thought to be common, this mechanism of initiating pairing independently of DSB formation is also found to hold true in Caenorhabditis elegans and Drosophila melanogaster (Dernburg et al. 1998; McKim and Hayashi-Hagihara 1998). In budding yeast, the majority of cells do not undergo pairing until DSBs are formed, and in the absence of DSBs, limited synaptonemal complex assembly occurs (Bhuiyan and Schmekel 2004). Even though some chromosomal pairing and synapsis may occur without meiotic DSBs, viable offspring are not produced due to random chromosome segregation in the absence of crossovers.
Homologous recombination (HR) begins when the topoisomerase-like protein, Spo11, is recruited to the DNA to initiate DSBs (Keeney et al. 1997). Spo11 is the catalytic subunit of a multi-subunit protein complex, as found in a number of studies in Saccharomyces cerevisiae (Table 5.1). Spo11 binds directly to the DNA, creates a DSB, and then remains covalently bound to the DSB until resection, the formation of ssDNA from dsDNA via nuclease activity, takes place (Neale et al. 2005). Spo11-induced DSBs are not random, as there are regions with little to no breaks (i.e., telomeres and centromeres in S. cerevisiae), while others are enriched with DSBs (“hot spots”) (Gerton et al. 2000). DSB formation is influenced by epigenetic marks that can determine DSB levels and/or positioning.
PRDM9 was identified as a gene essential for meiosis that regulates recombination hot spots (Baudat et al. 2010; Hayashi et al. 2005; Parvanov et al. 2010). Germ cells that lack PRDM9 have an aberrant DSB pattern with a very small effect on DSB levels (inferred directly or by ssDNA-binding protein localization) (Brick et al. 2012; Sun et al. 2015). Most mammals studied express the PRDM9 protein that recognizes a specific DNA sequence via its Zn-finger motif, both DNA sequences and the Zn finger motif are rapidly evolving (Berg et al. 2010; Ponting 2011; Ségurel et al. 2011). In humans and mice, allelic variants of PRDM9 Zn-finger motifs are associated with hot spot usage (Baudat et al. 2010). Another protein domain found in PRDM9 is an H3K4 trimethyltransferase (me3) domain (Hayashi et al. 2005, ix; Koh-Stenta et al. 2014). Hot spots have an open chromatin structure (Getun et al. 2010). PRDM9 me3 of H3K4 causes chromatin restructuring, which leaves a nucleosome-free region (Baker et al. 2014). Altogether, these data suggest that PRDM9-dependent H3K4me3 may expose a DNA region to make them more amenable to Spo11-induced DSBs that will otherwise be induced elsewhere, thus determining hot spot positioning. Mammalian PRDM9 sites are frequently found outside promoters in intragenic regions (Brick et al. 2012). Conversely, nonmammalian eukaryotes from yeast to birds have DSB and/or recombination hot spots that are normally located in promoter regions of genes (Lam and Keeney 2015; Singhal et al. 2015) or within genes (Adrian and Comeron 2013). These locations are under much greater evolutionary constraint; therefore, these hot spots evolve at a much slower rate (Lam and Keeney 2015).
Studies done in the yeast S. cerevisiae and in C. elegans agree with studies done in mice, showing that epigenetic marks play an important role in hot spot definition. The chromatin’s overall structure comprises a series of large loops attached at their base to the meiotic axis (Zickler and Kleckner 1999). DSBs are formed in the loop regions and are scarcely found in proximity to the axis (Blat et al. 2002). Based on the presence of “recombination nodules” (seen on EM images of meiotic chromosome spreads), it was suggested that the recombination process occurs in a location embedded in the synaptonemal complex (Carpenter 1975). These studies altogether suggest that DSBs formed in the loops are moved to the synaptonemal complex for repair. Spo11 in S. cerevisiae forms DSBs via interaction of proteins in the holocomplex with the axis as well as with Spp1, a member of the H3K4me3 Set1 complex (Acquaviva et al. 2013; Sommermeyer et al. 2013). Spp1 physically interacts with the promoter-enriched H3K4me3, bringing the DSB to the synaptonemal complex (Acquaviva et al. 2013; Sommermeyer et al. 2013). H3K4me3 seems important for DSB activity since methyl transferase mutants that result in reduction of H3K4me3 also result in reduced hot spot activity (Sollier et al. 2004). In C. elegans, the H3K4me3 was not shown to play a role in DSB formation, but other marks do appear to play an important role, through the action of proteins such as HIM-17 and XND-1. HIM-17 is required for H3K9 methylation in early meiotic prophase (Reddy and Villeneuve 2004). In the absence of HIM-17, DSB formation is decreased but not eliminated, as indicated by reduction in the number of bivalents formed (Saito et al. 2012; Tsai et al. 2008). Global histone acetylation levels are associated with changes in the frequency of DSB formation in C. elegans (Gao et al. 2015). This pathway likely involves CRA-1 and XND-1 that are suggested to promote crossover formation on the autosomes and the X chromosomes, respectively (Gao et al. 2015, Wagner et al. 2010).
5.2.2 DSB Processing
Once DSBs are formed, nucleolytic activity removes the covalently bound Spo11 and processes the DNA ends in a process termed resection. This leads to the formation of long single-stranded DNA at meiotic DSBs (Sun et al. 1991). These single-stranded DNA overhangs are required for the strand invasion step that follows. A key protein complex acting in this process is the MRN/X protein complex composed of Mre11, Rad50, and NBS1/Xrs2 (Usui et al. 1998). Homologs of Mre11 and Rad50 exist between species, despite varying levels of sequence similarity (Badugu et al. 2015; Hopfner and Tainer 2003). The third member of the MRN complex is NBS1 in mammals or Xrs2 in S. cerevisiae (Carney et al. 1998; Ivanov et al. 1992). NBS1/Xrs2 is the least conserved member of the MRN complex; the conservation between S. cerevisiae Xrs2 and human NBS1 is low and restricted to the N-terminus of the protein (Carney et al. 1998). C. elegans on the other hand lacks an identifiable NBS1/Xrs2 homolog.
The MRN/X complex is thought to hold the broken ends of the chromosome together through the long coiled-coil domains of Rad50, while Mre11 resects the 5′ end of the broken DNA to promote the formation of 3′ single-stranded DNA overhangs (Paull and Gellert 1998). In S. cerevisiae and C. elegans, MRE11 is required both for DSB formation and resection, while in mice, it is known to act only in DSB processing (Ajimura et al. 1993; Cherry et al. 2007; Johzuka and Ogawa 1995). DSB formation and resection are two separate activities of MRE11. For example, in C. elegans and in S. cerevisiae, mutations in MRE11 have been isolated that are proficient in DSB initiation but are unable to resect broken DNA (Nairz and Klein 1997; Yin and Smolikove 2013). Mre11 has both endo- and exonuclease activities (Furuse et al. 1998; Moreau et al. 1999; Paull and Gellert 1998). This nuclease activity of Mre11 is suggested to be sufficient for Spo11 removal, which is required for resection of meiotic DSBs (Moreau et al. 1999; Neale et al. 2005). Mre11 has an additional meiotic function outside of its role in nucleolytic activity: it promotes the assembly of the MRX complex by recruitment of polySUMO chains (Chen et al. 2016). NBS1/Xrs2 is primarily involved in protein–protein interactions. In mitotic cells, it is posttranslationally modified through phosphorylation, ubiquitination, and SUMOylation (Cremona et al. 2012; Falck et al. 2012; Stiff et al. 2006; Wu et al. 2012).
Another key nuclease that participates in DSB processing in meiosis is Ctp1/Sae2/CtIP/COM-1. CtIP is an endonuclease required for Spo11 removal from the ends of broken DNA (Prinz et al. 1997; Rothenberg et al. 2009). Following Spo11 removal, CtIP is required for further end resection, a function that is separable from its role in Spo11 removal (Ma et al. 2015). CtIP was also suggested to act by facilitating the nuclease activity of Mre11 (Cannavo and Cejka 2014). It has been observed that CtIP is phosphorylated and that this posttranslational modification is necessary for its recruitment to DSBs through CtIP binding to Nbs1 (Dodson et al. 2010).
Once initial resection is underway, other proteins assist in further DNA processing to reform long-range resection. Compared to non-meiotic DNA damage resection, meiotic resection produces much smaller regions of single-stranded DNA. In S. cerevisiae mitosis, Dna2 and Exo1 contribute to resection (Mimitou and Symington 2008; Zhu et al. 2008). In S. cerevisiae meiosis, Dna2 is also capable of meiotic resection, but only in the absence of the single-stranded DNA-binding protein Dmc1 (Manfrini et al. 2010). DNA-2 likely does not play a role in meiotic resection in C. elegans. In S. cerevisiae, Exo1 is a 5′–3′ exonuclease that collaborates with Mre11 in bidirectional resection following nick formation by Mre11 (Zakharyevich et al. 2010). In C. elegans, Exo-1 functions in resection only when Mre11 or COM-1 resection is impaired and NHEJ is inactive (Lemmens et al. 2013; Yin and Smolikove 2013).
Processive resection may also be involved in the repression of error-prone DSB repair pathways, such as nonhomologous end joining (NHEJ). In C. elegans, inhibiting DSB resection by com-1 deletion or an mre-11 separation of function allele permits the utilization of NHEJ as a DSB repair pathway (Yin and Smolikove 2013). In other organisms, regulation of NHEJ in meiosis is independent of resection; in S. cerevisiae, NHEJ proteins are exported out of the nucleus upon meiotic entry, and in mammals, NHEJ proteins are degraded, which effectively eliminates the possibility of NHEJ as a mode of repair during meiosis (Goedecke et al. 1999; Valencia et al. 2001).
5.2.3 Strand Invasion
ssDNA must be protected from both nucleolytic digest that would shorten its length and extensive resection that will increase the length of ssDNA. The first may lead to a loss of genetic information, while the latter may increase the chance of creating regions of microhomology that could be utilized for error-prone repair pathways resulting in deletions and/or translocations. In meiosis, three repair proteins are used to protect these ssDNA ends before homology search begins: RPA, Rad51, and Dmc1 (Bishop et al. 1992; Sung and Robberson 1995). The first protein complex to recognize and load onto the ssDNA is RPA, a three subunit protein complex: RPA70/RPA1, RPA32/RPA2, and RPA14/RPA3. Each RPA complex binds to approximately 20 or 30 nucleotides of ssDNA, and each ssDNA strand is coated by multiple RPA complexes forming a filament (Chen and Wold 2014). The RPA filament inhibits strand exchange, thus keeping premature recombination from taking place, but RPA is also required for recombination due to its role in preventing secondary structures of ssDNA (Gasior et al. 2001). Interestingly, RPA does not bind to DSBs when CtIP is depleted, indicating that RPA is in some way regulated by CtIP (Costelloe et al. 2012).
Once RPA is bound, Rad51 and Dmc1 are loaded onto the ssDNA (Nešić et al. 2004; Wang and Haber 2004). Rad51 is associated with both mitotic and meiotic HR, whereas Dmc1 is the meiosis-specific single-stranded binding protein and a member of the RecA/Rad51 gene family (Lin et al. 2006). Not all organisms have a Dmc1 homolog; both C. elegans and Drosophila are missing a homolog (Schurko and Logsdon 2008). The Rad51–Dmc1 filament nucleates from the center of the single-stranded DNA, and the two proteins elongate in opposing directions: Rad51 in the 3′–5′ direction and the Dmc1 filament in the 5′–3′ direction (Brown and Bishop 2015). Dmc1 has DNA strand exchange activity (Sehorn et al. 2004). The molecular mechanism behind how Dmc1 promotes strand exchange involves recognition of three bases at a time (Lee et al. 2015). This base triplet mechanism is highly similar to that of Rad51-based strand exchange (Lee et al. 2015; Qi et al. 2015). It has been shown that in meiosis, the filament formed can be composed of both Rad51 and Dmc1, when Dmc1 is found at a terminal position (Brown et al. 2015).
Dmc1 is required for proper completion of meiotic homologous recombination in most organisms. In S. cerevisiae, Dmc1 is essential for sporulation, and Rad51 plays a minor role in homologous recombination (Bishop et al. 1992; Cloud et al. 2012). In S. pombe, however, Rad51 rather than Dmc1 plays a more dominant role in homologous recombination (Fukushima et al. 2000). However, Rad51 can partially substitute for Dmc1; when S. cerevisiae Rad51 is overexpressed in cells that lack Dmc1, meiosis is partially restored (Tsubouchi and Roeder 2003). Crossover interference, the mechanism that prevents adjacent crossovers, is not restored by Rad51 overexpression in Dmc1 mutants, suggesting a role for Dmc1 in crossover interference (Tsubouchi and Roeder 2003). Interhomolog recombination is strongly favored during meiosis. This homolog bias is required to promote accurate chromosome segregation at MI via suppression of recombination between sister chromatids. Dmc1 and Rad51 are required for homolog bias; intersister recombination frequency is drastically increased in the absence of Dmc1 or Rad51 in S. cerevisiae (Cloud et al. 2012; Lao et al. 2013). Rad51 is inhibited in S. cerevisiae, inhibition of meiotic cells by Hed1, which restricts the access of Rad51 to Rad54 (Rad54 is a protein that facilitates D loop formation) (Busygina et al. 2008, 2012). Furthermore, Dmc1 is necessary for the proper localizations of the synaptonemal complex protein Red1 and the kinase Mek1 that may indirectly contribute to homolog bias in S. cerevisiae (Lao et al. 2013).
Mediator proteins are needed for Rad51/Dmc1 proteins to be loaded onto the DNA. The conserved Mei5-Sae3/Swi5 complex promotes Dmc1 loading in S. cerevisiae and Rad51 loading in S. pombe (Hayase et al. 2004). In S. cerevisiae, Rad52 is a primary mediator protein that assists with Rad51/Dmc1 loading onto the DNA, as well as playing a role in displacing RPA (Gasior et al. 1998; Sugiyama and Kowalczykowski 2002). Rad52 is conserved throughout eukaryotes with two known exceptions: C. elegans and Drosophila. These two organisms lack any known Rad52 homolog, which is intriguing due to its integral role in strand exchange in all other organisms studied to date. Vertebrates, as well as C. elegans, contain an alternative Rad51 loader: Brca2 (Sharan and Bradley 1997). Brca2 possesses Rad51 binding and DNA-binding sites that may act in Rad51 loading (Sharan et al. 1997). Brca2 is required for meiosis in mouse (Sharan et al. 2004). In C. elegans, the Brc2 homolog, BRC-2, functions in Rad51 loading and RPA displacement on Spo-11 generated DSBs (Martin et al. 2005).
Throughout eukaryote and archaea, Rad51/RecA contains multiple paralogs, in addition to eukaryotic Dmc1 (Lin et al. 2006). The C. elegans RFS-1/RIP-1 complex promotes Rad51 loading, and the rfs-1 mutant shows meiotic defects when combined with helicase helq-1 mutants (Taylor et al. 2015). Rad51C is found in mouse meiotic prophase nuclei and is required for meiosis (Kuznetsov et al. 2007; Liu et al. 2007). In Drosophila, RAD51C/SPN-D is required for DSB repair in meiosis (Joyce et al. 2012). The Rad55–Rad57 complex, other RAD51 paralogs, in S. cerevisiae promotes Rad51 assembly by preventing Rad51 filament disruption by anti-recombinase activity (Liu et al. 2011). Both Rad55 and Rad57 are required for meiosis in S. cerevisiae (Gasior et al. 1998; Lovett and Mortimer 1987), while in S. pombe, Rad55 has a small effect on meiotic recombination (Lorenz et al. 2014).
5.2.4 Crossover Formation
ssDNA invasion into the homologous sequences is followed by DNA displacement, creating a D-loop. DNA synthesis “captures” the loop leading to the formation of a double Holliday Junction (dHJ). The ZMM (ZIP–MSH–MER) proteins, identified in S. cerevisiae, promote the resolution of this structure into a crossover. ZMM proteins include synaptonemal complex proteins (as Zip1/2/3/4) and proteins directly involved in crossover formation (as Msh4/5) (Lynn et al. 2007). Msh4/5 creates a sliding clamp complex that utilizes ATP hydrolysis for movement (Snowden et al. 2004). In C. elegans, MSH-5 can be found uniformly throughout the chromatin, but once crossovers are formed in early to mid pachytene, MSH-5 begins to form distinct foci at the single interfering crossover on each chromosome (Yokoo et al. 2012). These two proteins are thought to be involved in physically holding the crossover intermediates until recombination is complete, and a loss of this function would affect the outcome of recombination. In S. cerevisiae, Msh4/5 mutants that are unable to interact with DNA have a decrease in the number of crossovers, whereas ATP hydrolysis defective Msh4 mutants have reduced crossover interference as well as crossover numbers (Krishnaprasad et al. 2015; Nishant et al. 2010; Novak et al. 2001; Rakshambikai et al. 2013). This indicates that the Msh4/5 complex in yeast may not only play a stabilizing role during recombination but also affect crossover numbers and interference via a distinct mechanism.
