
Abstract
LVNC is a relatively new, genetically heterogeneous, cardiomyopathy. Clinical presentation and prognosis range from asymptomatic disease with no or slow progression, to severe disabling, rapidly progressive cardiac failure. Initial presentation includes the triad of heart failure (potentially lethal) arrhythmias and/or thrombo-embolism. LVNC may occur at all ages, even prenatally. In childhood, clinical features are often more severe and LVNC is frequently associated with congenital heart defects. In adults, the majority of LVNC is isolated. The echocardiographic diagnostic criteria as proposed by Jenni et al. are currently the most widely applied. General cardiac guidelines for chronic heart failure and ICDs are applicable to the LVNC population. In approximately 40 % of isolated LVNC, molecular testing may yield a genetic (mostly sarcomere) defect, with MYH7 as the most prevalent disease gene. The nonisolated forms of LVNC are caused by a range of rare genetic defects. Until now, in half of familial isolated LVNC, the genetic defect remains unknown. Genetic defects in a large number of sarcomere and other cardiomyopathy genes and in genes primarily associated with skeletal myopathies indicate that LVNC may result from a wide range of pathophysiologic mechanisms. Shared genetic defects and familial aggregation of LVNC, HCM, and DCM indicates that LVNC may be part of a broad spectrum of cardiomyopathies. The genetic etiology of LVNC requires that patients and their relatives are offered genetic testing and counseling. This may include (predictive) molecular analysis of relatives, when applicable, and/or cardiac evaluation of at-risk relatives, even when they are as yet asymptomatic.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cardiac Resynchronization Therapy
- Implantable Cardioverter Defibrillator
- Implantable Cardioverter Defibrillator Therapy
- Implantable Cardioverter Defibrillator Implantation
- Limb Girdle Muscular Dystrophy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Noncompaction of the left ventricle or left ventricular noncompaction (LVNC) is a relatively new clinicopathologic entity, first described by Feldt et al. in 1969 [1]. LVNC is characterized by a prominent trabecular meshwork and deep intertrabecular recesses communicating with the left ventricular (LV) cavity, morphologically reminiscent of early cardiac development, and is therefore thought to be caused by an arrest of normal embryogenesis of the myocardium [2, 3]. Initial presentation includes congestive heart failure, thromboembolic events, and (potentially lethal) arrhythmias, including sudden cardiac death. LVNC may be a part of a more generalized cardiomyopathy, involving both the morphologically normal and the predominantly apical, abnormal LV segments. The cardiologic features of LVNC range from asymptomatic in adults to severe congenital forms [4–6]. LVNC was classified by the American Heart Association (AHA) as a separate primary, genetic cardiomyopathy, based on the predominant myocardial involvement and genetic etiology [7]. The European Society of Cardiology (ESC) considers LVNC as unclassified, due to the lack of consensus whether LVNC is a separate individual cardiomyopathy or a nonspecific morphological trait that can be found solitary or in combination with other forms of cardiomyopathy like hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), or with congenital heart disease [8]. The overlap in phenotypes raises the question whether LVNC is in fact a distinct cardiomyopathy or whether it is a morphological expression of different underlying diseases [9]. The majority of LVNC diagnosed in adults is isolated. Nonisolated forms of LVNC are more frequent in childhood and may co-occur with congenital heart malformations, or may be part of a malformation or chromosomal syndrome [6]. The combination of LVNC and neuromuscular disorders is observed in adults as well as in children.
The majority of LVNC, both isolated and nonisolated, is hereditary and LVNC appears to be genetically heterogenous. An important proportion of isolated LVNC in children and adults has been associated with mutations in the same sarcomere genes that are involved in HCM, DCM, and restrictive cardiomyopathy (RCM) [10]. Absence of an identifiable genetic defect does not preclude a genetic cause of LVNC. In approximately half of the familial LVNC, the genetic defect remains unknown [10]. Shared sarcomere defects and the occurrence of HCM and DCM in families with LVNC patients indicate that at least some forms of LVNC are part of a broader cardiomyopathy spectrum.
The literature differentially refers to this form of cardiomyopathy as left ventricular noncompaction (LVNC), noncompaction cardiomyopathy (NCCM), noncompaction of the left ventricular myocardium (NCLVM), left ventricular hypertrabeculation (LVHT), spongiform cardiomyopathy, embryonic myocardium, honeycombed myocardium, persisting myocardial sinusoids, myocardial dysgenesis, ventricular dysplasia, or spongy myocardium.
Definition
LVNC is defined by prominent trabeculations on the luminal surface of the LV apex, the lateral wall, and rarely the septum in association with deep recesses that extend into the ventricular wall, which do not communicate with the coronary circulation. It is associated with a clinical triad of heart failure, arrhythmias, and/or thromboembolic events [11, 12].
Epidemiology
Estimates of prevalence of LVNC were derived from large retrospective studies of patients referred for echocardiography. Population studies for LVNC have not been performed. In 1997 Ritter et al. identified LVNC in 17 of 37,555 (0.045 %) patients who had an echocardiographic examination [13]. Similarly, in 2006 Aras et al. reported a prevalence of 0.14 % in over 42,000 patients and in 2008 Sandhu identified definite or possible LVNC in 13/4929 (0.26 %) patients referred for echocardiography [14, 15]. Prevalence was much higher (3.7 %) in patients selected for a LV ejection fraction ≤45 % [14]. Depending on the diagnostic criteria applied, even higher prevalence of LVNC (15.8 % by Belanger; 23.6 % by Kohli) was reported recently, indicating that LVNC may be more prevalent than previously indicated [12, 16]. A substantial proportion of individuals is asymptomatic, suggesting that true prevalence of LVNC may be higher, because asymptomatic individuals may go unnoticed in the studies of cardiologic patients [10, 12]. In a large study on childhood cardiomyopathies, LVNC was the most frequent cardiomyopathy after DCM and HCM, with an estimated prevalence of 9 % in pediatric cardiomyopathies [17].
Clinical Aspects
Heart failure is among the most frequent presentations of LVNC, followed by supraventricular and ventricular arrhythmias, including sudden cardiac death, and thromboembolic events. However, as in other cardiomyopathies, there is a great variability in presentation, even within families, ranging from a fully asymptomatic course to severe heart failure necessitating cardiac transplantation. The age of presentation is also highly variable varying from prenatal and neonatal diagnosis to diagnosis at the age of 94 years [5, 18, 19]. Prenatal diagnostic imaging more often detects bilateral ventricular hypertrophy/hypertrabeculations than the typical left ventricular morphologic changes observed postnatally and in adults [20]. The fourth to fifth decade is the median age for diagnosis in adult isolated LVNC, constituting a relatively young population in adult cardiologic practice. Many patients remain asymptomatic and may be detected due to an asymptomatic heart murmur, or by chance by preoperative cardiac evaluation or medical assessment for insurance or jobs or because they participated in cardiologic family screening, after a relative had been diagnosed with LVNC. Symptomatic patients may present clinical symptoms of dyspnea, fatigue (atypical) chest pain, and/or (pre) syncope. LVNC may also present as a peripartum cardiomyopathy [4, 21]. Review of the literature revealed a male to female ratio of almost 2:1 [22]. This gender difference cannot be fully explained by the occurrence of X-linked forms of LVNC.
Different arrhythmias and conduction disorders may occur in LVNC patients (Table 7.1) [23]. None of these arrhythmias is characteristic or pathognomonic for LVNC. Thromboembolic events may include stroke (cerebrovascular event or transient ischemic attack), peripheral embolism, and mesenterial thrombosis.
Clinical Diagnosis
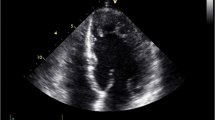
Diagnosis of LVNC is still a challenge and relies on two-dimensional transthoracic echocardiography and/or cardiac magnetic resonance imaging (MRI) (Table 7.2). Improvements in cardiac imaging techniques have led to increased recognition and diagnosis of LVNC. Figure 7.1 displays echocardiographic and cardiac MRI images of two LVNC patients, showing the abnormal segmental trabeculations as the hallmark of this entity.

(a, b) Cardiac MRI and echocardiography of a 43-year-old patient illustrating a two-layered myocardium with prominent intertrabecular recesses. (c, d) Cardiac MRI and echocardiography, four-chamber view each, of a 15-year-old patient with LVNC
Features of noncompaction observed in cardiologic patients and normal controls still illustrate the necessity of defining criteria in order to accurately differentiate between normal physiological trabecularization and LVNC [16].
In 1990, the first diagnostic criteria for LVNC by Chin et al. were derived from the observations made in eight LVNC patients [2]. These diagnostic criteria defined LVNC by the ratio of the distance from the epicardial surface to the trough of the trabecular recess (X) to the distance from the epicardial surface to the peak of the trabeculations (Y), with ratio X/Y ≤ 0.5.
More than a decade later, Jenni et al. proposed new diagnostic criteria for isolated LVNC, consisting of four echocardiographic features: (1) an excessively thickened LV myocardial wall with a two-layered structure consisting of a compact epicardial layer (C) and a noncompacted endocardial layer (NC) of prominent trabeculations and deep intertrabecular recesses; (2) a maximal end-systolic NC/C ratio > 2, measured at the parasternal short axis; (3) color-Doppler evidence of deeply perfused intertrabecular recesses; (4) absence of coexisting cardiac anomalies [11].
