
Abstract
More than 30 types of muscular dystrophies are included in the broader group of neuromuscular cardiomyopathies. Skeletal muscle weakness is a defining characteristic of muscular dystrophy, but as treatments improve and patients live longer, cardiomyopathy has emerged as the leading cause of death for patients with Duchenne or Becker muscular dystrophy. This chapter focuses on Duchenne and Becker muscular dystrophies, from their genetic origins, symptoms and disease progression to cardiac involvement and treatments, as well as research findings. Promising emerging therapies include exon skipping, dystrophin mini-gene replacement, sealants, and gene-editing strategies.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
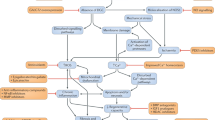


Keywords
- Neuromuscular cardiomyopathies
- Duchenne muscular dystrophy
- Becker muscular dystrophy
- Dystrophinopathies
- Exon skipping
- Dystrophin gene
- Gene editing
Neuromuscular disorders are a diverse and heterogeneous group of diseases that affect the nervous system and/or muscle function. More than 80 distinct neuromuscular disorders exist, which include the muscular dystrophies.
Muscular dystrophies comprise more than 30 genetic disorders characterized by progressive degradation of striated muscles. While many individual muscular dystrophies exist, with varying ages of onset, severity, and patterns of muscle weakness, they share a genetic and pathophysiological origin. That shared origin is a mutation in a gene encoding one of the many structural cytoskeletal proteins that play a critical role in muscle function.
The primary presenting symptom in muscular dystrophies is skeletal muscle weakness. However, cardiac muscle, another type of striated muscle, is similarly affected in many of the muscular dystrophies.
Cardiomyopathies associated with muscular dystrophies are an increasingly recognized manifestation of these neuromuscular disorders and play a significant role in morbidity and mortality in these disease processes. The most common muscular dystrophies to have cardiac involvement include the dystrophinopathies, Duchenne and Becker muscular dystrophies, and myotonic dystrophies.
Dystrophinopathies : A Historical Perspective
The earliest detailed descriptions of muscular dystrophies were by Meryon in England in 1852 and by Duchenne in France in 1868 [1, 2]. They identified a disease that affected young boys with severe muscular weakness and an abnormal increase in calf size. Duchenne obtained muscle biopsies of these boys and noted fatty-fibrous replacement of the skeletal muscle. For this contribution, the disease that he described was named after him: Duchenne muscular dystrophy (DMD ).
In 1886, Gowers described the way that a child affected with muscular dystrophy uses his arms to rise from the ground [3]. While DMD had been known as a clinical entity since the 1860s, its cause was not elucidated for more than 100 years. In a landmark discovery in 1986, Louis Kunkel’s laboratory identified the gene responsible for causing Duchenne muscular dystrophy and, in 1987, showed that mutations resulting in the absence of the 427-kDa rodlike protein dystrophin caused DMD [4, 5].
These major advancements enabled development of diagnostic and genetic testing as well as the establishment of disease models that have facilitated our knowledge of DMD. While a number of questions remain about the muscular dystrophies, the rich history of discovery has significantly accelerated our knowledge of DMD since the mid-1980s.
Dystrophinopathies : Inheritance
DMD is the most common form of muscular dystrophy , affecting 1 of every 5000 boys born in the United States [6–9]. It results from an inherited or spontaneous mutation of the dystrophin (DMD) gene. The Dystrophin (DMD) gene is a 2.5-Mb gene located on chromosome Xp21.1 [5, 7, 10]. Dystrophin is among the largest known genes, encoding a 14-kb transcript with 79 exons (◘ Fig. 12.1).

Dystrophin gene and protein. (a) The 79 exons of the full-length dystrophin gene with promoters for the alternatively spliced isoforms with red arrows and boxes specific for each promoter. Full-length dystrophin promoters include M, muscle promoter; B, brain promoter; and P, purkinje promoter. The isoforms for other shorter dystrophin isoforms include Dp260, which is expressed in the retina (R); Dp140, which is expressed in kidney (K) and brain (B); Dp116, which is expressed in Schwann cells (S); and the ubiquitously expressed Dp71. (b) The protein structure of dystrophin, which includes the actin-binding site at the N-terminus and the dystroglycan complex protein-binding site at the C-terminus. The rod domain contains 24 spectrin-like repeats with four hinge regions
The 11-kb cDNA encodes a 3685 amino acid dystrophin protein of 427 kDa. The full-length dystrophin gene has three gene promoters : (1) the M promoter produces the Dp427m isoform, which is located in the skeletal and cardiac muscle; (2) the B promoter produces Dp427c, which is located in the brain; and (3) the P promoter produces Dp427p, which is in the brain’s Purkinje cells [11–13]. Other non-full-length dystrophin isoforms are produced, including Dp260 (retina), Dp140 (brain and kidney), and Dp71 (ubiquitous) [14–17].
An out-of-frame mutation of one or several of the 79 exons in the full-length dystrophin gene results in an absence of a functional dystrophin protein, which is the hallmark finding in DMD. DMD is inherited in an X-linked recessive manner, where a female carrier with one X chromosome carrying the DMD mutation has a 50 % chance of passing on the mutated X chromosome to her son. Female carriers can also have symptoms based on their X inactivation. While the majority of DMD mutations are inherited, spontaneous mutations account for 30 % of DMD cases [18].
Dystrophin and DGC Organization
The 79 exons code for dystrophin, a 427-kDa protein that is expressed in the skeletal muscle, cardiomyocytes, and brain [7]. Dystrophin, a rod-shaped cytoplasmic protein, connects the dystroglycan complex (DGC) to the intracellular contractile apparatus and extracellular matrix (ECM) of the cell (◘ Fig. 12.2) [19]. Dystrophin is similar to spectrin and other structural proteins and consists of two ends separated by long, flexible rodlike regions. The N-terminus binds actin and the C-terminus binds to glycoproteins in the sarcolemma. The role of dystrophin is to stabilize the plasma membrane by transmitting forces generated by the sarcomeric contraction to the ECM.

Schematic diagram of the dystroglycan complex (DGC) . The DGC spans the sarcolemma and links the cytoplasmic actin cytoskeleton to the extracellular matrix via dystrophin. The transmembrane components of the DGC include sarcoglycans, beta-dystroglycan, and extracellular alpha-dystroglycan which binds laminin. The cytoplasmic components include dystrophin (or utrophin) which binds dystrobrevin, syntrophin, and nitric oxide synthase
The DGC is a multimeric complex composed of glycated integral membrane proteins and peripheral proteins that form a structural link between the f-actin cytoskeleton and the ECM in both the cardiac and skeletal muscle [20–23]. The DGC is comprised of cytoskeletal proteins, dystrophin, syntrophins, dystroglycans, sarcoglycans, DGC-associated proteins, neuronal nitric oxide synthetase, and dystrobrevin [24–26]. The fully nucleated DGC provides mechanical support to the skeletal or cardiac plasma membrane during contraction. Loss of function or absence of one or several of these DGC proteins leads to plasma membrane fragility.
DMD and Becker muscular dystrophy (BMD ) arise from mutations in dystrophin. Other forms of muscular dystrophies arise from mutations in the DGC components. In the skeletal muscle, the DGC is located at regular intervals in structures known as costameres, whereas in the cardiac muscle, the DGC is not located in discrete costameres [27]. The composition of the DGC in the skeletal muscle and cardiac myocytes may be different and differentially altered in dystrophin deficiency [28].
Pathogenesis and Physical Findings
The skeletal muscle and hearts lacking functional dystrophin are mechanically weak, and contraction of the cell (skeletal myocytes and cardiac myocytes) leads to membrane damage [29, 30]. Loss of membrane integrity leads to a cascade of increased calcium influx into the cell and eventual cell death.
Clinically, the loss of dystrophin manifests as progressive muscle weakness [19, 31]. Symptoms are first noted in early childhood and include calf pseudohypertrophy, an inability to stand without using the arms for assistance (Gowers sign), toe walking, and difficulty keeping up with peers. As the disease progresses, the skeletal muscle becomes increasingly weak, causing atrophy and contractures that subsequently result in the loss of ambulation by the age of 10–12. Patients with DMD have markedly elevated serum levels of creatinine kinase (CK), a muscle protein, due to ongoing muscle damage. Creatinine kinase levels may be 10–100 times the normal limit in patients with DMD, and elevated CK levels are a diagnostic sign [32]. DMD diagnosis can be confirmed by muscle biopsy demonstrating the absence of dystrophin (◘ Fig. 12.3) and by genetic testing for dystrophin mutations.

Absence of dystrophin in the DMD skeletal muscle. Photomicrographs of cross sections of the normal and DMD human skeletal muscle. (a) Normal muscle biopsy shows relatively uniform fiber diameter, peripherally located nuclei with no fiber degeneration, inflammation, or endomysial fibrosis. (b) DMD muscle biopsy shows varied fiber diameter, inflammation, and necrotic change with fat and fibrous replacement of the normal muscle tissue. (c) Normal muscle biopsy stained for dystrophin, which is located at the plasmalemma (brown). (d) DMD muscle biopsy stained for dystrophin, demonstrating the absence of dystrophin expression
Survival
Historically, this progressive muscle weakness resulted in the loss of ambulation by 10–12 years of age and death during the second decade of life mainly due to respiratory failure. However, with the advent of nocturnal ventilation, spinal surgery, and steroid treatment, the life expectancy of boys with DMD has increased to the late 20s to early 30s [33] (◘ Fig. 12.4). While respiratory problems used to be the major cause of death in DMD patients, cardiomyopathy now is the predominant cause of death in these patients [34].

Interventions that prolong survival in Duchenne muscular dystrophy. A marked increase in survival of DMD patients has been observed with addition of steroids, ventilation, and spinal surgery. Adapted from [33]
Treatments
Advances in management, namely, corticosteroid treatments, spinal stabilization, and improved pulmonary support, have significantly improved life expectancy of boys/men with DMD to the late 20s and early 30s [33]. Glucocorticoids are one of the mainstays of treatment for DMD and have been shown to improve muscle strength and function and pulmonary function in patients with DMD [35–39].
Corticosteroid Treatments
Glucocorticoids are recommended for patients with DMD who are age 5 or older who are not gaining motor skills or have a decline in motor skills [40]. While the precise mechanism of glucocorticoid therapy in preserving strength is not known, it is postulated that the mechanisms may include inhibition of muscle proteolysis, stimulation of myoblast proliferation, membrane stabilization, an increase in myogenic repair, and a reduction in inflammation [41–46]. The current glucocorticoid protocols that have been tested in randomized clinical trials include daily prednisone 0.75 mg/kg/day, intermittent prednisone 0.75 mg/kg/day 10 days on and 10 days off, and daily deflazacort 0.9 mg/kg/day [47].
Side effects with prednisone and prednisolone include weight gain, Cushingoid appearance, short stature, delayed puberty, and increased risk of osteoporosis and fractures. Patients prescribed steroids should receive preventive measures such as calcium and vitamin D supplementation to reduce bone loss.
Deflazacort is another steroid used with DMD patients; it may have a more limited side effect profile compared to prednisone [48]. At press time, deflazacort is not approved by the US Food and Drug Administration (FDA), so it is not available in the United States. The FDA granted it fast-track status in 2015. Steroid therapy has also been demonstrated to preserve cardiac function in DMD patients in a retrospective study [49].
While steroids are known to be beneficial, no consensus exists on the ideal dosing strategy, age of initiation, or duration of therapy for glucocorticoids in DMD. The FOR-DMD randomized control trial aims to answer the question of ideal dosing strategy by enrolling 300 DMD patients (4–7 years of age) who have not previously received steroids and randomizing them to one of the three dosing regimens listed above .
Spinal Stabilization
Scoliosis and kyphosis are common, progressive sequelae of DMD, occurring near the time the patient loses ambulation. Spinal deformities further decrease pulmonary function as measured by forced vital capacity intervention such as spinal stabilization surgery [50]. Thus, spinal stabilization surgeries using Harrington rods and other techniques have been used with DMD patients to help maintain or improve pulmonary function and improve patient comfort [51, 52].
Pulmonary Support
Respiratory complications occur frequently in patients with DMD as they lose respiratory muscle strength, leading to decreased ventilation and increased risk for pneumonia, atelectasis, and respiratory failure [53]. Before rigorous pulmonary screening and intervention, the majority of deaths in end-stage DMD occurred as a result of respiratory failure and infections.
It is recommended that patients with DMD see a pulmonologist and obtain pulmonary function testing by age 10 and then twice yearly after loss of ambulation or when the vital capacity is less than 80 % of predicted or after the age of 12 [54, 55]. Noninvasive nocturnal mechanical ventilation is offered first at night to treat sleep-related breathing problems and hypoventilation, which are common in patients with DMD [55, 56]. Nocturnal ventilation has improved survival in patients with DMD [57]. Once respiratory weakness progresses past nocturnal ventilation, full ventilator support can be initiated .
Cardiac Involvement in DMD
Nocturnal ventilation and spinal stabilization surgery have reduced respiratory-related deaths, but also increased DMD cardiomyopathy due to the advanced age of DMD patients [33, 57, 58]. Cardiac involvement is nearly ubiquitous in older DMD patients, as more than 90 % of young men over the age of 18 have evidence of cardiac dysfunction [34]. Dilated cardiomyopathy typically has an onset in the mid-teen years and progressively remodels and contributes to the demise of DMD patients [34, 59] (◘ Fig. 12.5). Distinct dystrophin mutations have been correlated to an increased incidence of cardiomyopathy and possible response to treatment [60].

DMD cardiac disease progression . DMD cardiomyopathy disease progression. Initially, DMD patients have structurally normal hearts. Subsequently, DMD patients develop fibrosis of the inferobasal wall as the earliest sign of myocardial involvement. Over time this leads to progressive fibrosis, left ventricular dysfunction, and dilatation leading to end-stage heart failure
Recognition of cardiomyopathy in DMD patients can be challenging due to physical inactivity and other respiratory complaints that can obscure the diagnosis [61]. Currently, clinical guidelines recommend initial cardiac screening at the time of diagnosis of DMD, and every 2 years until age 10, and then yearly thereafter [40].
Most DMD patients will have abnormal electrocardiographic tracings. The classical pattern demonstrates tall R waves and increased R/S amplitude in lead V1, Q waves in the left precordial leads, right axis deviation, or complete right bundle branch block [62–64] (◘ Fig. 12.6). These ECG findings correlate to the pathological studies that have shown a tendency to develop fibrosis in the heart’s basal posterior wall in patients with muscular dystrophies and may reflect reduced electrical activity in the inferobasal wall [65]. ECG findings are believed to precede echocardiographic findings of cardiomyopathy, but no correlation between ECG findings, and the presence of cardiomyopathy has yet been established [62, 66].

Electrocardiographic changes in DMD. Electrocardiogram (ECG) from a patient with Duchenne muscular dystrophy. The ECG demonstrates sinus tachycardia; Q waves in leads I, aVL, and V4-6; and large R waves in V1 and V2
Arrhythmias are a common cardiac involvement in patients with muscular dystrophies. Sinus tachycardia is a common finding in patients with DMD [67–69]. Dystrophic patients experience elevated heart rates even when compared to patients with other muscular dystrophies or deconditioned patients [69]. Pathological examination of the heart in dystrophic patients has shown fibrosis of the conduction system in addition to the myocardium [70], which may explain autonomic cardiac dysfunction in the DMD population [71, 72].
Additionally, sinus tachycardia may correlate with cardiac dysfunction in patients with DMD [73]. Dalmaz et al. reported that urinary catecholamines increased in DMD patients around the age of 10 years, which corresponded to the time of heart rate elevation and cardiac involvement [74]. In patients with DMD cardiomyopathy, atrial arrhythmias including atrial fibrillation, atrial flutter, and atrial tachycardias can occur. Ventricular tachycardia, premature ventricular contractions (PVCs), and other conduction abnormalities have been noted and are correlated with progressive ventricular dilation and dysfunction [75].
Cardiac Imaging in DMD
Cardiac imaging can be challenging in patients with end-stage DMD due to scoliosis, ventilation, and contractures. Imaging modalities commonly used include echocardiography and cardiac magnetic resonance imaging (MRI).
Echocardiography in DMD cardiomyopathy shows regional wall motion abnormalities in the posterior basal wall, left ventricular dilation, and overall reduced systolic function [76]. Current guidelines recommend obtaining an echocardiogram at the time of diagnosis or by age 6, with repeat echocardiograms every 1–2 years until age 10. After age 10, it is recommended that patients have an annual echocardiogram to assess left ventricular function [51]. While echocardiography is easily accessible, relatively quick, and a cost-effective imaging modality, it can be technically challenging in patients with DMD due to chest wall deformities, scoliosis, and respiratory dysfunction, thus limiting the diagnostic yield [77].
CMR imaging has been used with DMD patients and can provide one of the most accurate assessments of left ventricular size and function [78–80]. Silva et al. performed gadolinium contrast-enhanced CMR on ten patients with dystrophinopathies (eight DMD and two BMD patients) and were the first to demonstrate late gadolinium enhancement (LGE) by CMR in a dystrophic heart. They further reported that, using echocardiography, LGE was present even with normal left ventricular function [71].
Pulchalski et al. subsequently performed a study of 74 patients with DMD; the majority had LGE in the posterobasal region of the left ventricle in a subepicardial distribution [81]. This pattern of LGE in the basal inferior and inferolateral walls is consistent with the pathological findings of fibrosis in the inferior basal wall (◘ Fig. 12.7) [65, 82]. A large single-center study by Hor et al. retrospectively evaluated LGE in 314 DMD patients and demonstrated that LGE increased with age and with decreasing left ventricular ejection fraction [LVEF] [83]. Thus, CMR imaging may provide an earlier detection of cardiovascular involvement in DMD and enable accurate and reproducible quantification of left ventricular function and size and promote initiation of earlier cardioprotective treatment. While CMR has many imaging benefits for patients with DMD, it can also be challenging, especially in the pediatric population, due to the need for sedation, higher costs, and limited access to this type of specialized imaging .

Cardiac magnetic resonance imaging demonstrating dilated cardiomyopathy and fibrosis in a patient with DMD. Cardiac magnetic resonance delayed enhancement image in the basal short axis view performed on 1.5-T MRI. This image demonstrates near-transmural enhancement (gray) in the left ventricular basal-mid anterolateral, inferolateral, and lateral wall (area denoted by dashed white line) consistent with myocardial scar or fibrosis. This is in an 18-year-old male patient with Duchenne muscular dystrophy and exon 24 deletion with associated cardiomyopathy (LVEF 33 %)
Pharmacologic Treatment of Cardiomyopathy
Studies have shown the benefits of corticosteroid therapy and, as shown by echocardiography and cardiac MRI, support the notion that steroid treatment delays left ventricular dysfunction in DMD patients [59].
Duboc et al. evaluated the impact of angiotensin-converting enzyme (ACE) inhibitors , which have proven effective in asymptomatic adults with left ventricular dysfunction, and in patients with DMD with preserved left ventricular function [84]. The investigators randomized 57 children with DMD (mean age of 10.7 years) to the ACE inhibitor perindopril (2–4 mg/day) or placebo. At 3 years of follow-up, there was no significant difference in LV function between the children treated with perindopril or placebo. However, at the 3-year mark, all patients were switched to perindopril treatment and followed for an additional 2 years. After crossing over to perindopril at 2 years, there was no difference in mean LV function between those patients treated with perindopril initially versus those initially treated with placebo.
However, the initial placebo group had 8 of 29 patients with LVEF < 45 % and only 1 of 27 patients in the perindopril group (p = 0.02), which suggests that early treatment with perindopril was effective in preventing progression to left ventricular dysfunction in DMD. Subsequently, these patients were followed for 10 years, and in the initial placebo group, only 65 % of patients were alive versus 92.9 % in the initial perindopril group (p = 0.013), which emphasized that early use of an ACE inhibitor reduced mortality in patients with DMD [85].
Treatment guidelines recommend initiation of ACE inhibitors in patients with DMD only once LV dysfunction has developed [51], but based on the studies by Duboc et al., we recommend initiation of an ACE inhibitor before the development of left ventricular dysfunction in patients with DMD, as they are at high risk for developing left ventricular dysfunction (American College of Cardiology heart failure stages A and B) [84, 85]. Additionally, for those patients who are intolerant to ACE inhibitors, angiotensin receptor blockers (ARBs) can also be used. ARBs have been shown as effective as ACE inhibitors in DMD [86].
While the benefit of ACE inhibitors in DMD cardiomyopathy has been definitive, the efficacy of beta-blockers in DMD cardiomyopathy has been less clear. The use of carvedilol has been assessed in pediatric DMD patients with elevated atrial natriuretic peptide (ANP) or brain natriuretic peptide (BNP), and a low ejection fraction (EF < 40 %), as measured by echocardiography. No significant difference in carvedilol-treated patients was seen regarding symptoms of left ventricular dysfunction [87]. However, in a study by Rhodes et al., carvedilol was shown efficacious in patients with DMD cardiomyopathy [88].
When superimposed on background therapy of ACE inhibitors, the use of carvedilol in this patient population remains unclear. An analysis of 13 patients with DMD who were treated with ACE-I versus ACE-I and carvedilol revealed a beneficial effect of beta-blocker therapy in increasing left ventricular shortening and decreasing left ventricular end-diastolic dimensions, as shown by echocardiography [89]. In contrast, a recent study by Viollet et al. tested ACE inhibitor alone versus ACE inhibitor and metoprolol, but in this study, low-dose beta-blocker was added only for heart rates above 100 bpm or if arrhythmias occurred [90]. The results of this study showed an improvement from pretreatment LVEF in both groups, but no difference between the treatment groups. Further research with larger groups of patients and more robust trial designs are needed to definitively address the use of beta-blockers in DMD. Based on current ACC/American Heart Association/Heart Failure Society of America guidelines, we recommend that beta-blockers be initiated in DMD patients with left ventricular dysfunction [91].
Spironolactone , an aldosterone inhibitor, is a standard heart failure therapy and has also been demonstrated in the mouse model to improve cardiomyopathy [92]. In a recently completed randomized double-blinded clinic trial, eplerenone, an aldosterone receptor antagonist versus placebo, was added to the background therapy of ACE inhibitors or ARBs in DMD patients with normal LV function to assess the efficacy of eplerenone in preventing cardiomyopathy in DMD.
Twenty patients were randomized to eplerenone and 20 to placebo. They were followed for 6 and 12 months using cardiac MRI. The primary endpoint at 12 months was change in left ventricular circumferential strain, which is a marker of contractility. At 12 months, the decline in LV circumferential strain was lower in the group treated with eplerenone than the placebo group, although there was no overall change in left ventricular function in either group [93]. This study, while small, demonstrated attenuation of progressive left ventricular dysfunction that occurs in DMD. And while this study is positive, further studies to assess the impact of eplerenone in DMD survival are warranted.
Cardiac Transplantation and Left Ventricular Assist Devices
The gold standard for treating end-stage or advanced heart failure remains cardiac transplantation. When a patient has heart failure with multisystem organ involvement and is not expected to be able to rehabilitate after cardiac transplantation, a heart transplant is not advised. These contraindications have limited the applicability of heart transplants for patients with muscular dystrophy .
Rees et al. were the first to describe heart transplantation in patients with muscular dystrophies in a single German center [94]. Of the 582 transplants performed, three patients with DMD and one patient with BMD underwent cardiac transplantation, with a mean duration of follow-up of 40 months. They described that these patients tolerated immunosuppression, had no difference in postoperative intubation, and were able to be rehabilitated [94].
Ruiz-Cano et al. described a Spanish single-center experience with heart transplantation in three patients with BMD who underwent cardiac transplantation with a mean follow-up duration of 57 months. These investigators also demonstrated that BMD patients had an intraoperative and postoperative course comparable to nonmuscular dystrophy patients undergoing heart transplantation [95].
Patane et al. described a single case of successful transplantation in a patient with cardiomyopathy secondary to BMD [96]. We performed a more recent multicenter registry analysis of cardiac transplantation from the Cardiac Transplant Research Database and identified 29 patients with muscular dystrophies. Of those, 15 had BMD and three had DMD and underwent cardiac transplantation between 1995 and 2005. We compared them to 275 nonmuscular dystrophy, non-ischemic patients who were matched for age, body mass index, gender, and race [97] and found no significant difference in survival at 1 or 5 years, transplant rejection, infection, or allograft vasculopathy between the muscular dystrophy and nonmuscular dystrophy patients.
These studies described comparable outcomes of cardiac transplantation in a small and select group of patients with DMD and BMD with end-stage cardiomyopathy. However, the functional status of these patients prior to transplantation was not known, and these studies may have a selection bias. Further research regarding cardiac transplantation in patients with DMD and BMD with end-stage cardiomyopathy is needed.
Given the scarcity of organs for heart transplantation, left ventricular assist devices (LVADs) have been proven effective in treating patients with end-stage or advanced heart failure as well. These devices are also applicable to a larger population, including those with muscular dystrophies, as they can be used as destination therapy without the need for transplantation [98, 99].
Two groups recently reported cases of successful implantation of LVADs as destination therapy in DMD patients [100, 101]. Amedeo et al. were the first to describe LVAD implantation in two pediatric DMD patients [100]. These investigators implanted the Jarvik 2000 LVAD in a 15-year-old boy with DMD who had inotrope refractory heart failure and in a 14-year-old boy with DMD who was bridged from extracorporeal membrane oxygenation to a Jarvik 2000 LVAD. The first patient was discharged 3 months after LVAD implantation and the second patient 6 months after LVAD implantation.
Ryan et al. subsequently described the HeartMate II LVAD implantation in a 29-year-old male patient with DMD and end-stage heart failure and a HeartWare LVAD implantation in a 23-year-old female symptomatic DMD carrier with end-stage heart failure [101].
LVAD as destination therapy is a potentially promising therapy to address end-stage heart failure in patients with dystrophin-deficient heart failure. However, postoperative complications including respiratory failure, rehabilitation, bleeding, stroke, and arrhythmias need to be evaluated further in this population. Extensive preoperative and postoperative management in an experienced center would be necessary for LVAD implantation in the DMD population. Larger studies are needed to evaluate the efficacy and outcomes in this population.
Becker Muscular Dystrophy and Cardiomyopathy
In 1955, German physicians Becker and Kiener described an X-linked muscular dystrophy with a much milder clinical course than DMD, which is now known as Becker muscular dystrophy [102, 103]. In 1984, the gene responsible for BMD was located on the X chromosome [104] and, subsequently, that mutations in the DMD gene resulted in BMD [105]. BMD has an incidence of 1:19,000 and is an X-linked recessive disorder resulting from a mutation of the dystrophin gene [106]. Compared to patients with DMD, who have a complete absence of dystrophin, dystrophin mutations in BMD tend to be in-frame and result in misfolded or abnormal and less-functional protein [107].
Patients with BMD have an age of onset that is typically later than those with DMD [108] (◘ Table 12.1). While the muscular symptoms may be less severe, over 70 % of patients with BMD also develop cardiomyopathy [109, 110], and it is the leading cause of death in BMD patients [51].
Cardiomyopathy may be more severe in BMD patients than in DMD patients. This could be due to BMD patients being ambulatory much longer, placing greater stress on the heart [111–113]. The onset of cardiomyopathy is variable in BMD and is not correlated to skeletal muscle involvement [109, 114].
Cardiac magnetic resonance studies have shown a similar inferobasal fibrotic pattern in BMD and DMD patients [115]. Patients with BMD cardiomyopathy should receive standard medical heart failure therapy [91]. Given that BMD patients have a relatively milder skeletal muscle phenotype, several of these patients have received heart transplantation for severe cardiomyopathy with good outcomes (see Transplant/LVAD section) [94–97].
Carrier Status and Cardiomyopathy
While DMD and BMD affect males—given that they are inherited in an X-linked recessive manner—females can be carriers. The majority of DMD and BMD are inherited; thus, a significant number of females are carriers of DMD and BMD. Most female carriers of DMD and BMD are asymptomatic; however, female carriers can become symptomatic or become manifesting carriers. Manifesting carriers can have symptoms such as mild muscle weakness, elevated serum creatinine kinase, and, also, cardiomyopathy [116, 117]. Hoogerwaard et al. evaluated 90 women who were carriers of dystrophin mutations and identified 22 % with symptoms including muscle weakness and 18 % with evidence of a dilated left ventricle [118]. The age of onset for carriers is variable and the percent of symptomatic carriers has ranged from 2.5 to 22 % in prior studies [120].
X inactivation is the process by which one of the two X chromosomes in female cells randomly becomes transcriptionally inactive. It is postulated that carriers can become symptomatic based on the extent of random X inactivation of the normal X chromosome versus the dystrophic X chromosome. While DMD and BMD carriers have been identified to have cardiomyopathy, the prevalence and severity of disease are incompletely defined [118, 121–123]. We recommend that all female dystrophinopathy carriers be screened for cardiomyopathy .
Models of Muscular Dystrophy
Animal models of DMD have played an important role in elucidating mechanisms of dystrophin deficiency. The dystrophin-deficient mdx mouse, dystrophin/utrophin double knockout mouse (U-dko), and the golden retriever muscular dystrophy (GRMD) models are the most studied models regarding cardiac phenotype (◘ Fig. 12.8).

Animal models for muscular dystrophy . (a) Mdx and utrophin/dystrophin double knockout mice demonstrating that age- and sex-matched U-dko mice are smaller than mdx mice. (b) Golden retriever muscular dystrophy dog model at 3 months (top photo) and at 6 months (bottom photo) demonstrating severe muscle loss with contractures (adapted from [124, 125])
The mdx mice arise from a naturally occurring nonsense point mutation in exon 23 of the DMD gene. This mutation results in the formation of a stop codon and the absence of full-length dystrophin [126, 127]. The skeletal muscle phenotype in these animals has been well characterized and is relatively mild. Young mice do not typically have any cardiac involvement, but after 10 months of age, they begin to develop signs of cardiomyopathy, including poor contractility, myocardial necrosis, and fibrosis, as well as echocardiographic and electrocardiographic changes [128, 129].
While a significant cardiomyopathy is not seen in younger mdx mice under normal conditions, various forms of stress, such as beta-adrenergic stress or pressure overload, demonstrate a cardiomyopathic phenotype [130–132]. Thus, the mdx mouse shares a similar genotype and some features of cardiomyopathy that are seen in patients with DMD. The mdx mice, however, do not develop cardiomyopathy until relatively late in comparison to patients, although the mdx mice have a mildly reduced lifespan compared to normal control mice [133].
In a recent study, Long et al. used the emerging technology of genome editing to genetically correct the dystrophin mutation in the mdx mouse [134]. The genome-edited mdx mice had between 2 and 100 % correction of the gene with this strategy. Mosaic mdx mice, with only 17 % correction of dystrophin by gene editing, had a nearly normal muscle phenotype [134].
Mdx/utrophin double knockout mouse model : Utrophin is an autosomal paralogue of dystrophin, which is upregulated in the mdx mouse [135]. The upregulation of utrophin in the mdx mouse likely compensates for the loss of dystrophin, thus producing a less severe phenotype. The utrophin and dystrophin double knockout mouse was generated to evaluate the impact of utrophin upregulation. Compared to the mdx mouse, the double knockout mouse model has a much more severe phenotype [136, 137]. Double knockout mice are smaller, with progressive muscle atrophy, and die prematurely at about 10 weeks of age. The double knockout mouse also develops a severe cardiomyopathy by 8 weeks, which is comparable to that of an aged mdx mouse [136]. The double knockout mouse may more closely represent human DMD.
Canine models: The canine model of dystrophin deficiency is a large animal model of DMD that more closely resembles human disease. The golden retriever muscular dystrophy (GRMD) dog model is one of the best-characterized dog models of DMD [138]. In 1988, Valentine et al. described a family of golden retrievers with male members affected by a severe neurodegenerative disorder with muscle weakness and gait abnormalities, and absence of dystrophin on muscle biopsy, which paralleled human DMD [138]. Subsequently, the mutation was identified to be a single mutation in the consensus splice site of intron 6, which leads to an out-of-frame mutation and absence of dystrophin during RNA processing [139].
GRMD dogs develop significant muscle weakness by 2 months of age with progressive weakness and also have a reduced lifespan [140]. GRMD dogs also develop a severe cardiomyopathy [141]. GRMD is a useful model to better understand DMD cardiomyopathy and test new therapies; however, the larger size of the GRMD dogs can increase maintenance costs and restrict their use [142]. GRMD colonies operate in the United States at the University of North Carolina at Chapel Hill and the Fred Hutchinson Cancer Center in Seattle. Other colonies exist in Brazil, France, Japan, and the Netherlands.
Human cell-based models : Animal models have been useful to evaluate the pathophysiology of DMD; however, they have limitations and do not fully recapitulate the disease phenotype observed in human patients. Obtaining human tissues, especially cardiomyocytes, is challenging and poses a risk to the patient. The advent of human inducible pluripotent stem cells (hiPSCs) in 2007 has been a major scientific development [143, 144].
hiPSCs represent a novel tool to study human genetic diseases though cellular reprogramming of patient-specific cells that retain the genetic composition and are pluripotent. hiPSCs have been used to study various cardiac diseases including hypertrophic cardiomyopathy and long QT syndrome [145, 146]. hiPSC disease modeling is a promising method to study DMD cardiomyopathy. Several hiPSC lines from patients with various mutations have been derived [147, 148]. We and other groups have successfully differentiated the DMD patient-specific hiPSC to cardiomyocytes. Modeling DMD cardiomyopathy using hiPSC is a promising method to elucidate the pathophysiology of DMD cardiomyopathy and test novel therapies [149].
Emerging Therapies
A major challenge for treating DMD is finding therapies that address dystrophin deficiency and that target both the cardiac and skeletal muscle. A number of promising emerging therapies include exon skipping, dystrophin mini-gene replacement, sealants, and gene-editing strategies.
Exon Skipping
DMD mutations are heterogeneous, but the majority of DMD arises from an out-of-frame deletion or duplication of one or several of the 79 exons within the DMD gene. These mutations affect the normal transcription of dystrophin mRNA open reading frame and, subsequently, prevent the full-length dystrophin protein from being transcribed. An exciting and novel therapy for the treatment of DMD involves exon skipping within the central rod domain. This can restore the normal mRNA reading frame and convert an out-of-frame mutation to a less severe, in-frame mutation, comparable to those seen in Becker muscular dystrophy. The exon-skipping process uses antisense oligonucleotides (AONs), which are short, synthetic fragments of nucleic acids.
The AONs bind RNA sequences that regulate RNA splicing, which enable a smaller but functional mRNA to be produced that lacks the exon mutation (◘ Fig. 12.9). This strategy may be effective in 80 % of DMD mutations [150]. AONs for exon skipping target exon 51 of the DMD gene, which accounts for ~13 % of DMD mutations, including drisapersen (GSK-2402698, GlaxoSmithKline and Prosensa) and eteplirsen (AVI 4658, Sarepta Therapeutics), which are currently in clinical trials. These AONs have been tested in the mdx mouse and canine models. Intramuscular injections were found to be safe and restored dystrophin expression without significant side effects [151–155].

Schematic of exon skipping . Dystrophin exons are spliced to form mature messenger RNA (mRNA), which results in the translation of a full-length, functional dystrophin protein. However, in classic mutations, such as in exon 52, this results in skipping of exon 52 during splicing. This leads to a disrupted reading frame that produces a truncated, nonfunctional dystrophin protein. Introduction of antisense oligonucleotide that skips exon 51 restores the reading frame and produces a truncated but functional dystrophin protein
The initial phase I and II trials for both eteplirsen and drisapersen were promising in restoring dystrophin expression, but did not demonstrate a significant change in the primary endpoint of the 6-min walk test. Major side effects included renal toxicity with drisapersen and thrombocytopenia at high doses [156, 157]. These drugs are in ongoing phase III clinical trials .
Dystrophin Mini-gene Replacement
Given that dystrophin mutations lead to the complete absence of the functional dystrophin protein in DMD, one strategy to mitigate the disease sequelae is gene replacement therapy using recombinant adenoviral virus vectors (rAAVs ) . Compared to the exon-skipping strategy, currently available only for exon 51 skipping, gene replacement strategies can be beneficial for all patients with DMD regardless of their DMD mutation.
One limitation of the rAAV technology is the inability to deliver the large full-length dystrophin gene due to the limitations of packaging. The 2.4-Mb dystrophin gene is the largest single gene in the human genome, and as a result, cloning the 14-kb cDNA is challenging to introduce into delivery vectors [10, 158]. However, observing mildly affected BMD patients with very large dystrophin gene deletions led to the understanding that truncated dystrophins could be functional [105, 159]. This, in turn, led to the development of micro- or mini-dystrophins, which contain only the essential regions of the gene. They are functional and can be packaged within the rAAV [160] (◘ Fig. 12.10).

Full-length dystrophin, mini- and micro-dystrophin constructs. Full-length dystrophin has a 14-kb cDNA, which results in a 427-kDa protein. Full-length dystrophin has an N-terminal actin-binding domain, a rod domain with 24 spectrin-like repeats and four hinge domains, and a C-terminal region that has dystroglycan, syntrophin, and dystrobrevin-binding domains. Examples of a mini- and a micro-dystrophin with rod domain deletions that are able to be cloned into recombinant adeno-associated viral vectors
rAAV6 vectors containing microdystrophin have been administered to dystrophin-deficient mice and demonstrated restoration of dystrophin, improved muscular function, and increased lifespan [161–164]. Dystrophin gene replacement therapy has been effective in mouse models, but in large animal models such as those using dogs and nonhuman primates, gene therapy efficacy has been limited due to the host immune response [165–170].
In a human preclinical trial of rAAV-mediated mini-dystrophin gene transfer, six DMD patients received intramuscular injections in the biceps brachii muscle. None of the patients demonstrated significant levels of dystrophin and one patient had a microdystrophin T-cell response within 15 days of injection [168, 169]. In addition to the T-cell-mediated response, humoral or antibody-mediated immune responses are also observed in large animal models of rAAV dystrophin delivery [171]. Dystrophic muscles have two features that promote the induction of an immune response, including cellular contents exposed to the environment due to muscle necrosis along with elevated immune effector cells [172–174].
Regarding cardiac gene transfer, delivery has been achieved through transendocardial injection of rAAV vectors in a nonhuman primate model where rAAV6 was more effective than rAAV 8 or 9 [175]. rAAV8 delivery via the internal jugular vein also demonstrated target gene expression after 2.5 months of infusion [176]. Although limited by immune side effects that may be overcome by immunosuppressant regimens, gene replacement therapy remains a promising therapy for DMD, including the associated cardiomyopathy .
Sealants
Dystrophin deficiency can lead to increased membrane fragility and is exacerbated by any mechanical stress. Yasuda et al. demonstrated that isolated dystrophin-deficient cardiomyocytes, when stretched, have reduced compliance and increased susceptibility to calcium overload [130]. Thus, increased membrane stability is a potential pathway to modify the disease sequelae related to membrane fragility seen in DMD patients.
Poloxamer 188 (P-188) is a triblock copolymer that is able to insert into damaged lipid bilayers and repair membranes. It has been used to stabilize red blood cell membranes in sickle cell disease [177]. This concept of membrane stabilization repair was applied to the mdx mouse receiving adrenergic stimulation with either dobutamine or isoproterenol as a stressor, and administration of P-188, which demonstrated improved left ventricular function and increased survival [130, 178].
The use of P-188 as a cardioprotective agent was also tested in the more severely affected golden retriever muscular dystrophy model where the dogs received an 8-week infusion of P-188. This infusion resulted in decreased fibrosis and prevention of left ventricular dilatation [142].
While P-188 has been shown to be cardioprotective in animal models, the daily doses may be a limitation. Furthermore, the sealant (P-188) does not address the absence of dystrophin. Additionally, the impact on the skeletal muscle has not been well described.
In one recent study, mdx mice given either a one-time or daily P-188 injection had a decline in skeletal muscle force generation [179]. While cardiac benefits in animal models have been demonstrated, further testing of P-188 on both the skeletal and cardiac function is needed.
Gene Editing
A new promising technology for DMD is gene editing . Gene editing can be mediated with CRISPR (clustered regularly interspaced short palindromic repeat)/CRISPR-associated systems (Cas) to alter the genome [180–182]. The CRISPR/Cas9 system binds to the target gene and generates a double-strand DNA break. It can then be replaced by the corrected gene sequence (◘ Fig. 12.11). This system precisely removes the mutated gene of interest and replaces it with a functional copy of the gene.

