
Abstract
Depending on tumor type, stage, and genetic context, autophagy can play an opposite role in cancer by promoting tumor progression or regression. It is now well established that autophagy limits tumor initiation, however, it promotes the progression of well-established tumors. In the context of tumor progression and immune response, experimental evidence indicate that autophagy plays a key role in maintaining survival of tumor cells under stress condition such as hypoxia. Indeed, by activating autophagy, tumor cells are able to escape immunosurveillance by activating several overlapping mechanisms in cancer cells. Such findings have inspired significant interest to develop autophagy inhibitor molecules as an entirely new approach to cancer treatment. While much remains to be learned mechanistically, it is now widely established that modulation of this process will be an attractive avenue for future anticancer therapeutic approaches. In this chapter, we will briefly describe the role of autophagy in tumor regression in the context of inflammation, necrosis, oxidative stress and genomic instability. We will also focus on recent reports highlighting the role of autophagy in the impairment of the anti-tumor immune response. In keeping with this, we believe that targeting autophagy may represent a conceptual realm for new anti-tumor strategies aiming to block immune escape.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Autophagy Regulation in Physiological and Pathological Conditions
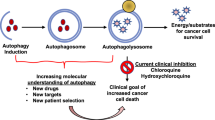
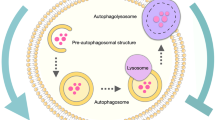
Autophagy acts as a catabolic process crucial for cellular homeostasis and maintenance of cell integrity under stressful conditions (Mizushima 2007; Yang and Klionsky 2010). Autophagy is a degradation mechanism of cell components which allows the recycling of essential amino acids, nucleotides, and fatty acids necessary for energy and macromolecule biosynthesis (Corcelle et al. 2009; Glick et al. 2010). During cancer progression , autophagy can be induced by different stresses, particularly hypoxia, nutrient deprivation, or extracellular matrix detachment (Rosenfeldt and Ryan 2009; Yang and Klionsky 2009). The autophagic process is characterized by the formation of phagophore or isolation membrane mainly dependent on Beclin-1 (BECN1) complexes . Following this so-called nucleation stage, the phagophore is elongated by several Autophagy-related proteins (ATG) and the Microtubule-associated protein 1A/1B-light chain 3 (LC3)-I is lipidated into LC3-II. Maturation of the phagophore, through the action of LC3-II and BECN1 proteins, enables the sequestration of cell constituents into well-characterized vesicles named autophagosomes. Fusion of autophagosomes with lysosomes leads to the formation of autolysosomes and the degradation of their contents by lysosomal hydrolases (Kang et al. 2011).
Under physiological conditions, autophagy is constitutively executed at basal level in all cells to promote cell homeostasis. However, in tumor cells autophagy is activated in response to various cellular stresses and environmental factors including hypoxia (Mathew and White 2011). Therefore, the major consensus that emerge is that autophagy can act as tumor suppressor and tumor promoter . Such opposite role of autophagy in cancer seems to be related to the stage of the tumor. In fact, autophagy clearly suppresses the initiation and the development of tumors, however, it is considered as a key survival pathway in response to stress, and many established tumors require autophagy to survive.
7.2 Autophagy as a Tumor Regression Mechanism
The role of autophagy in tumor suppression relies on its effect on several oncogenic pathways such as the activation of the PI3K/Akt pathway via activating PI3K mutations, AKT amplifications, or PTEN loss of function. (Guertin and Sabatini 2007). Moreover, the amplification of the apoptosis inhibitor Bcl-2 has been reported in some circumstances to inhibit autophagy through its binding to beclin1 (Sinha and Levine 2008; Maiuri et al. 2007). The involvement of p53 in the regulation of autophagy seems to be complex. Indeed, the activation of p53 by nutrient deprivation or genotoxic stress leads to the activation of autophagy through the inhibition of mTOR or by the activation of DRAM (damage-regulated autophagy modulator) (Balaburski et al. 2010; Crighton et al. 2006; Feng et al. 2005). However, consistent with the role of autophagy as tumor suppressor, the functional loss of p53 was expected to decrease autophagy or suppress basal autophagy. The later effect seems to depend on the cytoplasmic, not the nuclear, pool of p53 (Tasdemir et al. 2008).
In addition to the indirect evidence described above, several direct evidences support the tumor suppressing properties of autophagy. Indeed the autophagy execution protein Beclin1 is a haplo-insufficient tumor suppressor protein. Mono-allelic deletion of BECLIN1 are reported in sporadic human breast and ovarian carcinoma (Aita et al. 1999), and heterozygous deletion of BECLIN1 predisposed mice to a variety of tumors including mammary neoplastic lesions, lung adenocarcinomas, hepatocellular carcinomas and B cell lymphomas (Qu et al. 2003). These results indicate that functional autophagy may be constraining tumor initiation (Liang et al. 1999). Similarly, homozygote deletion of ATG5 was shown to predisposed mice specifically to liver tumors with high penetrance (Takamura et al. 2011). The tumor suppressive properties of autophagy have been extensively investigated. Below we will provide some mechanistic insights into the tumor-suppressive functions of autophagy.
7.2.1 Autophagy Inhibition Regulates Tumor Necrosis and Inflammation
It has been reported that autophagy can modulate the inflammatory microenvironment that play a major role in tumor development and considered as a common future of early cancer development. Thus, experimental evidence suggest that autophagy-deficient tumors displayed an increased level of necrosis and inflammation. The activation of autophagy in tumor cells inhibits necrotic cell death which subsequently stimulates a robust inflammatory response in vivo (Kono and Rock 2008). In addition, it has been proposed that the impairment of both apoptosis and autophagy promotes necrotic cell death, in vitro and in vivo, associated with an inflammatory response and an accelerated tumor growth (Degenhardt et al. 2006). These results highlight that autophagy regulates necrosis-induced cell death and inflammation. Furthermore, autophagy also prevents necroptosis which is a form of caspase-independent cell death mediated by cell death ligands (i.e. TNF-α and FasL) (Degterev and Yuan 2008; Shen and Codogno 2012). Indeed, autophagy is essential to overcome zVAD-induced necroptosis in L929 cells. Activation of PI3K-Akt-mTOR pathway, a well-known autophagy inhibitor pathway, can sensitize L929 cells to zVAD-induced necroptosis, while amino-acid and serum starvation protect these cells (Wu et al. 2009). Similarly, autophagy prevents poly-(ADP-ribose) polymerase (PARP)-mediated cell death . Such cytoprotective role of autophagy in PARP-mediated necrosis was illustrated by showing that DNA damages induced by doxorubicin in fibroblasts lead to PARP-1 activation and autophagy induction which protects cells against necrosis. Targeting autophagic genes ATG5 or BECLIN1, sensitizes cells to doxorubicin-induced necrotic cell death (Munoz-Gamez et al. 2009).
Autophagy is also a key process for the maintenance of intracellular ATP level required for the secretion of lysophosphatidylcholine (LPC) . Secretion of LPC is associated with the acute phase of the inflammatory response and is involved in the development of chronic inflammation. It has been shown that autophagy-deficient cells fail to generate phosphatidylserine on the outer membrane surface—an important anti-inflammatory pro-apoptotic marker. Such observation could explain why defect in autophagy stimulates inflammatory response subsequently to insufficient clearance of dead cells (Pierdominici et al. 2012).
Following autophagy inhibition, the accumulation of the autophagy cargo protein p62/SQSTM1 activates the pro-inflammatory transcription factor NF-kB and the stress-responsive transcription factor NRF2, thus favoring inflammation and tissue injury (Levine et al. 2011). The transcription factors NF-kB family members regulate the expression of a broad range of genes involved in development, proliferation, and survival of tumor cells. The activation of these transcription factors leads to the regulation of inflammation and innate and adaptive immune responses (Smale 2011). As the activation of NF-kB is mediated by the IkB kinase (IKK) complexes , it has been reported that IKK complexes are targets for degradation by autophagy when the heat shock protein 90 (Hsp90) function is inhibited (Xu et al. 2011). Another mechanism of regulation of NF-kB by autophagy is mediated by the Kelch-like ECH-associated protein 1 (Keap1) . Keap1 interacts with the kinase domain of IKKβ through its C-terminal domain. This domain is also required for the binding of Keap1 to the transcription factor NRF2, which controls the expression of some antioxidant genes. In response to tumor necrosis factor (TNF) , Keap1 negatively regulates the activation of NF-kB through inhibition of the IKKβ phosphorylation and induction of IKKβ degradation by autophagy pathway (Fan et al. 2010). The E3 ubiquitin ligase Ro52 is another signaling molecule that targets IKKβ for degradation through the autophagy pathway . In response to distinct stimuli, specific interactions of Hsp90, Keap1 and Ro52 with IKKs regulate NF-kB activity through their ability to activate or repress the degradation of IKKs by autophagy (Trocoli and Djavaheri-Mergny 2011). It has been suggested that the crosstalk between NF-kB and autophagy regulates inflammasome activity leading to the modulation of the activation of caspase-1 and subsequently the secretion of potent pro-inflammatory cytokines (Strowig et al. 2012). Based on the studies discussed above, it appears that autophagy is an important modulator of cancer pathogenesis through its ability to regulate inflammation.
7.2.2 Autophagy Prevents Oxidative Stress and Genomic Instability
The role of autophagy in cancer suppression has been reported by several in vivo studies (White et al. 2010). Thus, Beclin1-defective mice showed an increased susceptibility to develop cancer (Qu et al. 2003; Yue et al. 2003). This could be related to the involvement of autophagy in the management of oxidative stress and in the maintenance of the genomic integrity. In this regard, it has been described that autophagy can limit DNA damage, chromosomal instability and aneuploidy (Mathew et al. 2007). Several studies suggested that the ubiquitin- and LC3-binding protein p62 may play a determinant role (Komatsu et al. 2007; Mathew et al. 2009). Indeed, the inability of autophagy-deficient cells to degrade p62 lead to the aberrant accumulation of this protein , which is sufficient to promote tumorigenesis (Mathew et al. 2009). Thus, p62 activates the transcription factor NRF2 through the direct inhibition of Keap1 (Komatsu et al. 2010; Lau et al. 2010). However, the role of NRF2 in DNA damage promotion is not clearly understood so far. In addition, p62 may act as an important NF-kB modulator in tumorigenesis (Duran et al. 2008). This study highlights that the increase in DNA damage in autophagy-deficient cells is associated with high levels of damaged mitochondria and reactive oxygen species (ROS), accumulation of ER chaperones and protein disulfide isomerases. DNA alterations were suppressed by ROS scavengers, confirming the essential role of autophagy in oxidative stress management and, subsequently, in protein quality control (Mathew et al. 2009).
Excessive exposure to ROS alters the function of multiple cellular macromolecules by oxidation (e.g. nucleic acids, lipids, proteins). However, oxidative stress is closely linked to mitochondria dysfunction. Since autophagy is the only process allowing the mitochondrial turnover by a mechanism so-called mitophagy , preventing the accumulation of damaged mitochondria highly reduces the risk of oxidative stress. Moreover, mitochondria produce the bulk of ATP required for vital cellular functions (e.g. DNA replication, mitosis, transcription). In this regard, the ability of autophagy to control proteins/organelles quality and to maintain cellular energy homeostasis highlights its antitumorigenic activity (Jin 2006). Such a role has been demonstrated in autophagy-defective cells, where the presence of damaged proteins is crucial in DNA replication, mitosis or centrosome function. Moreover, autophagy defective cells displaying defect in mitochondrial clearance and subsequently an alteration in ATP production may also alter DNA replication or repair by affecting the arrest of the replication forks and the generation of breakage/fusion/bridge cycles responsible for gene amplification (Jin and White 2008). Finally, the implication of autophagy in the physiological protein turnover may also influence the occurrence of DNA damage. Indeed, cell cycle progression is driven by the periodic activity of proteins including Cyclin-dependent kinases (CDKs) , Cyclins, CDKs inhibitors. Therefore, it stands to reason that a deregulation in the physiological protein turnover in autophagy-deficient cells may alter the correct sequence of the cell cycle progression (Jin and White 2008). Taken together, it has become clear that autophagy helps normal cells to overcome several types of stresses (e.g. metabolic, oncogenic), that directly limits their oncogenic transformation. In contrast, such management of cellular stresses is also observed in cancer cells, and leads in this case to cancer promotion (Rosenfeldt and Ryan 2011).
Senescence is an irreversible cell cycle arrest associated with an active metabolism, which can limit the proliferation of abnormal cells. In this context, autophagy is also able to mitigate the accumulation of genomic alteration by inducing the mitotic senescence transition. Young et al. reported an accumulation of autophagosomes in Ras-induced IMR90 senescent fibroblasts, suggesting that autophagy is required for tumor senescence. In addition, targeting ATG5/7 delayed the senescent phenotype, while induction of autophagy clearly enhanced the protein turnover that contributed to synthesis of pro-senescence cytokines (e.g. IL-6, IL-8) (Young et al. 2009). This study suggests that autophagy not only facilitates the entry into senescence but also reinforces the senescent phenotype of cells.
7.2.3 Autophagy Contributes to Tumor Cell Death
The role of autophagy in promoting tumor cell death has been proposed based on the observation that apoptosis occurs concomitantly with features of autophagy (Kroemer and Levine 2008) and that prolonged stress and progressive autophagy can lead to cell death (Mathew and White 2007). Together with apoptosis (type I cell death) and necrosis (type III cell death) (Schweichel and Merker 1973), autophagy was first described as type II cell death. The relevance of autophagic cell death in development has been established in lower eukaryotes and invertebrates such as Dictyostelium discoideum and Drosophila melanogaster (Denton et al. 2009; Kosta et al. 2004). Evidence has been reported that mammalian development does not require autophagy, as newborn mice lacking essential autophagy genes show any anatomical or histological defects and no impairment of the cell death (Mizushima et al. 2008). This evidence is supported by the fact that the depletion of autophagy genes in human or mice mammalian cells induces apoptosis rather than protects cell against death induced by different stresses (Boya et al. 2005; Gonzalez-Polo et al. 2005). The role of autophagy in cell death induction is not clear, and needs further investigation. However, the more convincing evidence highlighting the role of autophagy in cell death has been reported in mammal's neuronal cells. Indeed, following insulin starvation, hippocampal neural stem cells undergo autophagic cell death, while targeting autophagy by silencing ATG7 blocks this process. It is worthy to note that autophagic cell death occurs only in cells with functional apoptosis and is caspase-independent (Yu et al. 2008). Currently, the majority of experimental evidence showing autophagic cell death in mammalian cells were mainly conducted in vitro and in cells defective in apoptosis machinery. It has been shown that DAPK (death associated protein kinase) plays an important role in the regulation of both autophagy and apoptosis. Indeed, DAPK induces autophagy by phosphorylation of Beclin1, and is associated with the induction of apoptosis. However this type of DAPK-dependent autophagic death is caspase dependent, and it remains to be elucidated whether DAPK-mediated cell death is a real autophagic cell death, or whether autophagy only assists in the apoptosis execution phase (Gozuacik et al. 2008). It has been proposed that cells rather die with autophagy, and not by autophagy as they showed that none of 1400 compounds, evaluated for their ability to induce autophagic puncta and increase autophagic flux, killed tumor cells through the induction of autophagy (Shen and Codogno 2012). Moreover a careful determination of the autophagic flux is needed to differentiate autophagic cell death from other forms of non-apoptotic programmed-cell death, such as necroptosis. These examples illustrate that autophagy may be involved in lethal signaling although the role of autophagy itself in cell killing remains unclear. Thus, further studies are required in order to define the exact role of autophagic cell death mechanism.
7.3 Autophagy Modulates the Anti-tumor Immune Response
Recently, autophagy has emerged as a new critical mechanism activated in tumor cells in hypoxic microenvironment that mediates tumor resistance to innate and adaptive anti-tumor immune responses. Several reports demonstrate that autophagy activation not only enables tumor cells to survive stress conditions during cancer development but also provides them an intrinsic resistance mechanism to escape anti-tumor immune response .
7.3.1 Role of Autophagy in Tumor Cell Resistance to CTL-Mediated Killing
The first evidence for such a role of autophagy was provided by Noman et al. who demonstrated that hypoxic lung carcinoma cells can evade cytolytic T lymphocyte (CTL) -mediated lysis through autophagy induction (Noman et al. 2011, 2012). Indeed, inhibition of autophagy using small interfering RNA (siRNA) directed against ATG5 or BECN1 restored tumor cells sensibility to CTL-mediated lysis which correlated with a decrease in hypoxia-dependent induction of the phosphorylation of Signal Transducer and Activator of Transcription (STAT)-3 . This result allowed the prediction that blocking autophagy would inhibit pSTAT3-dependent survival mechanism making tumor cells more susceptible to CTL attack under hypoxia. However, considering the degradation role of autophagy, it is difficult to perceive that autophagy is involved in the stabilization of pSTAT3 under hypoxia. Focusing on the crosstalk between the adaptor protein p62/SQSTM1, the ubiquitin-proteasome system (UPS) and autophagy, this study revealed that the induction of hypoxia inducible factor (HIF)-1α has two effects in tumor cells: (i) HIF-1α triggers the phosphorylation of Src which subsequently phosphorylates the tyrosine residue Y705 of STAT3 (ii) HIF-1α activates autophagy by a mechanism implicating the increased expression of BCL2/adenovirus E1B 19 kDa protein-interacting protein (BNIP)3/BNIP3L and the dissociation of the BECN1-BCL2 (B cell lymphoma 2) complex . Autophagy activation results in degradation of the p62 protein. Knowing that p62 is the receptor/adaptor protein responsible for targeting pSTAT3 to the UPS, the autophagy-dependent degradation of p62 leads to the accumulation of pSTAT3. When autophagy is inhibited in tumor cells, the degradation of p62 is blocked and therefore accumulates in tumor cells. This accumulation accelerates the UPS-dependent degradation of pSTAT3 (Noman et al. 2009) (Fig. 7.1a).

Autophagy activation in tumor cell acts as an intrinsic resistance mechanism against anti-tumor immune response . The tumor microenvironment and/or EMT program activate autophagy in target cells. The induction of autophagy operates as a cell resistance mechanism leading to tumor escape from CTL- or NK-mediated lysis. (a) Hypoxic stress leads to the accumulation of HIF-1α. HIF-1α activates autophagy and simultaneously increases the phosphorylation level of STAT3 at the Tyr705 residue. As an autophagic substrate, p62/SQSTM1 is degraded in the autophagosomes following their fusion with lysosomes. As p62 is involved in targeting pSTAT3 to the UPS, its degradation leads to the accumulation of pSTAT3 in cells and such accumulation constitutes a cell survival mechanism. In autophagy-defective cells, p62 is no longer degraded, and its accumulation accelerates the UPS-dependent degradation of pSTAT3 and thereby restores CTL-mediated tumor cell lysis. (b) The acquisition of an EMT phenotype confers resistance to CTL-mediated lysis through autophagy induction. The increase in mesenchymal markers following the activation of EMT program leads to the up-regulation of BECN1 by a yet undefined mechanism. Such upregulation induces autophagy and impairs CTL-mediated tumor cell lysis. In mesenchymal cells, targeting BECN1 is sufficient to restore CTL-mediated lysis. (c) Following the recognition of their targets, NK cells secrete cytotoxic granules containing PRF1, GZMB, and other hydrolytic enzymes that enter target cells, traffic to enlarged endosomes, and initiate tumor cell death. Under hypoxia, excessive autophagy in target cells leads to the fusion of autophagosomes with vesicles containing GZMB leading to its specific degradation by autophagy, thereby inhibiting NK-mediated lysis. Targeting autophagy prevents the degradation of GZMB and thereby restores NK-mediated tumor cell killing
Epithelial to mesenchymal transition (EMT) is a trans-differentiation process necessary for the morphogenesis of tissue during embryonic development (Nieto 2013). While its role in cancer cell invasion, metastasis and drug resistance is well established, recent report described that autophagy can be activated in tumor cells undergoing EMT and that such EMT-induced autophagy represents another mechanism of cancer cell resistance to CTL-mediated lysis (Akalay et al. 2013a, b). In this study, the authors showed that the induction of EMT program by overexpression of SNAI1 in breast cancer cells coincides with a drastic change in cell morphology and the activation of autophagy flux most likely through the overexpression of BECN1 in mesenchymal cells. Although the exact molecular mechanism by which the EMT affects the expression of BECN1 remained to be addressed, several lines of evidence indicate that this may be related to SNAI1- or EMT-dependent repression of microRNA(s) involved in modulation of BECN1 expression (Siemens et al. 2011; Yu et al. 2012). This result extended the role of SNAI1 as a regulator of autophagy and paves the way to investigate the functional role of EMT-induced autophagy in tumor cells. In this context, results described in this study showed that targeting BECN1 in mesenchymal cells was sufficient to restore CTL-mediated tumor cell lysis, without affecting the mesenchymal morphology and the expression of EMT markers. This finding implies that autophagy is a downstream target of the EMT program in breast cancer cells. Overall, this study suggests that EMT-induced autophagy is a novel mechanism by which tumor cells regulate CTL reactivity and impede their cytotoxic activity, and further points to the complex relationship between the tumor and the immune system (Fig. 7.1b).
7.3.2 Role of Autophagy in Tumor Cell Resistance to NK-Mediated Killing
It is now well established that several resistance mechanisms are regulated in tumor cells to escape immune surveillance in hypoxic tumor microenvironment. Recent evidence described how tumor cells can escape natural killer (NK)-mediated immune surveillance by activating autophagy under hypoxia (Baginska et al. 2013; Viry et al. 2014). Indeed, NK cells recognize and kill their targets by several mechanisms including the release of cytotoxic granules containing perforin (PRF1) and serine protease granzyme B (GZMB) . It has been recently proposed that PRF1 and GZMB enter target cells by endocytosis and traffic to large endosomes named “gigantosomes ” (Thiery et al. 2010, 2011). Subsequently, PRF1 is involved in the formation of pores in the membrane of the “gigantosome”, leading to the gradual release of GZMB and the initiation of apoptotic cell death. The formation of amphisomes following the fusion between autophagic vacuoles and early endosomes appears to be necessary in some cases for the generation of autolysosomes. In this report (Baginska et al. 2013), the authors described that the pro-apoptotic protein GZMB is selectively degraded upon autophagy activation in hypoxic cells, thereby blocking NK-mediated target cell apoptosis (Fig. 7.1c). In line with this, they showed that GZMB is detected in autophagosomes and provided evidence that GZMB level is significantly decreased in hypoxic compared to normoxic target cells. Furthermore, targeting autophagy genetically or inhibiting lysosomal hydrolases by pharmacological approaches restored GZMB level which ultimately leads to the recovery of hypoxic cells lysis by NK cells in vitro and in vivo. Based on these results, the authors stated that tumor regression can be achieved by inhibiting autophagy in hypoxic cancer cells, thus enabling their NK-mediated lysis (Baginska et al. 2013; Viry et al. 2014).
Overall, studies described above underline the activation of autophagy as a key mechanism in tumor escape from immune cell attack within the tumor microenvironment. However, an important issue that arises from these studies is whether hypoxia is the only microenvironmental factor involved in the induction of autophagy in tumor cells. An interesting recent report provided strong evidence that lymphoid effectors not only provide lytic signals but also promote autophagy in the remaining target cells, a process called cell-mediated autophagy (C-MA) (Buchser et al. 2012). Thus, C-MA has been reported in different human epithelial tumors after interaction with immune cells at high ratio of effectors to targets. Importantly, it has been showed that C-MA not only acts as a mechanism of resistance to immune cell-mediated lysis but also limits the cytotoxic activity of stress factors such as γ-radiation (Buchser et al. 2012).
These studies highlight that the activation of autophagy plays a critical role in tumor cell escape from both adaptive and innate immunity. Therefore, targeting autophagy has been proposed to improve CTL- and NK-based immunotherapy in experimental mouse model (Baginska et al. 2013; Noman et al. 2011, 2012). Intense research efforts are currently focusing on the development of autophagy inhibitors that could improve tumor immunotherapy.
7.4 Targeting Autophagy in Cancer Therapy
Currently, there are several clinical trials registered in the National Cancer Institute (www.cancer.gov/clinicaltrials) exploring anti-autophagy strategies in a variety of human cancers. Most of these trials are ongoing, with minimal published results available, and nearly all use Hydroxychloroquine (HCQ) or Chloroquine (CQ). It is worthy to note that CQ or HCQ are lysosomotropic agents that act at the level of the lysosome by inhibiting acidification, thereby impairing autophagosome degradation. These clinical trials were initiated based on the fact that autophagy is induced as a survival mechanism in a variety of tumor cells and preclinical models by several types of chemotherapeutic agents. Because only a subpopulation of tumor cells undergo autophagy, it is unlikely that autophagy inhibitors are used in cancer therapy as single agent. Indeed, most of these clinical trials used HCQ in combination with other anti-cancer therapies. While these preclinical data are generally supportive of incorporating anti-autophagy therapies in cancer treatment trials, it has been observed, in some circumstances, that inhibition of autophagy decreases therapeutic efficacy . Understanding the circumstances in which autophagy inhibition impairs the therapeutic effect will be of great importance. Importantly, while CQ and HCQ are effective inhibitors of autophagy in vitro, whether they will do so at doses used in current clinical trials is still uncertain. An important issue related to the use of these autophagy inhibitors concerns the micromolar concentration that is required to inhibit autophagy and show anti-tumor efficacy in preclinical models. While this is theoretically achievable at tolerated doses after prolonged dosing, it should be better optimized in clinic (Tett et al. 1993; Munster et al. 2002). Trials combining HCQ as neoadjuvant treatment will provide tumor tissues available for analysis both before and after HCQ treatment. However, the effectiveness of HCQ in the inhibition of autophagy still prove difficult, as HCQ is often combined with other therapies (chemotherapy and radiotherapy) that are also known to modulate autophagy. Alternative biomarkers to predict for autophagy activation as well as autophagy dependence are currently an area of intense investigation (Kimmelman 2011). A recently reported phase I trial of HCQ in combination with adjuvant temozolomide and radiation in patients with glioblastoma found that the maximum tolerated dose of HCQ was 600 mg per day, and this dose achieved concentrations of HCQ required for autophagy inhibition in preclinical studies. In this trial, investigators observed a dose-dependent inhibition of autophagy, as indicated by increases in autophagic vesicles (revealed by electron microscopy), and detected elevations in LC3-II in peripheral blood mononuclear cells. In addition, in a phase I trial of 2-deoxyglucose, an agent that blocks glucose metabolism, autophagy occurred in association with a reduction in p62/SQSTM1 in peripheral blood mononuclear cells (Stein et al. 2010). These data suggest the potential interest of such biomarkers in the evaluation of autophagy modulation during therapy and in the correlation with treatment outcome.
CQ inhibits the last step of autophagy at the level of the lysosome, thereby impacting lysosomal function. Therefore, its effects are not entirely specific to autophagy. Currently, there is a great deal of interest in developing new inhibitors of autophagy. In this regards, and given the complexity of the autophagic process, multiple proteins involved in this process could be good candidates for developing others autophagy inhibitors. It is likely that kinases would be prime candidates for inhibition such as Vps34, a class III PI3K, which has a critical early role in autophagosome development. This is particularly attractive, as there has been significant success in designing effective class I PI3K inhibitors (Wong et al. 2010). However, one potential issue which needs to be considered is that Vps34 has roles in other aspects of endosome trafficking, and this may lead to unwanted effects and toxicity (Backer 2008). The mammalian orthologs of yeast ATG1, ULK1/2, which acts downstream from AMPK and the TOR complex , have been recently shown as critical proteins for autophagy activation (Hara et al. 2008; Egan et al. 2011). Other potential targets for autophagy inhibitors would be LC3 proteases, such as ATG4b , which are necessary for LC3 processing. However, whichever approach is taken, the delicate balance between potency and toxicity must be determined to achieve a clinical success. While there are still uncertainties of how autophagy inhibition will fare as an anti-cancer therapy, the preclinical data generally support this approach. The current clinical trials will hopefully provide insight into whether this will be a viable therapeutic paradigm (Kimmelman 2011).
References
Aita, V. M., Liang, X. H., Murty, V. V., Pincus, D. L., Yu, W., Cayanis, E., et al. (1999). Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics, 59(1), 59–65. doi:10.1006/geno.1999.5851.
Akalay, I., Janji, B., Hasmim, M., Noman, M. Z., Andre, F., De Cremoux, P., et al. (2013a). Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Research, 73(8), 2418–2427. doi:10.1158/0008-5472.CAN-12-2432.
Akalay, I., Janji, B., Hasmim, M., Noman, M. Z., Thiery, J. P., Mami-Chouaib, F., et al. (2013b). EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy, 9(7), 1104–1106. doi:10.4161/auto.24728.
Backer, J. M. (2008). The regulation and function of Class III PI3Ks: Novel roles for Vps34. The Biochemical Journal, 410(1), 1–17. doi:10.1042/BJ20071427.
Baginska, J., Viry, E., Berchem, G., Poli, A., Noman, M. Z., van Moer, K., et al. (2013). Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proceedings of the National Academy of Sciences of the United States of America, 110(43), 17450–17455. doi:10.1073/pnas.1304790110.
Balaburski, G. M., Hontz, R. D., & Murphy, M. E. (2010). p53 and ARF: Unexpected players in autophagy. Trends in Cell Biology, 20(6), 363–369. doi:10.1016/j.tcb.2010.02.007.
Boya, P., Gonzalez-Polo, R. A., Casares, N., Perfettini, J. L., Dessen, P., Larochette, N., et al. (2005). Inhibition of macroautophagy triggers apoptosis. Molecular and Cellular Biology, 25(3), 1025–1040. doi:10.1128/MCB.25.3.1025-1040.2005.
Buchser, W. J., Laskow, T. C., Pavlik, P. J., Lin, H. M., & Lotze, M. T. (2012). Cell-mediated autophagy promotes cancer cell survival. Cancer Research, 72(12), 2970–2979. doi:10.1158/0008-5472.CAN-11-3396.
Corcelle, E. A., Puustinen, P., & Jaattela, M. (2009). Apoptosis and autophagy: Targeting autophagy signalling in cancer cells—‘Trick or treats’? FEBS Journal, 276(21), 6084–6096. doi:10.1111/j.1742-4658.2009.07332.x.
Crighton, D., Wilkinson, S., O’Prey, J., Syed, N., Smith, P., Harrison, P. R., et al. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell, 126(1), 121–134. doi:10.1016/j.cell.2006.05.034.
Degenhardt, K., Mathew, R., Beaudoin, B., Bray, K., Anderson, D., Chen, G., et al. (2006). Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell, 10(1), 51–64. doi:10.1016/j.ccr.2006.06.001.
Degterev, A., & Yuan, J. (2008). Expansion and evolution of cell death programmes. Nature Reviews. Molecular Cell Biology, 9(5), 378–390. doi:10.1038/nrm2393.
Denton, D., Shravage, B., Simin, R., Mills, K., Berry, D. L., Baehrecke, E. H., et al. (2009). Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Current Biology: CB, 19(20), 1741–1746. doi:10.1016/j.cub.2009.08.042.
Duran, A., Linares, J. F., Galvez, A. S., Wikenheiser, K., Flores, J. M., Diaz-Meco, M. T., et al. (2008). The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell, 13(4), 343–354. doi:10.1016/j.ccr.2008.02.001.
Egan, D., Kim, J., Shaw, R. J., & Guan, K. L. (2011). The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy, 7(6), 643–644.
Fan, W., Tang, Z., Chen, D., Moughon, D., Ding, X., Chen, S., et al. (2010). Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy, 6(5), 614–621. doi:10.4161/auto.6.5.12189.
Feng, Z., Zhang, H., Levine, A. J., & Jin, S. (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of the United States of America, 102(23), 8204–8209. doi:10.1073/pnas.0502857102.
Glick, D., Barth, S., & Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. The Journal of Pathology, 221(1), 3–12. doi:10.1002/path.2697.
Gonzalez-Polo, R. A., Boya, P., Pauleau, A. L., Jalil, A., Larochette, N., Souquere, S., et al. (2005). The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. Journal of Cell Science, 118(Pt 14), 3091–3102. doi:10.1242/jcs.02447.
Gozuacik, D., Bialik, S., Raveh, T., Mitou, G., Shohat, G., