
Abstract
The seven members of the signal transducer and activator of transcription (STAT) family of proteins are transcription factors that are activated in response to, and mediate signaling downstream of, growth factors and cytokines. STATs are dysregulated in a broad range of cancer types. Although the genes that encode STATs are rarely mutated in cancer, constitutive phosphorylation and hence activation of STATs, particularly STAT3, is a common alteration in cancer. STAT3 and STAT5 are considered to play primarily pro-tumorigenic roles in tumor cells and within the tumor microenvironment (TME), while STAT1 has been described as a tumor suppressor (although recent publications have also revealed pro-tumorigenic functions of STAT1). In this chapter, we survey STATs in cancer, providing a general overview of STAT function and regulation in tumor cells and in immune cells within the TME.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
The signal transducer and activator of transcription (STAT) family comprises seven structurally similar proteins (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) that can function as both signaling proteins and transcription factors. STAT5A and STAT5B are encoded by two different genes that generate highly homologous proteins [1, 2]. Although STAT5A and STAT5B are distinct proteins with overlapping but non-redundant functions, they are often referred to collectively as STAT5.
Each STAT protein consists of six functionally conserved domains, including an SH2 domain and the C-terminal transactivation domain (TAD), which can be phosphorylated on a conserved tyrosine residue (Tyr705 in STAT3) [3–6]. Tyrosine phosphorylation of STATs often occurs downstream of cytokine and growth factor receptors. STAT protein phosphorylation leads to STAT dimerization and translocation into the nucleus, where the STAT dimers can activate or repress transcription. Thus, phosphorylation of STATs links growth factor and cytokine signaling to gene expression.
Tyrosine phosphorylation of the TAD domain is the most well-characterized post-translational modification of STAT proteins. Serine phosphorylation of STATs also occurs and has been shown to be dysregulated in cancer [1, 4, 7–12]. Additional STAT regulatory mechanisms include ubiquitination, sumoylation, acetylation, and interactions with protein inhibitor of activated STAT (PIAS) proteins, which block STAT-DNA binding. This chapter will focus on the regulation of tyrosine phosphorylation of STATs in cancer. Recent reviews have addressed alternative STAT regulatory mechanisms [1, 3, 13–15].
2.2 Tyrosine Phosphorylation of STAT Proteins
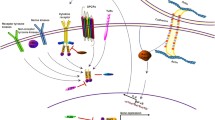
In normal (non-transformed) cells, tyrosine phosphorylation of STAT proteins is triggered by the binding of growth factors and cytokines to their cognate receptors. Though the precise mechanism of activation is specific to each ligand/receptor complex, a common mechanism of STAT phosphorylation downstream of these receptors is by members of the Janus kinase (JAK) family of non-receptor tyrosine kinases (JAK1, JAK2, JAK3, and TYK2) [4–7, 13, 16, 17] (Fig. 2.1).

IL-6-induced activation of JAK/STAT3 signaling and gene expression. STAT proteins are important mediators of signal transduction downstream of cytokine and growth factor receptors. Depicted here is STAT3-mediated IL-6 signaling. Binding of IL-6 to IL-6 receptor α (IL-6Rα) induces formation of the IL-6 receptor complex. This leads to activation of JAK family kinases (often JAK1, but also JAK2 or TYK2), which can subsequently phosphorylate several tyrosine residues on gp130. The SH2 domain of STAT3 can then bind to phosphorylated gp130, positioning STAT3 for phosphorylation by JAKs. This promotes STAT3 dimerization, which occurs via reciprocal interactions between the SH2 domain of one STAT3 molecule and the tyrosine-phosphorylated transactivation domain (TAD) of another STAT. STAT3 homodimers can be transported into the nucleus and promote expression of many genes. Shown are examples of STAT3 target genes that promote tumor cell proliferation (CCND1, MYC), protection from apoptosis (BCL2L1, BCL2), and immunosuppression in the TME (IL6). Notably, STAT3 induction of IL6 gene expression generates a feed-forward loop that further drives IL-6/JAK/STAT3 signaling. On the other hand, STAT3 also promotes expression of the gene encoding SOCS3 (an inhibitor of JAK1, JAK2, and TYK2). This generates a negative feedback loop that can be disrupted by hypermethylation of the SOCS3 promoter, which has been detected in several cancer types. IL-6 signaling can also lead to activation of STAT1, which can reduce STAT3 homodimerization by sequestering STAT3 molecules in STAT1:STAT3 heterodimers
Following receptor dimerization, JAKs are recruited to and phosphorylate intracellular tyrosine residues on these receptors [4–7, 13, 16, 17]. For some receptors, phosphorylation of these sites can also be accomplished by autophosphorylation. This creates docking sites for STAT proteins, as the SH2 domains of STATs can bind the phosphorylated residues and, in turn, become phosphorylated by JAKs at the conserved tyrosine residue within the TAD. Phosphorylation at this site promotes STAT homo- or heterodimerization via reciprocal interactions between the SH2 domain of one STAT molecule and the tyrosine-phosphorylated TAD of its dimerization partner. Phosphorylated STAT dimers can be recognized by importins and transported into the nucleus [3, 7, 18], where they can activate or repress gene expression. It should be noted that, while JAKs are the primary mediators of STAT tyrosine phosphorylation downstream of cytokine and growth factor receptors, other kinases have also been shown to phosphorylate STATs.
Given the importance of tyrosine phosphorylation for STAT function and the involvement of STATs in cellular processes that are often dysregulated in cancer, it is not surprising that aberrant phosphorylation of STATs has been observed in many cancer types. Constitutive phosphorylation of STAT proteins often occurs downstream of oncogenic proteins and/or as a result of increased secretion of cytokines or growth factors in the TME. Oncogenic proteins can drive STAT phosphorylation independent of extracellular ligands, uncoupling STAT protein phosphorylation from growth factor/cytokine signaling, while increased secretion of cytokines or growth factors in the TME can elicit STAT protein hyperphosphorylation by activating receptors upstream of these STATs [4, 7, 13, 19–22]. Notably, these secreted factors can induce phosphorylation of STATs not only in tumor cells, but also in stromal cells and tumor-infiltrating immune cells.
2.3 Negative Regulators of STAT Signaling
Spatial and temporal regulation of STAT protein phosphorylation is coordinated by a number of phosphatases. While some of these phosphatases act directly on STATs, phosphatases targeting upstream molecules can also elicit downregulation of STAT phosphorylation. Loss of expression or function of these phosphatases or other inhibitors of the JAK/STAT pathway can lead to constitutive activation of STAT proteins and contribute to the malignant phenotype [6, 19, 23–25].
Among the STAT pathway inhibitors that have been shown to be dysregulated in cancer are members of the protein tyrosine phosphatase (PTP) and suppressor of cytokine signaling (SOCS) families [3–6, 17, 19, 23–28]. Interestingly, several of the genes encoding SOCS proteins, which downregulate STAT signaling via inhibition of growth factor/cytokine receptors and members of the JAK family of protein tyrosine kinases, are STAT transcriptional targets [28–30]. This negative feedback loop is disrupted in malignant cells that exhibit hypermethylation of SOCS gene promoters [19, 25, 31].
2.4 STAT Function in the Nucleus
STAT protein dimers are transported into the nucleus by importins [3, 7, 18]. Once inside, STAT proteins can either promote or downregulate gene expression, often by cooperating with co-activators and co-repressors of transcription [1, 3, 12, 15]. Thus, STAT target gene expression can be shaped by not only the expression, phosphorylation, and nuclear translocation of STAT proteins themselves, but also by a cadre of transcriptional co-regulators.
It should be noted that, although tyrosine phosphorylation of STAT proteins plays a major role in STAT function, dimerization can occur independent of tyrosine phosphorylation, and unphosphorylated STAT proteins have also been shown to enter the nucleus and activate gene transcription, often in cooperation with other transcription factors [15, 19, 32, 33]. For example, unphosphorylated STAT3 can promote transcription of the oncogene MET in cooperation with nuclear factor kappa B (NF-KB) [32, 34].
2.5 STAT Proteins in Tumor-Infiltrating Immune Cells
The mechanisms that regulate STATs within tumor cells also govern their functions in immune cells, wherein STATs have been shown to play diverse roles in innate and adaptive immune cells in the TME. While STAT2 and STAT4 promote the anti-tumor immune response, STAT3 and STAT6 mediate immunosuppression in the TME, and STAT1 and STAT5 have been implicated in both activation and suppression of the anti-tumor immune response (Table 2.1). Thus, the roles of STAT proteins in cancer extend beyond their functions in tumor cells themselves. It is now well-established that immunosuppression in the TME contributes to tumor progression, and therapies that activate the anti-tumor immune response have demonstrated efficacy in a number of cancer types. The functions of STATs in tumor-infiltrating immune cells will be discussed alongside their tumor cell-intrinsic roles in the following sections.
2.6 STAT1
STAT1 was initially considered to function primarily as a tumor suppressor. Though studies continue to demonstrate tumor suppressive roles of STAT1, pro-tumorigenic roles of STAT1 have also been identified.
2.6.1 STAT1 Opposes Tumor Cell Proliferation and Survival
STAT1 can oppose cell proliferation through the activation of genes that promote growth arrest and through mechanisms independent of its role as a transcription factor. Several STAT1 target genes encode proteins that negatively regulate cell cycle progression, including the cyclin-dependent kinase (CDK) inhibitors p21Cip1/Waf1 (gene name: CDKN1A) and p27Kip1 (CDKN1B) [11, 27]. STAT1 can also promote stabilization of p27Kip1 through transcriptional repression of the gene encoding S-phase kinase-associated protein 2 (Skp2), a ubiquitin ligase that tags p27Kip1 for proteasomal degradation [35]. In addition, serine-phosphorylated STAT1 can block progression through G1 by interacting with the cyclin D1/CDK4 complex and inducing proteasome-mediated degradation of cyclin D1 [9].
STAT1 can inhibit proliferation by repressing transcription of the proto-oncogene MYC [12, 27]. It should be noted, however, that STAT1 was recently identified as a positive regulator of MYC transcription in serous papillary endometrial cancer (SPEC) and thus acted as a driver of tumor progression in this cancer type [36]. STAT1 can promote apoptosis by activating the expression of pro-apoptotic genes and inhibiting expression of pro-survival genes [27]. On the other hand, unphosphorylated STAT1 has been shown to protect cells from apoptosis by suppressing the expression of Fas and Bad [37].
2.6.2 STAT1 Can Promote or Inhibit the Anti-Tumor Immune Response
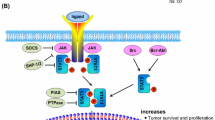
Additional pro- and anti-tumorigenic roles of STAT1 have emerged from studies on STAT1 in tumor-infiltrating immune cells and in modulation of the anti-tumor immune response by tumor cells (Table 2.1, Fig. 2.2). Many functions of STAT1 in cancer are linked to its role as a mediator of type I and type III interferon signaling.

Roles of STAT1 in tumor cells and immune cells within the TME. STAT1 is thought to act primarily as a tumor suppressor through its ability to inhibit growth and promote apoptosis of tumor cells and through its promotion of Th1-type anti-tumor immune responses (left side of figure). STAT1 can promote the activation of tumor cell-targeting Th1 cells by DCs and mediate type I interferon-induced activation of anti-tumor (M1) macrophages (MΦ) and CD8+ T cells. However, STAT1 can also promote expansion of immunosuppressive MDSCs and M2 polarization of MΦ (right side of figure), and can induce expression of PD-L1 on tumor cells, protecting them from T cell-mediated lysis
T helper 1 (Th1) immune responses are characterized by the activation of the Th1 subset of CD4+ T cells, which can drive anti-tumor immune responses by releasing pro-inflammatory cytokines such as interferon gamma (IFN-γ) that can mobilize anti-tumor macrophages and cytotoxic CD8+ T cells [13, 16, 38]. STAT1 is an important mediator of the Th1 immune response, as it promotes the expression of IL-12 (a cytokine that induces the polarization of naïve CD4+ T cells into Th1 cells) and mediates the expression of many IFN-γ-inducible genes [13, 16, 39]. Among these genes are those encoding class I major histocompatibility complex (MHC) and co-stimulatory molecules, which are required for effective antigen presentation to and activation of anti-tumor T cells by dendritic cells (DCs) [2, 7, 33]. This implicates STAT1 in the anti-tumor immune response.
STAT1 can antagonize the anti-tumor immune response by inducing expression of the gene encoding programmed death-ligand 1 (PD-L1), an immune checkpoint molecule [8, 40, 41]. PD-L1 expressed on tumor cells engages the inhibitory receptor programmed death-1 (PD-1) on activated natural killer (NK) and T cells in the tumor microenvironment, thereby protecting tumor cells from NK- and T-cell-mediated destruction [8, 41]. A recent study identified activation of the JAK2/STAT1 axis in response to epidermal growth factor (EGF) and interferon gamma (IFN-γ) in head and neck cancer cells [8]. In this system, inhibition of JAK2 abrogated STAT1-dependent expression of PD-L1 and enhanced the ability of NK cells to lyse tumor cells [8].
An additional mechanism by which STAT1 promotes tumor immune evasion is through the induction of myeloid-derived suppressor cells (MDSCs) [27, 42, 43]. MDSCs are a heterogeneous class of immature myeloid cells that share the ability to suppress both innate and adaptive immune cells, thereby impeding the anti-tumor immune response [42]. Immunosuppressive cytokines trigger the expansion of MDSCs, and STAT1 has been shown to promote their accumulation within tumors [27, 43].
