
Abstract
This chapter includes the advances achieved so far in the design and implementation of molecular biological tools (MBTs) for the assessment of hydrocarbon-degrading potential in microbial communities from coastal environments of Patagonia. A brief introduction on the role of hydrocarbon-degrading bacteria in marine environments follows the basic concepts of MBTs, methods, and applications. The review then focuses on studies performed on the Patagonian coast to identify functional biomarker genes associated with hydrocarbon biodegradation, with emphasis on polycyclic aromatic hydrocarbons (PAHs): (a) advances on determining the identity, abundance, and biogeographic distribution of dioxygenase gene variants from known obligate PAH-degrading marine bacteria as well as yet uncultured microorganisms; (b) testing of selected variants in experimental systems; and (c) results of recent metagenomic analyses revealing the genetic context and PAH-degrading capabilities of uncultured microorganisms from Patagonia carrying an ecologically relevant biomarker gene. Alkane biodegradation biomarker genes are also covered, as well as analyses based on phylogenetic biomarker genes. Obligate and specialized hydrocarbon degraders are identified in microbial communities from Patagonia by culture-independent approaches based on the 16S rRNA gene. Last, the design and testing of a community-level ecological indicator based on high-throughput sequencing of the 16S rRNA gene and perspectives on the use of MBTs in coastal regions of Argentinean Patagonia are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Molecular Biological Tools
Microorganisms have a key role in the fate of environmental pollutants, mainly participating in their removal through the use of these compounds as carbon and energy sources (Jeon and Madsen 2013). The assimilation of these compounds leads to a rapid increase in the abundance of these populations and higher biodegradation rates. Therefore, information of the presence, abundance, or activity of pollutant-degrading microbial populations in a polluted site could aid in decision making at each stage of the remediation process, as a complement of contaminant concentrations and geochemistry of the site (Lebron et al. 2011). By increasing the efficiency and predictability of the biodegradation process, this information could reduce the time and costs of bioremediation and accelerate the adoption of these technologies. Because it is impossible or difficult to culture most of the environmental microorganisms, traditional culture-dependent methods are not able to provide this information accurately and rapidly. Molecular biological tools (MBTs) targeting pollutant-degrading microorganisms , often used to increase our understanding of pollutant biodegradation processes, can also be adapted to develop novel tools for environmental site management (Interstate Technology and Regulatory Council Environmental Molecular Diagnostics 2011).
The MBTs target biomolecules of microbial populations that participate in pollutant biodegradation processes, such as nucleic acids, proteins, or lipids (Table 2.1). Each of these biomolecules will provide a different type of information, such as revealing the presence of microbial populations with degrading potential [e.g., polymerase chain reaction (PCR) , fingerprinting methods such as denaturing gradient gel electrophoresis (DGGE) , terminal restriction fragment length polymorphism (tRFLP) , clone library construction and sequencing], assessing the abundance of key microorganisms, and monitoring population growth [quantitative PCR (qPCR), next-generation sequencing-based fingerprinting such as pyrotags or I-tags, microarrays], providing evidence of pollutant biodegradation (SIP), or estimating the activity of specific microbial populations (RT-qPCR). As the microorganisms involved in the biodegradation process could vary among sites, even for the same type of environment (Lozada et al. 2014b), an in-depth knowledge of the system is required for the development of molecular diagnostic tools that can provide accurate information in bioremediation applications. In spite of the challenges for the development and implementation of MBTs, these tools are increasingly being used in multiple cleanup sites in various countries (Interstate Technology and Regulatory Council Environmental Molecular Diagnostics 2013), particularly when a limited number of pollutants and microorganism types are involved, such as the anaerobic biodegradation of chlorinated solvents (Lebron et al. 2011). The biodegradation of hydrocarbon pollutants in coastal environments represents one of the most challenging scenarios. It involves the presence of multiple chemical structures, varying with the contamination source, as well as dynamic environmental conditions that influence the structure and function of the involved microbial communities.