In 2002, Stollberger et al. proposed other diagnostic criteria for LVNC, wherein the diagnosis was a function of the number of trabeculations (>3) protruding from the LV wall, apically to the papillary muscles and visible in a single image plane with obligatory perfusion of the intertrabecular spaces from the ventricular cavity visualized on color-Doppler imaging [24].
More recently, MRI criteria for LVNC introduced by Petersen et al. indicated that a noncompacted/compacted ratio (NC/C) of >2.3, measured in end-diastole, can differentiate with sufficient sensitivity between the normal variation of noncompaction of the LV in the population, noncompaction in other cardiovascular disorders, and LVNC; the localization of noncompaction appeared to be more in the apical and lateral segments than in the basal and septal segments [49]. Jacquier et al. measured the trabeculated LV mass by MRI and postulated that a mass above 20 % is specific for the diagnosis of LVNC [50].
Belanger et al. proposed a classification system of LVNC by dividing noncompaction into four categories (none, mild, moderate, and severe) according to noncompaction to compaction ratio and the size of the noncompaction area [12]. This new classification scheme used the following criteria: (1) absence of congenital heart disease, hypertrophic or infiltrative cardiomyopathy, and coronary artery disease; (2) evidence of prominent trabeculations in the apex in any view (noncompacted to compacted ratio does not require to be >2); (3) concentration of the noncompacted area in the apex; (4) blood flow through the area of noncompaction.
The Jenni echo criteria have been the most convenient to work with in daily clinical practice and have been most widely applied in studies. However, further efforts to reach universal consensus with respect to the diagnosis of LVNC remain needed. A disparity in diagnosis has been observed when comparing the application of three different sets of LVNC criteria (Chin, Jenni, and Stollberger) in a cohort of 199 heart failure patients; 79 % fulfilled the Chin criteria, 64 % fulfilled the Jenni criteria, and 53 % fulfilled the criteria proposed by Stollberger. In only 30 % of patients, there was consensus among the three criteria on the diagnosis. Moreover, 8.3 % of normal controls fulfilled one or more criteria with a higher prevalence in black controls, and overdiagnosis is easily facilitated with the current diagnostic criteria [16, 51].
For now, it is disputable whether any of these diagnostic criteria are sufficiently sensitive to diagnose patients with mild noncompaction, and identify patients who may benefit from careful surveillance. For instance, in LVNC family studies, a substantial proportion of (mostly asymptomatic) relatives showed mild to moderate features of LVNC [10]. Longitudinal studies of mild forms of LVNC are required to determine whether the current diagnostic criteria are suitable for diagnosis of family members in familial LVNC, or should be adapted in analogy to the criteria proposed for diagnosis of attenuated forms of familial HCM in relatives.
Pathology
Macroscopy
The noncompacted endocardial layer of the myocardium comprises excessively numerous and prominent trabeculations with deep intertrabecular recesses that extend into the compacted myocardial layer. The apical and mid ventricular segments of the LV inferior and lateral wall are predominantly affected [52, 53]. In a pathoanatomical study of LVNC, Burke et al. described the morphology and microscopy of 14 pediatric LVNC cases. The macroscopic appearance varied from anastomosing trabeculae to a relatively smooth endocardial surface, with narrow openings of the recesses to the ventricular cavity. Three types of recess patterns were distinguished: (1) anastomosing broad trabeculae, (2) coarse trabeculae resembling multiple papillary muscles, and (3) interlacing smaller muscle bundles or relatively smooth endocardial surface with compressed invaginations, identified primarily microscopically (Fig. 7.2). In this study, no morphological differences were found between isolated and nonisolated LVNC [52].

LVNC gross pathology with a variety of LVNC patterns: (a) Anastomosing broad trabeculae. (b) Coarse trabeculae resembling multiple papillary muscles. (c) Interlacing smaller muscle bundles resembling a sponge. (d) Trabeculae viewed en face. (e) Subtle LVNC on gross section, requires histological confirmation (Reproduced from Burke et al. [52] with permission)
Jenni et al. described pathology of seven adult LVNC cases; the pathoanatomical localization of the noncompacted myocardium corresponded to the echocardiographic findings. Two patients also showed involvement of the right ventricular apex [11].
In a review of published pathology of LVNC, Stollberger et al. distinguished three particular morphologic features of LVNC in adults and children: (1) extensive spongiform transformation of the LV, (2) prominent coarse trabeculations and deep recesses, covered with endocardial tissue and not communicating with coronary arteries, and (3) dysplastic thinned myocardium with excessive trabeculations [22]. The first morphology was frequently associated with other cardiac malformations, compared to the second and third.
In 1987, in an autopsy study of 474 normal hearts of all ages, it was found that prominent trabeculations may be observed in as many as 68 % of the hearts, although more than three trabeculations were only identified in 3.4 % [54].
Microscopy
Two patterns of myocardial structure in the superficial noncompacted layer in LVNC have been described by Burke et al.: (1) anastomosing muscle bundles forming irregularly branching endocardial recesses with a staghorn-like appearance and (2) multiple small papillary muscles, resulting in an irregular surface appearance (Fig. 7.3). In most patients, these patterns overlapped. Endocardial fibrosis with prominent elastin deposition was found in all 14 cases, and subendocardial replacement fibrosis, consistent with microscopic ischemic infarcts, was present in 10; right ventricular involvement was identified in 6 cases [52].

Histological features in LVNC. The ratio of noncompact versus compact myocardium is larger than 2. (a) Relatively smooth endocardial surface (left) with anastomosing broad trabeculae. (b) Polypoid pattern of trabeculae; prominent fibrous band separating the noncompact from the compact myocardium (Reproduced from Burke et al. [52] with permission)
Histological examination in another study showed that ventricular endocardium covered the recesses in continuity with the LV cavity and identified ischemic lesions in the thickened endocardium and the prominent trabeculae. Interstitial fibrosis ranged from being absent to severe. No fiber disarray was identified in any of these cases. Signs of chronic inflammation and abnormalities of intramyocardial blood vessels were present in some patients [11].
Freedom et al. proposed two criteria for the pathological diagnosis of LVNC: (1) absence of well-formed LV papillary muscles and (2) histological verification of more than 50 % penetration of invaginated endocardial recesses toward the epicardial surface. The endothelium that covers the recesses extends close to the surface of the compact layer. The recesses neither communicate nor connect with the coronary circulation [55].
Differential Diagnosis
The definitive diagnosis of LVNC relies on the morphological features of the LV myocardium, as defined by an imaging modality, like echocardiography, MRI, CT, or LV angiography. The variability in the extent of physiological trabecularization may complicate distinction of LVNC from normal physiological LV trabeculations. Especially in the area around the base of the papillary muscles of the mitral valve, more trabeculations may be present. However, in the normal heart, there is no excessive segmental thickening (due to hypertrabeculation) like in LVNC and the thickness of these physiological trabeculations does not exceed the thickness of the compact layer. Also, the area of noncompaction is larger in LVNC than in physiological trabeculations [12].
Secondary forms of (acquired) LVNC may be the result of hypertension, chronic volume or pressure overload, ischemic heart disease or extreme physical activity (i.e., athletes), leading to LVNC-like abnormalities. These are referred to as pseudo-left ventricular noncompaction or LVNC look-alike. Hypertensive patients are diagnostically challenging, because of the occurrence of LV hypertrophy due to hypertension. Further studies are needed to confirm whether excessive trabeculation is more prevalent in specific ethnic groups, as suggested by one study [16].
Furthermore, dilated, hypertrophic, and ischemic cardiomyopathy may be mistaken for LVNC or vice versa, due to prominent trabeculations or abnormal myocardial thickening. Neuromuscular disorders, syndromes, and chromosomal abnormalities (Tables 7.3, 7.4, and 7.5) should be considered in the differential diagnosis of nonisolated LVNC, especially when LVNC occurs in patients with dysmorphism, growth retardation, or skeletal muscle weakness.
Work-Up, Therapy, Follow-Up, and Prognosis
Work-Up
Work-up of an LVNC patient should focus on identifying the underlying cause, either genetic or other (Table 7.3).
Therapy and Follow-Up
Current guidelines for heart failure, arrhythmias, cardiac resynchronization therapy, and ICD implantation for primary and secondary prevention are applied for LVNC [56–58]. β-Blockers and angiotensin-converting enzyme (ACE) inhibitors are the cornerstones of the treatment in the presence of LV dysfunction and/or arrhythmias. Establishing an expert consensus rapport, similar to HCM [59], based on case reports, small cohorts and clinical registries would be recommended since no randomized trials or studies on management of LVNC have been conducted, and clear-cut evidence-based clinical guidelines for this disorder are therefore missing. An important issue is the use of prophylactic anticoagulants, in view of frequent thromboembolic events. The early case reports and case series emphasized the high risk of thromboembolism and advised routine anticoagulation therapy. However, a review of 22 publications addressing the issue concluded that thromboembolic events are rare in LVNC [60]. Fazio et al. came to the same conclusion [61]. Therefore, anticoagulation therapy is advised only in patients with an ejection fraction less than 40 % (cutoff arbitrary), paroxysmal or persistent atrial fibrillation and/or previous thromboembolic events.