A schematic of CRISPR/Cas9-mediated genome editing. Guide RNA is fused with the DNA sequence targeting the host gene of interest. The guide RNA recognizes specific regions on the host RNA and complexes with Cas9, which recognizes the PAM sequence on the target and exerts its endonuclease function to cause double-stranded breaks in the DNA. This triggers repair mechanisms, which enable the donor DNA to be inserted at the break site, and results in targeted genome editing
Correction of DMD using gene editing targeted to exon 51 of dystrophin has been used to restore the dystrophin gene reading frame in DMD patient myoblasts [183]. The restored dystrophin reading frame enabled restoration of functional dystrophin expression in the DMD patient myoblasts [183]. In a recent study, gene editing using a CRISPR/Cas9 strategy was used in vivo to correct the DMD gene mutation in the germ line of mdx mice [134]. In the genome-edited animals, between 2 and 100 % of DMD gene correction was observed. In mosaic mdx mice with only 17 % correction of dystrophin by gene editing, a normal muscle phenotype was observed. This nascent technology has possible therapeutic benefits in patients with DMD. Further animal studies evaluating the cardiac function are needed .
Cell-based therapy is also another potential way to treat dystrophin deficiency. Therapeutic cells can be obtained from a donor bearing normal dystrophin or from the DMD patient, corrected ex vivo, reimplanted, and injected into the DMD patient. Skeletal muscle stem cells are known as satellite cells. They have been shown to respond to muscle injury by proliferating and restoring the muscle architecture [184]. Satellite cells are also easy to isolate and culture in vitro. Thus normal myoblast injections were first tested in mdx mice showing dystrophin-positive muscle cells [185, 186].
However, subsequent human clinical trials with myoblast transplantation have demonstrated only up to 10 % dystrophin-positive fibers [187–189]. Given the challenges with myoblast engraftment, survival, and migration, other cell types that have myogenic potential—including bone marrow-derived stem cells and CD133+ muscle stem cells—have been tested in DMD patients [190, 191]. However, further testing is required for these therapies. Vessel-derived stem cells have been used to treat mdx double knockout mice to prevent cardiomyopathy, but no human stem cell trials have been conducted to assess the efficacy of any stem cell therapies in DMD cardiomyopathy [192].
Myotonic Dystrophy
Myotonic dystrophies are among the most common inherited, adult-onset, muscular dystrophies that have multisystem impact. Myotonic dystrophies are characterized by myotonia or a delay in relaxation of muscle contraction. Myotonic dystrophy leads to skeletal muscle weakness and muscle wasting and, in end-stage disease, respiratory muscle weakness. Patients also have endocrine abnormalities that commonly include hypogonadism and insulin resistance. The gastrointestinal system is also impacted and dysphagia, reflux, and hypermotility are often noted. Central nervous system and ocular manifestations include cataracts, cognitive impairment, and hypersomnolence. Cardiac consequences of myotonic dystrophy are also prominent, and predominantly involve conduction abnormalities and arrhythmias, although cardiomyopathy is being increasingly identified [193].
In 1909, German physician Hans Steinert and English physicians Frederick Batten and H. P. Gibb described for the first time a muscular dystrophy that was characterized by myotonia, or involuntary muscle contraction and delayed relaxation, and muscle weakness [194, 195]. This disorder was initially called Steinert disease and, later, myotonic dystrophy.
In 1911, Griffith described a patient with myotonic dystrophy who had bradycardia with a heart rate of 30 bpm, and subsequently, other early clinical reports of cardiovascular abnormalities in muscular dystrophy were reported [196, 197]. Nearly 80 years later, the gene responsible for myotonic dystrophy was identified [198]. However, in 1994, a group of patients who had clinical features similar to what is now classified as myotonic dystrophy type 1, but without the gene mutation, were described [199, 200]. This was later classified as myotonic dystrophy type 2. The gene responsible for the pathogenesis was identified in 2001 [201]. The two genetically defined subtypes of myotonic dystrophies are myotonic dystrophy type 1 and myotonic dystrophy type 2 (◘ Table 12.2).
Myotonic Dystrophy Type 1
Myotonic dystrophy type 1 is also known as Steinert disease and has an incidence of 1:8000 [202]. Myotonic dystrophy type 1 is the most common adult form of muscular dystrophy. It is further subdivided into three subtypes based on age of onset and severity.
Congenital DM1 typically occurs before the first year of life and is the most severe. It occurs primarily due to maternal transmission. Classical myotonic dystrophy type 1 is divided into childhood or adult onset. Adult-onset disease typically manifests in the second through fourth decades [203]. Myotonic dystrophy type 1 is caused by autosomal dominant inheritance of an abnormal expansion of a cytosine-thymine-guanine (CTG) trinucleotide repeat in the DMPK gene in chromosome 19 encoding for the protein dystrophia myotonica protein kinase [204]. The DM protein kinase is expressed in skeletal, cardiac, and smooth muscle cells. Normally, less than 37 CTG repeats exist, but in patients with myotonic dystrophy type 1, more than 50 to thousands of repeats may exist.
A direct correlation exists between an increasing number of CTG repeats and the age of onset and severity of neuromuscular and cardiac disease in myotonic dystrophy type 1. CTG repeats typically expand as it is passed from parents to offspring, resulting in a more severe disease phenotype in a process known as genetic anticipation .
Myotonic Dystrophy Type 2
Myotonic dystrophy type 2 is less common, with an incidence of 13:100,000 people. It has a similar autosomal inheritance pattern as myotonic dystrophy type 1, but type 2 is a result of a tetranucleotide CCGT repeat in the gene ZNF9 gene on chromosome 3 coding for zinc finger protein 9 [205]. The CCGT repeats in myotonic dystrophy type 2 are typically in the thousands. Myotonic dystrophy type 2 is typically adult onset and is less severe than myotonic dystrophy type 1.
Pathogenesis
In both myotonic dystrophies, the underlying molecular mechanism is similar. The gene including the abnormal repeat is transcribed into RNA, but not translated. The abnormal RNA accumulates in the nucleus and disrupts splicing of pre-messenger RNA into mature RNA through affecting RNA-binding proteins [206]. This process subsequently results in abnormal function of different genes including chloride channels needed for muscle function, cardiac troponin, and insulin receptors [207].
Myotonic dystrophy type 1 typically presents with muscle weakness and myotonia of the distal muscles. Patients may have a characteristic long face, male pattern baldness, and atrophy of the facial and jaw muscles. Myotonic dystrophy type 2 also manifests with myotonia and weakness, as well as the other systemic sequelae but at a later age and with less severity .
Cardiac involvement in these patients involves fibrosis and degeneration—typically affecting the conduction system as well as the myocardial tissue—leading to arrhythmias and cardiomyopathy. Conduction abnormalities occur in 75–80 % of myotonic dystrophy type 1 patients with more than 65 % of patients having abnormal ECGs [208]. Atrial and ventricular arrhythmias are also common and affect 25 % of patients with myotonic dystrophy. The conduction abnormalities and arrhythmias can cause sudden cardiac death, which accounts for more than one-third of deaths in these patients.
In a study of myotonic dystrophy type 1 patients, a correlation between CTG length and presence of arrhythmias and conduction abnormalities was noted [209]. Patients with myotonic dystrophy are also more likely to develop cardiomyopathies, with 20 % of patients with myotonic dystrophy type 1 demonstrating left ventricular dilatation and 14 % of patients with left ventricular dysfunction [210, 211]. In patients who have left ventricular dysfunction, medical therapy including ACE inhibitors and beta-blockers should be used. These therapies are standard for systolic dysfunction, and while they have not been tested in myotonic patients, the reverse left ventricular remodeling and survival benefit have been extrapolated to this population as well.
Mexiletine, a class Ib antiarrhythmic agent and a sodium channel blocker, is being used more commonly in myotonic patients for treatment of myotonia without increasing QRS duration or changing cardiac conduction [212]. AV nodal blocking agents for the treatment of arrhythmias should be used cautiously, given the bradyarrhythmias and conduction blocks seen in these patients.
Pacemaker and implantable cardioverter-defibrillator (ICD) therapy guidelines have specifically included the myotonic dystrophy population. Permanent pacemaker implantation is a class IIa indication in myotonic patients with an HV interval >100 ms and class IIb indication for any degrees of atrioventricular (AV) block regardless of symptoms [213]. ICDs are indicated in myotonic patients with ventricular tachycardia, and a wider indication may exist in this patient population [214].
Cardiac Involvement in Other Muscular Dystrophies
While patients with DMD, BMD, and myotonic dystrophy type 1 and type 2 have a known high incidence of cardiovascular involvement, many other muscular dystrophies including certain limb-girdle muscular dystrophies have known cardiac involvement. Facioscapulohumeral muscular dystrophies and other similar muscular dystrophies have unknown cardiac involvement. ◘ Table 12.3 summarizes the major inherited muscular dystrophies and cardiac involvement. Cardiac screening in patients with inherited muscular dystrophies is warranted, as many muscular dystrophies do have a high incidence of cardiac involvement, and in others, cardiac involvement is less well known.
References
Meryon E. On granular and fatty degeneration of the voluntary muscles. Med Chir Trans. 1852;35:73–4.
Duchenne G. Recherches sur la paralysie musculaire pseudo-hypertrophique ou paralysie myo-sclerosique. Arch Gen Med. 1868;11:5–25.
Gowers WR. A manual of diseases of the nervous system, vol. 1. London: J&A Churchill; 1886. p. 391–4.
Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–50.
Hoffman EP, Brown Jr RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–28.
Emery AE. The muscular dystrophies. Lancet. 2002;359(9307):687–95.
Cox GF, Kunkel LM. Dystrophies and heart disease. Curr Opin Cardiol. 1997;12(3):329–43.
Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR. Gene therapy for duchenne muscular dystrophy: expectations and challenges. Arch Neurol. 2007;64(9):1236–41.
Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J, Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R, Weiss RB. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–13.
Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17.
Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3(4):283–91.
Nudel U, Zuk D, Einat P, Zeelon E, Levy Z, Neuman S, Yaffe D. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature. 1989;337(6202):76–8.
Gorecki DC, Monaco AP, Derry JM, Walker AP, Barnard EA, Barnard PJ. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet. 1992;1:505–10.
D’Souza VN, Nguyen TM, Morris GE, Karges W, Pillers DA, Ray PN. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum Mol Genet. 1995;4(5):837–42.
Lidov HG, Selig S, Kunkel LM. Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet. 1995;4(3):329–35.
Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature. 1990;348(6303):725–8.
Austin RC, Howard PL, D’Souza VN, Klamut HJ, Ray PN. Cloning and characterization of alternatively spliced isoforms of Dp71. Hum Mol Genet. 1995;4(9):1475–83.
Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, Hart KJ, Tuohy T, White S, den Dunnen JT, Weiss RB, Flanigan KM. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005;134(3):295–8.
Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150(5):1209–14.
Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122(4):809–23.
Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66(6):1121–31.
Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338(6212):259–62.
Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345(6273):315–9.
Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23(10):1456–71.
Burton EA, Davies KE. Muscular dystrophy--reason for optimism? Cell. 2002;108(1):5–8.
O’Brien KF, Kunkel LM. Dystrophin and muscular dystrophy: past, present, and future. Mol Genet Metab. 2001;74(1-2):75–88.
Klietsch R, Ervasti JM, Arnold W, Campbell KP, Jorgensen AO. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ Res. 1993;72(2):349–60.
Johnson EK, Zhang L, Adams ME, Phillips A, Freitas MA, Froehner SC, Green-Church KB, Montanaro F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS One. 2012;7(8):e43515.
Cox GA, Cole NM, Matsumura K, Phelps SF, Hauschka SD, Campbell KP, Faulkner JA, Chamberlain JS. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993;364(6439):725–9.
Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90(8):3710–4.
Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Mol Cell Biochem. 1998;179(1-2):111–23.
Konagaya M, Takayanagi T. Regularity in the change of serum creatine kinase level in Duchenne muscular dystrophy. A study with long-term follow-up cases. Jpn J Med. 1986;25(1):2–8.
Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12(10):926–9.
Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26(3):271–7.
Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve. 2007;36(4):424–35.
Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, King W, Signore L, Pandya S, Florence J, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320(24):1592–7.
Griggs RC, Moxley 3rd RT, Mendell JR, Fenichel GM, Brooke MH, Pestronk A, Miller JP. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch Neurol. 1991;48(4):383–8.
Griggs RC, Moxley 3rd RT, Mendell JR, Fenichel GM, Brooke MH, Pestronk A, Miller JP, Cwik VA, Pandya S, Robison J, et al. Duchenne dystrophy: randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology. 1993;43(3 Pt 1):520–7.
Backman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1995;5(3):233–41.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93.
Elia M, Carter A, Bacon S, Winearls CG, Smith R. Clinical usefulness of urinary 3-methylhistidine excretion in indicating muscle protein breakdown. Br Med J (Clin Res Ed). 1981;282(6261):351–4.
Rifai Z, Welle S, Moxley 3rd RT, Lorenson M, Griggs RC. Effect of prednisone on protein metabolism in Duchenne dystrophy. Am J Physiol. 1995;268(1 Pt 1):E67–74.
Jacobs SC, Bootsma AL, Willems PW, Bar PR, Wokke JH. Prednisone can protect against exercise-induced muscle damage. J Neurol. 1996;243(5):410–6.
Anderson JE, Weber M, Vargas C. Deflazacort increases laminin expression and myogenic repair, and induces early persistent functional gain in mdx mouse muscular dystrophy. Cell Transplant. 2000;9(4):551–64.
Kissel JT, Burrow KL, Rammohan KW, Mendell JR. Mononuclear cell analysis of muscle biopsies in prednisone-treated and untreated Duchenne muscular dystrophy. CIDD Study Group. Neurology. 1991;41(5):667–72.
Burrow KL, Coovert DD, Klein CJ, Bulman DE, Kissel JT, Rammohan KW, Burghes AH, Mendell JR. Dystrophin expression and somatic reversion in prednisone-treated and untreated Duchenne dystrophy. CIDD Study Group. Neurology. 1991;41(5):661–6.
Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;(1):CD003725.
Bonifati MD, Ruzza G, Bonometto P, Berardinelli A, Gorni K, Orcesi S, Lanzi G, Angelini C. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23(9):1344–7.
Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, Khairy P. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013;61(9):948–54.
Kurz LT, Mubarak SJ, Schultz P, Park SM, Leach J. Correlation of scoliosis and pulmonary function in Duchenne muscular dystrophy. J Pediatr Orthop. 1983;3(3):347–53.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C, DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9(2):177–89.
Do T. Orthopedic management of the muscular dystrophies. Curr Opin Pediatr. 2002;14(1):50–3.
Gozal D. Pulmonary manifestations of neuromuscular disease with special reference to Duchenne muscular dystrophy and spinal muscular atrophy. Pediatr Pulmonol. 2000;29(2):141–50.
Finder JD, Birnkrant D, Carl J, Farber HJ, Gozal D, Iannaccone ST, Kovesi T, Kravitz RM, Panitch H, Schramm C, Schroth M, Sharma G, Sievers L, Silvestri JM, Sterni L, American Thoracic Society. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. 2004;170(4):456–65.
Barbe F, Quera-Salva MA, McCann C, Gajdos P, Raphael JC, de Lattre J, Agusti AG. Sleep-related respiratory disturbances in patients with Duchenne muscular dystrophy. Eur Respir J. 1994;7(8):1403–8.
Smith PE, Edwards RH, Calverley PM. Ventilation and breathing pattern during sleep in Duchenne muscular dystrophy. Chest. 1989;96(6):1346–51.
Eagle M, Bourke J, Bullock R, Gibson M, Mehta J, Giddings D, Straub V, Bushby K. Managing Duchenne muscular dystrophy--the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord. 2007;17(6):470–5.
American Academy of Pediatrics Section on Cardiology and Cardiac Surgery. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy [comment]. Pediatrics. 2005;116(6):1569–73.
Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD, Cripe LH. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol. 2005;26(6):768–71.
Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112(18):2799–804.
Romfh A, McNally EM. Cardiac assessment in duchenne and becker muscular dystrophies. Curr Heart Fail Rep. 2010;7(4):212–8.
Thrush PT, Allen HD, Viollet L, Mendell JR. Re-examination of the electrocardiogram in boys with Duchenne muscular dystrophy and correlation with its dilated cardiomyopathy. Am J Cardiol. 2009;103(2):262–5.
Perloff JK, Roberts WC, de Leon Jr AC, O’Doherty D. The distinctive electrocardiogram of Duchenne’s progressive muscular dystrophy. An electrocardiographic-pathologic correlative study. Am J Med. 1967;42(2):179–88.
Sanyal SK, Johnson WW, Thapar MK, Pitner SE. An ultrastructural basis for electrocardiographic alterations associated with Duchenne’s progressive muscular dystrophy. Circulation. 1978;57(6):1122–9.
Frankel KA, Rosser RJ. The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Hum Pathol. 1976;7(4):375–86.
Markham LW, Michelfelder EC, Border WL, Khoury PR, Spicer RL, Wong BL, Benson DW, Cripe LH. Abnormalities of diastolic function precede dilated cardiomyopathy associated with Duchenne muscular dystrophy. J Am Soc Echocardiogr. 2006;19(7):865–71.
Boas EP, Lowenburg H. The heart rate in progressive muscular dystrophy. Arch Int Med. 1931;47(3):376–83.
Fitch CW, Ainger LE. The Frank vector cardiogram and the electrocardiogram in Duchenne progressive muscular dystrophy. Circulation. 1967;35(6):1124–40.
Kovick RB, Fogelman AM, Abbasi AD, Peter JB, Pearce ML. Echocardiographic evaluation of posterior left ventricular wall motion in muscular dystrophy. Circulation. 1975;52(3):447–54.
Nomura H, Hizawa K. Histopathological study of the conduction system of the heart in Duchenne progressive muscular dystrophy. Acta Pathol Jpn. 1982;32(6):1027–33.
Yotsukura M, Sasaki K, Kachi E, Sasaki A, Ishihara T, Ishikawa K. Circadian rhythm and variability of heart rate in Duchenne-type progressive muscular dystrophy. Am J Cardiol. 1995;76(12):947–51.
Yotsukura M, Fujii K, Katayama A, Tomono Y, Ando H, Sakata K, Ishihara T, Ishikawa K. Nine-year follow-up study of heart rate variability in patients with Duchenne-type progressive muscular dystrophy. Am Heart J. 1998;136(2):289–96.
Thomas TO, Morgan TM, Burnette WB, Markham LW. Correlation of heart rate and cardiac dysfunction in Duchenne muscular dystrophy. Pediatr Cardiol. 2012;33(7):1175–9.
Dalmaz Y, Peyrin L, Mamelle JC, Tuil D, Gilly R, Cier JF. The pattern of urinary catecholamines and their metabolites in Duchenne myopathy, in relation to disease evolution. J Neural Transm. 1979;46(1):17–34.
Corrado G, Lissoni A, Beretta S, Terenghi L, Tadeo G, Foglia-Manzillo G, Tagliagambe LM, Spata M, Santarone M. Prognostic value of electrocardiograms, ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. Am J Cardiol. 2002;89(7):838–41.
Rapezzi C, Leone O, Biagini E, Coccolo F. Echocardiographic clues to diagnosis of dystrophin related dilated cardiomyopathy. Heart. 2007;93(1):10.
Markham LW, Kinnett K, Wong BL, Woodrow Benson D, Cripe LH. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord. 2008;18(5):365–70.
Silva MC, Meira ZM, Gurgel Giannetti J, da Silva MM, Campos AF, Barbosa Mde M, Starling Filho GM, Ferreira Rde A, Zatz M, Rochitte CE. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J Am Coll Cardiol. 2007;49(18):1874–9.
Hagenbuch SC, Gottliebson WM, Wansapura J, Mazur W, Fleck R, Benson DW, Hor KN. Detection of progressive cardiac dysfunction by serial evaluation of circumferential strain in patients with Duchenne muscular dystrophy. Am J Cardiol. 2010;105(10):1451–5.
Hor KN, Wansapura J, Markham LW, Mazur W, Cripe LH, Fleck R, Benson DW, Gottliebson WM. Circumferential strain analysis identifies strata of cardiomyopathy in Duchenne muscular dystrophy: a cardiac magnetic resonance tagging study. J Am Coll Cardiol. 2009;53(14):1204–10.
Puchalski MD, Williams RV, Askovich B, Sower CT, Hor KH, Su JT, Pack N, Dibella E, Gottliebson WM. Late gadolinium enhancement: precursor to cardiomyopathy in Duchenne muscular dystrophy? Int J Cardiovasc Imaging. 2009;25(1):57–63.
Verhaert D, Richards K, Rafael-Fortney JA, Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging. 2011;4(1):67–76.
Hor KN, Taylor MD, Al-Khalidi HR, Cripe LH, Raman SV, Jefferies JL, O’Donnell R, Benson DW, Mazur W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: effect of age and left ventricular systolic function. J Cardiovasc Magn Reson. 2013;15:107.
Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45(6):855–7.
Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, Berard C, Vaksmann G, Weber S, Becane HM. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154(3):596–602.
Allen HD, Flanigan KM, Thrush PT, Dvorchik I, Yin H, Canter C, Connolly AM, Parrish M, McDonald CM, Braunlin E, Colan SD, Day J, Darras B, Mendell JR. A randomized, double-blind trial of lisinopril and losartan for the treatment of cardiomyopathy in duchenne muscular dystrophy. PLoS Curr. 2013;5.
Saito T, Matsumura T, Miyai I, Nozaki S, Shinno S. Carvedilol effectiveness for left ventricular-insufficient patients with Duchenne muscular dystrophy. Rinsho Shinkeigaku. 2001;41(10):691–4.
Rhodes J, Margossian R, Darras BT, Colan SD, Jenkins KJ, Geva T, Powell AJ. Safety and efficacy of carvedilol therapy for patients with dilated cardiomyopathy secondary to muscular dystrophy. Pediatr Cardiol. 2008;29(2):343–51.
Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, Osawa M, Nakanishi T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70(8):991–4.
Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol. 2012;110:98–102.
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith Jr SC, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B, American College of Cardiology; American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112(12):e154–235.
Rafael-Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfín DA, Janssen PM, Raman SV. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation. 2011;124(5):582–8.
Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael-Fortney JA, Lowe J, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(2):153–61.
Rees W, Schuler S, Hummel M, Hetzer R. Heart transplantation in patients with muscular dystrophy associated with end-stage cardiomyopathy. J Heart Lung Transplant. 1993;12(5):804–7.
Ruiz-Cano MJ, Delgado JF, Jimenez C, Jimenez S, Cea-Calvo L, Sanchez V, Escribano P, Gomez MA, Gil-Fraguas L, Saenz de la Calzada C. Successful heart transplantation in patients with inherited myopathies associated with end-stage cardiomyopathy. Transplant Proc. 2003;35(4):1513–5.
Patane F, Zingarelli E, Attisani M, Sansone F. Successful heart transplantation in Becker’s muscular dystrophy. Eur J Cardiothorac Surg. 2006;29(2):250.
Wu RS, Gupta S, Brown RN, Yancy CW, Wald JW, Kaiser P, Kirklin NM, Patel PC, Markham DW, Drazner MH, Garry DJ, Mammen PP. Clinical outcomes after cardiac transplantation in muscular dystrophy patients. J Heart Lung Transplant. 2010;29(4):432–8.
Rose EA, Gelijns AC, Moskowitz AJ, Heitjan DF, Stevenson LW, Dembitsky W, Long JW, Ascheim DD, Tierney AR, Levitan RG, Watson JT, Meier P, Ronan NS, Shapiro PA, Lazar RM, Miller LW, Gupta L, Frazier OH, Desvigne-Nickens P, Oz MC, Poirier VL, Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med. 2001;345(20):1435–43.
Slaughter MS, Rogers JG, Milano CA, Russell SD, Conte JV, Feldman D, Sun B, Tatooles AJ, Delgado 3rd RM, Long JW, Wozniak TC, Ghumman W, Farrar DJ, Frazier OH, HeartMate II Investigators. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med. 2009;361(23):2241–51.
Amodeo A, Adorisio R. Left ventricular assist device in Duchenne cardiomyopathy: can we change the natural history of cardiac disease? Int J Cardiol. 2012;161(3):e43.
Ryan TD, Jefferies JL, Sawnani H, Wong BL, Gardner A, Del Corral M, Lorts A, Morales DL. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with duchenne muscular dystrophy: lessons learned from the first applications. ASAIO J. 2014;60(2):246–8.
Becker PE, Kiener F. A new x-chromosomal muscular dystrophy. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1955;193(4):427–48.
Becker PE. Two families of benign sex-linked recessive muscular dystrophy. Rev Can Biol. 1962;21:551–66.
Kingston HM, Sarfarazi M, Thomas NS, Harper PS. Localisation of the Becker muscular dystrophy gene on the short arm of the X chromosome by linkage to cloned DNA sequences. Hum Genet. 1984;67(1):6–17.
Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, Meng G, Muller CR, Lindlof M, Kaariainen H, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45(4):498–506.
Worton R. Muscular dystrophies: diseases of the dystrophin-glycoprotein complex. Science. 1995;270(5237):755–6.
Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–5.
Bradley WG, Jones MZ, Mussini JM, Fawcett PR. Becker-type muscular dystrophy. Muscle Nerve. 1978;1(2):111–32.
Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, Angelini C, Dalla Volta S. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation. 1996;94(12):3168–75.
Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C, Vitiello L, Miorelli M, Buja GF, Mostacciuolo ML, et al. Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol. 1993;22(7):1927–34.
Finsterer J, Stollberger C. Cardiac involvement in Becker muscular dystrophy. Can J Cardiol. 2008;24(10):786–92.
Nigro G, Comi LI, Politano L, Limongelli FM, Nigro V, De Rimini ML, Giugliano MA, Petretta VR, Passamano L, Restucci B, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve. 1995;18(3):283–91.
Hoogerwaard EM, de Voogt WG, Wilde AA, van der Wouw PA, Bakker E, van Ommen GJ, de Visser M. Evolution of cardiac abnormalities in Becker muscular dystrophy over a 13-year period. J Neurol. 1997;244(10):657–63.
Nigro G, Politano L, Nigro V, Petretta VR, Comi LI. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul Disord. 1994;4(4):371–9.
Yilmaz A, Gdynia HJ, Baccouche H, Mahrholdt H, Meinhardt G, Basso C, Thiene G, Sperfeld AD, Ludolph AC, Sechtem U. Cardiac involvement in patients with Becker muscular dystrophy: new diagnostic and pathophysiological insights by a CMR approach. J Cardiovasc Magn Reson. 2008;10:50.
Norman A, Harper P. A survey of manifesting carriers of Duchenne and Becker muscular dystrophy in Wales. Clin Genet. 1989;36(1):31–7.
Comi LI, Nigro G, Politano L, Petretta VR. The cardiomyopathy of Duchenne/Becker consultants. Int J Cardiol. 1992;34(3):297–305.
Hoogerwaard EM, van der Wouw PA, Wilde AA, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, van Essen AJ, Leschot NJ, de Visser M. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1999;9(5):347–51.
Norman AM, Upadhyaya M, Thomas NS, Roberts K, Harper PS. Duchenne muscular dystrophy in Wales: impact of DNA linkage analysis and cDNA deletion screening. J Med Genet. 1989;26(9):565–71.
Hoogerwaard EM, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, Leschot NJ, Van Essen AJ, Brunner HG, van der Wouw PA, Wilde AA, de Visser M. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: a cohort study. Lancet. 1999;353(9170):2116–9.
Schade van Westrum SM, Hoogerwaard EM, Dekker L, Standaar TS, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, van Essen AJ, Leschot NJ, Wilde AA, de Haan RJ, de Visser M, van der Kooi AJ. Cardiac abnormalities in a follow-up study on carriers of Duchenne and Becker muscular dystrophy. Neurology. 2011;77(1):62–6.
Grain L, Cortina-Borja M, Forfar C, Hilton-Jones D, Hopkin J, Burch M. Cardiac abnormalities and skeletal muscle weakness in carriers of Duchenne and Becker muscular dystrophies and controls. Neuromuscul Disord. 2001;11(2):186–91.
Politano L, Nigro V, Nigro G, Petretta VR, Passamano L, Papparella S, Di Somma S, Comi LI. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA. 1996;275(17):1335–8.
Duan D. Challenges and opportunities in dystrophin-deficient cardiomyopathy gene therapy. Hum Mol Genet. 2006;15 Spec No 2:R253–61.
Kornegay JN, Sharp NJ, Schueler RO, Betts CW. Tarsal joint contracture in dogs with golden retriever muscular dystrophy. Lab Anim Sci. 1994;44(4):331–3.
Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81(4):1189–92.
Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–80.
Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14(8-9):491–6.
Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14(14):1921–33.
Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436(7053):1025–9.
Kamogawa Y, Biro S, Maeda M, Setoguchi M, Hirakawa T, Yoshida H, Tei C. Dystrophin-deficient myocardium is vulnerable to pressure overload in vivo. Cardiovasc Res. 2001;50(3):509–15.
Yue Y, Skimming JW, Liu M, Strawn T, Duan D. Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Hum Mol Genet. 2004;13(15):1669–75.
Chamberlain JS, Metzger J, Reyes M, Townsend D, Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;21(9):2195–204.
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345(6201):1184–8.
Pons F, Robert A, Fabbrizio E, Hugon G, Califano JC, Fehrentz JA, Martinez J, Mornet D. Utrophin localization in normal and dystrophin-deficient heart. Circulation. 1994;90(1):369–74.
Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–38.
Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90(4):717–27.
Valentine BA, Cooper BJ, de Lahunta A, O’Quinn R, Blue JT. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci. 1988;88(1-3):69–81.
Sharp NJ, Kornegay JN, Van Camp SD, Herbstreith MH, Secore SL, Kettle S, Hung WY, Constantinou CD, Dykstra MJ, Roses AD, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13(1):115–21.
Valentine BA, Winand NJ, Pradhan D, Moise NS, de Lahunta A, Kornegay JN, Cooper BJ. Canine X-linked muscular dystrophy as an animal model of Duchenne muscular dystrophy: a review. Am J Med Genet. 1992;42(3):352–6.
Valentine BA, Cummings JF, Cooper BJ. Development of Duchenne-type cardiomyopathy. Morphologic studies in a canine model. Am J Pathol. 1989;135(4):671–8.
Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120(4):1140–50.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20.
Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12(1):101–13.
Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schömig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(1):1397–409.
Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H, Hiramatsu K, Yoshino T, Kazuki K, Ishihara C, Takehara S, Higaki K, Nakagawa M, Takahashi K, Yamanaka S, Oshimura M. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol Ther. 2010;18(2):386–93.
Guan X, Mack DL, Moreno CM, Strande JL, Mathieu J, Shi Y, Markert CD, Wang Z, Liu G, Lawlor MW, Moorefield EC, Jones TN, Fugate JA, Furth ME, Murry CE, Ruohola-Baker H, Zhang Y, Santana LF, Childers MK. Dystrophin-deficient cardiomyocytes derived from human urine: new biologic reagents for drug discovery. Stem Cell Res. 2014;12(2):467–80.
