
Abstract
The cardiovascular system is designed to supply the body to the needs of metabolic demand. When cardiac output (CO) falls short or systemic metabolic demand is increased, the patient is in jeopardy. CO is greatly determined by the amount of venous return (VR) delivered to the right ventricle (RV). VR is dependent on the balance between unstressed and stressed volume, which portends the driving pressure for VR. Stressed volume can be calculated by measurement of mean systemic filling pressure (P ms). There are multiple (clinical and experimental) modalities to measure P ms. The net driving pressure estimated as the upstream pressure (P ms) relative to right atrial pressure (P ra), often deferred the driving pressure for venous return (P vr). Resistance to flow, which counts for both arterial vascular resistance and resistance to venous return (RVR), is calculated by various methods depending on the vascular circuit and flow characteristics each circuit creates. In conjunction to the systemic circulation, atria serve as a reservoir and conduit, where the right atrium particularly plays an important role in modulating venous return. Owing to this reservoir and conduit function, atrial contraction plays a lesser role in delivery of blood to the chambers. The ventricles, thanks to the Frank-Starling effect, increase contraction when supplied with this preload. A complex interplay of inotropy, lusitropy, and chronotropy comprising both systolic and diastolic function enable the heart to optimally transfer the blood it receives, thereby assisted by the concepts of ventricular interdependence, ventriculo-arterial coupling, the role of the pericardium, and coronary blood supply.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Cardiac
- Function
- Contractility
- Systolic
- Diastolic
- Inotrope
- Chronotropy
- Lusitropy
- Right
- Left
- Atrium
- Ventricle
- Ventricular
- Venous return
- Mean systemic filling pressure
- Stressed volume
- Unstressed volume
- Venous return
1 Introduction
The cardiovascular system subserves the metabolic needs of the body, which itself is highly variable and often unpredictable. Since the primary goal of the cardiovascular system is to sustain adequate oxygen delivery to the metabolically active tissues relative to their demand, the controls for this system are inherently complex yet simple at the same time. The heart serves as a central pumping function to deliver blood under low pressure to the pulmonary circulation and under very high pressures to the systemic circulation, while at all times keeping its filling pressure as low as possible as to maintain maximal filling and minimize tissue edema. The interactions between the right side of the heart, systemic venous return, pulmonary blood flow, the left side of the heart, pulmonary venous return, and left ventricular ejection are controlled to a large degree by intrinsic cellular mechanisms that require no external control or feedback. It is both the moment-to-moment changes in venous return induced by breathing efforts (referred to as heart-lung interactions) and the metabolic demands modified by muscular activity, digestion, and mentation that drive steady-state cardiac output. Historically, reviews of cardiovascular system control usually start with descriptions of ventricular pump function. However, except with severe right or left ventricular failure, resting cardiac output remains remarkably constant as cardiac reserve diminishes. In fact, cardiac impairment is assessed not by baseline measures of cardiac output or oxygen delivery to the tissues, but by its responsiveness to a defined exercise challenge, like walking or bicycle exercise. Clearly, when dealing with bedside treatment of critically ill patients in circulatory shock, a broader approach of cardiovascular (dys)function assessment, monitoring, and management is essential. This chapter summarizes in brief terms normal and dysfunctional cardiovascular physiology. The reader will note that though a circuit and closed, each component is uniquely designed and adapted to its specific role(s) which make their monitoring and specific treatment more relevant. Since inadequate oxygen delivery to the tissues represents the end stage of circulation insufficiency, understanding the components of oxygen delivery and metabolic demand becomes central to this analysis. Although increasing oxygen carrying capacity by increasing hemoglobin concentration and increasing its level of oxygen saturation reflect treatment options to increase oxygen delivery to the body, their impact is limited except in the negative. The primary means by which the body and by inference critical care physicians increase oxygen delivery to patients to reverse circulatory shock is by increasing tissue blood flow specifically and cardiac output (CO) in general. This point forms the basis for most hemodynamic monitoring and treatment decisions.
2 Cardiovascular Physiology
Under normal conditions the major determinants of CO are metabolic demand of the tissues which varies directly with whole-body oxygen consumption. However, in stressed states, wherein blood volume is diminished, ventricular function impaired or obstruction to flow occurs, and other forces become prominent in determining CO. Within this regard, the heart can only pump what blood it receives. Thus, the primary determinant of CO must be venous return (VR) from the body to the heart, which itself is a function of its developed downstream pressure, called mean systemic filling pressure (P ms). P ms shares a complex relation to total circulating blood volume, peripheral vasomotor tone, and blood flow distribution and will be later explained in detail. From the heart’s perspective, each given stroke volume will be a function of how much blood has entered the ventricular chamber during diastole, referred to as preload, heartbeat frequency, or heart rate; the arterial pressure opposing ventricular ejection, called afterload; the innate ability of the cardiac myocytes to vary their force of contraction in response to varying loads, called inotropy; their ability to actively relax during diastole, called lusitropy; as well as the impact that varying beat frequency has on intrinsic contractility, called chronotropy. Other determinants of cardiac function are architectural and geometrical variables including wall thickness, size, and shape of its cavity as well as global synchrony of myocardial contraction and relaxation [1].
2.1 Venous Return
The main function of the venous system is to serve as both the low pressure reservoir for capillary blood drainage and to serve as the upstream reservoir driving blood flow back to the right ventricle (RV) [2, 3]. The cardiovascular system can be artificially divided in systemic and pulmonary compartments, each with 80% and 20% of the blood volume, respectively. In the systemic circulation, the small veins and venules contain most of the systemic blood volume (upward of 75%). However, the amount of blood compartmentalized there is a function of blood flow distribution. If the splanchnic circulation has an increased metabolic demand, as what occurs postprandially, then a greater amount of blood pools in the splanchnic circulation because its outflow is impeded by hepatic parenchymal resistance, as all portal venous blood must traverse the liver before entering the systemic venous circulation. Thus, under resting conditions the splanchnic venous bed stores roughly one third of the blood volume in the venous compartment [3]. Therefore, the venous compartment serves as a capacitance reservoir, whereas the arterial compartment serves as a high-pressure conduit. Things are quite different on the pulmonary side. Although the pulmonary circulation contains only a small portion of the total blood volume, it receives exactly the same CO as the systemic circulation but maintains it in at a much lower pressure. The reason for this is twofold. First, since right atrial pressure (P ra) is the back pressure to venous return, keeping right atrial pressure at or below zero at all times maximizes the driving pressure for venous return. Second, since the primary role of the lung is to exchange oxygen and carbon dioxide between pulmonary capillaries and alveoli, keeping pulmonary capillary pressure as low as possible and minimizing baseline pulmonary vascular resistance allows for maximal capillary-alveolar surface area by minimizing pulmonary edema formation while simultaneously maximizing intrinsic hypoxic pulmonary vasoconstriction matching of blood flow to alveolar oxygen to optimize gas exchange.
Until very recently, most cardiologists and intensivists considered cardiovascular regulation strictly from the perspective of ventricular pump function. In fact, the most common adhered concept of determining CO is that it is only regulated by left ventricular (LV) characteristics [2, 4]. However, as noted above, in a steady state, the heart can only eject what it receives from the vasculature, and the circulation can only return the amount of blood it receives from the heart. Therefore, CO must equal VR, and the heart will always pump out exactly what amount of VR it receives. Thus, as Guyton et al. noted over 50 years ago, VR is the primary determinant of CO [4,5,6,7,8,9,10]. According to the Guytonian approach, VR plays a pivotal role in determining CO, and therefore the only thing that cardiac function can do to affect CO is to reduce it by failing to pump the blood it receives resulting in an increased P ra. However, this is an oversimplification of cardiovascular physiology. Beard et al. questioned Guyton’s view on the circulation [11]. They stated that not merely P ra, but flow (CO) itself was the true independent variable through the systemic circulation. This was emphasized by the fact that Guyton had used an artificial rotator pump to maintain flow. He then used a collapsible tube as Starling resistor to vary the right atrial inflow. Grodins et al. reproduced Guyton’s experiments without using a Starling resistor, which identified the variable speed pump (CO) as the causal variable [12]. However, this neglects the fact that VR is still key to supplying the heart with blood. Therefore, the truth probably lies somewhere between VR and LV contractility, with the fulcrum balanced more toward the venous return side as being dominant under most conditions and the heart under conditions of severe LV systolic or diastolic heart failure.
From a purely physical basis, there are two main factors regulating venous return: the pressure gradient for venous return, namely, the pressure difference between P ms and P ra, and the resistance to venous return (RVR), stressed volume (V s) and unstressed volume (V o) [2]. Each of these terms will be explained.
2.2 Mean Systemic Filling Pressure
Already in the 1890s, Bayliss and Starling observed that if blood flow would be stopped or arrested, arterial pressure would drop and venous pressure would rise, until an equilibrium was reached. The equilibrated pressure is what they termed “mean systemic filling pressure” (P ms). Furthermore, they reasoned that P ms would be the same average pressure throughout the whole circulation, independent of mean arterial pressure (MAP), since P ms could be measured during circulatory arrest or absence of pump function in both arteries and veins [13]. This pressure was presumed to be independent of cardiac function, because it was neither raised nor lowered during circulatory arrest. In their studies and others by Guyton et al. in anesthetized dogs, P ms was estimated to be only about 7 ± 2 mmHg (mean ± SD). P ms is lower than capillary pressure, almost equal to portal pressure and higher than P ra. This sequence of pressures is probably of evolutional importance since there is a need for venous compensation to maintain VR despite the orthostatic hypotensive effect of gravity. It is believed that reflex venoconstriction plays a key role in this mechanism; otherwise we would all faint when moving from a supine to standing position. Furthermore, the point of measurement of P ms had to be situated on the venous side because of the higher capacitance of this reservoir, allowing venous pressure to slightly increase during circulation arrest. P ms is regarded as the upstream pressure for VR and arterial constriction trapping arterial blood at a slightly higher value than P ms [14]. Later on, Guyton et al. further developed this concept in a series of experiments which formed the basis for our understanding of the circulation. The methods of these experiments have been elaborately described elsewhere [7,8,9,10]. In short, in open-chest anesthetized dogs, arterial pressure and P ra were measured through catheters inserted into the arterial and venous systems. A special cannula was inserted in the right atrium, and blood was pumped through an external circuit with a variable rate propulsion pump. Blood flow was measured with a flowmeter, and the blood was eventually redirected to the pulmonary artery across a Starling resistor such that at all times the maximal venous return was delivered into the pulmonary circulation. Thus, the system replaced the RV and was called a right heart bypass preparation. P ms was varied by raising or lowering an inflow site for the vascular pump thereby varying the P ra from low negative (−15 mmHg) to very high positive values (+20 mmHg). In different experiments multiple modalities of circulatory arrests, areflexia, administration of vasopressors, types of anesthesia, and hemodynamic circumstances (fluid resuscitation, hypovolemia, distributive shock) were simulated. Based on this massive body of work, a unified understanding of the systemic controls of venous return evolved.
During normovolemia stable dogs’ P ms values ranged from 7 to 12 mmHg. However, as expected P ms values will vary greatly and quickly in response to physiologic stresses. For example, slow initial increments of epinephrine infused caused P ms to increase to 16 mmHg, while stopping the heart by constricting the pulmonary artery caused stop-flow P ms to raise to 13 mmHg. Intravascular fluid infusion induced an increase in P ms proportional to total volume infused, consistent with the assumption that vascular capacitance is linear over the physiologic range. Importantly, large-volume fluid infusion increased P ra, P ms, and CO, but the increase in P ms was greater than the increase in P ra, such that the pressure gradient for venous return, namely, P ms − P ra, increased proportionally to the CO increase [7,8,9,10].
The compliance and resistance of the arterial and venous systems are markedly different and reflect their relative needs in sustaining blood flow. The compliance of the venous system is 30 times higher than the compliance of the arterial system and its resistance 40 times less. The splanchnic circulation receives approximately 20% of the cardiac output and comprises up to 30% of the total blood volume. Because of the high compliance of the venous system, changes in blood volume are associated with relatively small changes in venous transmural pressure. Veins are capable of accepting large volumes of blood with minimal change in P ms. In fact, in an otherwise stable patient, massive fluid infusion will not increase CO or P ms because the newly infused volume is distributed into expanding capacitance vessel’s distribution until such time as in the associated increased glomerular filtration rate increases urine output. Thus, the venous system serves as a reservoir for blood and is capable of accommodating large changes in blood volume if not needed to increase CO. Venous capacity is the amount of blood volume contained in a vein (or venous system) at a specific distending pressure [3]. Vascular capacitance is the relationship between total blood volume and distending pressure, whereas vascular compliance is the relation between changing blood volume and changing P ms. The relation between blood volume and P ms is not constant and is a function of blood volume, blood flow distribution among the various vascular beds, and venomotor tone. The veins, when collapsed, can be distended with a certain amount of volume before they start to resist future distention. Conformational changes in their shape as they move from collapsed to distended first passes through a zone in which their volume increases with any measurable change in distending pressure. The amount of blood volume needed to distend the venous capacitance vessels before any measurable increase in P ms occurs is referred to as the unstressed volume (V o). Under normal conditions approximately 60–70% of total circulating blood volume is in the unstressed volume. If intravascular volume is increased beyond this amount, P ms increases as the veins resist further distention. The volume above this unstressed volume is referred to as the stressed volume (V s) and defines the relation between changing blood volume and P ms [14]. Thus, capacitance vessels have both an unstressed volume and a stressed volume. Importantly, the absolute amount of potential unstressed volume in the venous circulation exceeds total circulating blood volume. Thus, under conditions of maximal vasodilation, P ms falls to zero and so does both VR and CO. This is what happens transiently during vasovagal syncope.
For purposes of physiologic definitions, then we can make the following definitions:
-
Vascular compliance is defined as the slope of change in intravascular volume and P ms (ΔV/ΔP ms). Under most conditions over the normal physiologic range, vascular compliance is linear.
-
Vascular distensibility/compliance is the fractional change in volume per unit change in pressure (ΔV/V oΔP).
-
Static vascular elasticity or elastance is the reciprocal of compliance: ∆P/∆V = slope.
-
Unstressed volume (V o) is the blood volume contained in a vessel without generating a measurable change in P ms. It is not possible to measure V o in the living human or animal, because values of zero distending pressure are not achievable since there will always be a certain part of distending pressure in the vascular tree. Instead, for both clinical and experimental purposes, it is possible to measure stressed volume (V s).
-
Stressed volume (V s) is the volume above V o generating intravascular (and thus distending) pressure thereby reflecting the upstream driving pressure. It is calculated as P ms times compliance (V s = C × P ms). V o is calculated by subtracting V s from the total blood volume (TBV) leading to the equation V o = TBV − V s [2,3,4,5,6,7,8,9,10, 14].
-
Vascular capacitance represents the overall circulating blood volume to P ms relation, which under most conditions is highly variable and of little clinical importance.
There are two ways that a subject can increase their CO in response to a metabolic stress. Either they decrease their V o by diverting blood flow away from high capacitance circuits, like the splanchnic circulation, and into circuits with low unstressed volume, like muscle vascular beds, or they decrease the resistance to venous return (RVR) by increasing the parallel number of venous conducting circuits draining the venous system or by vasodilating the large venous resistance vessels (e.g., vena cavae). In practice, they do both. The arterial circuit vasoconstricts blood flow to the splanchnic circulation thus decreasing V o while also vasodilating the large venous conduit vessels such as the inferior vena cava.
Several clinical examples illustrate these dynamic interactions. Non-specific arterial vasoconstriction will cause a decrease in arterial flow, decreasing venous blood volume and thus CO, as seen with phenylephrine infusion in normal adults. Whereas a combined increased arterial tone and contractility, as created by norepinephrine infusion, will result in the pooled central blood being ejected for completely increasing CO for the same increase in arterial pressure as induced by phenylephrine. When CO decreases due to ventricular failure, P ra also increases causing VR to decrease pooling blood in the venous reservoirs, increasing both venous vascular pressure and peripheral edema formation. With isolated LV failure causing decreased forward blood flow, baroreceptors induce a reactive vascular vasoconstriction. This causes LV afterload to increase further impeding LV ejection while also increasing P ra, if backward failure impairs RV function. Similarly, the increased sympathetic tone increased venomotor tone thereby decreases V o increasing P ms. To the extent that P ra can be kept low by decreasing LV afterload, then CO will not decrease and may even go up. An active response of capacitance vessels in response to smooth muscle activity (caused by reflexes or sympathetic nerve stimulation) causes a change in the venous pressure/volume (compliance). In times of increased sympathetic tone, V o decreases and for the same circulating blood volume causing P ms to increase. This dynamic process is the primary mechanism used to sustain CO as metabolic demand rapidly changes, as with sudden exercise [14].