Successful cardiac resynchronization therapy has been described in several LVNC patients, leading to LV reverse remodeling and an increase in LV function [20, 36, 62–64].
Heart transplantation has been performed in some LVNC patients with severe heart failure [23, 65–67]. LV restoration surgery has been reported successful in a single patient [68]. Treatment with an implantable cardioverter defibrillator (ICD) will be discussed further on.
The indication for cardiologic follow-up depends on individual symptoms and cardiac abnormalities. In asymptomatic patients with preserved LV function, annual or biannual cardiologic follow-up is recommended, including ECG and echocardiography. If necessary, these could be extended with 24-h-Holter monitoring and exercise-testing. When EF is below 50 %, β-blocker therapy and ACE inhibitors should be prescribed, especially when LVNC is accompanied by hypertension or arrhythmias.
Risk Stratification and Indication for ICD
Patients at the highest risk for sudden death are patients who previously experienced (aborted) cardiac arrest, ventricular fibrillation, and sustained VF. Family history of sudden death, unexplained syncope (especially during exercise), abnormal blood pressure response during exercise tests, frequent premature ventricular beats on the resting ECG, and/or nonsustained ventricular tachycardia on Holter monitoring and significantly impaired LV function may be considered risk factors. The results from longitudinal studies and the understanding of underlying disease mechanisms will hopefully help to gain more insight into the risk factors and allow more appropriate risk stratification.
Consensus and guidelines for prophylactic ICD treatment in LVNC patients are also needed. Regular ICD indications include primary and secondary prevention. For secondary prevention, that is, after a previous episode of aborted cardiac death or collapse due to sustained VT or VF, current ICD guidelines advise ICD implantation. In the Rotterdam LVNC cohort of 67 patients, an ICD was indicated in 42 % according to the current ICD guidelines (n = 28; 21 primary and 7 for secondary prevention). After a long-term follow-up, appropriate ICD therapy occurred only in patients with secondary prevention (n = 3). Inappropriate ICD therapy occurred in 33 % of the patients with primary prevention and in 29 % of the patients with secondary prevention [69]. In another study, a follow-up of 12 patients who received an ICD showed overall appropriate therapy in 42 % in primary and secondary prevention combined. In primary prevention, 25 % of ICD therapy was appropriate opposed to 50 % in secondary prevention [45]. This accentuates the need for further research of appropriate risk stratification of sudden cardiac death in patients with LVNC.
Prognosis
Initially, LVNC was reported to have a grave prognosis. However, the application of new imaging techniques allowing diagnosing LVNC in asymptomatic individuals suggests that the first observations were influenced by selection of the most severely affected individuals. In children, age is not a predictor of the outcome [70]. New York Heart Association Class III or higher and presence of cardiovascular complications do seem to be a strong predictor [71]. It has become clear that prognosis of LVNC is as variable as the prognosis in other cardiomyopathies. Even in those with presentation in early childhood, gradual improvement in cardiac function may be observed, although in others evolvement to severe heart failure requiring heart transplantation does occur. Similarly, in some adult patients a rapid deterioration of heart function occurs, whereas in others the disease remains stable up to old age. Malignant arrhythmias leading to sudden cardiac death and heart failure are the main indicators of poor prognosis, also in children [72]. The establishment of appropriate risk stratification will be an important issue in the near future in order to identify patients at risk and to help prevent sudden cardiac death.
Etiology and Molecular Genetics
The etiology of LVNC is rapidly being unraveled as more and more genetic defects in different genes are found, indicating that LVNC is genetically heterogeneous. Currently, genetic defects are identified in approximately 40 % of LVNC patients [10, 63]. Most genetic defects are inherited as autosomal dominant trait (Table 7.4), with exception of rare genetic causes of syndromal LVNC, predominantly diagnosed in children. However, absence of a genetic defect does not exclude a genetic etiology. By performing systematic cardiologic family studies, it was shown that no genetic defect could be found in approximately half of the familial forms of LVNC, indicating that further studies are needed to find additional genetic causes for LVNC [10].
There is evidence that some forms of LVNC are part of a spectrum of cardiomyopathies, including hypertrophic, dilated, and restrictive cardiomyopathy. A shared etiology consisting of genetic defects in the same sarcomere genes, sometimes even with identical mutations, has been found in these types of cardiomyopathy. Co-occurrence of LVNC, HCM, and DCM within families endorses a shared genetic susceptibility to these different forms of cardiomyopathy [10]. The phenotypic variability of cardiomyopathies within families, including variability in age at onset and severity of clinical features, might be explained by additional modifying factors, additional genetic variants or defects, or may depend on yet unidentified exogenous or systemic factors.
Molecular Defects in LVNC
Isolated LVNC has been associated with mutations in 20 different genes (Table 7.4). Defects in sarcomere genes have been identified to be the most prevalent genetic cause occurring in approximately 30 % of all patients with isolated LVNC [10, 63].
Over 40 different mutations in sarcomere genes encoding thick (MYH7), intermediate (MYBPC3), and thin filaments (TNNT2, TNNI3, TPM1, ACTC) have been described. In particular in MYH7, the most frequent LVNC-associated gene, accounting for up to 21 % of isolated LVNC [10, 63]. MYH7 mutations currently associated with LVNC cluster in the ATP-ase active site of the head region in the N-terminal part of MYH7. This is an evolutionary well-conserved region of MYH7. As the ATP-ase active site is required for normal force production, impaired force generation might play a role in the etiology of LVNC. Mutations in this region have been associated with LVNC with or without Ebstein anomaly [64, 81]. Other MYH7 mutations (30 %) were found in the C-terminal rod region of the MYH7 protein that plays an important role in the formation of the core of the thick filament. Mutations in this region of the gene are more commonly associated with skeletal myopathies. Relatively few cardiomyopathy mutations are situated in this region.
With the availability of targeted cardiomyopathy panels (next generation sequencing), more genes are and will be associated with LVNC, but as these data are still unpublished they are not mentioned here. Complex genotypes will become more common when more genes are analyzed per patient.
Multiple or compound/double heterozygous mutations were identified in 25 % of the children and in 10 % of the adult LVNC patients [10]. In hypertrophic cardiomyopathy, complex genotypes have been described in 7 % [94]. In HCM, double heterozygosity for truncating sarcomere mutations have been previously associated with severe congenital forms mostly inherited in an autosomal recessive mode [95–98]. Nonsarcomere genetic causes for isolated LVNC include mutations in the calcium-handling genes calsequestrin (CASQ2) and phospholamban (PLN), in taffazin (TAZ), α-dystrobrevin (DTNA), lamin A/C (LMNA) and LIM domain binding 3 (LDB3), potassium voltage-gated channel (KCNH2), and sodium channel type 5 (SCN5A) genes. However, mutations in these genes were only rare causes of LVNC in single families.
The absence of a mutation in approximately half of familial LVNC could be explained by phenotype assignment errors, the involvement of other yet unidentified genes, the presence of mutations in nonanalyzed gene sequences, and incomplete sensitivity of the methods used.
Pathogenesis
Mutations in different genes associated with LVNC affect different mechanisms in the cardiomyocyte leading to changes that may individually cause LVNC or lead to a common cellular disturbance resulting in LVNC. Cellular growth and differentiation signaling pathways are thought to be involved in LVNC pathogenesis [83, 99–101].
Mutations in sarcomere genes may have their effect through defective force generation (either by a dominant negative mechanism where the mutant protein acts as a “poison polypeptide” or by haploinsufficiency resulting in less protein); mutated cytoskeletal proteins may lead to a defective force transmission; myocardial energy deficits may be the result of mutations in ATP-regulatory genes and a fourth possible mechanism is abnormal calcium homeostasis due to either changes in calcium availability or myofibrillar sensitivity for calcium [102]. The development of LVNC features might be a compensatory response to dysfunction in one of these mechanisms.
The variable phenotypic expression of (sarcomere) gene mutations leading to different types of cardiomyopathy has not been explained. The localization of the mutations may partly explain phenotypic diversity. Another theory is “dose-effect”; the extent of the defective mechanism may determine which phenotype develops. Third, there might be independent pathways leading to the different cardiomyopathies. Finding identical mutations in different phenotypes suggests a role for additional factors, either environmental or molecular.
Isolated LVNC
The first hypothesis on the pathogenesis of LVNC stemmed from observations that the morphology of LVNC was reminiscent of the embryonic stages of cardiac development. Consequently, it was postulated that LVNC could be the result from an arrest of compaction of myocardial fibers [103]. Figure 7.4 illustrates the striking resemblance between LVNC and the physiological embryonic noncompaction in the eighth to tenth embryonic week. However, the possible mechanisms causing the arrest remain unclear. Epicardium-derived cells are thought to play an important role in myocardial architecture and in the development of noncompaction [104, 105]. Mutations in genes involved in myocardial genesis like peroxisome proliferator activator receptor binding protein (PBP), jumonji (JMJ), FK506 binding protein (FKBP12), transcription factor specificity protein (Sp3), homeobox factor NKX2.5, bone morphogenetic protein 10 (BMP10) lead to congenital LVNC in knock out mice [106–111]. However, apart from the NKX2.5 gene, in human LVNC, no mutations in these genes have been described.