2 Biodegradation of Hydrocarbons in Marine Environments
Although hydrocarbons are energy-rich compounds, their use requires the activation of a highly stable molecule. The most energetically efficient way to capture this energy is by its oxidation with molecular oxygen (Austin and Callaghan 2013). Aerobic hydrocarbon degradation initiates via enzymatic complexes, which include a terminal oxygenase and accompanying enzymes forming an electron transport chain (Peng et al. 2008; Rojo 2009). Marine microorganisms have been exposed to hydrocarbons during millions of years, such as in natural oil seeps. As a consequence, the capability of hydrocarbon biodegradation has been acquired rather frequently through evolution, and hydrocarbon-degrading microorganisms are widespread in nature (Prince et al. 2010). In the marine environment, important bacterial groups participating in hydrocarbon biodegradation are members of the Gammaproteobacteria (e.g., Pseudomonas, Alteromonas, Neptunomonas, Marinobacter, Alcanivorax , Cycloclasticus ) (Vila et al. 2015), the Alphaproteobacteria (the sphingomonads) (Kertesz and Kawasaki 2010, the Roseobacter clade (Kim and Kwon 2010), and the Actinobacteria (Rhodococcus, Mycobacterium, Nocardioides) (Vila et al. 2015). Of special interest is a group of marine microorganisms that have specialized exclusively in the utilization of hydrocarbons, or almost exclusively, named obligate hydrocarbonoclastic bacteria (OHCB) : these are represented mainly by members of the Gammaproteobacteria class (Yakimov et al. 2007). Examples are Cycloclasticus, which degrades polycyclic aromatic hydrocarbons, and Alcanivorax and Oleispira , which feed on alkanes. Oil-degrading microorganisms can be a small proportion of the bacterial community. However, when these compounds are available in the environment, these populations grow at their expense increasing their number, which allows their monitoring through space and time, if appropriate tools are used (Head et al. 2006). Moreover, these populations can be stimulated to increase biodegradation rates in the natural environment, which are often too slow to prevent damage to vulnerable ecosystems and human health (Lozada et al. 2014a; Ron and Rosenberg 2014).
When released to the marine environment, oil suffers a number of abiotic and biotic processes that are collectively called weathering , and include spreading, evaporation, photooxidation, emulsification, dissolution, sedimentation, adsorption, and biodegradation (McGenity et al. 2012). Among these processes, biodegradation is a major mechanism of hydrocarbon removal from the marine environment. Hydrocarbons vary in their biodegradability, as a result of differences in their physicochemical properties such as their solubility, hydrophobicity, and capacity to adsorb to matrices, which affect their bioavailability for microorganisms (McGenity et al. 2012). Bacteria have evolved strategies for efficient uptake of hydrocarbons, such as the modification of cell membranes and the release of biosurfactants (Harms et al. 2010). In addition, the presence of nitrogen and phosphorus nutrients in sufficient amounts is a key factor affecting the rate of biodegradation in natural conditions (Ron and Rosenberg 2014). Oxygen is another key factor driving this process. In contrast to seawater, which is mainly aerobic, sediments are aerobic only on their surface and become rapidly anoxic with depth, giving rise to steep redox gradients . Therefore, aerobic biodegradation processes only occur in the sediment surface, while in deeper layers anaerobic processes are predominant, coupled to nitrate, iron, and/or sulfate reduction (Acosta-González et al. 2013). The latter are less understood at the molecular level than aerobic processes, specially for PAHs (Estelmann et al. 2015). In fact, sediments constitute a complex matrix in which diverse and spatially structured microbial communities take part in biotic and abiotic interactions that ultimately affect the biodegradation process (Cravo-Laureau and Duran 2014).
Although individual hydrocarbon-degrading strains typically exhibit the ability to degrade only a limited number of hydrocarbons, a natural microbial community can display an important biodegradation potential through synthrophy (cross-feeding), making these communities a suitable target for the depuration of the complex mixture present in oil (McGenity et al. 2012). Moreover, in coastal sediments cells are more concentrated than in open waters, increasing the probability and complexity of their interactions (McGenity et al. 2012). The use of culture-independent approaches targeting phylogenetic (e.g., 16S rRNA gene) and/or functional (e.g., hydrocarbon-activating oxygenases) marker genes, has allowed the characterization of hydrocarbon-degrading communities in various marine environments. The catastrophic spill resulting from the Deepwater Horizon blowout in 2010 provided a unsought opportunity for the scientific community to develop multidisciplinary analysis tools, including chemical and environmental analyses coupled to targeted-gene surveys, metagenomics, and meta transcriptomics. This development has resulted in unprecedented knowledge regarding the response of marine microbial communities to oil input and their potential for remediation (Kimes et al. 2014). Future perspectives involve systems biology, which includes multiple-level assessment of microbial communities and the environmental factors controlling mass fluxes across its members (Roling and van Bodegom 2014). This knowledge will aid in gaining predictability on these systems and advance into the application of knowledge-based bioremediation tools (de Lorenzo 2008).