2.3 Pressure for Venous Return
An important concept of VR is the net driving pressure estimated as the upstream pressure (P ms) relative to P ra, often deferred the driving pressure for venous return (P vr). Steady-state CO is determined by factors collectively grouped into preload, heart rate, intrinsic contractility, and afterload. From this perspective, the only way that cardiac function can modulate VR is by changing P ra, which in turn changes P vr. P ra represents the downstream pressure to VR, whereas P ms is the upstream pressure [2,3,4]. As Funk et al., inspired by Bressack and co-workers, recently opined that P vr calculations can be deducted from the knowledge as to the V s value, P ms in turn can be calculated by subtracting V s from total blood volume, divided by systemic compliance (P ms = (V total − V s)/C) (derived from Hagen-Poiseuille’s equation Q (flow) = P 1 − P 2/R, where Q represents the flow, P 1 the upstream pressure, P 2 the downstream pressure, and R the resistance to flow). Under influence of vasomotor effects, catecholaminergic stress responses, intravascular fluid infusion, exsanguination, changes in autonomic tone, or endo-/exogenous vasoactive substances can all change both vascular compliance and V s [2]. Thus, VR can be increased or decreased by changing either P ms (either by influencing stressed volume) or P ra (as a measure of ventricular contractile reserve) or changes in RVR. VR can therefore be calculated as VR = (P ms − P ra)/RVR [14].
2.4 Resistance to Venous Return
Resistance to flow, which counts for both arterial vascular resistance RVR, is calculated by various methods depending on the vascular circuit and flow characteristics each circuit creates. Poiseuille’s Law states that the pressure drop across a length of conducting vessel will be a function of the length of blood vessels (l), π, and blood viscosity (η) while inversely related to the radius (r) of blood vessel, resulting in the equation [2]:
The net change of blood volume redistribution coincides with a change in venous resistance. This occurs because the change in volume is related to the second power of the vessel diameter, whereas a change in vascular resistance is related to the fourth power of diameter. Small veins and venules have a smaller radius, but with a very large cross-sectional area in comparison to the much larger (in diameter) vena cava and large veins, they serve as an excellent reservoir. The caval and larger veins on the other hand act primarily as conduits, but due to their larger diameters, according to the equation for resistance, they account for the majority of venous resistance. Also, the speed of flow through a vascular bed largely determines RVR. Specifically, lengthy vascular beds with slow blood flow account for a higher venous resistance than short beds with fast flow. Thus, when a given percentage of venous conduit vessels experience a decrease in vessel diameter, resistance to blood flow will increase to the fourth power of the mean radius. The increase in resistance will lead to a decrease in flow, but an increase in upstream pressure (P ms), accumulation of blood in the venous circuit, and peripheral edema (see also Fig. 5.1) [14].

Integration of venous return curve and cardiac output curve as exemplified by Guyton and co-workers. Flow is presented on the y-axis. Right atrial pressure is presented on the x-axis. Black solid lines tagged “normal” represent the venous return and cardiac output curve in steady-state condition. The intersection of both curves (black dot) represents the optimal integration of CO and VR (equilibrating point). When increasing P ms (upper dotted curve), the VR curve shifts upward, thereby also shifting upward along the CO curve in order to reach a new equilibrium with a higher CO as a result. The opposite occurs when decreasing P ms (lowest dash-dotted curve), reaching a new equilibrium at the cost of a lower CO. Changes in RVR are represented as a change in slope. An increase in RVR will limit VR, flattening the slope of the curve, without changing P ms (small dashed curve). In contrast, a decrease in RVR causes the slope to increase (large dashed curve). By stimulating contractility, use of positive inotropes or by decreasing ventricular afterload, the slope of the CO changes resulting into an increase in CO. Opposite effects occur when decreasing contractility, lowering inotropy dosage, or increasing ventricular afterload. When cardiac compliance decreases (i.e., ischemia, diastolic dysfunction), the CO curve shifts upward along the x-axis reflecting higher diastolic filling pressures at the cost of a lower CO, higher P ra and P ms
Another factor influencing RVR is respiratory cycling. Normally, P ra exceeds pleural pressure and serves as the downstream pressure opposing VR. However, during spontaneous inspiration intrathoracic pressure becomes negative which is transferred to the right heart. P ra progressively decreases, and at that point it becomes subatmospheric, thus making atmospheric pressure the major downstream pressure opposing VR. Venous pressures and P ra ultimately fall below the atmospheric pressure during forceful inspiration, causing the veins to collapse, limiting any further increase in flow despite further increases in P vr. This phenomenon is reversed during spontaneous expiration [14]. Downstream pressure cannot decrease below zero, because as external tissue pressure exceeds intravascular pressure causing the veins to collapse as they enter the thorax [7,8,9,10]. Guyton reasoned that due to vascular compliance, resistance decreased to only a small extent when P ms rose. Importantly, in life P ra cannot increase to a value greater than P ms because otherwise there would be total circulatory arrest. At best, when the circulation is halted and VR becomes zero, P ra and P ms also equal. Thus, stop-flow P ra if measured before reflex vasoconstriction or metabolically induced vasodilation is an accurate estimate of P ms. In practice, Guyton measured the equilibrium pressures of arterial and venous pressure as he stopped flow and then rapidly pumped blood into the veins from the arteries, which took about 3 s. Furthermore, P ms represents the upper limit of the P ra values possible [7,8,9,10]. Newer techniques show that one can wait ~20 s and still get an accurate estimate of P ms from venous pressure (infra vide).
A decrease in arterial flow (e.g., by increasing arterial resistance or cross clamping the aorta) leads to a decrease in volume within the venous system, shifting blood from the venous system to the heart and thereby instantaneously increasing VR and CO.
Venous compliance is a change in blood volume associated with a change in distending pressure in a vein of venous system. Venous compliance reflects the relation between changing venous pressure and blood volume, whereas venous capacity is a static measure of the total blood volume generating a certain P ms. An increase in effective circulating blood volume can be achieved by either a decrease in vascular capacity or a decrease in compliance or both. For example, pharmacologic venoconstrictors (i.e., alpha-adrenergic agonists) decrease venous capacity without changing compliance by decreasing unstressed volume [3]. The resistance to venous return can thus be considered to be the sum of the venous and arterial resistance, multiplied by the ratio of total arterial and venous capacitances (RVR = R v + R a C a/C t) where R v is venous resistance, R a is arterial resistance, C a is arterial capacitance, and C t is total vascular capacitance [11].
3 Integrating Mean Systemic Filling Pressure and Cardiac Output
The relationship between CO and P ms is complex. P ms is a variable rather than a parameter which is dependent on blood volume, vascular compliances, and (un)stressed volumes. Previous studies have found that a 1 mmHg increase in P ms is equivalent to 4% change in blood volume [2, 14]. CO further depends on cardiac contractility, heart rate (HR), and diastolic filling (influenced by P ms and venous resistance). Guyton et al. integrated the venous return curve and the cardiac output curve (Fig. 5.1) to show how they are linked [7,8,9,10].
4 Clinical Application of P ms Measurements
There are multiple ways of measuring P ms at the bedside, based on our understanding of the determinants of P ms described above. Logically, the methods developed by Guyton et al. of creating a right heart bypass system and measuring vascular equilibrium pressure during transient stop-flow conditions would be the most accurate of means, but except under conditions of on-pump cardiopulmonary bypass during cardiovascular surgery, these are not clinically feasible.