Human embryos at Carnegie stage 16 (a), stage 18 (b) and after closing of the embryonic interventricular foramen (c). During development, there is an extensive trabecular layer forming the greater part of the ventricular wall thickness compared to the extent of the compact layer. The trabecular layer becomes compacted and forms the papillary muscles of the atrioventricular valves (asterisks) (Reproduced from Freedom et al. [55] with permission)
Until now, there is very little insight into factors that influence the variability in age at onset and severity of symptoms of LVNC, or any other familial form of cardiomyopathy.
In the majority of patients, LVNC is diagnosed in adulthood, similar to HCM and DCM, which are rarely congenital. Of course, it could be that in LVNC the lesions detected in adult patients were present from birth on, but remained unnoticed until symptoms developed and high-resolution cardiac imaging techniques were applied. However, the detection of sarcomere defects in LVNC patients may suggest otherwise, since mutations in sarcomere genes are known to cause late-onset HCM and DCM. Similarly, sarcomere mutations might lead to late onset LVNC. Longitudinal cardiologic studies of unaffected carriers of pathogenic mutations are necessary to provide insight whether noncompaction may develop later in life. The pathogenetic mechanism(s) of sarcomere defects in cardiomyopathies are not fully understood. It is possible that the pathological myocardial changes in the adult onset sarcomere related cardiomyopathies are caused by a compensatory response to impaired myocyte function resulting from mutations in the sarcomere genes [102, 112].
Nonisolated LVNC
LVNC has been observed in a number of neuromuscular disorders, metabolic and mitochondrial disease, congenital malformations, and chromosomal syndromes.
Some of these disorders may share pathogenetic mechanisms with LVNC. Alternatively, LVNC might be secondary to other cardiac malformations or other malformations or even vice versa. Another possibility is that the co-occurrence is coincidental. Congenital heart malformations, for instance, are relatively frequent (birth prevalence 0.008) and may therefore occasionally coincide with LVNC without a mutual etiology.
Congenital Heart Disease
The co-occurrence of congenital heart disease and noncompaction is predominantly observed in children. Tsai et al. showed that 78 % of 46 children with LVNC had a congenital heart defect [6]. Nevertheless, congenital heart defects and LVNC also co-occur in adults [113]. The large number of structural heart malformations reported in association with noncompaction is presented in Table 7.5, indicating that septal defects, patent ductus arteriosus, and Ebstein’s anomaly are the most prevalent congenital heart defects in LVNC.
Increasingly, congenital cardiac malformations (septal defects, Ebstein anomaly, patent ductus arteriosus, Fallot’s tetralogy, aortic coarctation, and aortic aneurysms) are being reported in familial cardiomyopathies (HCM, DCM, and LVNC) linked to sarcomere mutations, suggesting that these specific sarcomere defects may have been involved in cardiac morphogenesis [10, 73, 81, 139–142]. But since there is rarely more than one patient with a congenital heart defect, even in families with multiple cardiomyopathy patients, the association of sarcomere defects and heart defects still demands further exploration.
Neuromuscular Disease
Similar to HCM and DCM, LVNC has been associated with neuromuscular disorders. Stollberger and Finsterer identified LVNC-like morphological features in Duchenne and Becker muscular dystrophy and in myotonic dystrophy (see section, “Neuromuscular Disorders”) [143–145]. The gene mutated in Duchenne and Becker muscular dystrophy is a part of the dystrophine complex, a complex of muscle membrane associated proteins, connecting the cytoskeleton to the surrounding extracellular matrix and may also play a role in cell signaling. The dystrophine gene is expressed in skeletal and cardiac myocytes. Other genes previously associated with neuromuscular disorders, like adult onset myofibrillar myopathy (LDB3 or Cypher/ZASP), limb girdle muscular dystrophy (LGMD) (LMNA), scapuloperoneal myopathy (MYH7), myosin storage distal myopathy (MYH7), and Barth syndrome (TAZ) have recently been associated with isolated LVNC (Table 7.4). ZASP, lamin A and C, β-myosin heavy chain, and taffazin are all expressed in cardiac and skeletal muscle tissue. ZASP has a function in cytoskeletal assembly. Mutations in ZASP can lead to DCM and to skeletal myopathy. Lamin A and C, proteins situated in the nuclear membrane, play an important role in maintaining nuclear architecture. LMNA mutations have been described in three LVNC patients [10, 79, 80]. In one of them, there was familial limb girdle muscular dystrophy (LGMD) as well as DCM [10]. Over 200 mutations have been described in LMNA, causing over 20 different phenotypes, including isolated DCM, LGMD, Emery–Dreifuss muscular dystrophy, Hutchinson–Gilford progeria, partial lipodystrophy, and peripheral neuropathy. For many of the phenotypes, there is no clear genotype–phenotype correlation, phenotypes may overlap, and different phenotypes are associated with single mutations. Up to 25 % of patients with an LMNA mutation may remain cardiologically asymptomatic [146].
Syndromes
LVNC can occur as part of a syndrome in combination with dysmorphic features and other congenital malformations. When there are other congenital defects or when there are dysmorphic features in a patient, one of the listed syndromes in Table 7.6 or one of the chromosomal defects in Table 7.7 could be considered in the differential diagnosis.
Mitochondrial
Mitochondrial disorders often lead to multiorgan disease, including the central and peripheral nervous system, eyes, heart, kidney, and endocrine organs. One of the cardiac features observed in mitochondrial disease is LVNC. Cardiac features may be the first or only feature in patients suffering from a mitochondrial disorder. In a study of 113 pediatric patients with mitochondrial disease, LVNC was identified in 13 % [171]. Pignatelli et al. showed that 5 of the 36 pediatric LVNC patients who underwent a skeletal muscular biopsy had morphologic and biochemical evidence for a mitochondrial defect, including a partial deficiency of complex I-III of the mitochondrial respiratory chain [149]. Mutations in mitochondrial DNA (mtDNA) and in nuclear DNA have been identified in the mitochondrial disorders associated with LVNC [172, 173].
Cardiogenetic Aspects
Molecular and Cardiologic Family Screening
Familial LVNC has been estimated to occur in 18–71 % of adults with isolated LVNC, mostly consistent with an autosomal dominant mode of inheritance, indicating the importance of informing and examining relatives of patients with isolated LVNC [2, 10, 15, 149, 174–177]. Since extensive family studies showed that the majority of affected relatives are asymptomatic, cardiologic evaluation should include all adult relatives irrespective of medical history. Obviously, taking a family history is by itself insufficient to identify familial disease, given the high frequency of asymptomatic disease in families [10]. In families where a pathogenic mutation has been identified, relatives can be offered predictive DNA analysis. In families without a pathogenic mutation, cardiac family screening remains the method of choice to identify relatives at risk of developing symptomatic cardiomyopathy, who may benefit from early treatment. In families where a variant (class 3 or 4) is identified, DNA analysis and cardiologic screening are advised as depicted in Fig. 7.5.


Flowchart for family screening in LVNC including *likely pathogenic variants (class 4), **variants of unknown significance (class 3) and ***no variants or class 1 or 2 variants; # if clinically indicated; @ core panel: ACTC1, ACTN2, ANKRD1, BAG3, CALR3, CAV3, CRYAB, CSRP3, CTNNA3, DES, DSC2, DSG2, DSP, EMD, FHL1, GLA, JPH2, JUP, LAMA4, LAMP2, LMNA, LDB3, MIB1, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYOZ2, MYPN, NEXN, PKP2, PLN, PRDM16, PRKAG2, RBM20, SCN5A, TAZ, TCAP, TMEM43, TNNC1, TNNI3, TNNT2, TPM1, TTN, TTR, VCL
Apart from LVNC, other cardiomyopathies may co-occur within families, like hypertrophic and dilated cardiomyopathy, so cardiac screening should aim at identifying all cardiomyopathies. Cardiac screening of relatives may show minor abnormalities not fulfilling LVNC criteria, which may be difficult to differentiate from normal physiologic trabecularization. Hypothetically, these minor abnormalities might develop into LVNC eventually. Longitudinal studies of patients with mild LVNC features are needed to investigate the natural history of these forms of noncompaction.
Genotype–Phenotype Correlations
Molecular studies of LVNC have thus far shown that there are few recurrent mutations. Therefore, it is difficult to establish genotype–phenotype correlations. Additionally, intrafamilial phenotypic variability complicates predictions based on an identified mutation. Te presence of multiple (truncating) sarcomere mutations in an individual appears to result in a more severe phenotype with childhood onset [10, 97]. Multiple mutations identified in adults mostly also comprise involvement of a nonsarcomere gene. Adult patients with multiple mutations seem to have more symptoms than adults with a single mutation [10].
The proposed strategies for the molecular and cardiologic evaluation of LVNC are depicted in the flowchart in Fig. 7.5. Extensive genetic screening, preferably with a targeted cardiomyopathy gene panel, may lead to the identification of a molecular defect in over 40 % of isolated LVNC patients and in half of these patients an MYH7 mutation is found [10].