3 Hydrocarbon-Degrading Bacteria on the Patagonian Coast
A series of studies have shown the presence of hydrocarbon pollution in several sites along the Patagonian coast, as a result of oil extraction and transportation activities, as well as port and vessel operations (Commendatore et al. 2000, 2012). Increasing our understanding of hydrocarbon biodegradation processes in the coastal environments of Patagonia, and in particular the identification of the microbial populations that are key for these processes, is essential for the design of MBTs specific for this region. A series of culture-independent approaches were used to characterize hydrocarbon-degrading bacteria in sediments from coastal environments of Patagonia (Lozada et al. 2008, 2014b; Marcos et al. 2009, 2012; Dionisi et al. 2011; Guibert et al. 2012, 2016; Loviso et al. 2015). These studies considered both spatial and temporal variations that could occur in these populations. PCR-based approaches targeted both functional and phylogenetic biomarker genes, which revealed the identity of hydrocarbon-degrading bacterial populations, as well as their abundance and biogeographic distribution. In addition, metagenomic approaches allowed the analysis of genome fragments from some of these microorganisms, exposing their particular adaptations to pollutant biodegradation (Loviso et al. 2015; Guibert et al. 2016).
3.1 Polycyclic Aromatic Hydrocarbons (PAHs)
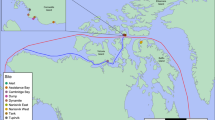
PAHs constitute the compounds of most concern among the components of crude oil or petroleum refined products due to their toxic and carcinogenic properties as well as their accumulation and persistence in the environment (Rojo-Nieto and Perales 2015). Various PAHs were identified in intertidal sediments of Patagonia, often exceeding the levels recommended for this matrix (Marcos et al. 2012). The biomarker gene most commonly used for the detection of bacterial populations with the potential to degrade PAHs encodes the large subunit of the terminal component of class A ring-hydroxylating oxygenases (RHOs) (Chakraborty et al. 2012). Using primer sets with different specificities, we amplified fragments of these genes from sediment DNA and cloned these amplification products to construct PCR clone libraries. This analysis revealed a high PAH-degrading potential in the sediments of polluted sites, as 25 distinct RHO α-subunit gene variants (sharing <80 % identity at the amino acid level) were detected in the PCR clone libraries (Lozada et al. 2008; Marcos et al. 2009; Dionisi et al. 2011; Loviso et al. 2015). Remarkably, 22 of the gene variants shared low or moderate identity values with previously identified genes. Most of the novel gene variants were only identified in sediments of South Patagonia, where they were found to be very abundant, outnumbering archetypical genes such as nahAc and phnAc (Marcos et al. 2012). These results suggest a biogeographic distribution of these populations restricted to cold environments, which was confirmed for some of these variants by the use of qPCR analyses (Fig. 2.1). In contrast, one of the gene variants identified in the sediments, named gene variant T, was found to have a distribution extending at least to North and South Patagonia (Loviso et al. 2015). The relative abundance of this gene was high in chronically polluted sediments, and further increased in experimental systems after PAH exposure in sediments of both regions. Archetypical marine PAH-degrading bacteria belonging to the genus Cycloclasticus were also identified in coastal sediments of North and South Patagonia, and their abundance in polluted sediments correlated with low molecular weight PAH concentrations (Marcos et al. 2012). These populations also increased their abundance after PAH exposure in experimental systems (Loviso et al. 2015). Overall, these results suggest that both Cycloclasticus and the uncultured populations carrying the gene variant T could be among the key players for PAH biodegradation in the coastal environments of Patagonia.

Map of the Patagonian coast showing the distribution of hydrocarbon-degrading microorganisms and related gene variants, as identified using different approaches. A, B, C, D, T novel RHO α-subunit gene variants quantified by qPCR in the sediments, phnA1 gene variant described in the obligate PAH-degrader Cycloclasticus spp.