COSA-1, a protein discovered in C. elegans, co-localizes to crossover sites during meiosis and acts in the same pathway as the MSH-4/5 complex (Yokoo et al. 2012). COSA-1 bears a close resemblance to the human Cyclin B1 protein based on structural modeling. COSA-1 homologs are widely conserved throughout eukaryotes, except in the Drosophila lineages in which Msh4, Msh5, and COSA-1 are all absent (Schurko and Logsdon 2008). The precise mechanism of COSA-1 function is still unknown; however, a model has been proposed consisting of a two-stage licensing process: starting with MSH-4/5 and COSA-1 localization, followed by COSA-1 partnering or interacting with an unknown CDK to phosphorylate MSH-4/5 which promotes recombination progression (Yokoo et al. 2012). The mammalian COSA-1 homolog, CNTD1, exhibits similar phenotypes as those of the C. elegans homolog, which includes normal progression of early meiosis but a failure of late meiotic recombination markers to appear or be removed from the chromatin, thus indicating defects in crossover formation. These findings lead to the model that a CDK partner may work with CNTD1 in mammals to ensure normal meiosis progression, likely via phosphorylation of specific target proteins (Holloway et al. 2014).
Crossover formation is also dependent upon proteins involved in posttranslational modifications. RNF212 in mammals, Zip3 in S. cerevisiae, and ZHP-3 in C. elegans are all homologous E3 ligase proteins. These proteins have been implicated in SUMOylation in yeast and possibly ubiquitination in other organisms (Reynolds et al. 2013). RNF212 is required for crossover formation in mammals; in the absence of RNF212, synapsis can still occur relatively normally; however, localization of crossover promoting protein such as MLH1 and MSH4 is reduced or eliminated, suggesting a role for RNF212 in stabilizing crossover precursors (Reynolds et al. 2013). In mammals, RNF212 localizes as puncta in early pachytene, which progressively become more selective and co-localize with the MSH4/5 complex (Reynolds et al. 2013). The ubiquitin ligase HEI10 has been implicated in this process as an antagonistic factor to RNF212 (Qiao et al. 2014; Ward et al. 2007). In HEI10-deficient mice, RNF212 localizes to crossovers but fails to be removed, preventing crossover formation and arresting meiosis (Qiao et al. 2014). HEI10 also localizes to crossovers in wild-type cells, suggesting a direct role in this pathway. These studies point to an interplay between SUMO and ubiquitin in order to promote the correct number of meiotic crossovers and noncrossovers. The RNF212 homolog in yeast, Zip3, has been implicated in both the SUMOylation and ubiquitination pathways (Cheng et al. 2006; Perry et al. 2005). Consistent with the findings from mice, the C. elegans RNF212 homolog, ZHP-3, is also important for crossover formation (Jantsch et al. 2004). Despite the fact that Zip3 was first implicated in assembly of the synaptonemal complex, a role in synaptonemal complex assembly was not identified for RNF212 (mice) or ZHP-3 (C. elegans) (Agarwal and Roeder 2000). Thus, although the role of RNF212/Zip3/ZHP-3 proteins in promoting crossover formation is conserved, their function in synaptonemal complex assembly is restricted to yeast.
A model based on studies in S. cerevisiae meiosis indicates that crossovers can arise as an outcome of two pathways: class I crossovers/interfering crossovers and class II crossovers/non-interfering crossovers. Class I crossovers make up the majority of meiotic crossovers and are promoted by the ZMM proteins. Class I and class II crossovers originate by the action of resolvases, structure-specific nucleases that can act on HJ. In S. cerevisiae, Mlh1 and Mlh3 form a heterodimer that acts to resolve dHJs in a pathway that leads exclusively to crossover formation (Hunter and Borts 1997; Wang et al. 1999). This pathway also involves the nuclease Exo1 (Zakharyevich et al. 2012). Similar observations were made in mouse: most meiotic crossovers arise by the action of the Mlh1–Mlh3 complex (Baker et al. 1996; Lipkin et al. 2002).
Class II crossovers originate from the combined activities of several endonucleases. These nucleases can resolve dHJs forming either crossovers or noncrossovers, although some resolvases can act on the D-loop intermediate as well and promote only crossovers (Osman et al. 2003; Svendsen and Harper 2010). Nuclease complexes that are involved in generation of class II crossovers include Mus81–Eme1/Mms4, Slx-1–Slx-4, Xpf1–ERCC1, and Gen1/Yen1 (Bailly et al. 2010; Boddy et al. 2001; Saito et al. 2012). These nuclease complexes exhibit partially redundant functions in HJ resolution, and the part each nuclease plays in HJ resolution varies from organism to organism. In S. cerevisiae, mms4 as well as mus81 mutants have a significant effect on meiosis, while yen1 (gen1 in mammals), slx1, and slx4 have no effect as shown in single mutant analysis (Mullen et al. 2001). The redundancy between these nucleases is revealed in double mutant analysis; combining mus81 with yen1, slx1, or slx4 deletion mutants exacerbates the mus81 mutant phenotype (Matos et al. 2011). A major role for these nucleases is observed in resolving unregulated joint molecules (e.g., HJ between sister or multiple partners), as found in sgs1 mutants (Oh et al. 2008; Zakharyevich et al. 2012). mus81 mutants produce no viable spores, suggesting that MUS81 has a key role in meiosis in S. pombe (Boddy et al. 2000). In mice, MUS81 appears to play a minor role; both BTBD12/slx4 and mus81 mutants have reduced fertility and meiotic progression defects, but cells that progress to diakinesis show no crossover defects (Holloway et al. 2011). The effect of mlh3 null mutant on crossover formation is enhanced (but not eliminated) by deleting mus81 in this background, an indication that Mus81 is required for crossover formation in the absence of Class I resolvases (Holloway et al. 2008). Moreover, MLH1 foci numbers are increased in mus81 mutants, likely indicating a cross talk between the class I and II crossover pathways (Holloway et al. 2008). Despite the fact that each of the three complexes (Mus81–Eme1, Slx-1–Slx-4, Xpf1–ERCC1) possesses a separable nuclease activity and frequently functions redundantly, evidence from mitotic cells suggests that in fact these complexes associate to form one multi-protein complex. In Drosophila, xpf1/Mei9-Ercc1 physically interacts with mus312 and slx4, and this interaction is mediated by Mei9 (Radford et al. 2005; Yildiz et al. 2002). In C. elegans, HIM18/Slx-4 interact with XPF-1, SLX-1, and MUS-81, while EME-1 and ERCC1 are brought to the complex by interaction with MUS-81 and XPF-1, respectively (Saito et al. 2013), and in human cells, a Mus81–Eme1–Slx-1–Slx-4 complex was identified to function in mitotic resolution (Wyatt et al. 2013).
Investigation of the role that structure-specific nucleases play in meiosis in C. elegans has added complexity to the picture. In C. elegans meiosis, all crossovers are MSH-4/5-dependent crossovers, suggesting that they are interfering (Kelly et al. 2000). Interestingly, however, the nucleases involved in each of these events utilize a class of nucleases named Class II in S. cerevisiae, challenging the classification of resolvases (Saito et al. 2013). In C. elegans, him-18/slx-4 mutants have reduced crossover numbers, bivalent stability is compromised, and bivalent differentiation is delayed (Saito et al. 2009). As found in S. cerevisiae, worm gen1 (yeast yen1) and slx-1 have mild effects on meiosis as single mutants (Bailly et al. 2010; Saito et al. 2012). However, double mutant analysis is consistent with a role in crossover resolution for all of these nucleases (Saito et al. 2013). These studies are consistent with a model for two main resolution pathways in C. elegans: one for MUS-81 and SLX-1 and one for XPF-1, with a very minor role for GEN-1. Also similarly to S. cerevisiae, C. elegans mus-81 and slx-1 mutant phenotypes are exacerbated in the absence of Sgs1/HIM-6 (Saito et al. 2013).
5.3 Chromosome Structure
5.3.1 Sister Chromatid Cohesion
As chromosomes enter meiosis, sister chromatids are connected to each other and held together by the sister chromatid cohesion complex (Fig. 5.3A). The composition of this complex resembles, but is not identical, to that of the mitotic sister chromatid cohesion complex. It is therefore not surprising that recruitment of mitotic cohesion to meiotic chromosomes is unable to compensate for the lack of meiotic cohesion (Yokobayashi et al. 2003). The cohesion complex is composed of four protein subunits: Smc1, Smc3, Scc3/SA/STAG, and Scc1/Rad21 (Klesin). In meiosis, Scc1 is replaced by meiotic specific subunit(s), and in some organisms other subunits are replaced as well. For example, Scc1 is replaced by Rec8, ubiquitously among organisms, while in mammals, Smc1 is replaced by Smc1β and Smc3/STAG1 is replaced by STAG3 (Pezzi et al. 2000; Revenkova et al. 2001; Watanabe and Nurse 1999). Cohesion protein components are evolutionarily conserved (Harvey et al. 2002; Koshland and Strunnikov 1996) and are related to other complexes that act in maintaining the function of chromosomes: the condensin complex (chromosome condensation), the Smc4/5 complex (DNA damage repair), and in Drosophila, the synaptonemal complex protein C(2)M shares sequence similarity to Scc1 (Heidmann et al. 2004).

The synaptonemal complex and sister chromatid cohesion. (A) Two sister chromatids (blue pair and orange pair) are held together by sister chromatid cohesion complexes. During pairing, axial elements (yellow) are first to bind and then central region proteins assemble to form the synaptonemal complex (red). In this context, axial elements are called lateral elements. (B) Close-up on the blue homolog region boxed in (A) of chromatin arranged in large loops. (C) Model one of the cohesion complex working during meiosis: a single cohesion complex consisting of Smc1, Smc3/Smc3β, Scc3/SA/STAG3, and Scc1/Rec8 forms a loop-like structure. Names of mitotic–meiotic subunits are in black and meiosis-specific subunits in blue. This single loop can fit two DNA strands within it, holding sisters together. (D) Model two: two complete cohesion complexes each have one strand of DNA within their loop and the two complexes interact with each other to hold the two DNA strands together. Both C and D are close-ups of black rectangle in B
The cohesion subunits assemble to form a ring structure (Anderson et al. 2002), most of which is accounted for by the Smc subunits, with each Smc subunit containing two long coiled-coil domains. A wide hole enclosed by the cohesion ring structure is compatible with different models explaining how sister chromatids are connected (Huang et al. 2005). Model 1 (Fig. 5.3C) suggests that the ~40 nM ring structure can fit two DNA molecules (10 nM fiber that contain nucleosomes) (Haering et al. 2002). This led to the proposal that the cohesion complex can connect two sister chromatids by passing the two DNA strands through a single ring in multiple positions along the chromosomes (Haering et al. 2002). An alternative model (model 2, Fig. 5.3D) suggests that each chromatid is captured by its own ring and two adjacent rings interact to establish cohesion (Zhang et al. 2008b). The establishment of sister chromatid cohesion typically occurs during S phase (Uhlmann and Nasmyth 1998). However, most of the cohesion complex is loaded onto the chromosomes prior to replication (Michaelis et al. 1997), which suggests that modification of the ring structure (opening/closing) is an important part of establishment of sister chromatid cohesion.
The variety in cohesion subunits suggests that multiple cohesion complexes may exist with specific functions. This is supported by a specific localization pattern temporally and spatially for some of the cohesion complexes. In the mouse, the Scc1/Rad21 meiotic subunit can be replaced not only by Rec8 but also Rad21L (Herrán et al. 2011; Ishiguro et al. 2011; Lee and Hirano 2011). In early prophase, Rad21L’s presence is predominant, compared to that of Rec8, and these two proteins occupy nonoverlapping positions on the chromosomes (Ishiguro et al. 2011; Lee and Hirano 2011). In C. elegans, five Scc1 subunits exist: COH-1, COH-2/SCC-1, COH-3, COH-4, and REC-8; all five subunits have suggested meiotic functions based on localization and/or mutant phenotypes (Severson and Meyer 2014). However, REC-8 and COH-3/4 seem to play the major role in meiotic cohesion (Pasierbek et al. 2001; Severson et al. 2009). The REC-8 and the COH-3/4 cohesin complexes play redundant roles, as breakdown of prophase cohesion requires depletion of both complexes (Severson et al. 2009). These functions are not identical as (1) REC-8 and COH-3/4 localize to different chromosomal domains on pre-metaphase chromosomes, (2) the COH-3/4 complex is able to bi-orient sister chromatids in the absence of REC-8, while REC-8 cannot bi-orient sisters in the absence of COH-3/4 (Severson and Meyer 2014; Severson et al. 2009). The picture became even more complex with the discovery that the mitotic cohesion complex may play a role in meiosis as well [Scc1/Rad21: Prieto et al. (2002), Smcα: Gutiérrez-Caballero et al. (2011), COH-1 and COH-2/SCC-1: Severson and Meyer (2014), STAG2: Prieto et al. (2002)]. However, in some cases, their roles in meiosis are debated. For example, despite the presence of Scc1/Rad21 on meiotic chromosomes, it is not required for meiotic division (Tachibana-Konwalski et al. 2010). Therefore, some cohesion proteins may “wait” on meiotic chromosomes to perform their role later on in mitotic divisions within the zygote.
Cohesin complexes are essential for chromosome function in meiosis. In early stages of meiosis, cohesion is essential for the formation and maintenance of the synaptonemal complex at the level of axis formation (the substructure of the synaptonemal complex, Fig. 5.3A, first to assemble). Yeast mutants that lack Rec8 do not form an axis (Klein et al. 1999). In organisms in which several cohesin complexes exist, partially overlapping function of cohesin complexes in synaptonemal complex assembly is revealed. In the absence of mouse Rec8 or Rad21L, synapsis is severely impaired but axes still form (Herrán et al. 2011; Xu et al. 2005). Loss of axis is observed only when both mouse Rec8 and Rad21L are depleted (Llano et al. 2012). Similarly to what is found in mice, in C. elegans mutants in which all meiotic cohesion is removed (rec-8, coh-3/4) the homologs are unable to form an axis. However, removing only one of these complexes restricts the effects only in to the central region of the synaptonemal complex (Severson et al. 2009). Effects on synaptonemal complex maintenance, as opposed to establishment, were also observed in Rad21L mutants in Drosophila and Rec8 mutants in Sordaria (Urban et al. 2014, Storlazzi et al. 2008). The role of cohesion in synaptonemal complex assembly may be indirect due to cohesion’s function in modifying chromosome structure or direct by interacting with synaptonemal complex proteins. The latter was demonstrated in Drosophila and mice: in Drosophila, mitotic cohesion protein Rad21 physically interacts with C(2)M, and in mouse, SMC1 and SMC3 interact with synaptonemal complex proteins SYC3 and SYC2 (Urban et al. 2014; Vallaster et al. 2011). In addition to its importance for synaptonemal complex assembly, cohesion plays a role in shaping the length of the meiotic axis. Increasing cohesion on chromosomes by inhibiting mechanisms for its removal shortens axis length in C. elegans and yeast (Challa et al. 2016; Crawley et al. 2016).
The key role of cohesion in synaptonemal complex assembly is likely to enable repair of meiotic DSBs, since the synaptonemal complex is required for and promotes interhomolog repair. However, cohesion may also control the repair of DSBs independently of its role in synaptonemal complex assembly. For example, cohesion is required for the repair of DSBs using sister chromatid recombination in the absence of the synaptonemal complex in C. elegans (Crawley et al. 2016; Smolikov et al. 2007). In C. elegans, defects in cohesion loading impair early steps in DSB processing (Lightfoot et al. 2011), whereas an increase in cohesion loading leads to a delay in DSB repair (Crawley et al. 2016). In yeast, increased cohesion also leads to DSB repair defects (Challa et al. 2016), and sister chromatid cohesion plays a role in implementing homolog bias in meiosis (Hong et al. 2013). DSB formation is also dependent on cohesion, as complete removal of the meiotic cohesion complexes prevents axis assembly which is required for DSB formation (Severson et al. 2009).
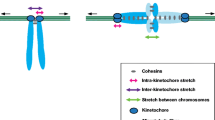
The most notable function of cohesion is in holding sister chromatids together, thus allowing proper chromosome segregation in mitosis and meiosis. Cohesion counteracts the action of forces generated by the spindle through the kinetochore that try to pull the chromatids apart. By this action, cohesion promotes proper alignment of chromosomes on the metaphase plate. Therefore, release of cohesion requires a regulated process: in mitosis, cohesion is released first from the chromosome arms (prophase to pre-metaphase, the “prophase pathway”) followed by release from centromeres (in anaphase) (Waizenegger et al. 2000). Release of meiotic cohesion occurs in a similar order, but the release of cohesion from chromosome arms occurs at MI releasing homologous chromosomes, while release of centromeric cohesion occurs at MII, releasing sister chromatids. Release of meiotic cohesion in anaphase of both meiotic divisions occurs by proteolytic cleavage of the cohesion ring by similar mechanisms to what is found in mitotic anaphase (Buonomo et al. 2000; Kudo et al. 2006; Salah and Nasmyth 2000). Protection of centromeric cohesion by Shugoshin and PP2A/Air2 prevents this mechanism from releasing centromeric cohesion in MI (Kitajima et al. 2004; Lee et al. 2008). Premature release of cohesion results in chromosome missegregation (as will be discussed in Sect. 5.3).
Many of the proteins that assist cohesion loading and establishment in mitosis have conserved functions in meiosis. These functions include the opening and closing of the cohesion ring without the need for proteolytic cleavage via mechanisms that are currently poorly understood. The NIPBL/Scc2-Mau2/Scc4 cohesion loader complex was suggested to act by a mechanism involving the ATP-dependent opening of the cohesin ring structure (Arumugam et al. 2003; Hu et al. 2011; Murayama and Uhlmann 2014; Unal et al. 2008). NIPBL/Nipped-B localizes to the synaptonemal complex axis in mouse, C. elegans, and Drosophila (Gause et al. 2008; Kuleszewicz et al. 2013; Lightfoot et al. 2011; Visnes et al. 2014), and Mau2/Scc4 additionally localizes to the synaptonemal complex in mouse (Visnes et al. 2014). Similar to its role in mitosis (Ciosk et al. 2000), NIPBL/Scc2 promotes sister chromatid cohesion in meiosis (Lin et al. 2011). However, in yeast, it was shown that at least part of the role that Scc2 plays in formation of meiotic cohesion is due to increasing transcription of the Rec8 gene (Lin et al. 2011). Nipped-B mutants in Drosophila have defects in the maintenance of cohesion (and the synaptonemal complex), while scc-2 mutants in C. elegans do not load cohesion and phenotypically resemble cohesion null mutants (Gause et al. 2008; Lightfoot et al. 2011). The protein Ctf7/Eco1 promotes cohesion establishment by acetylating Smc3 (Ivanov et al. 2002; Skibbens et al. 1999; Toth et al. 1999). RNAi for Arabidopsis Ctf7 in meiosis prevents Scc3 localization to the chromosome axis and impairs sister chromatid cohesion in meiosis (Singh et al. 2013). Pds5 is a cohesion-associated protein that promotes Eco1-mediated acetylation and thus is suggested to play a role in cohesion maintenance (Vaur et al. 2012). Pds5 is associated with the meiotic axis (Fukuda and Hoog 2010; Storlazzi et al. 2008; Zhang et al. 2005). Studies in several model organisms support an important, but not essential, role for pds5 in meiotic cohesion. The C. elegans pds5/evl-14 point mutant has a small effect on meiotic cohesion (Wang et al. 2003). In S. cerevisiae, Pds5 mutants show impaired loading of Rec8, but these defects are sufficient to prevent synaptonemal complex assembly (Jin et al. 2009; Zhang et al. 2005). In S. pombe and Sordaria, Rec8 loading is reduced but not eliminated in Pds5 mutants (Ding et al. 2006; Storlazzi et al. 2008). While in most organisms, the effects of cohesion loading on recombination can be explained as stemming from a function in synaptonemal complex assembly, studies in Arabidopsis suggest a direct role in recombination, as axis formation is not affected in Pds5 mutants but recombination is affected (Pradillo et al. 2015). Pds5 also affects chromosome structures in fission yeast, and Pds5 mutants in budding yeast lead to chromosomal hyper-condensation (Ding et al. 2016; Jin et al. 2009). One protein was shown to counteract cohesion maintenance in meiosis: WAPL is necessary for unloading the cohesins in mitosis. The activity of WAPL is regulated by Eco1-mediated acetylation of Smc3 (Rolef Ben-Shahar et al. 2008; Sutani et al. 2009; Unal et al. 2008). It is likely that WAPL plays a similar role in meiotic prophase I, although the biochemical mechanism is not yet resolved. WAPL localizes to the synaptonemal complex in mouse (Zhang et al. 2008a). In C. elegans, WAPL-1 regulates cohesion removal of the COH-3/4 complexes but not the REC-8 cohesion complex (Crawley et al. 2016). Drosophila wapl mutants lead to nondisjunction of achaismata chromosomes, likely via heterochromatin pairing (Vernì et al. 2000).
5.3.2 The Synaptonemal Complex: An Introduction
The synaptonemal complex, a multimeric protein complex, mediates the association of homologous chromosomes in meiotic prophase I (Fig. 5.3; Fawcett 1956; Moses 1956, 1958). The proteinaceous synaptonemal complex is composed of lateral elements which bind to the axis of each homologous chromosome pair and central region proteins that connect to the lateral elements. Together they form a tripartite, ladder-like structure which holds homologous chromosomes together (Zickler and Kleckner 1999). Components of the synaptonemal complex contain coiled-coil domains which are conserved across all Eukarya synaptonemal complex proteins, but the amino acid sequence of the proteins is highly variable across species (Fig. 5.4; Table 5.2; Fraune et al. 2012; Page and Hawley 2004). Complete formation of the synaptonemal complex occurs when central region proteins have loaded and form a bridge between the lateral element proteins; this is known as synapsis of homologous chromosomes. Full synapsis is important for stable pairing, which allows for the repair of DSBs and the formation of crossovers, one of the outcomes of this repair. Crossovers create a physical tether between homologous chromosomes. This tether is important for holding the homologous chromosomes together until segregation; therefore, complete synapsis is necessary for proper chromosome segregation during meiosis. Loss of the synaptonemal complex leads to unstable pairing and deficient homologous recombination (e.g., de Vries et al. 2005; MacQueen et al. 2002; Sym et al. 1993). Homologous recombination is crucial for proper segregation of homologous chromosomes, and improper segregation leads to the formation of aneuploid gametes.

Synaptonemal complex proteins vary among organisms. (A) SC in mouse consists of lateral proteins (yellow) and central region proteins, SYCP1 (red), TEX12 (orange), SYCE2 (orange/blue), and SYCE1 (blue). SYCP1 is the only central region protein to interact with the lateral elements, whereas TEX12, SYCE2, and SYCE1 interact only with each other and the opposite end of SYCP1. (B) SC in D. melanogaster consists of three proteins, C(3)G (red), Cona (blue), and Corola (blue). Cona and Corola form a complex in the middle of the central region and do not contact the lateral elements. C(3)G is the only protein to interact with lateral elements similar to mammalian SYCP1. (C) C. elegans SC is composed of four central region proteins: SYP-1 (red), SYP-2 (orange), SYP-3 (light blue), and SYP-4 (blue). Unlike other organisms, multiple C. elegans proteins interact with lateral elements, including SYP-1, SYP-3, and SYP-4
5.3.2.1 Synaptonemal Complex Assembly Dynamics, Pairing, and Signaling
Prior to synaptonemal complex assembly and complete synapsis along homologous chromosomes, the homologs need to pair. In general, the method by which the homologs find their partners is conserved across metazoans: chromosome movement prevents nonhomologous chromosome pairing while promoting homologs to pair, which acts through chromosome connections to the nuclear envelope and the cytoskeleton. Chromosomes are attached to the nuclear envelope through one or more ends in a configuration typically defined as a “bouquet” (Dresser and Giroux 1988; Harper 2004). Drosophila and C. elegans are exceptions in that they do not form a typical bouquet (Harper 2004). In C. elegans, initial pairing of chromosomes occurs at pairing centers, repetitive DNA sequences that are found on one end of each chromosome (Phillips et al. 2009). These sequences are recognized and bound by zinc-finger proteins, which are suggested to link the pairing centers of the chromosomes to the nuclear envelope (MacQueen et al. 2005; Phillips et al. 2005). In Drosophila, pairing initiates at centromeric locations, while in S. cerevisiae, pairing initiates at telomeres (Takeo et al. 2011; Tanneti et al. 2011; Trelles-Sticken et al. 2000). In the mouse, both telomeres and centromeres (telocentric in mouse) show significant levels of pairing in the initiation of meiosis (Boateng et al. 2013). SUN-KASH domain proteins were shown to be involved in the anchoring of chromosomes to the nuclear envelope in S. cerevisiae, S. pombe, C. elegans, and mouse model systems (Boateng et al. 2013; Conrad et al. 2008; Ding et al. 2007; Morimoto et al. 2012; Niwa et al. 2000). To disturb nonhomologous chromosome interactions, a whipping movement is created allowing for chromosomes to continue the search for homologs (Chikashige et al. 1994; Parvinen and Söderström 1976). In Drosophila and mice, a rotational movement is used to promote proper homologous chromosome pairing (Christophorou et al. 2015; Scherthan et al. 1996). In S. pombe, C. elegans, and Drosophila, the mechanism for homology search requires dynein-mediated movement of microtubules, while in S. cerevisiae, actin plays a role in the chromosomal movements in meiosis (Christophorou et al. 2015; Horn et al. 2013; Yamamoto et al. 1999).
Synaptonemal complex assembly initiates when the lateral elements (referred to as axial elements prior to synaptonemal complex assembly) bind to the chromosomes (Zickler and Kleckner 1999). The recruitment of axis proteins to chromosomes may be mediated by a direct interaction with the cohesion complex, providing a scaffold for synaptonemal complex assembly (Urban et al. 2014; Vallaster et al. 2011). Once chromosomes have found their homologs, central region protein nucleation occurs at the site of homolog pairing. In S. cerevisiae and Drosophila, the first synapsis initiation events were shown to coincide with centromeres, while in C. elegans, synapsis most likely initiates at pairing centers (Rog and Dernburg 2015). In mouse, however, centromeres are not the sites of synapsis initiation (Qiao et al. 2012). Once the central region proteins fully align along the chromosomes, the synaptonemal complex is considered to be fully synapsed. It is not clear yet if central region proteins interact directly with lateral elements or if the lateral elements create the right chromosomal environment for their assembly. The type and number of proteins that make up the two different parts of the synaptonemal complex (lateral and central elements) vary between organisms (see Table 5.2). Synaptonemal complex disassembly occurs at the end of prophase after DSB formation, repair, and the presence of crossovers that has now created a physical tether between homologs. Synaptonemal complex disassembly occurs in a stepwise manner, typically starting with the removal of central elements of the synaptonemal complex, followed by some elements of the axis (Eijpe et al. 2003; Nabeshima et al. 2005). Some synaptonemal complex proteins are retained on chromosomes in restricted locations and disassemble only during meiosis I, when homologous chromosomes segregate (Bisig et al. 2012; de Carvalho et al. 2008).
Just as the proteins of the synaptonemal complex vary from organism to organism, so does the interplay between recombination and the synaptonemal complex. In all cases, the synaptonemal complex is crucial for crossover formation between homologs. However, while in Drosophila and C. elegans, the synaptonemal complex is absolutely required for crossover formation, in mouse and in S. cerevisiae, the requirement is only partial (e.g., de Vries et al. 2005; MacQueen et al. 2002; Page and Hawley 2001; Sym et al. 1993). Another difference between organisms is in how much the synaptonemal complex requires DSBs for its formation. In C. elegans and Drosophila, the loss of DSB formation does not affect synaptonemal complex assembly, disassembly, or its structure (Colaiácovo et al. 2003; McKim et al. 1998). In mouse and in S. cerevisiae, in the absence of DSBs, the synaptonemal complex fails to properly form and very limited synapsis is observed (Bhuiyan and Schmekel 2004; Romanienko and Camerini-Otero 2000).
5.3.2.2 The Contribution of the Synaptonemal Complex to Recombination
The main role of the synaptonemal complex is considered to be to hold the homologous chromosomes together to stabilize the pairing interactions between them and to ensure crossover formation. Crossovers create a physical tether that holds together homologs at points where the synaptonemal complex disassembles. If the synaptonemal complex assembles improperly, such as partial assembly or aggregate formation, the ability to form crossovers is impaired and can lead to aneuploidy or cell death. The axial elements of the synaptonemal complex were shown to be required for DSB formation. In S. cerevisiae, both axis proteins Red1 and Hop1 are required for DSB formation (Mao-Draayer et al. 1996; Woltering et al. 2000; Xu et al. 1997), while in C. elegans, HTP-3 is required for DSB formation (Goodyer et al. 2008).
Other studies have suggested further roles for the synaptonemal complex in the regulation of the recombination process. The synaptonemal complex may promote the crossover/noncrossover decision by its specific effect on supporting crossovers. Mutants in the ZMM class of proteins in S. cerevisiae, which includes SC components, are required for crossover formation, but have no role in noncrossover formation (Börner et al. 2004). Axial element proteins of the synaptonemal complex act in ensuring homolog bias. Hop1 and Red1 from S. cerevisiae and SYCP3 from mouse impose homolog bias by serving as a barrier for sister chromatid recombination (Li et al. 2011), while in S. pombe, the axis imposes interhomolog recombination by promoting recombination with the homolog (Latypov et al. 2010). In C. elegans, reducing the level of synaptonemal complex central region proteins impairs interference (Libuda et al. 2013). However, it is not clear if the synaptonemal complex plays a role in crossover interference in other organisms since interference acts prior in S. cerevisiae (Bishop and Zickler 2004) or concurrently in Sordaria macrospora (Zhang et al. 2014) to synaptonemal complex assembly.
5.3.2.3 Posttranslational Modification
The proteins that make up the synaptonemal complex have been well studied, but how these proteins are regulated by posttranslational modifications is still not fully understood. One function of posttranslational modification is to regulate the assembly of the synaptonemal complex. S. cerevisiae provides the most complete understanding of synaptonemal complex assembly, a process which is regulated by SUMOylation in this organism. Zip3, one of the ZMM proteins, is an E3 SUMO ligase (Cheng et al. 2006). SUMO localizes to Zip1, and the absence of SUMO causes defects in synaptonemal complex assembly (Cheng et al. 2006; Hooker and Roeder 2006; Voelkel-Meiman et al. 2013). An additional component in this pathway includes UBC9, an E2 ligase that forms SUMO chains that are required for synaptonemal complex assembly (Klug et al. 2013). The C-terminal domains of the SC proteins Zip1 and Red1 contain SUMO-interacting domains (SIMs) (Cheng et al. 2006; Lin et al. 2010). Red1 is SUMOylated, in a Zip1-dependent manner, and SUMOylation facilitates the interaction between the two, promoting SC assembly (Eichinger and Jentsch 2010). A second step in SC assembly involves Emc11 SUMOylation (Humphryes et al. 2013; Leung et al. 2015; Voelkel-Meiman et al. 2013). SUMOylation of Emc11 promotes Zip1 assembly, via the central region of the SC, which is located at the Zip1 N-terminus (Leung et al. 2015). In C. elegans, synaptonemal complex aggregation is prevented by the activity of a neddylation regulated ubiquitin ligase, but it is still not clear if this effect on synaptonemal complex assembly is direct (Brockway et al. 2014). Posttranslational modification of synaptonemal complex proteins is also important for their disassembly. In M. musculus, PLK-1 localizes to and is required for the phosphorylation of SYCP1, TEX12, and SYCE1 in spermatocytes and is important for synaptonemal complex disassembly (Jordan et al. 2012). Phosphorylation of synaptonemal complex central region proteins also can initiate disassembly, as was shown for C. elegans SYP-2 (Nadarajan et al. 2016).
Another function of posttranslational modification of the synaptonemal complex is in regulation of recombination in meiosis. In S. cerevisiae, phosphorylation of the axial element, Hop1, is required for interhomolog recombination bias (Carballo et al. 2008), while the phosphorylation of Zip1, central region protein, promotes the formation of interfering crossovers as well as synaptonemal complex assembly (Chen et al. 2015). Axial element proteins of the synaptonemal complex are phosphorylated in a temporally regulated manner in mice, but the function of this phosphorylation is currently unknown (Fukuda et al. 2012). Since posttranslational modifications play an important role in the regulation of protein complexes, more in-depth understanding of how posttranslational protein modifications regulate the synaptonemal complex is required.
5.4 Signaling in Meiotic Prophase I
The accurate execution of meiotic events requires signaling pathways to communicate between different steps of the meiotic program. This communication is thought to occur between many different components of the meiotic program within each cell, although intercellular communication cannot be excluded. In terms of signaling related to recombination events, many of the pathway components are borrowed from DNA damage repair signaling. This is not surprising due to the similarity between meiotic and mitotic recombination. Pathways that regulate homologous chromosome pairing and synapsis are intertwined with recombination-related signaling, as expected from the interdependency between recombination and synapsis found in many organisms. Overall, meiotic signaling involves a combination of feedback loops acting within a pathway (e.g., DSB repair signals to DSB formation), with cross talk between pathways (e.g., crossover formation signals to chromosome pairing), and altogether regulating the complex molecular events that take place in meiosis.