Furthermore, the measures of P
ms, achieved during stop-flow preparations, are independent of the prior measures of P
ra before discontinuation of flow, which could trouble the interpretation of resistance to venous return [15]. In 1984 Pinsky postulated that, according to the concept of the VR curve (Fig. 5.1) and based on the assumption that changes in right ventricular (RV) stroke volume (SVRV) and VR are the same during small tidal volume (VT) breathing (VT < 10 mL/kg), gradually increased intrathoracic pressure applied by increasing positive-pressure ventilation resulted in an increase in P
ra without affecting P
ms. SVRV and P
ra were then plotted (P
ra x-axis), resulting in a relation with a negative slope, equal to the resistance to venous return with a positive-pressure SVRV intercept approximating P
ms [16]. Maas et al. applied older techniques validated in animals, as published by Versprille and Jansen in 1985 [17] in humans to assess P
ms, showing that transiently increasing P
ra using inspiratory breath-hold maneuvers of 10–15 s at different plateau airway pressures of 0, 2.5, 5, 7.5, and 10 cm H2O caused P
ra to increase and instantaneous CO, measured by minimally invasive arterial pulse pressure waveform analysis, to decrease. The resultant plot of these P
ra and CO values describes a VR curve, which when extrapolated to zero CO closely approximated P
ms [18, 19]. If the inspiratory hold maneuver is held just slightly longer (e.g., ~20 s), the resultant CO decrease is associated with an arterial pressure decrease because baroreceptor response is too slow to change arterial tone. One can then plot a MAP to CO graph similar to the P
ra and CO graph, because the CO is the same for both P
ra and MAP measures. The zero flow MAP to CO graph ends not at P
ms but at a pressure much higher than P
ms, reflecting the arterial critical closing pressure, otherwise referred to as a vascular waterfall, which will be described in greater detail below. Schipke et al. measured arterial and venous pressures in patients undergoing cardioverter/defibrillator implantation during cessation of flow caused by longer fibrillation/defibrillation sequences. Although this resembled the Guyton experiments, no equilibrium of pressures was achieved, probably due to the short cessation of circulation which kept the arterial critical closing pressure functional [20]. One step further, Repessé et al. measured venous and arterial pressures in critically ill patients who died at the ICU 1 min after circulation arrest. One-minute P
ms was 12.8 ± 5.6 mmHg with no difference in patient characteristics [21]. The group of Pinsky and Maas recently analyzed different bedside modalities of P
ms measurements in humans [18]. According to the hypothesis-generating work by Anderson, a stop-flow vascular pressure in a peripheral artery or vein could be used as an approximation of P
ms. The group of Maas compared transient stop-flow forearm arterial and venous equilibrium pressure (P
arm) with both P
ms achieved by inspiratory breath-hold maneuvers and an estimated value of P
ms calculated by a recent monitoring computerized algorithm developed by Parkin and Leaning which incorporates the Guytonian approach to measure P
ms. It assumes a lump sum arterial resistance 40 times greater than venous resistance and arterial compliance 20 times less than venous compliance. Thus, if one knows P
ra, MAP, and CO, P
ms is easily calculated. This value is referred to as P
ms analogue (P
msa). Although its clinical significance is still under investigation, it is a good example of how estimates of P
ms could be made in a minimal invasive clinical setting [22,23,24,25,26]. Maas et al. found that P
arm correlated well with P
ms as measured by inspiratory hold maneuvers, whereas P
msa had a systemic bias which could be accounted for by dividing P
msa by 0.7. Consequently, they compared changes in volume status by passive leg raising and volume infusion between P
ms, P
arm, and P
msa, which showed that all three measures of P
ms reliably tracked the changes in effective circulating blood volume status. Cecconi and co-workers used estimates of P
msa using this methodology to measure P
ms and P
vr in postsurgical patients in response to volume resuscitation. They observed that all volume challenges increased P
msa, but only in volume responders, defined as those whose CO increased by >15%, did P
vr also increase. In the non-volume responders, P
ra increased in proportion to the increase in P
msa. Thus, if a patient is given a fluid challenge and P
ra increases, that fluid challenge should be stopped and the patient reassessed because they are probably not volume responsive [27]. Another technique studied is the vascular stop-flow equilibrium method. If peripheral venous and arterial pressures are being simultaneously measured in the same arm and a vascular blood pressure cuff is rapidly inflated to greater than systolic arterial pressure, one sees the venous and arterial pressure merging toward a common value after about 20 s that closely tracks P
ms and its change in response to fluid loading. This stop-flow measure can be made from a radial arterial pressure catheter alone. Thus, there are several methods available to estimate P
ms at the bedside. Recently, clinical studies have shown that the changes in P
ms, P
vr, and RVR can be readily measured and performed in a fashion consistent with Guytonian physiological principals in postoperative cardiac surgery patients given norepinephrine to increase MAP and in septic patients during withdrawal from norepinephrine [19, 21].
5 Cardiac Physiology
5.1 Atrial Physiology in General
The atrium has five main functions. First of all, as described earlier, it has a major function as reservoir. Second, its presystolic contractile function optimizes emptying and thereby delivering blood into the chambers (atrial kick). Third, after opening of the atrioventricular (AV) valves, it functions as a conduit by transferring blood to the chambers down a pressure gradient. Fourth, it serves as a blood volume sensor which releases atrial natriuretic peptides during states of increased atrial wall stretch, thereby increasing diuresis and contributing to the restoration of normal intravascular volume state. And fifth, it contains receptors from afferent fibers answering to increased venous return (Brainbridge reflex) [28, 29].
Inspired by the experiments from Starling, Mitchell, Sarnoff, and Guyton, Anderson investigated in several experiments what he termed “the mechanical nature of the heart as a pump” [5, 13, 30]. With a similar extracorporeal circulatory design using subsequently a sucking and non-sucking pump in mongrel dogs, he found that the output of the sucking pump was dependent on the rate of the pump. Output of the non-sucking pump was more dependent on external (or likewise extra-cardiac) factors, e.g., flow rate to the heart. Changing pump rate in a non-sucking model merely changed the stroke volume but not the total (cardiac) output. Clamp-tubing experiments proved atrial function as (distensible) reservoirs to avoid stop flow during closure of AV valves. Anderson concluded that since the heart is non-sucking, continuous-inflow (atria) and intermittent-outflow pump atria function more as conduits at rest and supplemental pump during exercise [5]. Without atria the heart needs a four-time higher inflow pressure to achieve (venous pressure), but the atrial systole only attributes up to 15–25% of blood flow from the atria to the ventricles. This has been confirmed by many studies [13, 28, 31]. Additionally, atria in sinus rhythm are more effective because they do not contract during venous filling and do not interrupt inflow during ventricular systole, i.e., the atria remain distensible enough. Even during atrial contraction, their lumen never narrows to the extent in which venous resistance increases. Of note, atria never reach a negative pressure and are therefore to be considered non-suction pumps. On the contrary during atrial fibrillation and flutter, the atria prohibit atrial relaxation and thus venous inflow. This proves that atrial kick does not necessarily play a pivotal role in contributing to CO, but moreover the reservoir and conduit function, which fall short during atrial (tachy)arrhythmias.
6 Right Atrium
P ra should not be considered as the independent variable determining CO. It is clearly linked to RV function, stressed blood volume, and intrathoracic pressure. Still, P ra plays a pivotal interacting role in defining P vr and thus CO. There are three mechanisms which influence P ra. The first describes a decrease in P ra when CO increases, as a result from transferring blood from the venous part to the arterial tree. Second, relaxation of smooth muscle resulting in venodilation decreases P ms which eventually leads to a decrease in cardiac filling, reflected by a decrease in P ra, CO, and arterial pressure. Finally, a reduction of blood volume also leads to a decrease in P ra. Since P ra is influenced by cardiac function (i.e., the ability to transfer the amount of blood which it receives) and thus increases when CO diminishes or the opposite, it is not a good parameter of venous return and intravascular filling status [14]. The atria are relatively empty after ventricular diastole, combined with the atrial contraction, allowing continuously venous inflow. Therefore, the atria serve both as a reservoir and a conduit, but their contraction plays only a minor role in comparison to its capacitive function except under circulatory stress and exercise conditions. The fact that the atria are capable of distension during ventricular systole allows venous drainage into the atria to be continuous, despite only episodic ventricular filling [5].
7 Left Atrium
In addition, atrial remodeling (which occurs mainly on the left side of the heart because of the following processes) is a combination of ion, structural, contractile, and metabolic changes induced by elevated left atrial pressures in several disease states. For example, it occurs as a result of as well as induces atrial tachyarrhythmias. Left atrial stretch or enlargement either resulting from long-time left ventricular diastolic dysfunction, hypertrophy, or otherwise elevated LV pressures caused by systolic LV failure can lead to poor atrial contractile performance and increased initiation and perpetuation of atrial fibrillation [32].
7.1 Ventricular Physiology in General
Preload is the load present before contraction has started, usually defined by the end-diastolic pressure or end-diastolic volume. It could better be defined as wall stress at the end of diastole. Afterload is the load opposing the ventricle during ejection and is considered to be the wall stress during LV ejection. When preload increases, ventricular end-diastolic volume increases, thereby increasing contraction force according to the Frank-Starling mechanism. Preload and afterload are interlinked, meaning that preload is related to the extent to which myocardial fibers are stretched during end diastole and afterload is related to the wall stress generated by these fibers during systole. This tight linkage of end-diastolic myocardial fiber tension and developed pressure has greatly improved knowledge of ventricular function to date.
8 Frank-Starling Effect
In the 1890s Starling and co-workers observed that when myocardial muscle strips were stretched, the force of their contraction increased. Sudden increases in developed tension (caused by increased preload) induce an immediate increase in developed pressure. This is referred to as heterometric autoregulation, to accent the fact that the autoregulation is due to external forces (i.e., preload). Since end-diastolic myocardial fiber length is a major determinant of LV end-diastolic volume, the Frank-Starling relation has been simplified to state that sudden increases in LV end-diastolic volume can cause an immediate increase in LV force of contraction. This process is essential to match over short time periods RV and LV outputs so as to maintain central blood volume constant. Since venous return varies widely during positive-pressure breathing owing to the effect of intrathoracic pressure on P ra, RV filling and thus output also vary widely across the ventilatory cycle, and parallel changes in LV end-diastolic volume are caused with a two- to three-heartbeat delay. The Frank-Starling effect keeps these two ventricles cycling their stroke volumes to these dynamic changes in preload [13]. Interestingly, the exact mechanism(s) causing this effect is poorly understood but must relate in some way to conformational changes in myocyte orientation or calcium proximity to troponin, because the effect is instantaneous.
9 Anrep Effect
An acute increase in ventricular afterload causes intrinsic contractility to increase by complex cellular mechanisms associated with ischemia that includes phosphorylation of the calcium channel receptor activator protein increasing calcium transits during systole. This effect takes about 2–3 min to be fully developed, but once present results in a lower LV end-diastolic volume and filling pressure for a greater stroke volume. This process is referred to as the Anrep effect of homeometric autoregulation to contrast its Frank-Starling effect because it is intrinsic to the cardiac myocyte metabolic processes. Von Anrep proposed a sudden release of myocardial stores of catecholamines by mechanical stretch, although more recent data suggest that increased calcium transiting due to changes in T-tubal associated flux is a more likely mechanism [33]. The Anrep effect is an important mechanism to support increased contractility for short intervals of stress, such as exercise, but usually subsides after a few hours.
10 Ventricular Wall Stress
Wall stress, according to Laplace’s law, is created when tension is applied to a cross-sectional area, measured in unit force per unit area. For a sphere, wall stress is calculated as = (pressure × radius)/(2 × wall thickness). This portends two important features. The first is that the larger ventricular size and radius, the greater the wall stress. The second is that at any given radius, the greater the pressure generated by the ventricle, the greater the wall stress. Thus, an increase in either ventricular size or intraventricular pressure will increase wall stress and thereby myocardial oxygen uptake. Under normal conditions maximal LV wall stress occurs at the start of ejection when the aortic value initially opens because the product of diastolic arterial pressure and end-diastolic volume is maximal. During normal LV ejection as LV volume decreases, the radius decreases much more than systolic arterial pressure increases. Thus under normal conditions, the left ventricle unloads itself during ejection to end systole. A good example of adaptation of the heart to increased wall stress is that of ventricular concentric hypertrophy. However, hypertension increases wall stress across all LV ejection. Increased wall thickness in response to systemic hypertension counterbalances increased intraventricular pressure thereby compensating increased wall stress; the radius does not change. However, this compensatory mechanism holds up to a certain limit, after which, as known in hypertensive heart disease, the ventricle eventually starts to dilate. Ventricular dilation is a good example of increased wall stress, as the radius increases. In systolic heart failure, decreased contractile function is compensated by dilation, thereby increasing wall stress. However, in systolic heart failure, radius stays too high throughout the contractile cycle, leading to both elevated end-systolic and end-diastolic wall stress. This pathological process underlies the logic in afterload reduction to treat systolic heart failure, because wall stress remains high during ejection in the setting of LV dilation. Therefore, overall reduction in ventricular size reduces wall stress and improves ventricular function [28].
11 Chronotropy
Heart rate (HR) changes inversely alter diastolic filling time. The faster the HR, the less time in diastole and thus the lower the LV end-diastolic volume causing a proportional decrease in stroke volume without a change in CO. However, in heart failure states, wherein P ra is elevated and both RV and LV end-diastolic volumes limit filling such that SV is fixed, HR increases initially increase CO until P ra decreases to a point where it no longer allows for an increased P vr. Second, as HR increases from 90 to approximately 110 beats/minute, intrinsic contractility also increases (Treppe or Bowditch effect). This effect is usually what is referred to as chronotropy and has a maximum response at heart rates 150–180 beats/minute in young adults and probably at low beat frequencies in the elderly. However, the first effect secondary to decreasing P ra described above is usually lumped into the overall effect because it is not possible to separate the two processes from each other. The cause of the HR-induced increased force of contraction is still unclear but probably related to nonsteady-state increases in calcium availability for troponin coupling because it has not had sufficient enough time to return completely to the t-tubules during diastole.
12 Work of the Heart
The work of the heart (external work) is calculated as pressure x volume. Volume work generally demands less energy (oxygen) than pressure work. Stroke volume (or cardiac output) is the volume moved against blood pressure, whereas the pressure work consumes an increased pressure or heart rate. In this relationship three components can be regarded determining myocardial oxygen uptake: preload, afterload, and heart rate, usually defined in the formula minute work = systolic blood pressure (SBP) × stroke volume (SV) × heart rate (HR). Following this formula of the pressure-work index, the double product of SBP × HR and SV × HR (i.e., CO) reveals that a lot of myocardial oxygen demand is involved (up to 40%). See also Fig. 5.2.

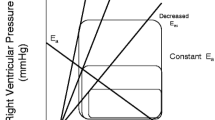
Graphical presentation of ventricular function and its pressure-volume relationship. The slope of the end-systolic pressure-volume relationship (E es) is displayed. Total external work of the heart is represented by the area of the pressure-volume curve. When cardiac output increases, the area of the external work curve becomes larger
13 Right Ventricle
13.1 Right Ventricular Anatomy
The RV is the cardiac chamber which is situated most anteriorly behind the sternum and marks the inferior border of the cardiac silhouette [34, 35]. It is defined by the presence of the annulus of the tricuspid valve and the pulmonary valve. There are three important RV anatomical compartments: (1) inflow tract (sinus), consisting of the tricuspid valve, chordae tendineae, and the papillary muscles; (2) the trabeculated apical myocardium; and (3) the infundibulum (conus), which represents the right ventricular outflow tract (RVOT). Furthermore, there are three muscular bands which characterize the right ventricle: (1) the parietal band, which, together with the infundibular septum, forms the crista supraventricularis, and (2) the septomarginal band, which continues into the moderator band (3) eventually attaching to the anterior papillary muscle. The ventriculo-infundibular fold, separating the tricuspid and pulmonary valves, is another specific marker of RV anatomy, as in contradiction to the LV, where fibrous continuity exists between the mitral and aortic valves. The RV has a triangular shape seen from aside but appears to have a half-moon shape on cross-sectional slides [34, 35]. The design of the RV is primarily aimed at volume capacity. It consists of coarse trabeculae (reaching from the sinus to the conus) which cause the RV to contract in an almost peristaltic pattern. The myofiber architecture of the RV consists of two layers (instead of three layers in the LV), where the superficial layer arrays more or less parallel to the atrioventricular groove, circumferentially surrounding the RV and eventually continuing into the superficial layer of the LV around the apex. Although the RV also has helical fibers (as is the case in the LV), it does not have an oblique middle layer of circumferential fibers (as present in the LV wall) and is therefore largely dependent on longitudinal shortening, in combination with torsion along with the LV. Furthermore, it moves along with the rotation of the LV apex relative to basal segments. The merged superficial layers of the RV and LV permit binding of the two ventricles together and forms the basis of ventricular interdependence (see further) [34].
13.2 Right Ventricular Physiology
In contrast to LV diastole, there is not an active RV diastolic relaxation-related ventricular sucking. Over 100 years ago, Starling demonstrated that the primary determinant effect of cardiac function on CO is how it inhibits venous return, not how contractile the myocardium is [3]. They and many others observed that the heart pumps out as much blood as it receives. Venous inflow (preload) or venous return, rather than P ra, determines CO. Starling made a striking comparison to a petrol engine when reviewing the function of the heart; the output only increased with an increase with venous inflow. Thus, the greater the volume of the heart, the more contractile the heart becomes [13]. This is not entirely true when it comes to RV physiology. In steady-state conditions, the RV can be ablated without causing a substantial increase in systemic venous pressures [36]. The primary role of the RV is to deliver all blood it receives into the pulmonary circulation. An acute increase in pulmonary vascular resistance (PVR) causes acute RV dilation and RV pump failure. A gradual increase in pulmonary artery pressure allows the RV to hypertrophy similar to LV remodeling due to an increase in systemic arterial pressure. Furthermore, Starling et al. observed that CO is independent of mean arterial pressure (MAP), albeit up to physiologic limits according to his experiments (>220 mmHg), after which the heart dilates and fails. Nevertheless, he also found that MAP is major determinant of coronary perfusion. Indeed, within physiological ranges of systemic arterial pressure, RV outflow is not impaired by changes in arterial pressure so long as venous return is sustained. Therefore, the right ventricle is the primary determinant of CO [14].
The primary function of the RV is to receive systemic venous return and pump it into the cardiopulmonary circulation. As explained earlier, RV function should be considered as the determinant of cardiac output, where LV function is a determinant of tissue organ perfusion, which will be focused on later on. RV contraction starts with contraction of the inlet and trabeculated myocardium followed by contraction of the infundibulum, the latter with a 25–50 ms later onset. The RV contraction pattern follows a three-step sequence, initiated by inward movement of the free wall (creating a bellowing effect), followed by contraction of the longitudinal fibers, shortening the long axis and drawing the tricuspid valve apparatus to the apex, and finally with traction on the free wall at the attachment points where RV is connected with the LV, secondary to its latter contraction [34].
RV contraction results in an early peaked and rapidly declining ventricular pressure curve, due to its low resistant and highly distensible pulmonary vascular tree into which blood is expelled. Also isovolumetric contraction phase is shorter than the LV, because RV systolic pressure rapidly exceeds the low pulmonary artery diastolic pressure earlier than LV systolic pressure exceeds aortic diastolic pressure.
Concerning afterload, the RV is more sensitive for afterload changes than the LV. RV stroke volume decreases almost 50 times faster with increasing intravascular pressure in comparison to the LV. In this light Guyton already questioned the flow-resistive properties of the pulmonary circulation, guided by the observation that a Fontan circulation falls short when pulmonary resistance increases too much. He wondered if it was purely an example of the RV falling short or that it could also be a lack of mean systemic filling pressure [37]. Although pulmonary vascular resistance (PVR) is the most commonly used parameter for RV afterload, a more complete model with both static and dynamic indices, as well as vascular impedance, intracavitary resistive properties, and valvular anomalies, would be more adept [34]. RV filling has an earlier onset in comparison to LV filling and finishes later. As with RV isovolumetric contraction phase, RV isovolumetric relaxation is shorter than LV, and RV filling pressures (E and A) are lower, with a lower E/A ratio. The influence of the respiratory variations on the RV filling is however higher than compared to LV.
14 Left Ventricle
14.1 Left Ventricular Anatomy
The design of the left ventricle is aimed at pressure capacity. Normal LV contraction is made possible by three key elements in LV construction. First, its helical layer of fibers which course between subendocardium and subepicardium determines both conical shape and movement pattern. However, this layer only contributes for 15% of length shortening in the LV contraction process and therefore contraction of LV oblique fibers further contributes to LV ejection. Furthermore, LV contains a middle layer consisting of circumferential constrictor fibers which cause the decrease of the internal diameter and thus being the driving force of the LV. The third important component of ejection is the rotation of the left ventricle from LV apex to the basal segments. LV contains three layers, obliquely oriented superficial myofibers, longitudinally oriented myofibers in the subendocardium, and circular fibers in between [1]. Its movement is a combination of torsion, translation, rotation, and thickening.
14.2 Left Ventricular Physiology
The basic hemodynamics of the heart are determined by three essential basic pump characteristics: (1) passive filling, (2) atrial effect (allows continuous uninterrupted venous inflow by dilation of the atria), and (3) intermittent or pulsatile outflow. With regard to the first characteristic, in vivo, this is shown in the fact that the heart is not sucking, though the chest is (raising negative pressure during forceful inspiration, creating negative intrathoracic pressure). Normally the end-systolic volume in the heart and the rapid filling from the normal inflow pressure never allow the heart to develop any negative pressure. The circulation rate is determined by extra-cardiac factors, as well as the balance between the systemic and pulmonary compartments. In addition, pulsatile outflow is shown to have a better diffuse perfusion of the body. Increases and decreases in diastolic pump filling correlate with elevations and decreases in arterial blood pressure. Vasopressors increase MAP by increasing P ms (as mentioned previously) and not by increasing resistance. Increased cardiac contraction in response to drugs, increased strength of myocardial contraction to increased diastolic filling (Starling), and increased heart rate are all observed in situations of increased cardiac output. They have been misinterpreted in the past as a cause of increased cardiac output, because they occurred concomitantly with an increase in cardiac output [5]. In this light, the primary function of the LV is to maintain arterial pressure (P sa) and flow, in order to guarantee organ perfusion [14].
14.3 Systolic Function
The systolic function of the left ventricle could be best explained by the pressure-volume relationship. The complex interaction between contractility, afterload, and preload of ventricular function (both RV and LV) is usually depicted in a pressure-volume relationship, with ventricular volume on the x-axis and ventricular pressure on the y-axis (Fig. 5.2).
It represents the ventricular pressure-volume changes within one cardiac cycle and can be divided into four phases:
-
1.
Filling phase. When ventricular pressures are at its minimum (end systole) and thus lower than atrial pressures, this allows for opening of the mitral/tricuspid valves and subsequently emptying of the atrial blood volume into the ventricles (diastole). This filling period consists thus of the early rapid filling phase, followed by diastasis and atrial systole. The slope of the passive ventricular distention is diastolic compliance. At the end of diastole, the pressure-volume relationship is at its minimum, and this point is considered to be a measure of diastolic compliance. However, it is influenced by extra-cardiac factors such as pericardial pressure, intrapulmonary pressure, and its neighboring ventricle based on the principle of interventricular dependence (see later section). As mentioned before, end-diastolic pressure is synonymous with preload.
-
2.
Isovolumetric contraction phase. When mechanical contraction occurs, intraventricular pressure increases, which allows the AV valves to close. Then the isovolumetric contraction phase follows, which is termed so because the volume of the ventricle does not change because all valves are closed.
-
3.
Ejection phase. When pressure further rises, the semilunar (aortic and pulmonary) valves open as soon as ventricular pressure exceeds aortic or pulmonary artery pressure. At this point, the curve represents maximal ventricular wall stress which is considered to be a measure of afterload (see earlier section). Under normal conditions maximal wall stress occurs at the beginning of ventricular ejection. The end-systolic pressure-volume relation, which can be approximated by a linear relationship with the slope of its relationship, is referred to as end-systolic elastance and is considered to be a good load-independent index of contractility (E es; end-systolic pressure-volume relationship) [28, 38, 39]. Time-varying elastance (E τ) is a marker of progressive stiffening of the ventricle during systole and its relaxation during diastole. It is calculated as a pair of isochronic pressure-volume relations during ejection and increases progressively from end diastole to end systole. Maximal elastance (E max) is the maximal LV pressure-volume ratio and usually occurs just after end systole [39].
-
4.
Isovolumetric relaxation phase. Once ejection has finished, ventricular relaxation commences. Diastolic relaxation or lusitropy, initiated by the isovolumetric pressure decline/isovolumetric relaxation phase, is an active process which requires energy. The semilunar valves close and ventricular pressures fall back to the diastolic pressure level.
14.4 Diastolic Function
Diastolic relaxation is an active process, which causes left ventricular intracavitary pressure to decrease fast and allow the ventricle to fill adequately without an abnormal increase in left atrial pressure. As mentioned before, the phases of diastole are isovolumic pressure decline and filling, of which the latter is divided into early rapid filling phase, followed by diastasis and atrial systole. The early rapid filling phase, driven by the LA-to-LV pressure gradient, contributes up to 80% of LV filling and decreases with aging. It is a complex combination of factors in which myocardial relaxation, LV elastic recoil, LV diastolic stiffness, LA pressures, ventricular interaction, pericardial constraint, pulmonary vein properties, and mitral orifice area play an important part. Diastasis contributes less than 5% to LV filling. Atrial systole only contributes up to 25% of LV filling; however, as mentioned before, its importance remains debated in the light of the reservoir and conduit function of the atria. Nevertheless, since the atrial kick is part of LV diastolic filling, it is worth mentioning that its function is dependent on the duration of the PR interval, atrial inotropic state, atrial preload, atrial afterload, autonomic tone, and heart rate. The process of LV filling consists of active relaxation and the recoil/suction, thereby “pulling” blood into the left ventricle. This enhances the LA-to-LV pressure gradient [40]. The isovolumic pressure decline can be quantitatively observed by measuring the peak rate of pressure fall (dP/dt.min) along with the time constant τ which is a marker of the rate of LV pressure decline during the isovolumetric relaxation process. A higher dP/dt.min signifies that relaxation rate is decreased and a larger value of τ implies it takes longer for the LV pressure to fall which is a sign of impaired relaxation. During systole potential kinetic energy is stored in the elastic elements of the myocytes, which is released during relaxation as the elastic element recoil and return to their original length and orientation. This greatly enhances LV pressure to fall which contributes to an early diastolic pressure gradient from LA to LV.
15 Ventricular Interdependence
Ventricular interdependence is characterized by the transmission of forces from one ventricle to another through the myocardium and pericardium. Systolic interaction refers to the positive contribution of both ventricles to each other [41]. The interventricular septum (IVS) plays a major role in modulating interventricular dependence in close conjunction with the pericardium. As mentioned in the anatomical section of both LV and RV, the latter is literally draped around the LV and has a more compliant character than the LV. Following contraction of the LV, the RV profits from its neighbor with a roughly 30% contribution to RV contraction and pulmonary flow [42]. This is explained by the concept of mechanical entrainment of both ventricles within the pericardium, but also LV contribution to systemic blood pressure, which is essential for RV coronary perfusion. Diastolic ventricular interaction accounts for the competition of space between the ventricles within the non-distensible pericardium. When LV end-diastolic volume (LVEDV) increases, this shifts the IVS toward the right, increasing RV end-diastolic pressure (RVEDP). Similarly, when RV end-diastolic volume (RVEDV) increases, this causes the IVS to shift leftward during diastole due to restrictions imposed by the pericardium on the RV cavity. This leftward shift impairs LV function by reducing LV volume, decreasing both LV filling and compliance which greatly decreases cardiac output eventually. It also increases muscle stiffness and LV wall tension [43].
16 Ventriculo-arterial Coupling
A proper physiologic function of the cardiovascular system is determined by the dynamic interaction between heart and systemic circulation. Both left ventricular systolic performance in terms of contractility and loading conditions (preload and afterload) and the arterial system (systemic arterial compliance, stiffness, and resistance) are in close conjunction. This phenomenon is termed ventriculo-arterial (VA) coupling. VA coupling can be defined as the ratio between arterial elastance and ventricular elastance (E es). An optimal E a/E es ratio implies an adequate left ventricular stroke volume at the lowest energy consumption. It is therefore an effective index of maximal cardiac performance and dynamic modulation of the arterial system. Also, at this point the lowest myocardial oxygen demand is required. As explained earlier, E es or the end-systolic pressure-volume relationship defines the intrinsic contractility of the heart. The steeper the slope, the greater contractility. E a consists of the total arterial afterload opposing the LV, taking into account systemic arterial compliance, stiffness, and resistance, which can be regarded as the ability of the vessels to process the increased pressure when LV stroke volume increases. It can be depicted as the slope from the LV end-diastolic volume to the LV end-systolic pressure as presented in Fig. 5.3 [44].

Left ventricular pressure-volume loop with LV ventricular elastance (E es) and arterial elastance (E a) slopes. Note the optimal point of VA coupling where both slopes intersect
17 Coronary Physiology
Myocardial oxygen uptake can be increased by preload, afterload, increased heart rate, or contractile function caused by beta-adrenergic stimulation. Both increased preload and afterload increase diastolic and systolic wall stress, respectively, which demands a higher oxygen uptake. Moreover, the concept of increased wall stress generating increased oxygen uptake is of crucial importance in relation to heart size, as a larger radius increases wall stress (see previous sections). Since LV coronary flow occurs only in diastole, the LV may be subjected to (subendocardial) ischemia with increased LV wall stress (i.e., with increased LV end-diastolic pressure, LVEDP). This is slightly different in the RV, where the myocardium is being perfused during both diastole and systole, because RV wall stress is usually low and can easily be overcome by coronary artery pressure. However, in pulmonary hypertension states, treatment must increase diastolic arterial pressure greater than pulmonary artery pressure to insure right ventricular blood flow and prevent acute cor pulmonale [45].
18 Pericardium
The most important function of the pericardium is its modulation of cardiac volume on which it has a restraining effect. This is obtained by the elasticity of the pericardium which reaches a limit at a certain point, thereby inhibiting the cardiac chambers from further dilating or overdistension. In this light, more specifically pericardial contact pressure, which is the pressure representing transmission of the pericardial pressure, determines to a great extent the upper limits of normal cardiac filling. This goes especially for the right-sided heart, since those filling pressure are lower than left-sided. Furthermore, it augments contractility of the RV through systolic ventricular interdependence and augments LV contractility by limiting energy loss by rotational changes during contraction. The normal pericardium also contributes to diastolic interaction between the ventricles via interventricular dependence. Thus it contributes to intracavitary filling pressures both through direct external contact pressure and because of increased diastolic interaction [46,47,48].
19 Concluding Remarks
In this chapter the interplay of the systemic circulation and the heart has been discussed. Clearly, there is a role for mean systemic filling pressure (P ms) in the determination of intravascular fluid status and its consequences for the amount of preload. Where it comes to the right side of the heart, preload is essential for its primary role which by its volume-pump function supplied by venous return greatly determines cardiac output. In between, the atrial function was highlighted, where it became clear that atrial kick does not portend the major contributor to cardiac output, but the atrial reservoir and conduit function do. The left ventricle on the contrary has a primary role in the determination of organ perfusion. Together, via its complex interaction of interventricular dependence and physiology of the pericardium, they determine total work of the heart in both steady state as during strenuous exercise and critical illness circumstances. Each of these parts in the chain plays their own major role, and failure of one of each has great implications for the performance and prognosis of the patient.
References
Katz AM, Rolett EL. Heart failure: when form fails to follow function. Eur Heart J. 2016;37:449–54.
Funk DJ, Jacobsohn E, Kumar A. The role of venous return in critical illness and shock–part I: physiology. Crit Care Med. 2013;41:255–62.
Gelman S. Venous function and central venous pressure—a physiologic story. Anesthesiology. 2008;108:735–48.
Bressack MA, Raffin TA. Importance of venous return, venous resistance and mean circulatory pressure in the physiology and management of shock. Chest. 1987;92:906–12.
Anderson RM, Fritz JM, O’Hare JE. The mechanical nature of the heart as a pump. Am Heart J. 1967;73(1):92–105.
Anderson RM. The gross physiology of the cardiovascular system. Tucson, AZ: Racquet Press; 1993.
Guyton AC, Abernathy B, Langston JB, Kaufmann BN, Fairchild HM. Relative importance of venous and arterial resistances in controlling venous return and cardiac output. Am J Physiol. 1954;179(2):261–7.
Guyton AC, Polizo D, Armstrong GG. Mean circulatory filling pressure measured immediately after cessation of heart pumping. Am J Physiol. 1959;196(5):1008–14.
Guyton AC, Lindsey AW, Kaufmann BN. Effect of mean circulatory filling pressure and other peripheral circulatory factors on cardiac output. Am J Physiol. 1955;180(3):463–8.
Guyton AC, Lindsey AW, Abernathy B, Richardson T. Venous return at various right atrial pressures and the normal venous return curve. Am J Physiol. 1957;189(3):609–15.
Beard DA, Feigl EO. Understanding Guyton’s venous return curves. Am J Physiol Heart Circ Physiol. 2011;301:629–33.
Grodins FS, Stuart WH, Veenstra RL. Performance characteristics of the right heart bypass preparation. Am J Physiol. 1960;198:552–60.
Starling EH. The Linacre Lecture on the Law of the Heart. London: Longmans, Green & Company; 1918.
Rothe CF. Mean circulatory filling pressure: its meaning and measurement. J Appl Physiol. 1993;74:499–509.
Pinsky MR. Mean systemic pressure monitoring. In: Cannesson M, Pearse R, editors. Perioperative hemodynamic monitoring and goal directed therapy. Cambridge: Cambridge University Press; 2014. p. 157–62.
Pinsky MR. Instantaneous venous return curves in an intact canine preparation. J Appl Physiol. 1984;56:765–71.
Versprille A, Jansen JR. Mean systemic filling pressure as a characteristic for venous return. Pflugers Arch. 1985;405:226–33.
Maas JJ, Pinsky MR, Geerts BF, de Wilde RB, Jansen JR. Estimation of mean systemic filling pressure in postoperative cardiac surgery patients with three methods. Intensive Care Med. 2012;38:1452–60.
Maas JJ, Geert BF, Van den Berg PC, Pinsky MR, Jansen JRC. Assessment of venous return curve and mean systemic filling pressure in postoperative cardiac surgery patients. Crit Care Med. 2009;37:912–8.
Schipke JD, Heusch G, Sanii AP, Gams E, Winter J. Static filling pressure in patients during induced ventricular fibrillation. Am J Physiol Heart Circ Physiol. 2003;285:2510–5.
Repessé X, Charron C, Fink J, Beauchet A, Deleu F, Slama M, Belliard G, Vieillard-Baron A. Value and determinants of the mean systemic filling pressure in critically ill patients. Am J Physiol Heart Circ Physiol. 2015;309:1003–7.
Parkin WG, Wright CA. Three dimensional closed loop control of the human circulation. J Clin Monit Comput. 1991;8:35–42.
Parkin WG, Wright CA, Bellomo R, Boyce N. Use of a mean systemic filling pressure analogue during the closed-loop control of fluid replacement in continuous hemodiafiltration. J Crit Care. 1994;9:124–33.
Parkin WG. Volume state control—a new approach. Crit Care Resusc. 1999;1:311–21.
Parkin WG, Leaning MS. Therapeutic control of the circulation. J Clin Monit Comput. 2008;22:391–400.
Pellegrino VA, Mudaliar Y, Gopalakrishnan M, Hortons MD, Killick CJ, Parkin WG, Playford HR, Raper RF. Computer based haemodynamic guidance system is effective and safe in management of postoperative cardiac surgery patients. Anaesth Intensive Care. 2011;39:191–201.
Cecconi M, Hollmann DA, Geisen M, Ebm C, Fletcher N, Grounds RM, Rhodes A. Changes in the mean systemic filling pressure during a fluid challenge in postsurgical intensive care patients. Intensive Care Med. 2013;39:1299–305.
Opie LH, Bers DM. Mechanisms of cardiac contraction and relaxation. In: Libby P, Bonow RO, Mann DL, Zipes DP, editors. Heart disease: a textbook of cardiovascular medicine. 10th ed. Philadelphia: WB Saunders; 2015. p. 429–53.
Pagel PS, Kehl F, Gare M, et al. Mechanical function of the left atrium: new insights based on analysis of pressure-volume relations and Doppler echocardiography. Anesthesiology. 2003;98:975.
Mitchell JH, Gilmore JP, Sarnoff SJ. The transport function of the atrium. Factors influencing the relation between mean left atrial pressure and left ventricular end-diastolic pressure. Am J Cardiol. 1962;9:237.
Mitchell JH, Gupta DN, Payne RM. Influence of atrial systole on effective ventricular stroke volume. Circ Res. 1965;17:11.
Grandi E, Pandit SV, Voigt N, et al. Human atrial action potential and Ca2+ model: Sinus rhythm and chronic atrial fibrillation. Circ Res. 2011;109:1055.
Von Anrep G. On the part played by the suprarenals in the normal vascular reactions of the body. Am J Physiol. 1912;45:307–17.
Haddad, et al. Right ventricular function in cardiovascular disease, part I. Anatomy, physiology, aging and functional assessment of the right ventricle. Circulation. 2008;117:1436–48.
Ho SY, Nihoyannopoulos P. Anatomy, echocardiography, and normal right ventricular dimensions. Heart. 2006;92(Suppl 1):i2–i13.
Starr I, Jeffers WA, Meade RH. The absence of conspicuous increments of venous pressure after severe damage to the right ventricle of the dog, with a discussion of the relation between clinical congestive failure and heart disease. Am Heart J. 1943;26:291–301.
Guyton AC, Lindsey AW, Gilluly JJ. The limits of right ventricular compensation following acute increase in pulmonary circulatory resistance. Circ Res. 1954;4:326–32.
Suga H, Sugawa K. Instantaneous pressure–volume relationships and their ratio in the excised supported canine left ventricle. Circ Res. 1974;35:117.
Pinsky MR. Circulation and circulatory support in the critically ill. In: Warrell DA, Cox TM, Firth JD, editors. Oxford textbook of medicine. 5th ed. Oxford: Oxford University Press; 2015.
Little WC. Diastolic dysfunction beyond distensibility: adverse effects of ventricular dilatation. Circulation. 2005;112:2888.
Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmüller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013;128:2016–25.
Damiano RJ Jr, La Follette P Jr, Cox JL, Lowe JE, Santamore WP. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261:H1514–24.
Barthélémy R, Gayat E, Mebazaa A. Pathophysiology and clinical assessment of the cardiovascular system (including pulmonary artery catheter). In: Tubaro M, Vranckxx P, Price S, Vrints C, editors. The ESC textbook of intensive and acute cardiovascular care. 2nd ed. Oxford: Oxford University Press; 2015. p. 101–10.
Guarracino F, Baldassarri R, Pinsky MR. Ventriculo-arterial decoupling in acutely altered hemodynamic states. In: Vincent JL, editor. Yearbook of intensive care and emergency medicine. Heidelberg: Springer; 2013. p. 225–36.
Feigl EO. Coronary physiology. Physiol Rev. 1983;63:1.
Johnson D. The pericardium. In: Standring S, et al., editors. Gray’s anatomy. New York: Elsevier Churchill Livingstone; 2005. p. 995–6.
Jöbsis PD, Ashikaga H, Wen H, et al. The visceral pericardium: macromolecular structure and contribution to passive mechanical properties of the left ventricle. Am J Physiol. 2007;293:H3379.
Eeffe O’Rourke RA, Dell’Italia LJ. Diagnosis and management of right ventricular myocardial infarction. Curr Probl Cardiol. 2004;29:6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Meijs, L.P.B., Bindels, A.J.G.H., Bakker, J., Pinsky, M.R. (2018). Cardiac Function (Cardiac Output and Its Determinants). In: Pinto Lima, A., Silva, E. (eds) Monitoring Tissue Perfusion in Shock. Springer, Cham. https://doi.org/10.1007/978-3-319-43130-7_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-43130-7_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43128-4
Online ISBN: 978-3-319-43130-7
eBook Packages: MedicineMedicine (R0)