PCR-based approaches are useful for the recovery of biomarker genes from microbial populations associated to specific metabolic processes. However, as this approach can only recover fragments of the targeted genes, the complete sequence of these genes and their genomic context are lost. Furthermore, it is very difficult to infer the potential host of the identified gene fragments, which precludes linking the structure with the function of the microbial community. Metagenomic libraries, the cloning of fragments of environmental DNA into appropriate vectors, can provide this type of information. We constructed a metagenomic library from chronically polluted sediments retrieved near an oil jetty in Ushuaia Bay, in South Patagonia (Loviso et al. 2015). The screening of the library with a broad specificity primer set previously used to construct PCR clone libraries allowed the identification of a fosmid clone (M117) containing a 37-kb fragment with a gene almost identical to gene variant T. This fragment could only be affiliated to the Proteobacteria phylum, as it is highly divergent from the genome sequences of currently described strains. Besides this gene, this metagenomic fragment contained five additional RHO α-subunit sequences, which indicates the high aromatic hydrocarbon-degrading potential of this microorganism (Loviso et al. 2015). These sequences shared low identity values at the protein level (19.4–42.5 %), and were most closely related to sequences identified in Cycloclasticus strains. Five of these genes had a codirectional β-subunit sequence, characteristic of RHOs with a α n β n hetero-multimeric structure (Chakraborty et al. 2012). Several highly specialized PAH-degrading microorganisms, such as Cycloclasticus strains, carry multiple genes coding for α- and β-subunits of RHOs (Lai et al. 2012; Cui et al. 2013). The analysis of the phylogenetic relationship of the identified metagenomic sequences with α-subunit sequences identified in complete genomes from three Cycloclasticus strains (Cycloclasticus sp. P1, Cycloclasticus pugetii PS-1, and Cycloclasticus zancles 7-ME) showed that the metagenomic sequences were clearly divergent from dioxygenases from these highly specialized organisms, also identified in coastal sediments of Patagonia (Fig. 2.2). Three of the oxygenases contained in the metagenomic fragment were classified as class A RHOs, which include enzymes that catalyze the dioxygenation of PAHs (Loviso et al. 2015). The modeling of the three-dimensional structures of these enzymes suggested that this microorganism could degrade high molecular weight PAHs, as two of these modeled proteins presented a catalytic pocket able to accommodate large PAH molecules. In particular, the catalytic cavity dimensions of one of the proteins (modeled from sequence M117-38; Fig. 2.2) were comparable with the ones from NidAB from Mycobacterium vanbaalenii PYR-1, which presents pyrene as the preferential substrate (Kweon et al. 2010).

Phylogenetic tree of RHO α-subunit sequences identified in fosmid M117 and in Cycloclasticus spp. genomes. The neighbor-joining tree includes metagenomic (in gray) and genomic deduced amino acid sequences. Sequence number within each genome (NCBI numbering) and strain name (in brackets) is indicated in each case. RHO classification according to the scheme proposed by Chakraborty et al. (2012) is indicated on the right. The phylogenetic tree was built with Mega 6 (Tamura et al. 2013) using the Jones-Taylor-Thornton (JTT) substitution model. Bootstrap values were calculated as percentage of 1000 repetitions, with only values ≥50 % indicated in the figure (black circles, >75 %; white circles, 50–75 %). Bar represents inferred amino acid changes per position
Overall, these analyses showed the presence of a large diversity of microorganisms with PAH-degrading potential in chronically polluted sediments of Patagonia, and allowed the identification of genes from key bacterial populations that could be used as targets for field assessment. In particular, two of the designed qPCR assays , targeting gene variants T (from an uncultured proteobacterium: Loviso et al. 2015) and phnA1 gene (from Cycloclasticus spp.: Marcos et al. 2012) are promising MBTs that could next be validated in a large-scale field study.
3.2 Aliphatic Hydrocarbons
Although not as toxic as aromatic hydrocarbons, alkanes are a major component of crude oil (Head et al. 2006). Their low water solubility and poor reactivity, and their tendency to accumulate on sediments, constitute a challenge for their effective removal (Rojo 2009). Various microbial strategies, however, have evolved to access these compounds, even the longer molecules that are solid at room temperature (paraffins) (Wentzel et al. 2007). In addition to genes coding PAH-degrading enzymes, we have analyzed the functional biomarker gene alkB, which codes for the initial oxygenase of aliphatic hydrocarbons (alkane-1-monooxygenase). AlkB is an integral-membrane non-heme di-iron monooxygenase, which hydroxylates the alkane molecule in the terminal position. It requires two electron transport proteins, a rubredoxin and a rubredoxin reductase. AlkB family enzymes are highly extended among oil-degrading bacteria (van Beilen and Funhoff 2007). Using a PCR-based method with a broad-specificity primer set targeting conserved regions of the gene, we could uncover a remarkable diversity of AlkB sequences in South Patagonian sediments (Guibert et al. 2012). The majority of the sequences were found to be related to uncultured microorganisms from cold marine sediments or soils from high-latitude regions, suggesting a major role of temperature in the selection of bacterial populations with this capability. The analysis of experimental systems constructed from these sediments and amended with crude oil showed the specific enrichment of various alkB variants, related to genes described in members of the Gammaproteobacteria and Actinobacteria. The majority of these variants were also detected in the corresponding environmental samples, highlighting their ecological relevance (Guibert et al. 2012). A complementary approach involving large-scale sequencing of 16S rRNA gene amplicons also allowed the identification of various genera that could not be targeted by the functional gene approach. For example, the obligate oil-degrading genus Oleispira , probably psychrophilic Oleispira antarctica , was detected in South Patagonian sediments by this method (Guibert et al. 2012). More recently, the analysis of a metagenomic shotgun sequencing dataset involving more than 6000 putative AlkB sequences from subtidal sediments from this site evidenced that AlkB diversity could be more than one order of magnitude higher than estimated by PCR-based methods (Guibert et al. 2016). The AlkB sequences identified by metagenomics exhibited high phylogenetic diversity, spanning the whole AlkB phylogenetic tree known up to date. Furthermore, completely novel sequences only moderately related to putative AlkBs from genomes of Bacteroidetes and Alphaproteobacteria were found in high abundance. Not all these enzymes have been characterized , and to date, the role of these bacterial groups in aliphatic hydrocarbon biodegradation has been largely underestimated. In the same work, the previously mentioned metagenomic library was also analyzed to search for alkB genes using a molecular approach. This analysis rendered two fosmid clones containing genomic fragments from uncultured bacteria belonging to the phylum Planctomycetes, allowing the description for the first time of alkane-degrading potential in members of this group (Guibert et al. 2016). These results highlight the power of metagenomic approaches for uncovering new potential features of microbial communities, as they are not as dependent on previous knowledge as the PCR-based methods. However, it must be noted that this approach still presents challenges related to functional annotation of metagenomic sequences as a result of the still-remaining knowledge gaps (Temperton and Giovannoni 2012).
3.3 Ecological Index of Hydrocarbon Exposure
When we identify microorganisms based on a phylogenetically informative gene (e.g., 16S rRNA gene), we normally infer their metabolic capabilities by searching the characteristics reported for the genus or species in the scientific literature. This type of inference is performed either advertently or inadvertently. Moreover, when we analyze the taxonomic composition of a microbial community, we draw conclusions about its metabolic capabilities and the potential processes occurring in the analyzed environment. For example, the presence of genera known to harbor sulfate-reducing strains is evidence that this process is probably occurring. An example of this inference carried out in a systematic manner is the picrust program, a software that predicts the metagenome of a microbial community based on the information obtained with the phylogenetic marker gene 16S rRNA (Langille et al. 2013). As it relies upon the existing information of microbial genomes for the prediction, the method is specially effective when the microbial community contains genera for which many sequenced genomes are available (e.g., the human microbiome) (Langille et al. 2013).
In the marine environment, hydrocarbonoclastic bacteria such as Alcanivorax were detected through their phylogenetic marker genes, allowing the inference of hydrocarbon-degrading processes following a pollution event (Kostka et al. 2011). This detection was possible because of the narrow substrate preferences of these microorganisms, which allowed the rapid linking of phylogenetic information to function. Interestingly, there are a number of well-described genera, which can be easily detected with community-level approaches, known to carry out biodegradation of hydrocarbons in the marine environment, allowing the monitoring of the whole process at the microbiological level (Dubinsky et al. 2013). Based on the fact that, in theory, these genera would normally be present at low abundances but that will increase with pollution if optimal conditions are met (Head et al. 2006), they can constitute good indicators of the environmental state of a certain site. We therefore developed an ecological indicator , the Ecological Index of Hydrocarbon Exposure (EIHE ) , which is defined as the sum of the relative abundances (proportion with respect to total community members) of potential hydrocarbon-degrading genera in a certain sample (Lozada et al. 2014b). This index involves an arbitrary list of genera harboring hydrocarbon-degrading strains normally encountered associated with biodegradation in the marine environment (Lozada et al. 2014b). A high EIHE value would be evidence of a previous exposure to pollutants, whereas a low value would be typical of a non-impacted site. Moreover, the variation of the index could be followed in polluted sites to monitor the growth of the populations in the presence of environmental constraints, for example, nutrient limitations. The estimation of the abundances should be ideally performed by high coverage techniques such as large-scale sequencing of 16S rRNA gene amplicons, which allow the rapid identification of a number of genera in relatively low abundance in the context of a diverse community, a condition that is found, for example, in sediments (Lozada et al. 2014b). The taxonomic information derived from the bioinformatic analysis of sequences is extracted automatically and processed to calculate the EIHE (Lozada et al. 2014b) (Fig. 2.3). A strength of this approach is that different populations of degrading bacteria arising in different environments or conditions can be detected, thus contributing to the index value (Fig. 2.3). This indicator has the potential to be utilized as a MBT, as it relies on standardized procedures during the whole process and has the possibility of high-throughput analysis of various samples .