When no targeted panel is available, MYH7 gene sequencing should be considered as an initial approach, being the most prevalent cause for LVNC in adults and children. Further molecular analyses of the other genes within the LVNC spectrum, which quantitatively have a relatively modest contribution to LVNC morbidity, may be considered when no mutation in MYH7 can be identified. Sarcomere gene analysis is also warranted in pediatric patients, given the high percentage of sarcomere mutations in this group. When an adult or pediatric patient is severely affected, screening for a second molecular defect is advised, given the high frequency of multiple mutations in LVNC.
Summary
LVNC is a relatively new, genetically heterogeneous, cardiomyopathy. Clinical presentation and prognosis range from asymptomatic disease with no or slow progression to severe disabling, rapidly progressive cardiac failure. Initial presentation includes the triad of heart failure (potentially lethal) arrhythmias and/or thromboembolism. In adults, the majority of LVNC is isolated.
The first clinical presentation of LVNC may occur at all ages, even prenatally. In childhood, clinical features are often more severe and LVNC is frequently associated with congenital heart defects. The echocardiographic diagnostic criteria as proposed by Jenni et al. are convenient in daily practice and currently the most widely applied. The general cardiac guidelines for chronic heart failure and ICDs are suitable and applicable to the LVNC population.
In as much as 40 % of isolated LVNC, molecular testing may yield a genetic defect, mostly in sarcomere genes. The MYH7 gene is the most prevalent disease gene. The nonisolated forms of LVNC are caused by a range of different (rare) genetic defects. Until now, in half of familial isolated LVNC, the genetic defect remains unknown. Genetic defects in a large number of sarcomere and other cardiomyopathy genes and in genes primarily associated with skeletal myopathies indicate that LVNC may result from a wide range of pathophysiologic mechanisms.
Shared genetic defects and familial aggregation of LVNC, HCM, and DCM indicate that LVNC may be part of a broad spectrum of cardiomyopathies.
The genetic etiology of LVNC requires that patients and their relatives are offered genetic testing and counseling. This may include (predictive) molecular analysis of relatives, when applicable, and/or cardiac evaluation of at-risk relatives, even when they are as yet asymptomatic.
Take Home Messages
LVNC is a difficult (clinical) diagnosis and is genetic/hereditary in the majority of cases.
Sarcomere gene defects (especially in MYH7) are the most frequent cause of genetic isolated LVNC.
Treatment consists of standard heart failure care and prevention of arrhythmia.
Prognosis is highly variable, even within families.
Relatives at risk may be asymptomatic, warranting active screening and a follow-up of first-degree relatives.
References
Feldt RH, Rahimtoola SH, Davis GD, Swan HJ, Titus JL. Anomalous ventricular myocardial patterns in a child with complex congenital heart disease. Am J Cardiol. 1969;23(5):732–4.
Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82(2):507–13.
Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36(2):493–500.
Hoedemaekers YM, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, Witsenburg M, et al. Cardiac {beta}-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J. 2007;28(22):2732–7.
Moura C, Hillion Y, Daikha-Dahmane F, Eydoux P, Fallet C, Oury JF, et al. Isolated non-compaction of the myocardium diagnosed in the fetus: two sporadic and two familial cases. Cardiol Young. 2002;12(3):278–83.
Tsai SF, Ebenroth ES, Hurwitz RA, Cordes TM, Schamberger MS, Batra AS. Is left ventricular noncompaction in children truly an isolated lesion? Pediat Cardiol. 2009.
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–16.
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270–6.
Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J. 2011;32(12):1446–56. Epub 2011/02/03.
Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet. 2010;3(3):232–9. Epub 2010/06/10.
Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86(6):666–71.
Belanger AR, Miller MA, Donthireddi UR, Najovits AJ, Goldman ME. New classification scheme of left ventricular noncompaction and correlation with ventricular performance. Am J Cardiol. 2008;102(1):92–6.
Ritter M, Oechslin E, Sutsch G, Attenhofer C, Schneider J, Jenni R. Isolated noncompaction of the myocardium in adults. Mayo Clin Proc. 1997;72(1):26–31.
Sandhu R, Finkelhor RS, Gunawardena DR, Bahler RC. Prevalence and characteristics of left ventricular noncompaction in a community hospital cohort of patients with systolic dysfunction. Echocardiography (Mount Kisco, NY). 2008;25(1):8–12.
Aras D, Tufekcioglu O, Ergun K, Ozeke O, Yildiz A, Topaloglu S, et al. Clinical features of isolated ventricular noncompaction in adults long-term clinical course, echocardiographic properties, and predictors of left ventricular failure. J Card Fail. 2006;12(9):726–33.
Kohli SK, Pantazis AA, Shah JS, Adeyemi B, Jackson G, McKenna WJ, et al. Diagnosis of left-ventricular non-compaction in patients with left-ventricular systolic dysfunction: time for a reappraisal of diagnostic criteria? Eur Heart J. 2008;29(1):89–95.
Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348(17):1639–46.
Ozkutlu S, Bostan O, Karagoz T, Deren O, Tekinalp G. Prenatal diagnosis of isolated non-compaction of the ventricular myocardium: study of six cases. Pediatr Int. 2007;49(2):172–6.
Sato Y, Matsumoto N, Matsuo S, Yoda S, Iida K, Kunimasa T, et al. Isolated noncompaction of the ventricular myocardium in a 94-year-old patient: depiction at echocardiography and magnetic resonance imaging. Int J Cardiol. 2007;119(1):e32–4.
Hoedemaekers YM, Cohen-Overbeek TE, Frohn-Mulder IM, Dooijes D, Majoor-Krakauer DF. Prenatal ultrasound diagnosis of MYH7 non-compaction cardiomyopathy. Ultrasound Obstet Gynecol. 2013;41(3):336–9. Epub 2012/08/04.
Bahl A, Swamy A, Sharma Y, Kumar N. Isolated noncompaction of left ventricle presenting as peripartum cardiomyopathy. Int J Cardiol. 2006;109(3):422–3.
Stollberger C, Finsterer J. Left ventricular hypertrabeculation/noncompaction. J Am Soc Echocardiogr. 2004;17(1):91–100.
Steffel J, Duru F. Rhythm disorders in isolated left ventricular noncompaction. Ann Med. 2012;44(2):101–8. Epub 2011/06/07.
Stollberger C, Finsterer J, Blazek G. Left ventricular hypertrabeculation/noncompaction and association with additional cardiac abnormalities and neuromuscular disorders. Am J Cardiol. 2002;90(8):899–902.
Fazio G, Pipitone S, Iacona MA, Marchi S, Mongiovi M, Zito R, et al. The noncompaction of the left ventricular myocardium: our paediatric experience. J Cardiovasc Med (Hagerstown, MD). 2007;8(11):904–8.
Sajeev CG, Francis J, Shanker V, Vasudev B, Abdul Khader S, Venugopal K. Young male with isolated noncompaction of the ventricular myocardium presenting with atrial fibrillation and complete heart block. Int J Cardiol. 2006;107(1):142–3.
Enriquez SG, Entem FR, Cobo M, Olalla JJ. Uncommon etiology of syncope in a patient with isolated ventricular noncompaction. Pacing Clin Electrophysiol. 2007;30(4):577–9.
Ozkutlu S, Ayabakan C, Celiker A, Elshershari H. Noncompaction of ventricular myocardium: a study of twelve patients. J Am Soc Echocardiogr. 2002;15(12):1523–8.
Celiker A, Kafali G, Dogan R. Cardioverter defibrillator implantation in a child with isolated noncompaction of the ventricular myocardium and ventricular fibrillation. Pacing Clin Electrophysiol. 2004;27(1):104–8.
Taniguchi M, Hioka T, Maekawa K, Takagagi K, Shoji K, Yoshida K. Adult case of isolated ventricular noncompaction discovered by complete atrioventricular block. Circ J. 2004;68(9):873–5.
Dagdeviren B, Eren M, Oguz E. Noncompaction of ventricular myocardium, complete atrioventricular block and minor congenital heart abnormalities: case report of an unusual coexistence. Acta Cardiol. 2002;57(3):221–4.
Okubo K, Sato Y, Matsumoto N, Kunimasa T, Kasama S, Sano Y, et al. Cardiac resynchronization and cardioverter defibrillation therapy in a patient with isolated noncompaction of the ventricular myocardium. Int J Cardiol. 2009;136(3):e66–8.
Caliskan K, Ujvari B, Bauernfeind T, Theuns DA, Van Domburg RT, Akca F, et al. The prevalence of early repolarization in patients with noncompaction cardiomyopathy presenting with malignant ventricular arrhythmias. J Cardiovasc Electrophysiol. 2012;23(9):938–44. Epub 2012/05/17.
Zhou Y, Zhang P, Zhou Q, Guo J, Xu Y, Li X. Giant P waves and focal atrial tachycardia in a patient with ventricular noncompaction. Int J Cardiol. 2008;123(2):210–2.
Ogawa K, Nakamura Y, Terano K, Ando T, Hishitani T, Hoshino K. Isolated non-compaction of the ventricular myocardium associated with Long QT Syndrome. Circ J. 2009.
Oginosawa Y, Nogami A, Soejima K, Aonuma K, Kubota S, Sato T, et al. Effect of cardiac resynchronization therapy in isolated ventricular noncompaction in adults: follow-up of four cases. J Cardiovasc Electrophysiol. 2008;19(9):935–8.