Dick E, Kalra S, Anderson D, George V, Ritso M, Laval SH, Barresi R, Aartsma-Rus A, Lochmuller H, Denning C. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013;22(20):2714–24.
Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, den Dunnen JT. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30:293–9.
Goyenvalle A, Babbs A, Powell D, Kole R, Fletcher S, Wilton SD, Davies KE. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol Ther. 2010;18(1):198–205.
Mann CJ, Honeyman K, Cheng AJ, Ly T, Lloyd F, Fletcher S, Morgan JE, Partridge TA, Wilton SD. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci U S A. 2001;98(1):42–7.
Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65(6):667–76.
Yokota T, Duddy W, Echigoya Y, Kolski H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: too many mutations, too few patients? Expert Opin Biol Ther. 2012;12(9):1141–52.
Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, Garralda ME, Rutherford M, McCulley C, Popplewell L, Graham IR, Dickson G, Wood MJ, Wells DJ, Wilton SD, Kole R, Straub V, Bushby K, Sewry C, Morgan JE, Muntoni F. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918–28. Erratum in: Lancet Neurol. 2009;8(12):1083.
Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364(16):1513–22. Erratum in: N Engl J Med. 2011;365(14):1361.
Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605.
Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, Takeda S, Wilton SD, Wolff JA, Wooddell CI, Xiao X, Tremblay JP. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19(5):830–40.
England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, Bulman DE, Harris JB, Davies KE. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343(6254):180–2.
Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8(3):253–61.
Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, Finn E, Adams ME, Froehne SC, Murry CE, Chamberlain JS. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12(7):787–9.
Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther. 2008;16(4):657–64.
Banks GB, Chamberlain JS, Froehner SC. Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci. 2009;40(4):433–41.
Banks GB, Combs AC, Chamberlain JR, Chamberlain JS. Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum Mol Genet. 2008;17(24):3975–86.
Ohshima S, Shin JH, Yuasa K, Nishiyama A, Kira J, Okada T, Takeda S. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol Ther. 2009;17(1):73–80.
Wang Z, Allen JM, Riddell SR, Gregorevic P, Storb R, Tapscott SJ, Chamberlain JS, Kuhr CS. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum Gene Ther. 2007;18(1):18–26.
Yuasa K, Yoshimura M, Urasawa N, Ohshima S, Howell JM, Nakamura A, Hijikata T, Miyagoe-Suzuki Y, Takeda S. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 2007;14(17):1249–60.
Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363(15):1429–37.
Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, Li J, Wang B, Monahan PE, Rabinowitz JE, Grieger JC, Govindasamy L, Agbandje-McKenna M, Xiao X, Samulski RJ. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther. 2012;20(2):443–55.
Flanigan KM, Campbell K, Viollet L, Wang W, Gomez AM, Walker CM, Mendell JR. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24(9):797–806.
Shin JH, Pan X, Hakim CH, Yang HT, Yue Y, Zhang K, Terjung RL, Duan D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol Ther. 2013;21(4):750–7.
Hartigan-O’Connor D, Kirk CJ, Crawford R, Mule JJ, Chamberlain JS. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol Ther. 2001;4(6):525–33.
Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. V: identification and quantitation of T8+ cytotoxic and T8+ suppressor cells. Ann Neurol. 1988;23(5):493–9.
Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. IV: cell-mediated cytotoxicity and muscle fiber necrosis. Ann Neurol. 1988;23(2):168–73.
Gao G, Bish LT, Sleeper MM, Mu X, Sun L, Lou Y, Duan J, Hu C, Wang L, Sweeney HL. Transendocardial delivery of AAV6 results in highly efficient and global cardiac gene transfer in rhesus macaques. Hum Gene Ther. 2011;22(8):979–84.
Pan X, Yue Y, Zhang K, Lostal W, Shin JH, Duan D. Long-term robust myocardial transduction of the dog heart from a peripheral vein by adeno-associated virus serotype-8. Hum Gene Ther. 2013;24(6):584–94.
Ballas SK, Files B, Luchtman-Jones L, Benjamin L, Swerdlow P, Hilliard L, Coates T, Abboud M, Wojtowicz-Praga S, Grindel JM. Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin. 2004;28(2):85–102.
Spurney CF, Guerron AD, Yu Q, Sali A, van der Meulen JH, Hoffman EP, Nagaraju K. Membrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord. 2011;11:20.
Terry RL, Kaneb HM, Wells DJ. Poloxomer 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS One. 2014;9(3):e91221.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6.
Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, Qin X, Fedrigo O, Mouly V, Tremblay JP, Gersbach CA. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther. 2013;21:1718–26. doi:10.1038/mt.2013.229. Erratum in: Mol Ther. 2013;21(11):2130.
Peault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, Gussoni E, Kunkel LM, Huard J. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15(5):867–77.
Morgan JE, Coulton GR, Partridge TA. Mdx muscle grafts retain the mdx phenotype in normal hosts. Muscle Nerve. 1989;12(5):401–9.
Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 1989;337(6203):176–9.
Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Benson S, McAndrew PE, Rice R, et al. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med. 1995;333(13):832–8.
Miller RG, Sharma KR, Pavlath GK, Gussoni E, Mynhier M, Lanctot AM, Greco CM, Steinman L, Blau HM. Myoblast implantation in Duchenne muscular dystrophy: the San Francisco study. Muscle Nerve. 1997;20:469–78.
Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard JP, Roy R, Dugre FJ, Sylvain M, Lachance JG, Deschenes L, Senay H, Tremblay JP. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65:371–86.
Sharma A, Sane H, Badhe P, Gokulchandran N, Kulkarni P, Lohiya M, Biju H, Jacob VC. A clinical study shows safety and efficacy of autologous bone marrow mononuclear cell therapy to improve quality of life in muscular dystrophy patients. Cell Transplant. 2013;22 Suppl 1:S127–38.
Torrente Y, Belicchi M, Marchesi C, D’Antona G, Cogiamanian F, Pisati F, Gavina M, Giordano R, Tonlorenzi R, Fagiolari G, Lamperti C, Porretti L, Lopa R, Sampaolesi M, Vicentini L, Grimoldi N, Tiberio F, Songa V, Baratta P, Prelle A, Forzenigo L, Guglieri M, Pansarasa O, Rinaldi C, Mouly V, Butler-Browne GS, Comi GP, Biondetti P, Moggio M, Gaini SM, Stocchetti N, Priori A, D’Angelo MG, Turconi A, Bottinelli R, Cossu G, Rebulla P, Bresolin N. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007;16(6):563–77.
Chun JL, O’Brien R, Song MH, Wondrasch BF, Berry SE. Injection of vessel-derived stem cells prevents dilated cardiomyopathy and promotes angiogenesis and endogenous cardiac stem cell proliferation in mdx/utrn-/- but not aged mdx mouse models for duchenne muscular dystrophy. Stem Cells Transl Med. 2013;2(1):68–80. Erratum in: Stem Cells Transl Med. 2013;2(2):following 158.
Pelargonio G, Dello Russo A, Sanna T, De Martino G, Bellocci F. Myotonic dystrophy and the heart. Heart. 2002;88(6):665–70.
Steinert H. Myopathologische Beitrage. I. Uber das klinische und anatomische Bild des Muskelschwunds der Myotoniker. Deutsche Zeitung für Nervenheilkunde. 1909;37:58–104.
Batten FE, Gibb HP. Two Cases of Myotonia Atrophica, showing a peculiar Distribution of Muscular Atrophy. Proc R Soc Med. 1909;2:32–3.
Griffith TW. On Myotonia. Quart J Med. 1911;5:229.
Waring JJ, Ravin A, Walker C. Studies in Dystrophia myotonica: II. Clinical features and treatment. Arch Int Med. 1940;65(4):763–99.
Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808.
Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, Moxley 3rd RT. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44(8):1448–52.
Thornton CA, Griggs RC. Plasma exchange and intravenous immunoglobulin treatment of neuromuscular disease. Ann Neurol. 1994;35(3):260–8.
Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293(5531):864–7.
Day JW, Ranum LP. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr Neurol Neurosci Rep. 2005;5(1):55–9.
Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11(10):891–905.
Fu YH, Pizzuti A, Fenwick Jr RG, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255(5049):1256–8.
Dalton JC, Ranum LPW, Day JW. Myotonic dystrophy type 2. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington; 1993.
Cooper TA. A reversal of misfortune for myotonic dystrophy? N Engl J Med. 2006;355(17):1825–7.
Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–77.
Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, Marashdeh MM, Zipes DP, Pascuzzi RM. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358(25):2688–97.
Groh WJ, Lowe MR, Zipes DP. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophysiol. 2002;13(5):444–8.
Wahbi K, Meune C, Becane HM, Laforet P, Bassez G, Lazarus A, Radvanyi-Hoffman H, Eymard B, Duboc D. Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: a case control study. Neuromuscul Disord. 2009;19(7):468–72.
Bhakta D, Lowe MR, Groh WJ. Prevalence of structural cardiac abnormalities in patients with myotonic dystrophy type I. Am Heart J. 2004;147(2):224–7.
Logigian EL, Martens WB, Moxley 4th RT, McDermott MP, Dilek N, Wiegner AW, Pearson AT, Barbieri CA, Annis CL, Thornton CA, Moxley 3rd RT. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 2010;74(18):1441–8.
Epstein AE, Dimarco JP, Ellenbogen KA, Estes 3rd NA, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, Hlatky MA, Newby LK, Page RL, Schoenfeld MH, Silka MJ, Stevenson LW, Sweeney MO, American College of Cardiology/American Heart Association Task Force on Practice; American Association for Thoracic Surgery; Society of Thoracic Surgeons. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: executive summary. Heart Rhythm. 2008;5(6):934–55.
Bhakta D, Shen C, Kron J, Epstein AE, Pascuzzi RM, Groh WJ. Pacemaker and implantable cardioverter-defibrillator use in a US myotonic dystrophy type 1 population. J Cardiovasc Electrophysiol. 2011;22(12):1369–75.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Kamdar, F., Mammen, P.P.A., Garry, D.J. (2017). Neuromuscular Cardiomyopathies. In: Garry, D., Wilson, R., Vlodaver, Z. (eds) Congestive Heart Failure and Cardiac Transplantation. Springer, Cham. https://doi.org/10.1007/978-3-319-44577-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-44577-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44575-5
Online ISBN: 978-3-319-44577-9
eBook Packages: MedicineMedicine (R0)