Schematic representation of the Ecological Index of Hydrocarbon Exposure (EIHE ) index concept and method. Three different samples are analyzed: two from different polluted sites, and an unpolluted sample. In the samples, the microbial communities are composed of hydrocarbon degraders (A to E) and non-degraders (x to z), of different types and abundances according to the history and environmental state of the site. The DNA is extracted, a hypervariable region of the phylogenetic marker gene (16S rRNA) is amplified with conserved primers and subjected to large-scale sequencing, and sequences are processed through bioinformatic methods, and taxonomically assigned to the lowest possible level relying on public databases for this gene. The taxonomic assignment of each sample is matched against the “EIHE ” list to extract the abundances of potential hydrocarbon-degrading genera. The abundances are summed and expressed as proportion of the total bacterial community (total sequences, N), which is the EIHE value. Note that the individual genera contributing to EIHE may vary in different polluted samples
The preliminary evaluation of this tool was performed in samples from the Patagonian coast, as well as in public sequence datasets from other oil-exposed marine samples, including sediments and seawater from experimental systems and field studies. In all cases, the EIHE was significantly higher in oiled than in unpolluted samples (Lozada et al. 2014b). Moreover, in sediment samples from acute pollution events such as the Deepwater Horizon spill , the EIHE index reached a value of approximately 30, which means that 30 % of the bacterial community was composed of potential hydrocarbon degraders; for a nearly non-impacted site, values remained as low as 2 (Lozada et al. 2014b). Our results suggest that this ecological indicator could be a promising tool for environmental diagnostics in marine systems. The next steps involve its assessment at field scale, in samples from the Patagonian coast subjected to chronic oil pollution .
4 Perspectives on the Use of MBTs in the Patagonian Coastal Region
The identified functional biomarker genes probably do not represent the complete diversity of hydrocarbon-degrading bacteria present in this region, as methodological challenges still limit the access to all the microorganisms present in an environmental sample. Through the use of multiple approaches, we identified potential key members of hydrocarbon biodegradation processes, which allowed the design of MBTs able to detect these microbial populations, estimate their abundance, and evaluate changes as a result of pollution events or the use of bioremediation technologies. The next steps are the validation of these tools and the development of standardized guidelines for their application in polluted sites. We envision the use of these tools to generate baseline information on hydrocarbon exposure in marine environments before offshore oil exploitation, as well as to evaluate hydrocarbon-degrading potential and the dynamics of key bacterial populations during natural attenuation of chronically polluted sites or after an accidental spill. There is also a need to increase the awareness in stakeholders such as government agencies and companies operating ports and oil terminals on the Patagonian coast on the availability of these tools. Many highly vulnerable environments can be threatened with an oil spill similar to that which occurred in Cordova Cove in December 2008, which affected 4 km of the coastline of an inlet used for recreation and for artisanal fisheries. Although enhanced bioremediation is not an alternative currently considered in these accidents, a more active evaluation of natural biodegradation processes and eventually the implementation of these technologies are necessary to limit the damage to human populations and environmental health in coastal environments of Patagonia .
References
Acosta-González A, Rosselló-Móra R, Marqués S (2013) Characterization of the anaerobic microbial community in oil-polluted subtidal sediments: aromatic biodegradation potential after the Prestige oil spill. Environ Microbiol 15:77–92
Austin RN, Callaghan AV (2013) Microbial enzymes that oxidize hydrocarbons. Front Microbiol 4:338
Chakraborty J, Ghosal D, Dutta A, Dutta TK (2012) An insight into the origin and functional evolution of bacterial aromatic ring-hydroxylating oxygenases. J Biomol Struct Dyn 30:419–436
Commendatore MG, Esteves JL, Colombo JC (2000) Hydrocarbons in coastal sediments of Patagonia, Argentina: levels and probable sources. Mar Pollut Bull 40:989–998
Commendatore MG, Nievas ML, Amin O, Esteves JL (2012) Sources and distribution of aliphatic and polyaromatic hydrocarbons in coastal sediments from the Ushuaia Bay (Tierra del Fuego, Patagonia, Argentina). Mar Environ Res 74:20–31
Cravo-Laureau C, Duran R (2014) Marine coastal sediments microbial hydrocarbon degradation processes: contribution of experimental ecology in the ‘omics’ era. Front Microbiol 5:39
Cui Z, Xu G, Li Q, Gao W, Zheng L (2013) Genome sequence of the pyrene-and fluoranthene-degrading bacterium Cycloclasticus sp. strain PY97M. Genome Announc 1:e00536-13
de Lorenzo V (2008) Systems biology approaches to bioremediation. Curr Opin Biotechnol 19:579–589
Dionisi HM, Lozada M, Marcos MS, Di Marzio WD, Loviso CL (2011) Aromatic hydrocarbon degradation genes from chronically polluted subantarctic marine sediments. In: de Bruijn FJ (ed) Handbook of molecular microbial ecology. II: Metagenomics in different habitats. Wiley, Hoboken, NJ, pp 461–473
Dubinsky EA, Conrad ME, Chakraborty R, Bill M, Borglin SE, Hollibaugh JT, Mason OU, Piceno YM, Reid FC, Stringfellow WT, Tom LM, Hazen TC, Andersen GL (2013) Succession of hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ Sci Technol 47:10860–10867
Estelmann S, Blank I, Feldmann A, Boll M (2015) Two distinct old yellow enzymes are involved in naphthyl ring reduction during anaerobic naphthalene degradation. Mol Microbiol 95:162–172
Guibert LM, Loviso CL, Marcos MS, Commendatore MG, Dionisi HM, Lozada M (2012) Alkane biodegradation genes from chronically polluted subantarctic coastal sediments and their shifts in response to oil exposure. Microb Ecol 64:605–616
Guibert L, Loviso C, Borglin S, Jansson J, Dionisi H, Lozada M (2016) Diverse bacterial groups contribute to the alkane degradation potential of chronically polluted subantarctic coastal sediments. Microb Ecol 71:100–112
Harms H, Smith KEC, Wick LY (2010) Microorganism–hydrophobic compound interactions. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 1479–1490
Head IM, Jones DM, Röling WFM (2006) Marine microorganisms make a meal of oil. Nat Rev Microbiol 4:173–182
Interstate Technology and Regulatory Council (ITRC) Environmental Molecular Diagnostics (EMD) (2011) Environmental Molecular Diagnostics Fact Sheets
Interstate Technology and Regulatory Council (ITRC) Environmental Molecular Diagnostics (EMD) (2013) Technical and Regulatory Guidance
Jeon CO, Madsen EL (2013) In situ microbial metabolism of aromatic-hydrocarbon environmental pollutants. Curr Opin Biotechnol 24:474–481
Kertesz M, Kawasaki A (2010) Hydrocarbon-degrading sphingomonads: Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis. In: Timmis K (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 1694–1705
Kim SJ, Kwon KK (2010) Marine, hydrocarbon-degrading Alphaproteobacteria. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 1707–1714
Kimes NE, Callaghan AV, Suflita JM, Morris PJ (2014) Microbial transformation of the Deepwater Horizon oil spill: past, present, and future perspectives. Front Microbiol 5:603
Kostka JE, Prakash O, Overholt WA, Green SJ, Freyer G, Canion A, Delgardio J, Norton N, Hazen TC, Huettel M (2011) Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl Environ Microbiol 77:7962–7974
Kweon O, Kim S-J, Freeman JP, Song J, Baek S, Cerniglia CE (2010) Substrate specificity and structural characteristics of the novel rieske nonheme iron aromatic ring-hydroxylating oxygenases NidAB and NidA3B3 from Mycobacterium vanbaalenii PYR-1. Mol Microbiol 1:e00135-10
Lai Q, Li W, Wang B, Yu Z, Shao Z (2012) Complete genome sequence of the pyrene-degrading bacterium Cycloclasticus sp. strain P1. J Bacteriol 194:6677
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821