Saito K, Ibuki K, Yoshimura N, Hirono K, Watanabe S, Watanabe K, et al. Successful cardiac resynchronization therapy in a 3-year-old girl with isolated left ventricular non-compaction and narrow QRS complex. Circ J. 2009.
Kubota S, Nogami A, Sugiyasu A, Kasuya K. Cardiac resynchronization therapy in a patient with isolated noncompaction of the left ventricle and a narrow QRS complex. Heart Rhythm. 2006;3(5):619–20.
El Menyar AA, Gendi SM. Persistent atrial standstill in noncompaction cardiomyopathy. Pediatr Cardiol. 2006;27(3):364–6. Epub 2006/03/28.
Ozkutlu S, Onderoglu L, Karagoz T, Celiker A, Sahiner UM. Isolated noncompaction of left ventricular myocardium with fetal sustained bradycardia due to sick sinus syndrome. Turk J Pediatr. 2006;48(4):383–6.
Schweizer PA, Schroter J, Greiner S, Haas J, Yampolsky P, Mereles D, et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol. 2014;64(8):757–67. Epub 2014/08/26.
Celiker A, Ozkutlu S, Dilber E, Karagoz T. Rhythm abnormalities in children with isolated ventricular noncompaction. Pacing Clin Electrophysiol. 2005;28(11):1198–202.
Wessels MW, De Graaf BM, Cohen-Overbeek TE, Spitaels SE, de Groot-de Laat LE, Ten Cate FJ, et al. A new syndrome with noncompaction cardiomyopathy, bradycardia, pulmonary stenosis, atrial septal defect and heterotaxy with suggestive linkage to chromosome 6p. Hum genet. 2008;122(6):595–603.
Milano A, Vermeer AM, Lodder EM, Barc J, Verkerk AO, Postma AV, et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol. 2014;64(8):745–56. Epub 2014/08/26.
Kobza R, Jenni R, Erne P, Oechslin E, Duru F. Implantable cardioverter-defibrillators in patients with left ventricular noncompaction. Pacing Clin Electrophysiol. 2008;31(4):461–7.
Fazio G, Corrado G, Pizzuto C, Zachara E, Rapezzi C, Sulafa AK, et al. Supraventricular arrhythmias in noncompaction of left ventricle: is this a frequent complication? Int J Cardiol. 2008;127(2):255–6.
Sato Y, Matsumoto N, Takahashi H, Imai S, Yoda S, Kasamaki Y, et al. Cardioverter defibrillator implantation in an adult with isolated noncompaction of the ventricular myocardium. Int J Cardiol. 2006;110(3):417–9.
Sato Y, Matsumoto N, Matsuo S, Imai S, Yoda S, Tani S, et al. Subendomyocardial perfusion abnormality and necrosis detected by magnetic resonance imaging in a patient with isolated noncompaction of the ventricular myocardium associated with ventricular tachycardia. Cardiovasc Revasc Med. 2009;10(1):66–8.
Petersen SE, Selvanayagam JB, Wiesmann F, Robson MD, Francis JM, Anderson RH, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005;46(1):101–5.
Jacquier A, Thuny F, Jop B, Giorgi R, Cohen F, Gaubert JY, et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur Heart J. 2010;31(9):1098–4. Epub 2010/01/22.
Niemann M, Stork S, Weidemann F. Left ventricular noncompaction cardiomyopathy: an overdiagnosed disease. Circulation. 2012;126(16):e240–3. Epub 2012/10/17.
Burke A, Mont E, Kutys R, Virmani R. Left ventricular noncompaction: a pathological study of 14 cases. Hum Pathol. 2005;36(4):403–11.
Hughes SE, McKenna WJ. New insights into the pathology of inherited cardiomyopathy. Heart. 2005;91(2):257–64.
Boyd MT, Seward JB, Tajik AJ, Edwards WD. Frequency and location of prominent left ventricular trabeculations at autopsy in 474 normal human hearts: implications for evaluation of mural thrombi by two-dimensional echocardiography. J Am Coll Cardiol. 1987;9(2):323–6.
Freedom RM, Yoo SJ, Perrin D, Taylor G, Petersen S, Anderson RH. The morphological spectrum of ventricular noncompaction. Cardiol Young. 2005;15(4):345–64.
Hunt SA. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure). J Am Coll Cardiol. 2005;46(6):e1–82.
Hunt SA, Baker DW, Chin MH, Cinquegrani MP, Feldman AM, Francis GS, et al. ACC/AHA Guidelines for the Evaluation and Management of Chronic Heart Failure in the Adult: Executive Summary A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1995 Guidelines for the Evaluation and Management of Heart Failure): Developed in Collaboration With the International Society for Heart and Lung Transplantation; Endorsed by the Heart Failure Society of America. Circulation. 2001;104(24):2996–3007.
Nieminen MS, Bohm M, Cowie MR, Drexler H, Filippatos GS, Jondeau G, et al. Executive summary of the guidelines on the diagnosis and treatment of acute heart failure: the Task Force on Acute Heart Failure of the European Society of Cardiology. Eur Heart J. 2005;26(4):384–416.
Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42(9):1687–713.
Stollberger C, Finsterer J. Thrombi in left ventricular hypertrabeculation/noncompaction--review of the literature. Acta Cardiol. 2004;59(3):341–4.
Fazio G, Corrado G, Zachara E, Rapezzi C, Sulafa AK, Sutera L, et al. Anticoagulant drugs in noncompaction: a mandatory therapy? J Cardiovasc Med (Hagerstown, MD). 2008;9(11):1095–7.
Battaglia A. Del 1p36 syndrome: a newly emerging clinical entity. Brain Dev. 2005;27(5):358–61. Epub 2005/07/19.
Probst S, Oechslin E, Schuler P, Greutmann M, Boye P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4(4):367–74. Epub 2011/05/10.
Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117(22):2893–901.
Williams T, Machann W, Kuhler L, Hamm H, Muller-Hocker J, Zimmer M, et al. Novel desmoplakin mutation: juvenile biventricular cardiomyopathy with left ventricular non-compaction and acantholytic palmoplantar keratoderma. Clin Res Cardiol. 2011;100(12):1087–93. Epub 2011/07/27.
Luxan G, Casanova JC, Martinez-Poveda B, Prados B, D’Amato G, MacGrogan D, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19(2):193–201. Epub 2013/01/15.
Kovacevic-Preradovic T, Jenni R, Oechslin EN, Noll G, Seifert B, Attenhofer Jost CH. Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience. Cardiology. 2009;112(2):158–64.
Shimamoto T, Marui A, Yamanaka K, Shikata N, Tambara K, Ikeda T, et al. Left ventricular restoration surgery for isolated left ventricular noncompaction: report of the first successful case. J Thorac Cardiovasc Surg. 2007;134(1):246–7.
Caliskan KTD, Hoedemaekers YM, Ten Cate FJ, Jordaens L, Szili TT. Implantable cardioverter-defibrillators for primary and secondary prevention in patients with noncompaction cardiomyopathy. J Am Coll Card. 2009;53(10, supplement 1):):A136.
Zuckerman WA, Richmond ME, Singh RK, Carroll SJ, Starc TJ, Addonizio LJ. Left-ventricular noncompaction in a pediatric population: predictors of survival. Pediatr Cardiol. 2011;32(4):406–12. Epub 2010/12/29.
Greutmann M, Mah ML, Silversides CK, Klaassen S, Attenhofer Jost CH, Jenni R, et al. Predictors of adverse outcome in adolescents and adults with isolated left ventricular noncompaction. Am J Cardiol. 2012;109(2):276–81. Epub 2011/11/01.
Brescia ST, Rossano JW, Pignatelli R, Jefferies JL, Price JF, Decker JA, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation. 2013;127(22):2202–8. Epub 2013/05/02.
Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28(16):1953–61.
Bagnall RD, Molloy LK, Kalman JM, Semsarian C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med Genet. 2014;15:99. Epub 2014/09/17.
Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001;103(9):1256–63.
Xing Y, Ichida F, Matsuoka T, Isobe T, Ikemoto Y, Higaki T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88(1):71–7.
Marziliano N, Mannarino S, Nespoli L, Diegoli M, Pasotti M, Malattia C, et al. Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am J Med Genet. 2007;143(9):907–15.
Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42(11):2014–27.
Hermida-Prieto MML, Castro-Beiras A, et al. Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with Lamin A/C gene mutations. Am J Cardiol. 2004;94:50–4.
Rankin J, Auer-Grumbach M, Bagg W, Colclough K, Nguyen TD, Fenton-May J, et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet. 2008;146A(12):1530–42.
Budde BS, Binner P, Waldmuller S, Hohne W, Blankenfeldt W, Hassfeld S, et al. Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta-myosin heavy chain gene. PLoS One. 2007;2(12):e1362.
Postma AV, van Engelen K, van de Meerakker J, Rahman T, Probst S, Baars MJ, et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ Cardiovasc Genet. 2011;4(1):43–50. Epub 2010/12/04.
Zhang W, Chen H, Qu X, Chang CP, Shou W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C: Semin Med Genet. 2013;163C(3):144–56. Epub 2013/07/12.
Arndt AK, Schafer S, Drenckhahn JD, Sabeh MK, Plovie ER, Caliebe A, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet. 2013;93(1):67–77. Epub 2013/06/19.
Shan L, Makita N, Xing Y, Watanabe S, Futatani T, Ye F, et al. SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol Genet Metab. 2008;93(4):468–74.
Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12(4):385–9.
Bleyl SB, Mumford BR, Brown-Harrison MC, Pagotto LT, Carey JC, Pysher TJ, et al. Xq28-linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet. 1997;72(3):257–65.
Bleyl SB, Mumford BR, Thompson V, Carey JC, Pysher TJ, Chin TK, et al. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet. 1997;61(4):868–72.
Brady AN, Shehata BM, Fernhoff PM. X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn. 2006;26(5):462–5.
Chen R, Tsuji T, Ichida F, Bowles KR, Yu X, Watanabe S, et al. Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Mol Genet Metab. 2002;77(4):319–25.
Cortez-Dias N, Varela MG, Sargento L, Brito D, Almeida A, Cerqueira R, et al. Left ventricular non-compaction: a new mutation predisposing to reverse remodeling? Rev Port Cardiol. 2009;28(2):185–94.
Kenton AB, Sanchez X, Coveler KJ, Makar KA, Jimenez S, Ichida F, et al. Isolated left ventricular noncompaction is rarely caused by mutations in G4.5, alpha-dystrobrevin and FK Binding Protein-12. Mol Genet Metab. 2004;82(2):162–6.
Yen TY, Hwu WL, Chien YH, Wu MH, Lin MT, Tsao LY, et al. Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur J Pediatr. 2008;167(8):941–4.
Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358(18):1899–908.
Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006.
Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(9):1903–10.
Wessels MW, Herkert JC, Frohn-Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, et al. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet. 2015;23(7):922–8. Epub 2014/10/23.
Zahka K, Kalidas K, Simpson MA, Cross H, Keller BB, Galambos C, et al. Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart. 2008;94(10):1326–30.
Chen H, Zhang W, Li D, Cordes TM, Mark Payne R, Shou W. Analysis of ventricular hypertrabeculation and noncompaction using genetically engineered mouse models. Pediatr Cardiol. 2009;30(5):626–34. Epub 2009/04/28.
Yang J, Bucker S, Jungblut B, Bottger T, Cinnamon Y, Tchorz J, et al. Inhibition of Notch2 by Numb/Numblike controls myocardial compaction in the heart. Cardiovasc Res. 2012;96(2):276–85. Epub 2012/08/07.
Chen H, Zhang W, Sun X, Yoshimoto M, Chen Z, Zhu W, et al. Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development. 2013;140(9):1946–57. Epub 2013/04/11.
Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82(4):945–80.
Sedmera D, Pexieder T, Vuillemin M, Thompson RP, Anderson RH. Developmental patterning of the myocardium. Anat Rec. 2000;258(4):319–37.
Lie-Venema H. The role of epicardium-derived cells (EPDCs) in the development of non-compaction cardiomyopathy. Florence International Course on Advances in Cardiomyopathies – 5th meeting of the European Myocardial and Pericardial Disease WG of the ESC; 22/24 May 2008; Florence Italy 2008.
Lie-Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, et al. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. TheScientificWorldJOURNAL. 2007;7:1777–98.
Breckenridge RA, Anderson RH, Elliott PM. Isolated left ventricular non-compaction: the case for abnormal myocardial development. Cardiol Young. 2007;17(2):124–9.
Crawford SE, Qi C, Misra P, Stellmach V, Rao MS, Engel JD, et al. Defects of the heart, eye, and megakaryocytes in peroxisome proliferator activator receptor-binding protein (PBP) null embryos implicate GATA family of transcription factors. J Biol Chem. 2002;277(5):3585–92.
Lee Y, Song AJ, Baker R, Micales B, Conway SJ, Lyons GE. Jumonji, a nuclear protein that is necessary for normal heart development. Circ Res. 2000;86(9):932–8.
Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, et al. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391(6666):489–92.
van Loo PF, Mahtab EA, Wisse LJ, Hou J, Grosveld F, Suske G, et al. Transcription factor Sp3 knockout mice display serious cardiac malformations. Mol Cell Biol. 2007;27(24):8571–82.
Tian T, Liu Y, Gao L, Wang J, Sun K, Zou Y, et al. Isolated left ventricular noncompaction: clinical profile and prognosis in 106 adult patients. Heart Vessel. 2014;29(5):645–52. Epub 2013/10/03.
Lombardi R, Betocchi S. Aetiology and pathogenesis of hypertrophic cardiomyopathy. Acta Paediatr Suppl. 2002;91(439):10–4.
Stahli BE, Gebhard C, Biaggi P, Klaassen S, Valsangiacomo Buechel E, Attenhofer Jost CH, et al. Left ventricular non-compaction: prevalence in congenital heart disease. Int J Cardiol. 2013;167(6):2477–81. Epub 2012/06/19.
Tunaoglu FS, Kula S, Olgunturk R, Ozturk G. Noncompaction with arcus aorta anomalies. Turk J Pediatr. 2003;45(4):363–6.
Niwa K, Ikeda F, Miyamoto H, Nakajima H, Ando M. Absent aortic valve with normally related great arteries. Heart Vessel. 1987;3(2):104–7.
Ali SK. Unique features of non-compaction of the ventricular myocardium in Arab and African patients. Cardiovasc J Afr. 2008;19(5):241–5.
Vijayalakshmi IB, Chitra N, Prabhu Deva AN. Use of an Amplatzer duct occluder for closing an aortico-left ventricular tunnel in a case of noncompaction of the left ventricle. Pediatr Cardiol. 2004;25(1):77–9.
Tatu-Chitoiu A, Bradisteanu S. A rare case of biventricular non-compaction associated with ventricular septal defect and descendent aortic stenosis in an young man. Eur J Echocardiogr. 2006.
Song ZZ. A combination of right ventricular hypertrabeculation/noncompaction and atrial septal defect. Int J Cardiol. 2009.
Salazar Gonzalez JJ, Rite Montanes S, Asso Abadia A, Pueo Crespo E, Salazar Gonzalez E, Placer Peralta LJ. [Isolated non-compaction of the ventricular myocardium] Miocardio ventricular no compacto aislado. Anales espanoles de pediatria. 2002;57(6):570-573.
Cavusoglu Y, Ata N, Timuralp B, Gorenek B, Goktekin O, Kudaiberdieva G, et al. Noncompaction of the ventricular myocardium: report of two cases with bicuspid aortic valve demonstrating poor prognosis and with prominent right ventricular involvement. Echocardiography (Mount Kisco NY). 2003;20(4):379–83.
Cavusoglu Y, Aslan R, Birdane A, Ozbabalik D, Ata N. Noncompaction of the ventricular myocardium with bicuspid aortic valve. Anadolu Kardiyol Derg. 2007;7(1):88–90.
Sato Y, Matsumoto N, Yoda S, Inoue F, Kunimoto S, Fukamizu S, et al. Left ventricular aneurysm associated with isolated noncompaction of the ventricular myocardium. Heart Vessel. 2006;21(3):192–4.
Cavusoglu Y, Tunerir B, Birdane A, Timuralp B, Ata N, Gorenek B, et al. Transesophageal echocardiographic diagnosis of ventricular noncompaction associated with an atrial septal aneurysm in a patient with dilated cardiomyopathy of unknown etiology. Can J Cardiol. 2005;21(8):705–7.
Unlu M, Ozeke O, Kara M, Yesillik S. Ruptured sinus of Valsalva aneurysm associated with noncompaction of the ventricular myocardium. Eur J Echocardiogr. 2008;9(2):311–3.
Friedman MA, Wiseman S, Haramati L, Gordon GM, Spevack DM. Noncompaction of the left ventricle in a patient with dextroversion. Eur J Echocardiogr. 2007;8(1):70–3.
Gorgulu S, Celik S, Eksik A, Tezel T. Double-orifice mitral valve associated with nonisolated left ventricular noncompaction--a case report. Angiology. 2004;55(6):707–10.
Sugiyama H, Hoshiai M, Toda T, Nakazawa S. Double-orifice mitral valve associated with noncompaction of left ventricular myocardium. Pediatr Cardiol. 2006;27(6):746–9.
Wang XX, Song ZZ. A combination of left ventricular noncompaction and double orifice mitral valve. Cardiovasc Ultrasound. 2009;7:11.
Betrian Blasco P, Gallardo Agromayor E. Ebstein’s anomaly and left ventricular noncompaction association. Int J Cardiol. 2007;119(2):264–5.
Ilercil A, Barack J, Malone MA, Barold SS, Herweg B. Association of noncompaction of left ventricular myocardium with Ebstein’s anomaly. Echocardiography (Mount Kisco, NY). 2006;23(5):432–3.
Sinkovec M, Kozelj M, Podnar T. Familial biventricular myocardial noncompaction associated with Ebstein’s malformation. Int J Cardiol. 2005;102(2):297–302.
Arslan S, Gurlertop HY, Gundogdu F, Senocak H. Left ventricular noncompaction and mid-caviter narrowing associated with Ebstein’s anomaly: three-dimensional transthoracic echocardiographic image. Int J Cardiol. 2007;115(1):e52–5.
Attenhofer Jost CH, Connolly HM, Warnes CA, O’Leary P, Tajik AJ, Pellikka PA, et al. Noncompacted myocardium in Ebstein’s anomaly: initial description in three patients. J Am Soc Echocardiogr. 2004;17(6):677–80.
Bagur RH, Lederlin M, Montaudon M, Latrabe V, Corneloup O, Iriart X, et al. Images in cardiovascular medicine. Ebstein anomaly associated with left ventricular noncompaction. Circulation. 2008;118(16):e662–4.
Friedberg MK, Ursell PC, Silverman NH. Isomerism of the left atrial appendage associated with ventricular noncompaction. Am J Cardiol. 2005;96(7):985–90.
Vanpraagh R, Ongley PA, Swan HJ. Anatomic types of single or common ventricle in man. Morphologic and geometric aspects of 60 necropsied cases. Am J Cardiol. 1964;13:367–86.
Dogan R, Dogan OF, Oc M, Duman U, Ozkutlu S, Celiker A. Noncompaction of ventricular myocardium in a patient with congenitally corrected transposition of the great arteries treated surgically: case report. Heart Surg Forum. 2005;8(2):E110–3.
Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–93.
Wessels MW, Willems PJ. Mutations in sarcomeric protein genes not only lead to cardiomyopathy but also to congenital cardiovascular malformations. Clin Genet. 2008;74(1):16–9.
Xin B, Puffenberger E, Tumbush J, Bockoven JR, Wang H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet. 2007;143A(22):2662–7.
Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38(3):343–9.
Finsterer J, Stollberger C. Spontaneous left ventricular hypertrabeculation in dystrophin duplication based Becker’s muscular dystrophy. Herz. 2001;26(7):477–81.
Finsterer J, Stollberger C, Feichtinger H. Noncompaction in Duchenne muscular dystrophy: frustrated attempt to create a compensatory left ventricle? Cardiology. 2006;105(4):223–5.
Finsterer J, Stollberger C, Wegmann R, Jarius C, Janssen B. Left ventricular hypertrabeculation in myotonic dystrophy type 1. Herz. 2001;26(4):287–90.
Malhotra R, Mason PK. Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr Opin Cardiol. 2009;24(3):203–8.
D’Adamo P, Fassone L, Gedeon A, Janssen EA, Bione S, Bolhuis PA, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet. 1997;61(4):862–7.
Stollberger C, Finsterer J. Noncompaction in Melnick Fraser syndrome. Pacing Clin Electrophysiol. 2007;30(8):1047 .author reply 8
Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108(21):2672–8.
Matsumoto T, Watanabe A, Migita M, Gocho Y, Hayakawa J, Ogawa S, et al. Transient cardiomyopathy in a patient with congenital contractural arachnodactyly (Beals syndrome). J Nippon Med Sch = Nihon Ika Daigaku zasshi 2006;73(5):285-288.
Limongelli G, Pacileo G, Marino B, Digilio MC, Sarkozy A, Elliott P, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007;100(4):736–41.
Wong JA, Bofinger MK. Noncompaction of the ventricular myocardium in Melnick-Needles syndrome. Am J Med Genet. 1997;71(1):72–5.
Finsterer J, Stollberger C, Kopsa W. Noncompaction on cardiac MRI in a patient with nail-patella syndrome and mitochondriopathy. Cardiology. 2003;100(1):48–9.
Amann G, Sherman FS. Myocardial dysgenesis with persistent sinusoids in a neonate with Noonan’s phenotype. Pediatr Pathol. 1992;12(1):83–92.
Mandel K, Grunebaum E, Benson L. Noncompaction of the myocardium associated with Roifman syndrome. Cardiol Young. 2001;11(2):240–3.
Happle R, Daniels O, Koopman RJ. MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea): an X-linked phenotype distinct from Goltz syndrome. Am J Med Genet. 1993;47(5):710–3.
Kherbaoui-Redouani L, Eschard C, Bednarek N, Morville P. [Cutaneous aplasia, non compaction of the left ventricle and severe cardiac arrhythmia: a new case of MLS syndrome (microphtalmia with linear skin defects)]. Arch Pediatr. 2003;10(3):224-226. Aplasie cutanee congenitale, defaut de compaction du ventricule gauche et troubles du rythme cardiaque graves : un nouveau cas de syndrome MLS (microphtalmia with linear skin defects).
Battaglia A, Hoyme HE, Dallapiccola B, Zackai E, Hudgins L, McDonald-McGinn D, et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121(2):404–10.
Cremer K, Ludecke HJ, Ruhr F, Wieczorek D. Left-ventricular non-compaction (LVNC): a clinical feature more often observed in terminal deletion 1p36 than previously expected. Eur J Med Genet. 2008;51(6):685–8.
Saito S, Kawamura R, Kosho T, Shimizu T, Aoyama K, Koike K, et al. Bilateral perisylvian polymicrogyria, periventricular nodular heterotopia, and left ventricular noncompaction in a girl with 10.5–11.1 Mb terminal deletion of 1p36. Am J Med Genet. 2008;146A(22):2891–7.
Thienpont B, Mertens L, Buyse G, Vermeesch JR, Devriendt K. Left-ventricular non-compaction in a patient with monosomy 1p36. Eur J Med Genet. 2007;50(3):233–6.
Digilio MC, Bernardini L, Gagliardi MG, Versacci P, Baban A, Capolino R, et al. Syndromic non-compaction of the left ventricle: associated chromosomal anomalies. Clin Genet. 2013;84(4):362–7. Epub 2012/12/06.
Kanemoto N, Horigome H, Nakayama J, Ichida F, Xing Y, Buonadonna AL, et al. Interstitial 1q43-q43 deletion with left ventricular noncompaction myocardium. Eur J Med Genet. 2006;49(3):247–53.
Pauli RM, Scheib-Wixted S, Cripe L, Izumo S, Sekhon GS. Ventricular noncompaction and distal chromosome 5q deletion. Am J Med Genet. 1999;85(4):419–23.
De Rosa G, Pardeo M, Bria S, Caresta E, Vasta I, Zampino G, et al. Isolated myocardial non-compaction in an infant with distal 4q trisomy and distal 1q monosomy. Eur J Pediatr. 2005;164(4):255–6.
McMahon CJ, Chang AC, Pignatelli RH, Miller-Hance WC, Eble BK, Towbin JA, et al. Left ventricular noncompaction cardiomyopathy in association with trisomy 13. Pediatr Cardiol. 2005;26(4):477–9.
Wang JC, Dang L, Mondal TK, Khan A. Prenatally diagnosed mosaic trisomy 22 in a fetus with left ventricular non-compaction cardiomyopathy. Am J Med Genet. 2007;143A(22):2744–6.
Altenberger H, Stollberger C, Finsterer J. Isolated left ventricular hypertrabeculation/noncompaction in a Turner mosaic with male phenotype. Acta Cardiol. 2009;64(1):99–103.
van Heerde M, Hruda J, Hazekamp MG. Severe pulmonary hypertension secondary to a parachute-like mitral valve, with the left superior caval vein draining into the coronary sinus, in a girl with Turner’s syndrome. Cardiol Young. 2003;13(4):364–6.
Sasse-Klaassen S, Probst S, Gerull B, Oechslin E, Nurnberg P, Heuser A, et al. Novel gene locus for autosomal dominant left ventricular noncompaction maps to chromosome 11p15. Circulation. 2004;109(22):2720–3.
Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–31.
Finsterer J, Bittner R, Bodingbauer M, Eichberger H, Stollberger C, Blazek G. Complex mitochondriopathy associated with 4 mtDNA transitions. Eur Neurol. 2000;44(1):37–41.
Finsterer J, Stollberger C, Schubert B. Acquired left ventricular hypertrabeculation/noncompaction in mitochondriopathy. Cardiology. 2004;102(4):228–30.
Espinola-Zavaleta N, Soto ME, Castellanos LM, Jativa-Chavez S, Keirns C. Non-compacted cardiomyopathy: clinical-echocardiographic study. Cardiovasc Ultrasound. 2006;4:35.
Ichida F, Hamamichi Y, Miyawaki T, Ono Y, Kamiya T, Akagi T, et al. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34(1):233–40.
Lofiego C, Biagini E, Pasquale F, Ferlito M, Rocchi G, Perugini E, et al. Wide spectrum of presentation and variable outcomes of isolated left ventricular non-compaction. Heart. 2007;93(1):65–71.
Murphy RT, Thaman R, Blanes JG, Ward D, Sevdalis E, Papra E, et al. Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J. 2005;26(2):187–92.
Acknowledgments
The authors thank Kadir Calsikan and Danielle Majoor-Krakauer for their work on the first edition of this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hoedemaekers, Y.M., Klaassen, S. (2016). Left Ventricular Noncompaction. In: Baars, H., Doevendans, P., Houweling, A., van Tintelen, J. (eds) Clinical Cardiogenetics. Springer, Cham. https://doi.org/10.1007/978-3-319-44203-7_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-44203-7_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44202-0
Online ISBN: 978-3-319-44203-7
eBook Packages: MedicineMedicine (R0)