DSB formation and repair are linked by signaling mechanisms that ensure when recombination proceeds normally, DSB formation is halted. ATM/Tel1 and ATR/Mec1 are two related protein kinases that play an important role in mitotic DSB repair (Guleria and Chandna 2016). In Drosophila and mouse meiosis, DSB formation is downregulated by ATM, while in S. cerevisiae, ATM and ATR are both involved in downregulating DSB formation (Joyce et al. 2011; Lange et al. 2011; Zhang et al. 2011). ATM and ATR’s regulation of DSB formation may involve phosphorylation of members of the Spo11 complex, as was shown for Rec114 in S. cerevisiae (Carballo et al. 2013). This mechanism of downregulating DSB formation may be controlling interference at the level of DSB formation, as in S. cerevisiae ATM preferentially downregulates proximal DSBs, preventing the positioning of adjacent DSBs (Garcia et al. 2015). ATM and ATR are also involved in regulating DSB repair. In S. cerevisiae, activation of ATM/Tel1 depends on Mre11, a key player in DSB resection (Usui et al. 2001). However, ATM is not only activated by Mre11 but also positively regulates resection with ATR (Cartagena-Lirola et al. 2006; Terasawa et al. 2008; Usui et al. 2001). ATM and ATR promote differential timing of resection on a different subset of DSBs; ATM regulation is earlier and acts upon a few DSBs, whereas ATR regulates the late and abundant DSBs (Joshi et al. 2015). The synaptonemal complex axis is also a target of ATM and ATR. In mouse, axis and cohesion proteins are phosphorylated, in an ATM- and ATR-dependent manner, in chromosomal regions not yet synapsed, suggesting a role in silencing of unsynapsed chromatin (Fukuda et al. 2012; Royo et al. 2013). In S. cerevisiae, ATM/ATR phosphorylates Hop1 to promote interhomolog recombination (Carballo et al. 2008; Penedos et al. 2015). Mutating a predicted ATM/ATR phosphorylation site on Zip3 reduces crossover levels in S. cerevisiae (Serrentino et al. 2013). These effects on crossover levels are in agreement with the role for ATM and (more so) for ATR in imposing homolog bias (Joshi et al. 2015). ATR1/Mec1 also mediates Zip1 phosphorylation, but this event is specifically required for the early meiotic role of Zip1 in binding centromeres (centromere coupling) (Falk et al. 2010). In C. elegans, the early meiotic role for ATM/ATR has not been described as these proteins function mainly in the context of externally induced DNA damage and the mitotic germ line (Garcia-Muse and Boulton 2005; Stergiou et al. 2007). In C. elegans meiosis, ATM is involved in the restoration of chromosomal structure after DNA damage is induced in meiotic nuclei (Couteau and Zetka 2011).
Similarly to ATM and ATR, CHK2/Mek1 acts primarily in the DNA damage response in mitotic cells (Zannini et al. 2014). In mice ovaries, Chk2 is regulated by ATM and in S. pombe, CHK2 is a target of ATR, thus linking the two DNA damage signaling pathways (Miles et al. 2010; Tougan et al. 2010). The activation of CHK2 in mouse ovaries is essential for DNA damage-induced apoptosis of oocytes (Bolcun-Filas et al. 2014). Work done in S. cerevisiae and S. pombe is consistent with a role for Chk2 in the DNA damage repair pathway, but in a different step: promoting meiotic DSB repair (Niu et al. 2007; Tougan et al. 2010; Xu et al. 1997). In S. cerevisiae, Chk2 was shown to be involved in inhibiting sister chromatid recombination (Niu et al. 2007). In C. elegans, CHK-2 acts both in promoting DSB formation and homologous chromosome pairing. These pathways add to the growing list of noncanonical functions of CHK2 beyond DNA damage signaling (Zannini et al. 2014). CHK-2 promotes the formation of meiotic DSBs by controlling the recruitment of proteins required for DSB formation (DSB-1 and DSB-2) to chromosomes (Rosu et al. 2013; Stamper et al. 2013). In its second meiotic function, CHK-2 is required for localization of most pairing center proteins, promoting both chromosome pairing and synapsis (MacQueen and Villeneuve 2001; Phillips and Dernburg 2006). These mechanisms involve the phosphorylation of SUN-1 (part of the protein complex tethering chromosomes to the nuclear envelope) by CHK-2 (Penkner et al. 2009). CHK-2 also acts as a “sensor” protein. Defects in chromosome synapsis or recombination are relayed to CHK-2 via proteins forming the synaptonemal complex axis; this process promotes prolongation of CK activities (Kim et al. 2015). Chk2 may have a meiotic function outside DSB repair in mouse as well, as it is required in spermatogenesis for a specific H3 phosphorylation mark (Govin et al. 2010).
Polo-like kinases (PLKs) are involved in the regulation of cell division in both mitosis and meiosis that includes a wide variety of cellular functions (Archambault et al. 2015). In C. elegans, two PLKs act in meiosis: the meiotic functioning PLK-2 and the mainly mitotic functioning PLK-1 which can partially substitute for PLK-2 (Chase et al. 2000; Harper et al. 2011). In C. elegans, SUN-1 is targeted by PLK-2 (and CHK-2, see above). PLK-2 localizes to SUN-1 and phosphorylates it, promoting pairing and inhibiting nonhomologous synapsis (Harper et al. 2011; Labella et al. 2011). PLK-2’s localization to SUN-1 patches is partially dependent upon the phosphorylation of SUN-1, suggesting a positive feedback loop between the two proteins (Woglar et al. 2013). In S. cerevisiae, the polo-like kinase homolog Cdc5 appears to perform a later meiotic function. Cdc5 is required for pachytene exit and synaptonemal complex disassembly and is the target of the transcriptional regulator Ndt80 (Sourirajan and Lichten 2008). Ndt80 was also suggested to be part of the signal transduction pathway signaling between the recruitment of the Spo11 complex to the MI division (Malone et al. 2004).
The MAP kinase (MAPK) pathway plays an important role in oocyte maturation, allowing for the transition from diplotene arrest to MI in vertebrates (Fan and Sun 2004; Fan et al. 2012; Sato 2015). In C. elegans, where cells do not normally arrest at diplotene, MAPK signaling is required for meiotic progression and the development of functional oocytes (Church et al. 1995; Lee et al. 2007). MAPK plays a role in meiotic DSB repair by serving as a signal for the transition from the meiotic repair mechanism to a mitotic-like repair pathway (Hayashi et al. 2007). The effect of the MAPK pathway on DSB repair may even take place prior to crossover formation, as defects in the timing of MPK activation are correlated with an alteration in the timing of key DSB repair events (Yin et al. 2016). Moreover, the MAPK pathway may regulate DSB repair directly, since proteins required for homologous recombination repair are among the suggested MAPK targets (Arur et al. 2009). MAPK inactivation is also required for the disassembly of synaptonemal complex central region proteins, thus acting to preserve synaptonemal complex structure until crossovers are formed (Nadarajan et al. 2016). Altogether, these studies show that the MAPK pathway may serve to connect the various molecular processes taking place in the pachytene stage and coordinate them with meiotic progression.
5.5 Defects Originating in Prophase that Lead to Aneuploidy
5.5.1 Meiosis and Aneuploidy
The inheritance of an abnormal number of chromosomes is termed aneuploidy. In humans, an aneuploid embryo with only one autosomal chromosome will die before a pregnancy is recognized, while a trisomic embryo will either end as a miscarriage or lead to the birth of a child with developmental disabilities (Herbert et al. 2015). Human oocytes show a high rate of aneuploidy [reviewed in Nagaoka et al. (2012)]: for example, 54 % of MII oocytes from young donors with no known fertility issues, and 62 % of oocytes from advanced maternal age in vitro fertilization donors show aneuploidy (Garcia-Cruz et al. 2010; Ottolini et al. 2015). These levels of aneuploidy are much higher than what is observed for oocytes in model organisms [despite varying levels of aneuploidy between studies in the same model organism, Drosophila: Traut (1980; Traut and Schröder (1978) and mice: (Hodges et al. (2005)]. Another feature of human meiosis is that aneuploidy rates increase as women age (Hassold and Chiu 1985; Pellestor et al. 2003). Oocytes of model organisms also exhibit a mild age-dependent increase in aneuploidy (Traut 1980; Traut and Schröder 1978). It was shown that this age effect is stronger for a long-lived mouse model (Lister et al. 2010). These findings suggest that the exceptional increase in aneuploidy in humans as they age is revealed as an outcome of the extension of reproductive age that follows the increase in life span of humans in the last century.
Missegregation of chromosomes during meiotic divisions is a leading cause for aneuploidy. It is conceivable that aneuploidy could originate from mitotic errors. However, studies of all three meiotic products of human oocyte meiosis establish that aneuploidy is exclusively caused by meiotic errors (Ottolini et al. 2015). Aneuploidy can arise from chromosome missegregation (nondisjunction) at MI or MII. In the first meiotic division, homologous chromosomes segregate away from each other. Since meiotic prophase establishes the crossovers connecting homologous chromosomes, aneuploidy arising from prophase I defects is more likely to lead to chromosome missegregation in the first meiotic division. However, missegregation of a whole chromosome is less common than predicted, and defects leading to premature separation of sister chromatids are common (Angell 1991). These events include reverse segregation and precocious separation of sister chromatids (PSSCs) and mostly occur in MI (Ottolini et al. 2015). In advanced maternal age women, single chromatid nondisjunction, including MII errors, is more frequently found (Pellestor et al. 2003) (Fragouli et al. 2011, 2013). MI versus MII origins of aneuploidy are also dependent on the chromosome analyzed, for example, chromosome 16 aneuploidy involves mainly MI events, while chromosome 18 aneuploidy involves mainly MII errors (Bugge et al. 1998). Analysis of chromosomal rates of nondisjunction, prior to implantation, indicates that smaller chromosomes (15, 16, 19, 21, and 22) are more likely to missegregate (Fragouli et al. 2013). As found in human oocytes, smaller chromosomes of mice are more likely to lack chiasma (Hodges et al. 2005). Although aneuploidy is the most common form of meiotic chromosomal aberration, partial losses of chromosome segments (large deletions) were also observed in low frequency in human oocytes (Fragouli et al. 2011).
Unlike sperm cells, oocytes are arrested in meiotic prophase I during embryonic development, when resumption of meiosis occurs following ovulation (MI at ovulation and MII at fertilization). Thus, in humans, oocytes arrest at a stage following crossover formation and prior to MI division for decade(s). Autosomal aneuploidy is predominant in oogenesis, as compared to spermatogenesis (Martin et al. 1991; Pacchierotti et al. 2007), suggesting that the causes of infertility may be connected to the unique properties of female meiosis. The fact that aneuploidy is increased as women age may suggest that the process that is delayed in female meiosis is the one sensitive to perturbation. These observations suggest that an early meiotic event that is maintained up until fertilization (pre-diplotene) is “time sensitive” and perturbed as oocytes age. The fact that chromosomes that undergo nondisjunction have altered recombination rates and patterns (see below) led to the proposal that aneuploidy arises from a “two hit” model: one hit, originating at embryonic development and a second hit that is the “age-dependent” component (Lamb et al. 1996). There are likely many causes for aneuploidy, but here we will focus on the connection between the proper execution of prophase events and aneuploidy.
5.5.2 Crossover Formation and Aneuploidy
Studies in model organisms establish that a lack of crossovers leads to missegregation of chromosomes in MI. Some exceptions to this rule, however, exist. For example, (a) backup mechanisms of achiasmatic chromosome segregation and (b) meiotic programs that do not rely on crossovers (Wolf 1994). Current models hold that crossovers are a prerequisite for ensuring that homologous chromosomes will segregate away from each other to opposing poles. It was therefore suggested that oocyte nondisjunction might arise from an inability to form crossovers on a particular chromosome.
Both the reduction and absence of recombination is found in trisomic chromosomes. However, the magnitude of the effect varies between chromosomes and studies. Analysis of spontaneous abortions due to trisomy 16 or 21 showed 30 % and 16–40 % reduction in map length on respective chromosomes (Hassold et al. 1995; Lamb et al. 2005). Chromosomes 21 and 22, which are prone to nondisjunction, also show reduced recombination rate by cytological analysis of fetal oocytes [~5 % and 6 % of oocytes with no MLH1 foci, respectively (Cheng et al. 2009)]. 25–47 % of the trisomies involve achiasmate bivalents for the chromosome effected [13: Bugge et al. (2007), Hall et al. (2007a); 15: Robinson et al. (1998); 18: Bugge et al. (1998); 21: Lamb et al. (1997, 2005); 22: Hall et al. (2007b); X: Thomas et al. (2001)]. However, despite the fact that the frequency of trisomy 21 increases with maternal age (Penrose 2009), oocytes isolated from advanced maternal age individuals are as likely to have achiasmatic chromosomes as younger donors (Lamb et al. 1997). Moreover, trisomies for chromosome 15 are less likely to show noncrossovers when originating in older mothers (Robinson et al. 1998). These findings do not exclude the possibility that achiasmatic chromosomes may contribute to the high baseline level of aneuploidy found in humans.
Studies in oocytes from advanced maternal age donors indicate that recombination rate is a factor that accounts for 18 % of the variation in the incidence of aneuploidy (Ottolini et al. 2015). These aged oocytes have ~6 less crossovers overall compared to euploid oocytes (Ottolini et al. 2015). It is intriguing that the overall reduction of crossover rates over the whole genome, and not just on chromosomes 21, predisposes trisomy 21 (Brown et al. 2000). Oocytes that enter meiosis earlier in development will ovulate earlier as well (Polani and Crolla 1991), which leads to the hypothesis that global rates of recombination are lower in oocytes from late fetal stage, accounting for the age-dependent effect on aneuploidy. This “production line” model is likely incorrect, as oocytes from early and late fetal stage have similar recombination rates [MLH1 foci counts (Rowsey et al. 2014)].
A single crossover may not be sufficient to promote proper chromosome segregation. Analysis of fetal oocytes indicate that chromosomes 21 and 22 are less likely to contain crossover markers on both p and q arms of the chromosome [MLH1 foci (Cheng et al. 2009)], and chromosome 16 is less likely to have multiple crossover events (Garcia-Cruz et al. 2010). It is possible that a single crossover will be sufficient for proper segregation; however, analysis of all meiotic products has indicated that two crossovers involving all sister chromatids are required for proper segregation (Ottolini et al. 2015). According to this study, nonrecombinant chromatids are more likely to be subjected to precocious separation of sister chromatids (at MI) or missegregation at MII, even if they were originally part of a bivalent (Ottolini et al. 2015).
5.5.3 Crossover Distribution and Aneuploidy
Crossover positioning plays an important role in chromosome missegregation. Terminal crossovers on chromosome 21 are more likely to be found on chromosomes that have undergone MI nondisjunction, whereas pericentromeric crossovers are more likely to be found on MII nondisjunction chromosomes (Lamb et al. 1997, 2005). Analysis of spontaneous abortions due to trisomy 16 showed that most of the reduction in map length was within the pericentromeric regions (Hassold et al. 1995). Oocytes from advanced maternal age donors (obtained from in vitro fertilization) show variation in the positions of the crossover events, which in some oocytes were pericentromeric, predisposing these oocytes for segregation defects (Ottolini et al. 2015). Maternal age also affects the distribution of crossovers on chromosomes with nondisjunction events (Ghosh et al. 2009; Oliver et al. 2008, 2012). Analysis of recombination maps of chromosomes 21 from Down syndrome patients revealed an increase in telomeric-proximal recombination in all age groups (Oliver et al. 2008, 2012). These events are found in a higher proportion in children with chromosome 21 trisomies born to young mothers (Oliver et al. 2008). These studies suggest that the distal crossovers may contribute to the basal high levels of human nondisjunction, regardless of mother’s age. However, the effect on MII was predominant in older mothers and mainly involve a centromeric exchange (Oliver et al. 2008), indicating that centromeric-proximal crossovers are subjected to the age-dependent effect on nondisjunction. Despite this correlation between altered recombination patterns and nondisjunction of autosomes, the X chromosome did not show this same association, which may indicate that the causes for nondisjunction vary between autosomes and the X chromosome (Thomas et al. 2001).
5.5.4 Cohesion Maintenance and Aneuploidy
Crossovers link homologous chromosomes only as long as sister chromatid cohesion is maintained. The age-related component of oocyte aneuploidy acts upon processes that take place after crossover formation, suggesting that compromised sister chromatid cohesion leads to a loss of chiasmata and the breakdown of the bivalent. Immunostainings for cohesins reveal a decreased association with chromosomes in aged mice oocytes (Liu and Keefe 2008; Tsutsumi et al. 2014). In humans, meiotic, but not mitotic, cohesion protein levels are reduced with age, as measured by immunofluorescence staining, despite having no effect on mRNA levels (Garcia-Cruz et al. 2010; Tsutsumi et al. 2014). Several studies, both in Drosophila and mouse, addressed this model by examining whether loss of cohesion predisposes chromosomes to nondisjunction in an age-dependent manner. Drosophila females with reduced levels of Ord (a protein required for meiotic cohesion) show an increase in nondisjunction which is notably augmented in aged oocytes (Jeffreys et al. 2003). Mice with a point mutation in the meiotic-specific cohesion complex subunit Smc1β show a severe age-dependent increase in the proportion of homologs with univalents, including a premature onset of these defects (Hodges et al. 2005). Altogether, these studies suggest that a reduction in cohesion levels contributes to the maternal-age effect on chromosome missegregation. This effect may be chromosomal region-dependent [centromeric-proximal but not telomeric-proximal crossovers are associated with age-dependent nondisjunction (Oliver et al. 2008)], suggesting that cohesion erosion in pericentromeric regions sensitizes the chromosome to age-dependent nondisjunction.
Cohesion proteins are loaded onto chromosomes prior and during meiotic prophase I; however, it is plausible that they will be replenished later, during the prolonged post-pachytene arrest. However, despite the expression of meiotic cohesion complex proteins in oocytes during their arrest, they do not turn over and their expression during the arrest is not required for maintenance of the chiasma (Tachibana-Konwalski et al. 2010). These studies suggest that the cohesion complexes loaded prior to oocyte arrest, during embryonic development, are the crucial proteins required for chiasma maintenance. How cohesion function is impaired is not yet clear. In addition to a direct effect on cohesion stability/structure, deregulation of pathways that regulate cohesion removal may come into play (e.g., the cohesion protector Sgo2).
5.5.5 Models Suggesting How Prophase I Defects Lead to Aneuploidy
As discussed above, the synaptonemal complex is required for the formation of all obligatory crossover events and may regulate their distribution. Therefore, it is conceivable that defects in the formation of the synaptonemal complex may lead to bivalents lacking a chiasma or having a non-favorable chiasma positioning. Defects in synapsis of chromosomes activate checkpoints that lead to cell death. If defects in synapsis are ignored, this may lead to the formation of oocytes predisposed to aneuploidy. Oocytes were shown to be less sensitive to the activation of these checkpoints compared to spermatocytes; similar synapsis defects will trigger apoptosis in all sperm cells, while a significant proportion of oocytes will survive (Morelli and Cohen 2005). Therefore, it is possible that a “weak checkpoint” may contribute to aneuploidy.
There are several possible explanations as to how crossover positioning may affect chromosome segregation. The crossover between homologous chromosomes in the bivalent structure is maintained by sister chromatid cohesion. Sister chromatid cohesion is established between the crossover site and each telomere, creating two cohesion domains in the presence of a single crossover. Therefore, cohesion loss on either of these domains is sufficient to break the bivalent structure into two univalents (separated homologous chromosomes; Fig. 5.5). In a bivalent with a terminal crossover, the segment between the crossover and the telomere region is shorter than a bivalent with a more central crossover. Assuming the amount of cohesion lost with age is similar, a bivalent with a terminal crossover will be more likely to lose all its cohesion between the crossover site and the telomere, increasing the likelihood of MI nondisjunction. Another possibility for terminal crossovers to cause MI nondisjunction is that the terminal crossover may compromise bi-orientation of kinetochores at MI. The contribution of pre-centromeric region crossovers to missegregation may also stem from interrupting centromere–spindle attachments and may interfere with centromere separation (Fig. 5.6). This can lead to sister chromatid nondisjunction at MI or MII. MI sister chromatid nondisjunction may contribute to errors associated with reverse meiosis [separation of sisters at MI, homologs at MII, Fig. 5.6B (Ottolini et al. 2015)].

Crossover position relative to the telomere and cohesion are both critical to proper meiotic segregation. A–C and E–G represent the bivalent prior to the meiotic divisions (chromosomes in orange and blue, cohesion in green, centromeres in black, and pulling forces in gray, arrows), while D and H are the outcomes of both meiotic divisions (chromosomes in oocyte and polar bodies). (A–C) In aged meiotic nuclei, sister chromatid synapsis can be lost prematurely before MI segregation (green region is depleted over time, A–C are sequentially older oocytes). If the crossover is far from the telomere region, even if cohesion deteriorates, some protein complexes are likely to be left on both sides of the crossover holding the sisters together. This reduction in cohesion will still permit proper segregation. (D) Four possible outcomes after MII: big circle represents the oocyte and the two smaller circles represent the two polar bodies. The circle on the top is polar body I (PBI) extruded after MI and the small one on the side is the second polar body (PBII) extruded after fertilization. (E–G) Aged meiotic nuclei that have crossovers closer to telomeres may lose all synapsis on one side of the crossover early, leading to missegregation (gray arrows—direction of pulling forces at MI). Resulting oocytes can become trisomic after fertilization. By chance, 50 % of segregation events of that chromosome may be “normal,” leading to segregation patterns as in D

Aneuploidy as a result of centromere–spindle attachment defects. A, C, and E represent the bivalent prior to the meiotic divisions (chromosomes in orange and blue, cohesion in green, centromeres in black, and pulling forces in gray, arrows), while B, D, and F are the outcomes of both meiotic divisions (chromosomes in oocyte and polar bodies). (A) Normal segregation is shown where centromere–spindle attachments correctly separate the homologs at the end of MI, and then sister chromatids separate after MII. Gray arrows point in the direction to which separating homologs are moving. (B) Four possible outcomes after MII: big circle represents the oocyte and the two smaller circles represent the two polar bodies. The small circle on the top is polar body I (PBI) extruded after MI and the small circle on the top right is the second polar body (PBII) extruded after fertilization. (C) If there are multiple spindles attaching to a single centromere, reverse meiosis can occur, in which sister chromatids separate at MI instead of MII. (D) Non-sister chromatids are extruded in PBI (small circle with two chromatids), as opposed to sister chromatids making up one homolog being in PBI normally. (E) If there is precocious separation of sister chromatids based on weak or unbalanced separation during MI, then aneuploidy can occur. (F) After MII, the oocyte can end up with either two, one, or no chromatids and polar bodies can contain up to three chromatids or as few as zero. This defect may be associated with centromeric-proximal crossovers (C and E). The schematics represent only one type of aberrant attachment, but others may be just as likely (e.g., recombined orange chromosomes pulling away from blue and non-recombined orange chromosome pulling in the other direction)
References
Acquaviva L, Székvölgyi L, Dichtl B, Dichtl BS, de La Roche Saint-André C, Nicolas A, Géli V (2013) The COMPASS subunit Spp1 links histone methylation to initiation of meiotic recombination. Science 339:215–218
Adrian AB, Comeron JM (2013) The Drosophila early ovarian transcriptome provides insight to the molecular causes of recombination rate variation across genomes. BMC Genomics 14:794
Agarwal S, Roeder GS (2000) Zip3 provides a link between recombination enzymes and synaptonemal complex proteins. Cell 102:245–255
Ajimura M, Leem SH, Ogawa H (1993) Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics 133:51–66
Anderson DE, Losada A, Erickson HP, Hirano T (2002) Condensin and cohesin display different arm conformations with characteristic hinge angles. J Cell Biol 156:419–424
Angell RR (1991) Predivision in human oocytes at meiosis I: a mechanism for trisomy formation in man. Hum Genet 86:383–387
Archambault V, Lépine G, Kachaner D (2015) Understanding the Polo Kinase machine. Oncogene 34:4799–4807
Arumugam P, Gruber S, Tanaka K, Haering CH, Mechtler K, Nasmyth K (2003) ATP hydrolysis is required for cohesin’s association with chromosomes. Curr Biol 13:1941–1953
Arur S, Ohmachi M, Nayak S, Hayes M, Miranda A, Hay A, Golden A, Schedl T (2009) Multiple ERK substrates execute single biological processes in Caenorhabditis elegans germ-line development. Proc Natl Acad Sci USA 106:4776–4781
Badugu SB, Nabi SA, Vaidyam P, Laskar S, Bhattacharyya S, Bhattacharyya MK (2015) Identification of Plasmodium falciparum DNA repair protein Mre11 with an evolutionarily conserved nuclease function. PLoS One 10(5):e0125358. PMID: 25938776
Bailly AP, Freeman A, Hall J, Déclais A-C, Alpi A, Lilley DMJ, Ahmed S, Gartner A (2010) The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS Genet 6:e1001025
Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A et al (1996) Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet 13:336–342
Baker CL, Walker M, Kajita S, Petkov PM, Paigen K (2014) PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res 24:724–732
Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, De Massy B (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327:836–840
Berg IL, Neumann R, Lam K-WG, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ (2010) PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet 42:859–863
Bhuiyan H, Schmekel K (2004) Meiotic chromosome synapsis in yeast can occur without spo11-induced DNA double-strand breaks. Genetics 168:775–783
Bishop DK, Zickler D (2004) Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell 117:9–15
Bishop DK, Park D, Xu L, Kleckner N (1992) DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69:439–456
Bisig CG, Guiraldelli MF, Kouznetsova A, Scherthan H, Hoog C, Dawson DS, Pezza RJ (2012) Synaptonemal complex components persist at centromeres and are required for homologous centromere pairing in mouse spermatocytes. PLoS Genet 8:e1002701
Blat Y, Protacio RU, Hunter N, Kleckner N (2002) Physical and functional interactions among basic chromosome organizational features govern early steps of meiotic chiasma formation. Cell 111:791–802
Boateng KA, Bellani MA, Gregoretti IV, Pratto F, Camerini-Otero RD (2013) Homologous pairing preceding SPO11-mediated double-strand breaks in mice. Dev Cell 24:196–205
Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR 3rd, Russell P (2000) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107(4):537–548
Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR, Russell P (2001) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107:537–548
Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC (2014) Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 343:533–536
Börner GV, Kleckner N, Hunter N (2004) Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117:29–45
Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV (2012) Genetic recombination is directed away from functional genomic elements in mice. Nature 485:642–645
Brockway H, Balukoff N, Dean M, Alleva B, Smolikove S (2014) The CSN/COP9 signalosome regulates synaptonemal complex assembly during meiotic prophase I of Caenorhabditis elegans. PLoS Genet 10:e1004757
Brown MS, Bishop DK (2015) DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb Perspect Biol 7:a016659
Brown AS, Feingold E, Broman KW, Sherman SL (2000) Genome-wide variation in recombination in female meiosis: a risk factor for non-disjunction of chromosome 21. Hum Mol Genet 9:515–523
Brown MS, Grubb J, Zhang A, Rust MJ, Bishop DK (2015) Small Rad51 and Dmc1 complexes often co-occupy both ends of a meiotic DNA double strand break. PLoS Genet 11:e1005653
Bugge M, Collins A, Petersen MB, Fisher J, Brandt C, Hertz JM, Tranebjaerg L, de Lozier-Blanchet C, Nicolaides P, Brøndum-Nielsen K et al (1998) Non-disjunction of chromosome 18. Hum Mol Genet 7:661–669
Bugge M, Collins A, Hertz JM, Eiberg H, Lundsteen C, Brandt CA, Bak M, Hansen C, Delozier CD, Lespinasse J et al (2007) Non-disjunction of chromosome 13. Hum Mol Genet 16:2004–2010
Buonomo SB, Clyne RK, Fuchs J, Loidl J, Uhlmann F, Nasmyth K (2000) Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 103:387–398
Busygina V, Sehorn MG, Shi IY, Tsubouchi H, Roeder GS, Sung P (2008) Hed1 regulates Rad51-mediated recombination via a novel mechanism. Genes Dev 22:786–795
Busygina V, Saro D, Williams G, Leung WK, Say AF, Sehorn MG, Sung P, Tsubouchi H (2012) Novel attributes of Hed1 affect dynamics and activity of the Rad51 presynaptic filament during meiotic recombination. J Biol Chem 287:1566–1575
Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514:122–125
Carballo JA, Johnson AL, Sedgwick SG, Cha RS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132:758–770
Carballo JA, Panizza S, Serrentino ME, Johnson AL, Geymonat M, Borde V, Klein F, Cha RS (2013) Budding yeast ATM/ATR control meiotic double-strand break (DSB) levels by down-regulating Rec114, an essential component of the DSB-machinery. PLoS Genet 9:e1003545
Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, Hays L, Morgan WF, Petrini JH (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93:477–486
Carpenter AT (1975) Electron microscopy of meiosis in Drosophila melanogaster females. I. Structure, arrangement, and temporal change of the synaptonemal complex in wild-type. Chromosoma 51:157–182
Cartagena-Lirola H, Guerini I, Viscardi V, Lucchini G, Longhese MP (2006) Budding yeast Sae2 is an in vivo target of the Mec1 and Tel1 checkpoint kinases during meiosis. Cell Cycle 5:1549–1559
Challa K, Lee M-S, Shinohara M, Kim KP, Shinohara A (2016) Rad61/Wpl1 (Wapl), a cohesin regulator, controls chromosome compaction during meiosis. Nucleic Acids Res. doi:10.1093/nar/gkw034
Chase D, Serafinas C, Ashcroft N, Kosinski M, Longo D, Ferris DK, Golden A (2000) The polo-like kinase PLK-1 is required for nuclear envelope breakdown and the completion of meiosis in Caenorhabditis elegans. Genesis 26:26–41
Chen R, Wold MS (2014) Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 36:1156–1161
Chen X, Suhandynata RT, Sandhu R, Rockmill B, Mohibullah N, Niu H, Liang J, Lo H-C, Miller DE, Zhou H et al (2015) Phosphorylation of the synaptonemal complex protein Zip1 regulates the crossover/noncrossover decision during yeast meiosis. Plos Biol 13:e1002329
Chen Y-J, Chuang Y-C, Chuang C-N, Cheng Y-H, Chang C-R, Leng C-H, Wang T-F (2016) S. cerevisiae Mre11 recruits conjugated SUMO moieties to facilitate the assembly and function of the Mre11-Rad50-Xrs2 complex. Nucleic Acids Res. doi:10.1093/nar/gkv1523
Cheng C-H, Lo Y-H, Liang S-S, Ti S-C, Lin F-M, Yeh C-H, Huang H-Y, Wang T-F (2006) SUMO modifications control assembly of synaptonemal complex and polycomplex in meiosis of Saccharomyces cerevisiae. Genes Dev 20:2067–2081
Cheng EY, Hunt PA, Naluai-Cecchini TA, Fligner CL, Fujimoto VY, Pasternack TL, Schwartz JM, Steinauer JE, Woodruff TJ, Cherry SM et al (2009) Meiotic recombination in human oocytes. PLoS Genet 5:e1000661
Cherry SM, Adelman CA, Theunissen JW, Hassold TJ, Hunt PA, Petrini JHJ (2007) The Mre11 complex influences DNA repair, synapsis, and crossing over in murine meiosis. Curr Biol 17:373–378
Chikashige Y, Ding DQ, Funabiki H, Haraguchi T, Mashiko S, Yanagida M, Hiraoka Y (1994) Telomere-led premeiotic chromosome movement in fission yeast. Science 264:270–273
Christophorou N, Rubin T, Bonnet I, Piolot T, Arnaud M, Huynh J-R (2015) Microtubule-driven nuclear rotations promote meiotic chromosome dynamics. Nat Cell Biol 17:1388–1400
Church DL, Guan KL, Lambie EJ (1995) Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121:2525–2535
Ciosk R, Shirayama M, Shevchenko A, Tanaka T, Toth A, Nasmyth K (2000) Cohesin’s binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell 5:243–254
Cloud V, Chan Y-L, Grubb J, Budke B, Bishop DK (2012) Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337:1222–1225
Colaiácovo MP, MacQueen AJ, Martinez-Perez E, McDonald K, Adamo A, La Volpe A, Villeneuve AM (2003) Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev Cell 5:463–474
Conrad MN, Lee C-Y, Chao G, Shinohara M, Kosaka H, Shinohara A, Conchello J-A, Dresser ME (2008) Rapid telomere movement in meiotic prophase is promoted by NDJ1, MPS3, and CSM4 and is modulated by recombination. Cell 133:1175–1187
Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S et al (2012) The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489:581–584
Couteau F, Zetka M (2011) DNA damage during meiosis induces chromatin remodeling and synaptonemal complex disassembly. Dev Cell 20:353–363