STAT1 has also been implicated in immune suppression mediated by another subset of cells of the myeloid lineage: tumor-associated macrophages (TAMs). Macrophages in the tumor microenvironment tend to be polarized toward the immunosuppressive type 2 (M2) phenotype. These TAMs oppose the anti-tumor immune response and are associated with poor prognosis in cancer [44–46]. STAT1 has been implicated in the expansion of M2-polarized macrophages in mouse mammary tumors [44] and in the immunosuppressive functions of M2 TAMs [47]. STAT1 has also been shown to promote M1 macrophage polarization, which is thought to promote the anti-tumor immune response [45, 46].
Overall, the evidence suggests that whether STAT1 functions as a tumor promoter or suppressor is context-specific [27, 33]; i.e., while STAT1 functions as a tumor suppressor by inhibiting tumor cell proliferation and survival in many cancer types, tumor-promoting roles of STAT1 have also been identified (for example, in serous papillary endometrial cancer) [36]. In addition, while STAT1 is a critical mediator of the Th1 response and thereby promotes anti-tumor immunity, it can also effect immunosuppression through expansion of MDSCs and upregulation of the immune checkpoint molecule PD-L1 on tumor cells.
2.7 STAT3
In contrast to STAT1, the functions of STAT3 identified in cancer thus far have been almost exclusively pro-tumorigenic. STAT3 is well-established as a proto-oncogene [3, 48], and constitutive activation of STAT3 has been observed in a broad range of cancer types. In addition, ample evidence implicates STAT3 in suppression of the anti-tumor immune response.
2.7.1 STAT3 Promotes Tumor Cell Proliferation, Survival, Invasion, and Metastasis
Like the other STAT proteins, STAT3 is rarely mutated in cancer. However, STAT3 is phosphorylated downstream of a number of oncogenes, including EGFR [49–51], Src [19, 51, 52], and c-MET [19, 51]. Secretion of STAT3-activating growth factors and cytokines, such as IL-6, and hypermethylation of or loss-of-function mutations in the genes encoding negative regulators of STAT3 signaling, such as SOCS3 or the phosphatases PTPRD and PTPRT, are additional mechanisms by which STAT3 can be constitutively phosphorylated in cancer [4, 23, 24, 31].
The pro-tumorigenic functions of STAT3 stem in part from its ability to activate genes that promote proliferation, protect cells from apoptosis, stimulate angiogenesis, and drive invasion and metastasis [3, 13, 22, 33]. STAT3 target genes that induce cell proliferation include those encoding cyclin D1 (CCND1) and c-Myc (MYC) [4, 13, 15, 32, 33, 48, 53, 54]. Tumor cell survival can be enhanced by STAT3-mediated expression of the genes BCL2, BCL2L1, and BIRC5, which encode the anti-apoptotic proteins Bcl-2, Bcl-xL and Survivin, respectively [4, 5, 10, 13, 15, 16, 32, 33, 54]. STAT3 promotes angiogenesis in part by activating transcription of the gene encoding vascular endothelial growth factor (VEGF). VEGF, in turn, can promote activation of STAT3 [4, 10, 15, 55, 56]. Additional mediators of STAT3-induced angiogenesis are the matrix metalloproteinases MMP-2, MMP-7, and MMP-9, which degrade the extracellular matrix and basement membrane, facilitating angiogenesis and tumor cell invasion and metastasis [13, 16, 33, 56, 57]. STAT3 also induces epithelial–mesenchymal transition (EMT), a transdifferentiation program that has been shown to enable metastasis, by promoting expression of the EMT-associated transcription factors Snail (SNAI1), Twist (TWIST1), and ZEB1 (ZEB1) [56, 58–62].
Another key function of STAT3 is mediating resistance to cancer therapy, including, but certainly not limited to, the EGFR-targeted monoclonal antibody cetuximab [63], the Src-family kinase inhibitor dasatinib [64], and chemotherapy [20, 65]. In a recent paper, feedback activation of STAT3 was found to mediate resistance to a number of oncogene-targeted therapies [66]. The authors first identified a STAT3-activating feedback loop in an EGFR-mutant non-small cell lung cancer (NSCLC) cell line (PC-9) treated with the EGFR tyrosine kinase inhibitor (TKI) erlotinib. In these cells, erlotinib treatment led to the secretion of molecules that induced tyrosine phosphorylation of STAT3. Exposing erlotinib-naïve PC-9 cells to conditioned medium from erlotinib-treated cells could induce resistance to erlotinib, and knockdown of STAT3 abrogated this effect, demonstrating that inhibition of EGFR could paradoxically drive STAT3 activation and induce STAT3-mediated drug resistance through secretion of STAT3-activating factors. Feedback activation of STAT3 via this mechanism was subsequently observed in many other oncogene-addicted cancer cell lines treated with an inhibitor targeting their driver oncogene. Thus, cumulative evidence supports activation of STAT3 as a common mechanism of resistance to cancer therapy and suggests that targeting STAT3 is a rational strategy to overcome resistance, as has been suggested previously [63, 65, 66].
The STAT3-activating feedback loop reported by Lee and colleagues was identified in the absence of immune cells, but highlights the paradigm of secreted factors in the tumor microenvironment inducing STAT3 phosphorylation within tumor cells [25, 67]. These secreted factors, which may be tumor-, stroma-, and/or immune cell-derived, can also effect STAT3 activation in tumor-infiltrating immune cells, thereby promoting tumor immune evasion.
2.7.2 Activation of STAT3 in Immune Cells in the TME Dampens the Anti-Tumor Immune Response
Activation of STAT3 in tumor cells can promote expression of the genes encoding the immunosuppressive cytokines IL-6, IL-10, and vascular endothelial growth factor (VEGF), which can promote the continued activation of STAT3 in tumor cells in an autocrine or paracrine manner [16, 55, 68]. These cytokines can also drive activation of STAT3 within tumor-infiltrating innate and adaptive immune cells, thereby promoting immunosuppression in the TME [16, 68] (Table 2.1, Fig. 2.3).

Roles of STAT3 in tumor cells and immune cells within the TME. Activation of STAT3 in tumor cells promotes proliferation, survival, and secretion of the immunosuppressive cytokines IL-6, IL-10, and VEGF. These cytokines can feed back to tumor cells in an autocrine or paracrine manner to further activate STAT3 in tumor cells. In addition, these cytokines can induce phosphorylation of STAT3 in innate and adaptive immune cells in the TME. Activation of STAT3 in MDSCs promotes their expansion and their ability to secrete immunosuppressive enzymes such as arginase-I and IDO. STAT3 promotes M2 polarization of macrophages (MΦ) and inhibits maturation of DCs. STAT3 can also promote differentiation of Th17 and Treg cells and mediate their secretion of IL-17 and IL-22, and IL-10 and TGF-β, respectively. Collectively, activation of STAT3 in tumor-infiltrating immune cells facilitates immunosuppression in the TME
Like STAT1, STAT3 can promote the expansion of MDSCs in the TME [16, 42, 68]. Tumor-derived S100A9 protein, the expression of which is promoted by STAT3, drives accumulation of MDSCs [42, 69]. Moreover, STAT3 mediates the immunosuppressive functions of MDSCs by inducing their production of the T cell-suppressive enzymes arginase-I and indoleamine 2,3-dioxygenase (IDO) [70, 71]. STAT3 has also been shown to mediate the secretion of pro-angiogenic factors by MDSCs [16, 22].
STAT3 further promotes immunosuppression in the TME by driving M2 polarization of TAMs and inhibiting dendritic cell (DC) maturation. Activation of STAT3 in TAMs inhibits secretion of pro-inflammatory cytokines and promotes secretion of immunosuppressive cytokines (such as IL-6 and IL-10) that activate STAT3 in DCs [13, 16, 21, 45, 68, 72–74]. STAT3 inhibits the functional maturation of DCs, impeding their ability to activate T cells to mount an effective anti-tumor immune response [13, 21, 45, 68, 72–75].
Activation of STAT3 in naïve CD4+ T cells can promote their differentiation into Th17 cells, a T-cell population associated with tumor progression [33, 68, 76, 77]. In addition, STAT3 is implicated in the expansion and immunosuppressive functions of regulatory T cells (Tregs) [78]. STAT3 mediates expression of immunosuppressive cytokines in both Tregs (which produce IL-10 and transforming growth factor (TGF)-β) and Th17 cells (IL-17 and IL-22) [16, 77]. Secretion of these cytokines can further facilitate immunosuppression in the TME [16].
2.8 STAT5
STAT5 is often implicated in hematologic malignancies, where it is activated downstream of the oncogenic fusion protein BCR-ABL (in chronic myelogenous leukemia (CML)) and as a result of activating mutations in JAK proteins [1, 16, 18, 19]. In solid tumors, cytokines often drive activation of STAT5 [19].
Compared to STAT3, relatively little is known about the role of STAT5 in the anti-tumor immune response. While expression of a constitutively active STAT5 mutant in CD8+ T cells was shown to promote their ability to lyse tumor cells in an immunocompetent mouse model of melanoma [79], suggesting that STAT5 can promote the anti-tumor immune response, STAT5 can also mediate IL-2-induced differentiation of Tregs, known antagonists of the anti-tumor immune response [13, 16, 78].
2.9 STAT2, STAT4 and STAT6
The remaining STAT proteins (STAT2, STAT4, and STAT6) have not been as extensively studied in the context of cancer, but functions for each of these proteins in tumor cells and/or immune cells in the TME have nonetheless been identified.
STAT2 and STAT4 participate in Th1 anti-tumor immune responses. STAT4 mediates IL-12-induced expression of IFN-γ [16, 80], while STAT2, operating as a heterodimer with STAT1, promotes expression of IFN-γ-stimulated genes [16, 81].
Evidence suggests that STAT6 primarily mediates pro-tumorigenic functions through its promotion of tumor cell proliferation and survival, particularly in hematologic malignancies [16, 82, 83], and through suppression of the anti-tumor immune response. STAT6 is activated in response to the cytokines IL-4 and IL-13 and mediates the immunosuppressive effects of these cytokines [82]. STAT6 promotes M2 polarization of macrophages and the expansion of MDSCs in the TME [16, 42, 84, 85]. In addition, STAT6 impairs CD8+ T cell tumor infiltration by inducing downregulation of very late antigen-4 (VLA-4, or integrin α4β1), which mediates migration of T cells into tumors [16, 86].
2.10 Conclusion
STAT biology is complex, and both pro- and anti-tumorigenic effects have been described for each STAT protein. STATs play roles in tumor cells as well as other cells in the TME, including tumor-infiltrating immune cells. As such, any attempt to utilize STAT inhibitors must consider the effects of these inhibitors on immune cells as well as on the tumor cells. Modulation of STAT activity in tumor-infiltrating immune cells does not appear to be a side effect of STAT inhibitors; rather, this may be critical for their anti-tumor efficacy. For example, STAT3 inhibitors would be predicted to exert their anti-tumor effects by both abrogating expression of STAT3-regulated genes in tumor cells themselves and antagonizing STAT3-mediated immunosuppression in the TME. Indeed, the anti-tumor efficacy of the STAT3 antisense oligonucleotide AZD9150 is currently thought to stem primarily from its ability to enhance the anti-tumor immune response [87]. STAT5 inhibitors are also in development for cancer treatment, and the bromodomain and extra-terminal (BET) family bromodomain inhibitor JQ1, which inhibits STAT5, has been shown to impact both tumor and immune cells [88–90]. Thus, administering STAT inhibitors, particularly inhibitors of STAT3, may be a promising way to target both tumor cells and the TME and elicit an effective anti-tumor therapeutic response.
References
Rani A, Murphy JJ (2015) STAT5 in cancer and immunity. J Interferon Cytokine Res 36(4):226–237
Li HS, Watowich SS (2013) Diversification of dendritic cell subsets: emerging roles for STAT proteins. JAKSTAT 2:e25112
Santos CI, Costa-Pereira AP (2011) Signal transducers and activators of transcription—from cytokine signalling to cancer biology. Biochim Biophys Acta—Rev Cancer 1816:38–49
Costa-Pereira AP, Bonito NA, Seckl MJ (2011) Dysregulation of Janus kinases and Signal transducers and activators of transcription in cancer. Am J Cancer Res 1:806–816
Dorritie KA, Redner RL, Johnson DE (2014) STAT transcription factors in normal and cancer stem cells. Adv Biol Regul 56:30–44
Xu D, Qu C-K (2008) Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci 13:4925–4932
Yang J, Stark GR (2008) Roles of unphosphorylated STATs in signaling. Cell Res 18:443–451
Concha-Benavente F, Srivastava RM, Trivedi S et al (2016) Identification of the cell-intrinsic and extrinsic pathways downstream of EGFR and IFN that induce PD-L1 expression in head and neck cancer. Cancer Res 76:1031–1043
Dimco G, Knight RA, Latchman DS, Stephanou A (2010) STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 9:4638–4649
Quesnelle KM, Boehm AL, Grandis JR (2007) STAT-mediated EGFR signaling in cancer. J Cell Biochem 102:311–319
Wang S, Raven JF, Durbin JE, Koromilas AE (2008) Stat1 phosphorylation determines Ras oncogenicity by regulating p27Kip1. PLoS One 3:1–14
Ramana CV, Grammatikakis N, Chernov M et al (2000) Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J 19:263–272
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7:41–51
Abroun S, Saki N, Ahmadvand M et al (2015) STATs: an old story, yet mesmerizing. Cell J 17:395–411
Timofeeva OP, Tarasova NI (2012) Alternative ways of modulating JAK-STAT pathway: looking beyond phosphorylation. JAKSTAT 1:274–284
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Böhmer F-D, Friedrich K (2014) Protein tyrosine phosphatases as wardens of STAT signaling. JAKSTAT 3:e28087
Berger A, Sexl V, Valent P, Moriggl R (2014) Inhibition of STAT5: a therapeutic option in BCR-ABL1-driven leukemia. Oncotarget 5:9564–9576
Buchert M, Burns CJ, Ernst M (2016) Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 35(8):939–951
Barré B, Vigneron A, Perkins N et al (2007) The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol Med 13:4–11
Albesiano E, Davis M, See AP et al (2010) Immunologic consequences of signal transducers and activators of transcription 3 activation in human squamous cell carcinoma. Cancer Res 70:6467–6476
Kujawski M, Kortylewski M, Lee H et al (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 118:3367–3377
Peyser ND, Du Y, Li H et al (2015) Loss-of-function PTPRD mutations lead to increased STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. PLoS One 10:e0135750
Peyser ND, Freilino M, Wang L et al (2016) Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 35(9):1163–1169
Chang Q, Bournazou E, Sansone P et al (2013) The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 15:848–862
Yoshikawa H, Matsubara K, Qian GS et al (2001) SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet 28:29–35
Meissl K, Macho-Maschler S, Müller M, Strobl B (2015) The good and the bad faces of STAT1 in solid tumours. Cytokine (in press: doi:10.1016/j.cyto.2015.11.011)
Babon JJ, Varghese LN, Nicola NA (2014) Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol 26:13–19
Lesina M, Kurkowski MU, Ludes K et al (2011) Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19:456–469
Croker BA, Krebs DL, Zhang J-G et al (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4:540–545
Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A (2013) SOCS, inflammation, and cancer. JAKSTAT 2:e24053
Yang J, Liao X, Agarwal MK et al (2007) Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkB. Genes Dev 21:1396–1408
Avalle L, Pensa S, Regis G et al (2012) STAT1 and STAT3 in tumorigenesis. JAKSTAT 1:65–72
Yang J, Chatterjee-Kishore M, Staugaitis SM et al (2005) Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 65:939–947
Wang S, Raven JF, Koromilas AE (2010) STAT1 represses Skp2 gene transcription to promote p27Kip1 stabilization in Ras-transformed cells. Mol Cancer Res 8:798–805
Kharma B, Baba T, Matsumura N et al (2014) STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res 74:6519–6530
Zimmerman MA, Rahman N-T, Yang D et al (2012) Unphosphorylated STAT1 promotes sarcoma development through repressing expression of Fas and Bad and conferring apoptotic resistance. Cancer Res 72:4724–4732
Knutson KL, Disis ML (2005) Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother 54:721–728
Ramana CV, Gil MP, Schreiber RD, Stark GR (2002) Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol 23:96–101
Loke P, Allison JP (2003) PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A 100:5336–5341
Bellucci R, Martin A, Bommarito D et al (2015) Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 4:e1008824
Ostrand-Rosenberg S, Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182:4499–4506
Hix LM, Karavitis J, Khan MW et al (2013) Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem 288:11676–11688
Tymoszuk P, Evens H, Marzola V et al (2014) In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur J Immunol 44:2247–2262
Sica A, Bronte V (2007) Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 117:1155–1166
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795
Kusmartsev S, Gabrilovich DI (2005) STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol 174:4880–4891
Bromberg JF, Wrzeszczynska MH, Devgan G et al (1999) Stat3 as an oncogene. Cell 98:295–303
Grandis JR, Drenning SD, Chakraborty A et al (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest 102:1385–1392
Grandis JR, Drenning SD, Zeng Q et al (2000) Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A 97:4227–4232
Harada D, Takigawa N, Kiura K (2014) The role of STAT3 in non-small cell lung cancer. Cancers (Basel) 6:708–722
Xi S, Zhang Q, Dyer KF et al (2003) Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem 278:31574–31583
Kiuchi N, Nakajima K, Ichiba M et al (1999) STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med 189:63–73
Yu H, Lee H, Herrmann A et al (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14:736–746
Niu G, Wright KL, Huang M et al (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000–2008
Devarajan E, Huang S (2009) STAT3 as a central regulator of tumor metastases. Curr Mol Med 9:626–633
Fukuda A, Wang SC, Morris JP et al (2011) Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 19:441–455
Teng Y, Ross JL, Cowell JK (2014) The involvement of JAK-STAT3 in cell motility, invasion, and metastasis. JAKSTAT 3:e28086
Wendt MK, Balanis N, Carlin CR, Schiemann WP (2014) STAT3 and epithelial–mesenchymal transitions in carcinomas. JAKSTAT 3:e28975
Xiong H, Hong J, Du W et al (2012) Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial–mesenchymal transition. J Biol Chem 287:5819–5832
Cheng GZ, Zhang W, Sun M et al (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 283:14665–14673
Yadav A, Kumar B, Datta J et al (2011) IL-6 promotes head and neck tumor metastasis by inducing epithelial–mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res 9:1658–1667
Sen M, Joyce S, Panahandeh M et al (2012) Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res 18:4986–4996
Byers LA, Sen B, Saigal B et al (2009) Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res 15:6852–6861
Poli V, Camporeale A (2015) STAT3-mediated metabolic reprograming in cellular transformation and implications for drug resistance. Front Oncol 5:1–9
Lee HJ, Zhuang G, Cao Y et al (2014) Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26:207–221
Wan S, Zhao E, Kryczek I et al (2014) Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 147:1393–1404
Lee H, Kumar Pal S, Reckamp K et al (2011) STAT3: a target to enhance antitumor immune response. Curr Top Microbiol Immunol 344:41–59
Cheng P, Corzo CA, Luetteke N et al (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–2249
Vasquez-Dunddel D, Pan F (2013) STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest 123:1580–1589
Yu J, Wang Y, Yan F et al (2014) Noncanonical NF-kB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol 193:2574–2586
Kortylewski M, Kujawski M, Wang T et al (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11:1314–1321
Park S-J, Nakagawa T, Kitamura H et al (2004) IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 173:3844–3854
Nefedova Y, Huang M, Kusmartsev S et al (2004) Hyperactivation of STAT3 Is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172:464–474
Wang T, Niu G, Kortylewski M et al (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10:48–54
Kryczek I, Wei S, Zou L et al (2007) Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol 178:6730–6733
Kortylewski M, Xin H, Kujawski M et al (2009) Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15:114–123
Zorn E, Nelson EA, Mohseni M et al (2006) IL-2 regulates FOXP3 expression in human CD4 + CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108:1571–1579
Grange M, Buferne M, Verdeil G et al (2012) Activated STAT5 promotes long-lived cytotoxic CD8+ T cells that induce regression of autochthonous melanoma. Cancer Res 72:76–87
Morinobu A, Gadina M, Strober W et al (2002) STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci U S A 99:12281–12286
Yue C, Xu J, Tan Estioko MD et al (2014) Host STAT2/type I interferon axis controls tumor growth. Int J Cancer 126:117–126
Bruns HA, Kaplan MH (2006) The role of constitutively active Stat6 in leukemia and lymphoma. Crit Rev Oncol Hematol 57:245–253
Skinnider BF, Kapp U, Mak TW (2002) The role of interleukin 13 in classical Hodgkin lymphoma. Leuk Lymphoma 43:1203–1210
Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 174:636–645
Kapoor N, Niu J, Saad Y et al (2015) Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J Immunol 194:1–13
Sasaki K, Zhao X, Pardee AD et al (2008) Stat6 signaling suppresses VLA-4 expression by CD8+ T cells and limits their ability to infiltrate tumor lesions in vivo. J Immunol 181:104–108
McCoon PE, Woessner R, Grosskurth S et al. (2015) Clinical and pre-clinical evidence of an immune modulating role for STAT3-targeting ASO AZD9150 and potential to enhance clinical responses to anti-PDL1 therapy. In: AACR 106th Annual Meeting, Philadelphia, PA, 18–22 April 2015
Liu S, Walker SR, Nelson EA et al (2014) Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Mol Cancer Ther 13:1194–1205
Toniolo PA, Liu S, Yeh JE et al (2015) Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation. J Immunol 194:3180–3190
Filippakopoulos P, Qi J, Picaud S et al (2010) Selective inhibition of BET bromodomains. Nature 468:1067–1073
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
O’Keefe, R.A., Grandis, J.R. (2016). STAT Proteins in Cancer. In: Ward, A. (eds) STAT Inhibitors in Cancer. Cancer Drug Discovery and Development. Humana Press, Cham. https://doi.org/10.1007/978-3-319-42949-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-42949-6_2
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-42947-2
Online ISBN: 978-3-319-42949-6
eBook Packages: MedicineMedicine (R0)