Lebron C, Petrovskis E, Loffler F, Henn K (2011) Application of nucleic acid-based tools for monitoring monitored natural attenuation (MNA), biostimulation and bioaugmentation at chlorinated solvent sites. DTIC Document, Port Hueneme, California
Loviso CL, Lozada M, Guibert LM, Musumeci MA, Sarango Cardenas S, Kuin RV, Marcos MS, Dionisi HM (2015) Metagenomics reveals the high polycyclic aromatic hydrocarbon-degradation potential of abundant uncultured bacteria from chronically polluted subantarctic and temperate coastal marine environments. J Appl Microbiol 119:411–424
Lozada M, Mercadal JPR, Guerrero LD, Di Marzio WD, Ferrero MA, Dionisi HM (2008) Novel aromatic ring-hydroxylating dioxygenase genes from coastal marine sediments of Patagonia. BMC Microbiol 8:50
Lozada M, Marcos M, Dionisi H (2014a) La Biorremediación de ambientes costeros contaminados con hidrocarburos. Fondo Editorial Provincial, Provinica del Chubut, Argentina
Lozada M, Marcos MS, Commendatore MG, Gil MN, Dionisi HM (2014b) The bacterial community structure of hydrocarbon-polluted marine environments as the basis for the definition of an ecological index of hydrocarbon exposure. Microbes Environ 29:269–276
Marcos MS, Lozada M, Dionisi HM (2009) Aromatic hydrocarbon degradation genes from chronically polluted subantarctic marine sediments. Lett Appl Microbiol 49:602–608
Marcos MS, Lozada M, Di Marzio WD, Dionisi HM (2012) Abundance, dynamics, and biogeographic distribution of seven polycyclic aromatic hydrocarbon dioxygenase gene variants in coastal sediments of Patagonia. Appl Environ Microbiol 78:1589–1592
McGenity TJ, Folwell BD, McKew BA, Sanni GO (2012) Marine crude-oil biodegradation: a central role for interspecies interactions. Aquat Biosyst 8:1–19
Peng R-H, Xiong A-S, Xue Y, Fu X-Y, Gao F, Zhao W, Tian Y-S, Yao Q-H (2008) Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev 32:927–955
Prince RC, Gramain A, McGenity TJ (2010) Prokaryotic hydrocarbon degraders. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 1671–1692
Rojo F (2009) Degradation of alkanes by bacteria. Environ Microbiol 11:2477–2490
Rojo-Nieto E, Perales JA (2015) Estimating baseline toxicity of PAHs from marine chronically polluted sediments and bioaccumulation in target organs of fish hypothetically exposed to them: a new tool in risk assessment. Environ Sci Process Impacts 17:1331–1339
Roling WFM, van Bodegom PM (2014) Toward quantitative understanding on microbial community structure and functioning: a modeling-centered approach using degradation of marine oil spills as example. Front Microbiol 5:125
Ron EZ, Rosenberg E (2014) Enhanced bioremediation of oil spills in the sea. Curr Opin Biotechnol 27:191–194
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Temperton B, Giovannoni SJ (2012) Metagenomics: microbial diversity through a scratched lens. Curr Opin Microbiol 15:605–612
van Beilen JB, Funhoff EG (2007) Alkane hydroxylases involved in microbial alkane degradation. Appl Microbiol Biotechnol 74:13–21
Vila J, Tauler M, Grifoll M (2015) Bacterial PAH degradation in marine and terrestrial habitats. Curr Opin Biotechnol 33:95–102
Wentzel A, Ellingsen TE, Kotlar H-K, Zotchev SB, Throne-Holst M (2007) Bacterial metabolism of long-chain n-alkanes. Appl Microbiol Biotechnol 76:1209–1221
Yakimov MM, Timmis KN, Golyshin PN (2007) Obligate oil-degrading marine bacteria. Curr Opin Biotechnol 18:257–266
Acknowledgments
M.L. and H.D. are staff members of the Argentinean National Research Council (CONICET). The work at the Environmental Microbiology Laboratory (Laboratorio de Microbiología Ambiental, CESIMAR, CONICET) has been supported by grants from CONICET, the National Agency for Science and Technology Promotion of Argentina (ANPCYT), The Secretary of Science, Technology and Innovation from the Chubut Province, Argentina, and the Community Sequencing Program, Joint Genome Institute (JGI-DOE, USA).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Lozada, M., Dionisi, H.M. (2016). Molecular Biological Tools for the Assessment of Hydrocarbon-Degrading Potential in Coastal Environments. In: Olivera, N., Libkind, D., Donati, E. (eds) Biology and Biotechnology of Patagonian Microorganisms. Springer, Cham. https://doi.org/10.1007/978-3-319-42801-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-42801-7_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42799-7
Online ISBN: 978-3-319-42801-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)