Crawley O, Barroso C, Testori S, Ferrandiz N, Silva N, Castellano-Pozo M, Jaso-Tamame AL, Martinez-Perez E (2016) Cohesin-interacting protein WAPL-1 regulates meiotic chromosome structure and cohesion by antagonizing specific cohesin complexes. eLife 5:563
Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, Zhao X (2012) Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell 45:422–432
de Carvalho CE, Zaaijer S, Smolikov S, Gu Y, Schumacher JM, Colaiácovo MP (2008) LAB-1 antagonizes the Aurora B kinase in C. elegans. Genes Dev 22:2869–2885
de Vries FAT, de Boer E, van den Bosch M, Baarends WM, Ooms M, Yuan L, Liu J-G, van Zeeland AA, Heyting C, Pastink A (2005) Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev 19:1376–1389
Dernburg AF, McDonald K, Moulder G, Barstead R, Dresser M, Villeneuve AM (1998) Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell 94:387–398
Ding D-Q, Sakurai N, Katou Y, Itoh T, Shirahige K, Haraguchi T, Hiraoka Y (2006) Meiotic cohesins modulate chromosome compaction during meiotic prophase in fission yeast. J Cell Biol 174:499–508
Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M (2007) SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 12:863–872
Ding D-Q, Matsuda A, Okamasa K, Nagahama Y, Haraguchi T, Hiraoka Y (2016) Meiotic cohesin-based chromosome structure is essential for homologous chromosome pairing in Schizosaccharomyces pombe. Chromosoma 125(2):205–214
Dodson GE, Limbo O, Nieto D, Russell P (2010) Phosphorylation-regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double-strand breaks. Cell Cycle 9:1516–1522
Dresser ME, Giroux CN (1988) Meiotic chromosome behavior in spread preparations of yeast. J Cell Biol 106:567–573
Eichinger CS, Jentsch S (2010) Synaptonemal complex formation and meiotic checkpoint signaling are linked to the lateral element protein Red1. Proc Natl Acad Sci USA 107:11370–11375
Eijpe M, Offenberg H, Jessberger R, Revenkova E, Heyting C (2003) Meiotic cohesin REC8 marks the axial elements of rat synaptonemal complexes before cohesins SMC1beta and SMC3. J Cell Biol 160:657–670
Falck J, Forment JV, Coates J, Mistrik M, Lukas J, Bartek J, Jackson SP (2012) CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep 13:561–568
Falk JE, Chan AC-H, Hoffmann E, Hochwagen A (2010) A Mec1- and PP4-dependent checkpoint couples centromere pairing to meiotic recombination. Dev Cell 19:599–611
Fan H-Y, Sun Q-Y (2004) Involvement of mitogen-activated protein kinase cascade during oocyte maturation and fertilization in mammals. Biol Reprod 70:535–547
Fan H-Y, Liu Z, Mullany LK, Richards JS (2012) Consequences of RAS and MAPK activation in the ovary: the good, the bad and the ugly. Mol Cell Endocrinol 356:74–79
Fawcett DW (1956) The fine structure of chromosomes in the meiotic prophase of vertebrate spermatocytes. J Biophys Biochem Cytol 2:403–406
Fragouli E, Alfarawati S, Goodall N-N, Sánchez-García JF, Colls P, Wells D (2011) The cytogenetics of polar bodies: insights into female meiosis and the diagnosis of aneuploidy. Mol Hum Reprod 17:286–295
Fragouli E, Alfarawati S, Spath K, Jaroudi S, Sarasa J, Enciso M, Wells D (2013) The origin and impact of embryonic aneuploidy. Hum Genet 132:1001–1013
Fraune J, Schramm S, Alsheimer M, Benavente R (2012) The mammalian synaptonemal complex: protein components, assembly and role in meiotic recombination. Exp Cell Res 318:1340–1346
Fukuda T, Hoog C (2010) The mouse cohesin-associated protein PDS5B is expressed in testicular cells and is associated with the meiotic chromosome axes. Genes (Basel) 1:484–494
Fukuda T, Pratto F, Schimenti JC, Turner JMA, Camerini-Otero RD, Hoog C (2012) Phosphorylation of chromosome core components may serve as axis marks for the status of chromosomal events during mammalian meiosis. PLoS Genet 8:e1002485
Fukushima K, Tanaka Y, Nabeshima K, Yoneki T, Tougan T, Tanaka S, Nojima H (2000) Dmc1 of Schizosaccharomyces pombe plays a role in meiotic recombination. Nucleic Acids Res 28:2709–2716
Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K (1998) Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J 17:6412–6425
Gao J, Kim H-M, Elia AE, Elledge SJ, Colaiácovo MP (2015) NatB domain-containing CRA-1 antagonizes hydrolase ACER-1 linking acetyl-CoA metabolism to the initiation of recombination during C. elegans meiosis. PLoS Genet 11:e1005029
Garcia V, Gray S, Allison RM, Cooper TJ, Neale MJ (2015) Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature 520:114–118
Garcia-Cruz R, Casanovas A, Brieno-Enriquez M, Robles P, Roig I, Pujol A, Cabero L, Durban M, Garcia Caldes M (2010) Cytogenetic analyses of human oocytes provide new data on non-disjunction mechanisms and the origin of trisomy 16. Hum Reprod 25:179–191
Garcia-Muse T, Boulton SJ (2005) Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J 24:4345–4355
Gasior SL, Wong AK, Kora Y, Shinohara A, Bishop DK (1998) Rad52 associates with RPA and functions with rad55 and rad57 to assemble meiotic recombination complexes. Genes Dev 12:2208–2221
Gasior SL, Olivares H, Ear U, Hari DM, Weichselbaum R, Bishop DK (2001) Assembly of RecA-like recombinases: distinct roles for mediator proteins in mitosis and meiosis. Proc Natl Acad Sci USA 98:8411–8418
Gause M, Webber HA, Misulovin Z, Haller G, Rollins RA, Eissenberg JC, Bickel SE, Dorsett D (2008) Functional links between Drosophila Nipped-B and cohesin in somatic and meiotic cells. Chromosoma 117:51–66
Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO, Petes TD (2000) Inaugural Article: Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 97:11383–11390
Getun IV, Wu ZK, Khalil AM, Bois PRJ (2010) Nucleosome occupancy landscape and dynamics at mouse recombination hotspots. EMBO Rep 11:555–560
Ghosh S, Feingold E, Dey SK (2009) Etiology of Down syndrome: evidence for consistent association among altered meiotic recombination, nondisjunction, and maternal age across populations. Am J Med Genet 149A:1415–1420
Goedecke W, Eijpe M, Offenberg HH, van Aalderen M, Heyting C (1999) Mre11 and Ku70 interact in somatic cells, but are differentially expressed in early meiosis. Nat Genet 23:194–198
Goodyer W, Kaitna S, Couteau F, Ward JD, Boulton SJ, Zetka M (2008) HTP-3 links DSB formation with homolog pairing and crossing over during C. elegans meiosis. Dev Cell 14:263–274
Govin J, Dorsey J, Gaucher J, Rousseaux S, Khochbin S, Berger SL (2010) Systematic screen reveals new functional dynamics of histones H3 and H4 during gametogenesis. Genes Dev 24:1772–1786
Guleria A, Chandna S (2016) ATM kinase: much more than a DNA damage responsive protein. DNA Repair 39:1–20
Gutiérrez-Caballero C, Herrán Y, Sánchez-Martín M, Suja JA, Barbero JL, Llano E, Pendás AM (2011) Identification and molecular characterization of the mammalian α-kleisin RAD21L. Cell Cycle 10:1477–1487
Haering CH, Löwe J, Hochwagen A, Nasmyth K (2002) Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell 9:773–788
Hall HE, Chan ER, Collins A, Judis L, Shirley S, Surti U, Hoffner L, Cockwell AE, Jacobs PA, Hassold TJ (2007a) The origin of trisomy 13. Am J Med Genet 143A:2242–2248
Hall HE, Surti U, Hoffner L, Shirley S, Feingold E, Hassold T (2007b) The origin of trisomy 22: evidence for acrocentric chromosome-specific patterns of nondisjunction. Am J Med Genet 143A:2249–2255
Harper L (2004) A bouquet of chromosomes. J Cell Sci 117:4025–4032
Harper NC, Rillo R, Jover-Gil S, Assaf ZJ, Bhalla N, Dernburg AF (2011) Pairing centers recruit a Polo-like kinase to orchestrate meiotic chromosome dynamics in C. elegans. Dev Cell 21:934–947
Harvey SH, Krien MJE, O’Connell MJ (2002) Structural maintenance of chromosomes (SMC) proteins, a family of conserved ATPases. Genome Biol 3:REVIEWS3003
Hassold T, Chiu D (1985) Maternal age-specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum Genet 70:11–17
Hassold T, Merrill M, Adkins K, Freeman S, Sherman S (1995) Recombination and maternal age-dependent nondisjunction: molecular studies of trisomy 16. Am J Hum Genet 57:867–874
Hayase A, Takagi M, Miyazaki T, Oshiumi H, Shinohara M, Shinohara A (2004) A protein complex containing Mei5 and Sae3 promotes the assembly of the meiosis-specific RecA homolog Dmc1. Cell 119:927–940
Hayashi K, Yoshida K, Matsui Y (2005) A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438:374–378
Hayashi M, Chin GM, Villeneuve AM (2007) C. elegans germ cells switch between distinct modes of double-strand break repair during meiotic prophase progression. PLoS Genet 3:e191
Heidmann D, Horn S, Heidmann S, Schleiffer A, Nasmyth K, Lehner CF (2004) The Drosophila meiotic kleisin C(2)M functions before the meiotic divisions. Chromosoma 113:177–187
Herbert M, Kalleas D, Cooney D, Lamb M, Lister L (2015) Meiosis and maternal aging: insights from aneuploid oocytes and trisomy births. Cold Spring Harb Perspect Biol 7:a017970
Herrán Y, Gutiérrez-Caballero C, Sánchez-Martín M, Hernández T, Viera A, Barbero JL, de Álava E, de Rooij DG, Suja JA, Llano E et al (2011) The cohesin subunit RAD21L functions in meiotic synapsis and exhibits sexual dimorphism in fertility. EMBO J 30:3091–3105
Hill WG, Robertson A (2007) The effect of linkage on limits to artificial selection. Genet Res 89:311–336
Hodges CA, Revenkova E, Jessberger R, Hassold TJ, Hunt PA (2005) SMC1β-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat Genet 37:1351–1355
Holloway JK, Booth J, Edelmann W, McGowan CH, Cohen PE (2008) MUS81 generates a subset of MLH1-MLH3-independent crossovers in mammalian meiosis. PLoS Genet 4:e1000186
Holloway JK, Mohan S, Balmus G, Sun X, Modzelewski A, Borst PL, Freire R, Weiss RS, Cohen PE (2011) Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genet 7:e1002094
Holloway JK, Sun X, Yokoo R, Villeneuve AM, Cohen PE (2014) Mammalian CNTD1 is critical for meiotic crossover maturation and deselection of excess precrossover sites. J Cell Biol 205:633–641
Hong S, Sung Y, Yu M, Lee M, Kleckner N, Kim KP (2013) The logic and mechanism of homologous recombination partner choice. Mol Cell 51:440–453
Hooker GW, Roeder GS (2006) A role for SUMO in meiotic chromosome synapsis. Curr Biol 16:1238–1243
Hopfner K-P, Tainer JA (2003) Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr Opin Struct Biol 13:249–255
Horn HF, Kim DI, Wright GD, Wong ESM, Stewart CL, Burke B, Roux KJ (2013) A mammalian KASH domain protein coupling meiotic chromosomes to the cytoskeleton. J Cell Biol 202:1023–1039
Hu B, Itoh T, Mishra A, Katoh Y, Chan K-L, Upcher W, Godlee C, Roig MB, Shirahige K, Nasmyth K (2011) ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr Biol 21:12–24
Huang CE, Milutinovich M, Koshland D (2005) Rings, bracelet or snaps: fashionable alternatives for Smc complexes. Philos Trans R Soc Lond B Biol Sci 360:537–542
Humphryes N, Leung W-K, Argunhan B, Terentyev Y, Dvorackova M, Tsubouchi H (2013) The Ecm11-Gmc2 complex promotes synaptonemal complex formation through assembly of transverse filaments in budding yeast. PLoS Genet 9:e1003194
Hunter N, Borts RH (1997) Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev 11:1573–1582
Ishiguro K-I, Kim J, Fujiyama-Nakamura S, Kato S, Watanabe Y (2011) A new meiosis-specific cohesin complex implicated in the cohesin code for homologous pairing. EMBO Rep 12:267–275
Ivanov EL, Korolev VG, Fabre F (1992) XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics 132:651–664
Ivanov D, Schleiffer A, Eisenhaber F, Mechtler K, Haering CH, Nasmyth K (2002) Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr Biol 12:323–328
Jantsch V, Pasierbek P, Mueller MM, Schweizer D, Jantsch M, Loidl J (2004) Targeted gene knockout reveals a role in meiotic recombination for ZHP-3, a Zip3-related protein in Caenorhabditis elegans. Mol Cell Biol 24:7998–8006
Jeffreys CA, Burrage PS, Bickel SE (2003) A model system for increased meiotic nondisjunction in older oocytes. Curr Biol 13:498–503
Jin H, Guacci V, Yu H-G (2009) Pds5 is required for homologue pairing and inhibits synapsis of sister chromatids during yeast meiosis. J Cell Biol 186:713–725
Johzuka K, Ogawa H (1995) Interaction of Mre11 and Rad50: two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics 139:1521–1532
Jordan PW, Karppinen J, Handel MA (2012) Polo-like kinase is required for synaptonemal complex disassembly and phosphorylation in mouse spermatocytes. J Cell Sci 125:5061–5072
Joshi N, Brown MS, Bishop DK, Börner GV (2015) Gradual implementation of the meiotic recombination program via checkpoint pathways controlled by global DSB levels. Mol Cell 57:797–811
Joyce EF, Pedersen M, Tiong S, White-Brown SK, Paul A, Campbell SD, McKim KS (2011) Drosophila ATM and ATR have distinct activities in the regulation of meiotic DNA damage and repair. J Cell Biol 195:359–367
Joyce EF, Paul A, Chen KE, Tanneti N, McKim KS (2012) Multiple barriers to nonhomologous DNA end joining during meiosis in Drosophila. Genetics 191:739–746
Keeney S, Giroux CN, Kleckner N (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88:375–384
Kelly KO, Dernburg AF, Stanfield GM, Villeneuve AM (2000) Caenorhabditis elegans msh-5 is required for both normal and radiation-induced meiotic crossing over but not for completion of meiosis. Genetics 156:617–630
Kim Y, Kostow N, Dernburg AF (2015) The chromosome axis mediates feedback control of CHK-2 to ensure crossover formation in C. elegans. Dev Cell 35:247–261
Kitajima TS, Kawashima SA, Watanabe Y (2004) The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature 427:510–517
Klein F, Mahr P, Galova M, Buonomo SB, Michaelis C, Nairz K, Nasmyth K (1999) A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell 98:91–103
Klug H, Xaver M, Chaugule VK, Koidl S, Mittler G, Klein F, Pichler A (2013) Ubc9 sumoylation controls SUMO chain formation and meiotic synapsis in Saccharomyces cerevisiae. Mol Cell 50:625–636
Koh-Stenta X, Joy J, Poulsen A, Li R, Tan Y, Shim Y, Min J-H, Wu L, Ngo A, Peng J et al (2014) Characterization of the histone methyltransferase PRDM9 using biochemical, biophysical and chemical biology techniques. Biochem J 461:323–334
Kolodkin AL, Klar AJ, Stahl FW (1986) Double-strand breaks can initiate meiotic recombination in S. cerevisiae. Cell 46:733–740
Koshland D, Strunnikov A (1996) Mitotic chromosome condensation. Annu Rev Cell Dev Biol 12:305–333
Krishnaprasad GN, Anand MT, Lin G, Tekkedil MM, Steinmetz LM, Nishant KT (2015) Variation in crossover frequencies perturb crossover assurance without affecting meiotic chromosome segregation in Saccharomyces cerevisiae. Genetics 199:399–412
Kudo NR, Wassmann K, Anger M, Schuh M, Wirth KG, Xu H, Helmhart W, Kudo H, Mckay M, Maro B et al (2006) Resolution of chiasmata in oocytes requires separase-mediated proteolysis. Cell 126:135–146
Kuleszewicz K, Fu X, Kudo NR (2013) Cohesin loading factor Nipbl localizes to chromosome axes during mammalian meiotic prophase. Cell Div 8:12
Kuznetsov S, Pellegrini M, Shuda K, Fernandez-Capetillo O, Liu Y, Martin BK, Burkett S, Southon E, Pati D, Tessarollo L et al (2007) RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J Cell Biol 176:581–592
Labella S, Woglar A, Jantsch V, Zetka M (2011) Polo kinases establish links between meiotic chromosomes and cytoskeletal forces essential for homolog pairing. Dev Cell 21:948–958
Lam I, Keeney S (2015) Nonparadoxical evolutionary stability of the recombination initiation landscape in yeast. Science 350:932–937
Lamb NE, Freeman SB, Savage-Austin A, Pettay D, Taft L, Hersey J, Gu Y, Shen J, Saker D, May KM et al (1996) Susceptible chiasmate configurations of chromosome 21 predispose to non-disjunction in both maternal meiosis I and meiosis II. Nat Genet 14:400–405
Lamb NE, Feingold E, Savage A, Avramopoulos D, Freeman S, Gu Y, Hallberg A, Hersey J, Karadima G, Pettay D et al (1997) Characterization of susceptible chiasma configurations that increase the risk for maternal nondisjunction of chromosome 21. Hum Mol Genet 6:1391–1399
Lamb NE, Yu K, Shaffer J, Feingold E, Sherman SL (2005) Association between maternal age and meiotic recombination for trisomy 21. Am J Hum Genet 76:91–99
Lange J, Pan J, Cole F, Thelen MP, Jasin M, Keeney S (2011) ATM controls meiotic double-strand-break formation. Nature 479:237–240
Lao JP, Cloud V, Huang C-C, Grubb J, Thacker D, Lee C-Y, Dresser ME, Hunter N, Bishop DK (2013) Meiotic crossover control by concerted action of Rad51-Dmc1 in homolog template bias and robust homeostatic regulation. PLoS Genet 9:e1003978
Latypov V, Rothenberg M, Lorenz A, Octobre G, Csutak O, Lehmann E, Loidl J, Kohli J (2010) Roles of Hop1 and Mek1 in meiotic chromosome pairing and recombination partner choice in Schizosaccharomyces pombe. Mol Cell Biol 30:1570–1581
Lee J, Hirano T (2011) RAD21L, a novel cohesin subunit implicated in linking homologous chromosomes in mammalian meiosis. J Cell Biol 192:263–276
Lee MH, Ohmachi M, Arur S, Nayak S, Francis R, Church D, Lambie E, Schedl T (2007) Multiple functions and dynamic activation of MPK-1 extracellular signal-regulated kinase signaling in Caenorhabditis elegans germline development. Genetics 177:2039–2062
Lee J, Kitajima TS, Tanno Y, Yoshida K, Morita T, Miyano T, Miyake M, Watanabe Y (2008) Unified mode of centromeric protection by shugoshin in mammalian oocytes and somatic cells. Nature 10:42–52
Lee JY, Terakawa T, Qi Z, Steinfeld JB, Redding S, Kwon Y, Gaines WA, Zhao W, Sung P, Greene EC (2015) DNA recombination. Base triplet stepping by the Rad51/RecA family of recombinases. Science 349:977–981
Lemmens BBLG, Johnson NM, Tijsterman M (2013) COM-1 promotes homologous recombination during Caenorhabditis elegans meiosis by antagonizing Ku-mediated non-homologous end joining. PLoS Genet 9:e1003276
Leung W-K, Humphryes N, Afshar N, Argunhan B, Terentyev Y, Tsubouchi T, Tsubouchi H (2015) The synaptonemal complex is assembled by a polySUMOylation-driven feedback mechanism in yeast. J Cell Biol 211:785–793
Li XC, Bolcun-Filas E, Schimenti JC (2011) Genetic evidence that synaptonemal complex axial elements govern recombination pathway choice in mice. Genetics 189:71–82
Libuda DE, Uzawa S, Meyer BJ, Villeneuve AM (2013) Meiotic chromosome structures constrain and respond to designation of crossover sites. Nature 502:703–706
Lightfoot J, Testori S, Barroso C, Martinez-Perez E (2011) Loading of meiotic cohesin by SCC-2 is required for early processing of DSBs and for the DNA damage checkpoint. Curr Biol 21:1421–1430
Lin Z, Kong H, Nei M, Ma H (2006) Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc Natl Acad Sci USA 103:10328–10333
Lin F-M, Lai Y-J, Shen H-J, Cheng Y-H, Wang T-F (2010) Yeast axial-element protein, Red1, binds SUMO chains to promote meiotic interhomologue recombination and chromosome synapsis. EMBO J 29:586–596
Lin W, Jin H, Liu X, Hampton K, Yu H-G (2011) Scc2 regulates gene expression by recruiting cohesin to the chromosome as a transcriptional activator during yeast meiosis. Mol Biol Cell 22:1985–1996
Lipkin SM, Moens PB, Wang V, Lenzi M, Shanmugarajah D, Gilgeous A, Thomas J, Cheng J, Touchman JW, Green ED et al (2002) Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet 31:385–390
Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y et al (2010) Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol 20:1511–1521
Liu L, Keefe DL (2008) Defective cohesin is associated with age-dependent misaligned chromosomes in oocytes. Reprod Biomed Online 16:103–112
Liu Y, Tarsounas M, O’regan P, West SC (2007) Role of RAD51C and XRCC3 in genetic recombination and DNA repair. J Biol Chem 282:1973–1979
Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer W-D (2011) Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479:245–248
Llano E, Herrán Y, García-Tuñón I, Gutiérrez-Caballero C, de Álava E, Barbero JL, Schimenti J, de Rooij DG, Sánchez-Martín M, Pendás AM (2012) Meiotic cohesin complexes are essential for the formation of the axial element in mice. J Cell Biol 197:877–885
Lorenz A, Mehats A, Osman F, Whitby MC (2014) Rad51/Dmc1 paralogs and mediators oppose DNA helicases to limit hybrid DNA formation and promote crossovers during meiotic recombination. Nucleic Acids Res 42:13723–13735
Lovett ST, Mortimer RK (1987) Characterization of null mutants of the RAD55 gene of Saccharomyces cerevisiae: effects of temperature, osmotic strength and mating type. Genetics 116:547–553
Lynn A, Soucek R, Börner GV (2007) ZMM proteins during meiosis: crossover artists at work. Chromosome Res 15:591–605
Ma L, Milman N, Nambiar M, Smith GR (2015) Two separable functions of Ctp1 in the early steps of meiotic DNA double-strand break repair. Nucleic Acids Res 43:7349–7359
MacQueen AJ, Villeneuve AM (2001) Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev 15:1674–1687
MacQueen AJ, Colaiácovo MP, McDonald K, Villeneuve AM (2002) Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev 16:2428–2442
MacQueen AJ, Phillips CM, Bhalla N, Weiser P, Villeneuve AM, Dernburg AF (2005) Chromosome sites play dual roles to establish homologous synapsis during meiosis in C. elegans. Cell 123:1037–1050
Malone RE, Haring SJ, Foreman KE, Pansegrau ML, Smith SM, Houdek DR, Carpp L, Shah B, Lee KE (2004) The signal from the initiation of meiotic recombination to the first division of meiosis. Eukaryot Cell 3:598–609
Manfrini N, Guerini I, Citterio A, Lucchini G, Longhese MP (2010) Processing of meiotic DNA double strand breaks requires cyclin-dependent kinase and multiple nucleases. J Biol Chem 285:11628–11637
Mao-Draayer Y, Galbraith AM, Pittman DL, Cool M, Malone RE (1996) Analysis of meiotic recombination pathways in the yeast Saccharomyces cerevisiae. Genetics 144:71–86
Martin RH, Ko E, Rademaker A (1991) Distribution of aneuploidy in human gametes: comparison between human sperm and oocytes. Am J Med Genet 39:321–331
Martin JS, Winkelmann N, Petalcorin MIR, McIlwraith MJ, Boulton SJ (2005) RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol 25:3127–3139
Matos J, Blanco MG, Maslen S, Skehel JM, West SC (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147:158–172
McKim KS, Hayashi-Hagihara A (1998) mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: evidence that the mechanism for initiating meiotic recombination is conserved. Genes Dev 12:2932–2942
McKim KS, Green-Marroquin BL, Sekelsky JJ, Chin G, Steinberg C, Khodosh R, Hawley RS (1998) Meiotic synapsis in the absence of recombination. Science 279:876–878
Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91:35–45
Miles DC, van den Bergen JA, Sinclair AH, Western PS (2010) Regulation of the female mouse germ cell cycle during entry into meiosis. Cell Cycle 9:408–418
Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774
Moreau S, Ferguson JR, Symington LS (1999) The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol 19:556–566
Morelli MA, Cohen PE (2005) Not all germ cells are created equal: aspects of sexual dimorphism in mammalian meiosis. Reproduction 130:761–781
Morimoto A, Shibuya H, Zhu X, Kim J, Ishiguro K-I, Han M, Watanabe Y (2012) A conserved KASH domain protein associates with telomeres, SUN1, and dynactin during mammalian meiosis. J Cell Biol 198:165–172
Moses MJ (1956) Chromosomal structures in crayfish spermatocytes. J Biophys Biochem Cytol 2:215–218
Moses MJ (1958) The relation between the axial complex of meiotic prophase chromosomes and chromosome pairing in a salamander (Plethodon cinereus). J Biophys Biochem Cytol 4:633–638
Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ (2001) Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157:103–118
Murayama Y, Uhlmann F (2014) Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505:367–371
Nabeshima K, Villeneuve AM, Colaiácovo MP (2005) Crossing over is coupled to late meiotic prophase bivalent differentiation through asymmetric disassembly of the SC. J Cell Biol 168:683–689
Nadarajan S, Mohideen F, Tzur YB, Ferrandiz N, Crawley O, Montoya A, Faull P, Snijders AP, Cutillas PR, Jambhekar A et al (2016) The MAP kinase pathway coordinates crossover designation with disassembly of synaptonemal complex proteins during meiosis. eLife 5:e12039
Nagaoka SI, Hassold TJ, Hunt PA (2012) Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 13:493–504
Nairz K, Klein F (1997) mre11S--a yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes Dev 11:2272–2290
Neale MJ, Pan J, Keeney S (2005) Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436:1053–1057
Nešić D, Hsu Y, Stebbins CE (2004) Assembly and function of a bacterial genotoxin. Nature 429:429–433
Nishant KT, Chen C, Shinohara M, Shinohara A, Alani E (2010) Genetic analysis of baker’s yeast Msh4-Msh5 reveals a threshold crossover level for meiotic viability. PLoS Genet 6:e1001083
Niu H, Li X, Job E, Park C, Moazed D, Gygi SP, Hollingsworth NM (2007) Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol Cell Biol 27:5456–5467
Niwa O, Shimanuki M, Miki F (2000) Telomere-led bouquet formation facilitates homologous chromosome pairing and restricts ectopic interaction in fission yeast meiosis. EMBO J 19:3831–3840
Novak JE, Ross-Macdonald PB, Roeder GS (2001) The budding yeast Msh4 protein functions in chromosome synapsis and the regulation of crossover distribution. Genetics 158:1013–1025
Oh SD, Lao JP, Taylor AF, Smith GR, Hunter N (2008) RecQ helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Mol Cell 31:324–336
Oliver TR, Feingold E, Yu K, Cheung V, Tinker S, Yadav-Shah M, Masse N, Sherman SL (2008) New insights into human nondisjunction of chromosome 21 in oocytes. PLoS Genet 4:e1000033–e1000039
Oliver TR, Tinker SW, Allen EG, Hollis N, Locke AE, Bean LJH, Chowdhury R, Begum F, Marazita M, Cheung V et al (2012) Altered patterns of multiple recombinant events are associated with nondisjunction of chromosome 21. Hum Genet 131:1039–1046
Osman F, Dixon J, Doe CL, Whitby MC (2003) Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol Cell 12:761–774
Ottolini CS, Newnham LJ, Capalbo A, Natesan SA, Joshi HA, Cimadomo D, Griffin DK, Sage K, Summers MC, Thornhill AR et al (2015) Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nature 47:727–735
Pacchierotti F, Adler ID, Eichenlaub-Ritter U, Mailhes JB (2007) Gender effects on the incidence of aneuploidy in mammalian germ cells. Environ Res 104:46–69
Page SL, Hawley RS (2001) c(3)G encodes a Drosophila synaptonemal complex protein. Genes Dev 15:3130–3143
Page SL, Hawley RS (2004) The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol 20:525–558
Parvanov ED, Petkov PM, Paigen K (2010) Prdm9 controls activation of mammalian recombination hotspots. Science 327:835
Parvinen M, Söderström KO (1976) Chromosome rotation and formation of synapsis. Nature 260:534–535
Pasierbek P, Jantsch M, Melcher M, Schleiffer A, Schweizer D, Loidl J (2001) A Caenorhabditis elegans cohesion protein with functions in meiotic chromosome pairing and disjunction. Genes Dev 15:1349–1360
Paull TT, Gellert M (1998) The 3″ to 5″ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell 1:969–979
Pellestor F, Andréo B, Arnal F, Humeau C, Demaille J (2003) Maternal aging and chromosomal abnormalities: new data drawn from in vitro unfertilized human oocytes. Hum Genet 112:195–203
Penedos A, Johnson AL, Strong E, Goldman AS, Carballo JA, Cha RS (2015) Essential and checkpoint functions of budding yeast ATM and ATR during meiotic prophase are facilitated by differential phosphorylation of a meiotic adaptor protein, Hop1. PLoS One 10:e0134297
Penkner AM, Fridkin A, Gloggnitzer J, Baudrimont A, Machacek T, Woglar A, Csaszar E, Pasierbek P, Ammerer G, Gruenbaum Y et al (2009) Meiotic chromosome homology search involves modifications of the nuclear envelope protein Matefin/SUN-1. Cell 139:920–933
Penrose LS (2009) The relative effects of paternal and maternal age in mongolism. 1933. J Genet 88:9–14
Perry J, Kleckner N, Börner GV (2005) Bioinformatic analyses implicate the collaborating meiotic crossover/chiasma proteins Zip2, Zip3, and Spo22/Zip4 in ubiquitin labeling. Proc Natl Acad Sci USA 102:17594–17599
Pezzi N, Prieto I, Kremer L, Pérez Jurado LA, Valero C, Del Mazo J, Martínez-A C, Barbero JL (2000) STAG3, a novel gene encoding a protein involved in meiotic chromosome pairing and location of STAG3-related genes flanking the Williams-Beuren syndrome deletion. FASEB J 14:581–592
Phillips CM, Dernburg AF (2006) A family of zinc-finger proteins is required for chromosome-specific pairing and synapsis during meiosis in C. elegans. Dev Cell 11:817–829
Phillips CM, Wong C, Bhalla N, Carlton PM, Weiser P, Meneely PM, Dernburg AF (2005) HIM-8 binds to the X chromosome pairing center and mediates chromosome-specific meiotic synapsis. Cell 123:1051–1063
Phillips CM, Meng X, Zhang L, Chretien JH, Urnov FD, Dernburg AF (2009) Identification of chromosome sequence motifs that mediate meiotic pairing and synapsis in C. elegans. Nat Cell Biol 11:934–942
Polani PE, Crolla JA (1991) A test of the production line hypothesis of mammalian oogenesis. Hum Genet 88:64–70
Ponting CP (2011) What are the genomic drivers of the rapid evolution of PRDM9? Trends Genet 27:165–171
Pradillo M, Knoll A, Oliver C, Varas J, Corredor E, Puchta H, Santos JL (2015) Involvement of the cohesin cofactor PDS5 (SPO76) during meiosis and DNA repair in Arabidopsis thaliana. Front Plant Sci 6:1034
Prieto I, Pezzi N, Buesa JM, Kremer L, Barthelemy I, Carreiro C, Roncal F, Martinez A, Gomez L, Fernandez R et al (2002) STAG2 and Rad21 mammalian mitotic cohesins are implicated in meiosis. EMBO Rep 3:543–550
Prinz S, Amon A, Klein F (1997) Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics 146:781–795
Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, Greene EC (2015) DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160:856–869
Qiao H, Chen JK, Reynolds A, Hoog C, Paddy M, Hunter N (2012) Interplay between synaptonemal complex, homologous recombination, and centromeres during mammalian meiosis. PLoS Genet 8:e1002790
Qiao H, Prasada Rao HBD, Yang Y, Fong JH, Cloutier JM, Deacon DC, Nagel KE, Swartz RK, Strong E, Holloway JK et al (2014) Antagonistic roles of ubiquitin ligase HEI10 and SUMO ligase RNF212 regulate meiotic recombination. Nature 46:194–199
Radford SJ, Goley E, Baxter K, McMahan S, Sekelsky J (2005) Drosophila ERCC1 is required for a subset of MEI-9-dependent meiotic crossovers. Genetics 170:1737–1745
Rakshambikai R, Srinivasan N, Nishant KT (2013) Structural insights into Saccharomyces cerevisiae Msh4-Msh5 complex function using homology modeling. PLoS One 8:e78753
Reddy KC, Villeneuve AM (2004) C. elegans HIM-17 links chromatin modification and competence for initiation of meiotic recombination. Cell 118:439–452
Revenkova E, Eijpe M, Heyting C, Gross B, Jessberger R (2001) Novel meiosis-specific isoform of mammalian SMC1. Mol Cell Biol 21:6984–6998
Reynolds A, Qiao H, Yang Y, Chen JK, Jackson N, Biswas K, Holloway JK, Baudat F, de Massy B, Wang J et al (2013) RNF212 is a dosage-sensitive regulator of crossing-over during mammalian meiosis. Nat Genet 45:269–278
Robinson WP, Kuchinka BD, Bernasconi F, Petersen MB, Schulze A, Brøndum-Nielsen K, Christian SL, Ledbetter DH, Schinzel AA, Horsthemke B et al (1998) Maternal meiosis I non-disjunction of chromosome 15: dependence of the maternal age effect on level of recombination. Hum Mol Genet 7:1011–1019
Rodman TC (1971) Chromosomes of the first polar body in mammalian meiosis. Exp Cell Res 68:205–210
Rog O, Dernburg AF (2015) Direct visualization reveals kinetics of meiotic chromosome synapsis. Cell Rep 10:1639–1645
Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F (2008) Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321:563–566
Romanienko PJ, Camerini-Otero RD (2000) The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 6:975–987
Rosu S, Zawadzki KA, Stamper EL, Libuda DE, Reese AL, Dernburg AF, Villeneuve AM (2013) The C. elegans DSB-2 protein reveals a regulatory network that controls competence for meiotic DSB formation and promotes crossover assurance. PLoS Genet 9:e1003674
Rothenberg M, Kohli J, Ludin K (2009) Ctp1 and the MRN-complex are required for endonucleolytic Rec12 removal with release of a single class of oligonucleotides in fission yeast. PLoS Genet 5:e1000722
Rowsey R, Gruhn J, Broman KW, Hunt PA, Hassold T (2014) Examining variation in recombination levels in the human female: a test of the production-line hypothesis. Am J Hum Genet 95:108–112
Royo H, Prosser H, Ruzankina Y, Mahadevaiah SK, Cloutier JM, Baumann M, Fukuda T, Hoog C, Tóth A, de Rooij DG et al (2013) ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes Dev 27:1484–1494
Saito TT, Youds JL, Boulton SJ, Colaiácovo MP (2009) Caenorhabditis elegans HIM-18/SLX-4 interacts with SLX-1 and XPF-1 and maintains genomic integrity in the germline by processing recombination intermediates. PLoS Genet 5:e1000735
Saito TT, Mohideen F, Meyer K, Harper JW, Colaiácovo MP (2012) SLX-1 is required for maintaining genomic integrity and promoting meiotic noncrossovers in the Caenorhabditis elegans germline. PLoS Genet 8:e1002888
Saito TT, Lui DY, Kim H-M, Meyer K, Colaiácovo MP (2013) Interplay between structure-specific endonucleases for crossover control during Caenorhabditis elegans meiosis. PLoS Genet 9:e1003586
Salah SM, Nasmyth K (2000) Destruction of the securin Pds1p occurs at the onset of anaphase during both meiotic divisions in yeast. Chromosoma 109:27–34
Sato K-I (2015) Transmembrane signal transduction in oocyte maturation and fertilization: focusing on Xenopus laevis as a model animal. Int J Mol Sci 16:114–134
Scherthan H, Weich S, Schwegler H, Heyting C, Härle M, Cremer T (1996) Centromere and telomere movements during early meiotic prophase of mouse and man are associated with the onset of chromosome pairing. J Cell Biol 134:1109–1125
Schurko AM, Logsdon JM (2008) Using a meiosis detection toolkit to investigate ancient asexual “scandals” and the evolution of sex. Bioessays 30:579–589
Ségurel L, Leffler EM, Przeworski M (2011) The case of the fickle fingers: how the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. Plos Biol 9:e1001211
Sehorn MG, Sigurdsson S, Bussen W, Unger VM, Sung P (2004) Human meiotic recombinase Dmc1 promotes ATP-dependent homologous DNA strand exchange. Nature 429:433–437
Serrentino ME, Chaplais E, Sommermeyer V, Borde V (2013) Differential association of the conserved SUMO ligase Zip3 with meiotic double-strand break sites reveals regional variations in the outcome of meiotic recombination. PLoS Genet 9:e1003416
Severson AF, Meyer BJ (2014) Divergent kleisin subunits of cohesin specify mechanisms to tether and release meiotic chromosomes. eLife 3:e03467
Severson AF, Ling L, van Zuylen V, Meyer BJ (2009) The axial element protein HTP-3 promotes cohesin loading and meiotic axis assembly in C. elegans to implement the meiotic program of chromosome segregation. Genes Dev 23:1763–1778
Sharan SK, Bradley A (1997) Murine Brca2: sequence, map position, and expression pattern. Genomics 40:234–241
Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A (1997) Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386:804–810
Sharan SK, Pyle A, Coppola V, Babus J, Swaminathan S, Benedict J, Swing D, Martin BK, Tessarollo L, Evans JP et al (2004) BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development 131:131–142
Singh DK, Andreuzza S, Panoli AP, Siddiqi I (2013) AtCTF7 is required for establishment of sister chromatid cohesion and association of cohesin with chromatin during meiosis in Arabidopsis. BMC Plant Biol 13:117
Singhal S, Leffler EM, Sannareddy K, Turner I, Venn O, Hooper DM, Strand AI, Li Q, Raney B, Balakrishnan CN et al (2015) Stable recombination hotspots in birds. Science 350:928–932
Skibbens RV, Corson LB, Koshland D, Hieter P (1999) Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev 13:307–319
Smolikov S, Eizinger A, Hurlburt A, Rogers E, Villeneuve AM, Colaiacovo MP (2007) Synapsis-defective mutants reveal a correlation between chromosome conformation and the mode of double-strand break repair during Caenorhabditis elegans meiosis. Genetics 176:2027–2033
Snowden T, Acharya S, Butz C, Berardini M, Fishel R (2004) hMSH4-hMSH5 recognizes Holliday Junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol Cell 15:437–451
Sollier J, Lin W, Soustelle C, Suhre K, Nicolas A, Géli V, de La Roche Saint-André C (2004) Set1 is required for meiotic S-phase onset, double-strand break formation and middle gene expression. EMBO J 23:1957–1967
Sommermeyer V, Béneut C, Chaplais E, Serrentino ME, Borde V (2013) Spp1, a member of the Set1 complex, promotes meiotic DSB formation in promoters by tethering histone H3K4 methylation sites to chromosome axes. Mol Cell 49:43–54
Sourirajan A, Lichten M (2008) Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev 22:2627–2632
Stamper EL, Rodenbusch SE, Rosu S, Ahringer J, Villeneuve AM, Dernburg AF (2013) Identification of DSB-1, a protein required for initiation of meiotic recombination in Caenorhabditis elegans, illuminates a crossover assurance checkpoint. PLoS Genet 9:e1003679
Stergiou L, Doukoumetzidis K, Sendoel A, Hengartner MO (2007) The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ 14:1129–1138
Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P, O’Driscoll M, Jeggo PA (2006) ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J 25:5775–5782
Storlazzi A, Tesse S, Ruprich-Robert G, Gargano S, Pöggeler S, Kleckner N, Zickler D (2008) Coupling meiotic chromosome axis integrity to recombination. Genes Dev 22:796–809
Sugiyama T, Kowalczykowski SC (2002) Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem 277:31663–31672
Sun H, Treco D, Szostak JW (1991) Extensive 3′-overhanging, single-stranded DNA associated with the meiosis-specific double-strand breaks at the ARG4 recombination initiation site. Cell 64:1155–1161
Sun F, Fujiwara Y, Reinholdt LG, Hu J, Saxl RL, Baker CL, Petkov PM, Paigen K, Handel MA (2015) Nuclear localization of PRDM9 and its role in meiotic chromatin modifications and homologous synapsis. Chromosoma 124:397–415
Sung P, Robberson DL (1995) DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 82:453–461
Sutani T, Kawaguchi T, Kanno R, Itoh T, Shirahige K (2009) Budding yeast Wpl1(Rad61)-Pds5 complex counteracts sister chromatid cohesion-establishing reaction. Curr Biol 19:492–497
Svendsen JM, Harper JW (2010) GEN1/Yen1 and the SLX4 complex: solutions to the problem of Holliday junction resolution. Genes Dev 24:521–536
Sym M, Engebrecht JA, Roeder GS (1993) ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72:365–378
Tachibana-Konwalski K, Godwin J, van der Weyden L, Champion L, Kudo NR, Adams DJ, Nasmyth K (2010) Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev 24:2505–2516
Takeo S, Lake CM, Morais-de-Sá E, Sunkel CE, Hawley RS (2011) Synaptonemal complex-dependent centromeric clustering and the initiation of synapsis in Drosophila oocytes. Curr Biol 21:1845–1851
Tamaki H (1965) Chromosome behaviour at meiosis in Saccharomyces cerevisiae. J Gen Microbiol 41:93–98
Tanneti NS, Landy K, Joyce EF, McKim KS (2011) A pathway for synapsis initiation during zygotene in Drosophila oocytes. Curr Biol 21:1852–1857
Taylor MRG, Spirek M, Chaurasiya KR, Ward JD, Carzaniga R, Yu X, Egelman EH, Collinson LM, Rueda D, Krejci L et al (2015) Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell 162:271–286
Terasawa M, Ogawa T, Tsukamoto Y, Ogawa H (2008) Sae2p phosphorylation is crucial for cooperation with Mre11p for resection of DNA double-strand break ends during meiotic recombination in Saccharomyces cerevisiae. Genes Genet Syst 83:209–217
Thomas NS, Ennis S, Sharp AJ, Durkie M, Hassold TJ, Collins AR, Jacobs PA (2001) Maternal sex chromosome non-disjunction: evidence for X chromosome-specific risk factors. Hum Mol Genet 10:243–250
Toth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, Nasmyth K (1999) Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev 13:320–333
Tougan T, Kasama T, Ohtaka A, Okuzaki D, Saito TT, Russell P, Nojima H (2010) The Mek1 phosphorylation cascade plays a role in meiotic recombination of Schizosaccharomyces pombe. Cell Cycle 9:4688–4702
Traut H (1980) X-chromosomal nondisjunction induced by aging oocytes of Drosophila melanogaster: the special susceptibility of mature eggs. Can J Genet Cytol 22:433–437
Traut H, Schröder FJ (1978) The increase in the frequency of X-chromosomal aneuploidy in Drosophila melanogaster as a consequence of suppressed oviposition. Mutat Res 49:225–232
Trelles-Sticken E, Dresser ME, Scherthan H (2000) Meiotic telomere protein Ndj1p is required for meiosis-specific telomere distribution, bouquet formation and efficient homologue pairing. J Cell Biol 151:95–106
Tsai CJ, Mets DG, Albrecht MR, Nix P, Chan A, Meyer BJ (2008) Meiotic crossover number and distribution are regulated by a dosage compensation protein that resembles a condensin subunit. Genes Dev 22:194–211
Tsubouchi H, Roeder GS (2003) The importance of genetic recombination for fidelity of chromosome pairing in meiosis. Dev Cell 5:915–925
Tsutsumi M, Fujiwara R, Nishizawa H, Ito M, Kogo H, Inagaki H, Ohye T, Kato T, Fujii T, Kurahashi H (2014) Age-related decrease of meiotic cohesins in human oocytes. PLoS One 9:e96710
Uhlmann F, Nasmyth K (1998) Cohesion between sister chromatids must be established during DNA replication. Curr Biol 8:1095–1101
Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE (2008) A molecular determinant for the establishment of sister chromatid cohesion. Science 321:566–569
Urban E, Nagarkar-Jaiswal S, Lehner CF, Heidmann SK (2014) The cohesin subunit Rad21 is required for synaptonemal complex maintenance, but not sister chromatid cohesion, during Drosophila female meiosis. PLoS Genet 10:e1004540
Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell 95:705–716
Usui T, Ogawa H, Petrini JH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7:1255–1266
Valencia M, Bentele M, Vaze MB, Herrmann G, Kraus E, Lee SE, Schär P, Haber JE (2001) NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature 414:666–669
Vallaster M, Vallaster CD, Wu SM (2011) Epigenetic mechanisms in cardiac development and disease. Acta Biochim Biophys Sin 44:92–102
Vaur S, Feytout A, Vazquez S, Javerzat JP (2012) Pds5 promotes cohesin acetylation and stable cohesin-chromosome interaction. EMBO Rep 13:645–652
Vernì F, Gandhi R, Goldberg ML, Gatti M (2000) Genetic and molecular analysis of wings apart-like (wapl), a gene controlling heterochromatin organization in Drosophila melanogaster. Genetics 154:1693–1710
Visnes T, Giordano F, Kuznetsova A, Suja JA, Lander AD, Calof AL, Ström L (2014) Localisation of the SMC loading complex Nipbl/Mau2 during mammalian meiotic prophase I. Chromosoma 123:239–252
Voelkel-Meiman K, Taylor LF, Mukherjee P, Humphryes N, Tsubouchi H, MacQueen AJ (2013) SUMO localizes to the central element of synaptonemal complex and is required for the full synapsis of meiotic chromosomes in budding yeast. PLoS Genet 9:e1003837
Waizenegger IC, Hauf S, Meinke A, Peters JM (2000) Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103:399–410
Wang X, Haber JE (2004) Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. Plos Biol 2:E21
Wang TF, Kleckner N, Hunter N (1999) Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci USA 96:13914–13919
Wang F, Yoder J, Antoshechkin I, Han M (2003) Caenorhabditis elegans EVL-14/PDS-5 and SCC-3 are essential for sister chromatid cohesion in meiosis and mitosis. Mol Cell Biol 23:7698–7707
Wagner CR, Kuervers L, Baillie DL, Yanowitz JL (2010) xnd-1 regulates the global recombination landscape in Caenorhabditis elegans. Nature 467(7317):839–843. PMID: 20944745
Ward JO, Reinholdt LG, Motley WW, Niswander LM, Deacon DC, Griffin LB, Langlais KK, Backus VL, Schimenti KJ, O’Brien MJ et al (2007) Mutation in mouse hei10, an e3 ubiquitin ligase, disrupts meiotic crossing over. PLoS Genet 3:e139
Watanabe Y, Nurse P (1999) Cohesin Rec8 is required for reductional chromosome segregation at meiosis. Nature 400:461–464
Woglar A, Daryabeigi A, Adamo A, Habacher C, Machacek T, La Volpe A, Jantsch V (2013) Matefin/SUN-1 phosphorylation is part of a surveillance mechanism to coordinate chromosome synapsis and recombination with meiotic progression and chromosome movement. PLoS Genet 9:e1003335
Wolf KW (1994) How meiotic cells deal with non-exchange chromosomes. Bioessays 16:107–114
Woltering D, Baumgartner B, Bagchi S, Larkin B, Loidl J, de los Santos T, Hollingsworth NM (2000) Meiotic segregation, synapsis, and recombination checkpoint functions require physical interaction between the chromosomal proteins Red1p and Hop1p. Mol Cell Biol 20:6646–6658
Wu J, Zhang X, Zhang L, Wu C-Y, Rezaeian AH, Chan C-H, Li J-M, Wang J, Gao Y, Han F et al (2012) Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol Cell 46:351–361
Wyatt HDM, Sarbajna S, Matos J, West SC (2013) Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol Cell 52:234–247
Xu L, Weiner BM, Kleckner N (1997) Meiotic cells monitor the status of the interhomolog recombination complex. Genes Dev 11:106–118
Xu H, Beasley MD, Warren WD, van der Horst GTJ, McKay MJ (2005) Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev Cell 8:949–961
Yamamoto A, West RR, McIntosh JR, Hiraoka Y (1999) A cytoplasmic dynein heavy chain is required for oscillatory nuclear movement of meiotic prophase and efficient meiotic recombination in fission yeast. J Cell Biol 145:1233–1249
Yildiz O, Majumder S, Kramer B, Sekelsky JJ (2002) Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI-9 to generate meiotic crossovers. Mol Cell 10:1503–1509
Yin Y, Smolikove S (2013) Impaired resection of meiotic double-strand breaks channels repair to nonhomologous end joining in Caenorhabditis elegans. Mol Cell Biol 33:2732–2747
Yin Y, Donlevy S, Smolikove S (2016) Coordination of recombination with meiotic progression in the Caenorhabditis elegans germline by KIN-18, a TAO kinase that regulates the timing of MPK-1 signaling. Genetics 202:45–59
Yokobayashi S, Yamamoto M, Watanabe Y (2003) Cohesins determine the attachment manner of kinetochores to spindle microtubules at meiosis I in fission yeast. Mol Cell Biol 23:3965–3973
Yokoo R, Zawadzki KA, Nabeshima K, Drake M, Arur S, Villeneuve AM (2012) COSA-1 reveals robust homeostasis and separable licensing and reinforcement steps governing meiotic crossovers. Cell 149:75–87
Zakharyevich K, Ma Y, Tang S, Hwang PY-H, Boiteux S, Hunter N (2010) Temporally and biochemically distinct activities of Exo1 during meiosis: double-strand break resection and resolution of double Holliday junctions. Mol Cell 40:1001–1015
Zakharyevich K, Tang S, Ma Y, Hunter N (2012) Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 149:334–347
Zannini L, Delia D, Buscemi G (2014) CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 6:442–457
Zhang Z, Ren Q, Yang H, Conrad MN, Guacci V, Kateneva A, Dresser ME (2005) Budding yeast PDS5 plays an important role in meiosis and is required for sister chromatid cohesion. Mol Microbiol 56:670–680
Zhang J, Håkansson H, Kuroda M, Yuan L (2008a) Wapl localization on the synaptonemal complex, a meiosis-specific proteinaceous structure that binds homologous chromosomes, in the female mouse. Reprod Domest Anim 43:124–126
Zhang N, Kuznetsov SG, Sharan SK, Li K, Rao PH, Pati D (2008b) A handcuff model for the cohesin complex. J Cell Biol 183:1019–1031
Zhang L, Kim KP, Kleckner NE, Storlazzi A (2011) Meiotic double-strand breaks occur once per pair of (sister) chromatids, and via Mec1/ATR and Tel1/ATM, once per quartet of chromatids. Proc Natl Acad Sci USA 108:20036–20041
Zhang L, Espagne E, De Muyt A, Zickler D, Kleckner NE (2014) Interference-mediated synaptonemal complex formation with embedded crossover designation. Proc Natl Acad Sci USA 111:E5059–E5068
Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134:981–994
Zickler D, Kleckner N (1999) Meiotic chromosomes: integrating structure and function. Annu Rev Genet 33:603–754
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Reichman, R., Alleva, B., Smolikove, S. (2017). Prophase I: Preparing Chromosomes for Segregation in the Developing Oocyte. In: Arur, S. (eds) Signaling-Mediated Control of Cell Division . Results and Problems in Cell Differentiation, vol 59. Springer, Cham. https://doi.org/10.1007/978-3-319-44820-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-44820-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44819-0
Online ISBN: 978-3-319-44820-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)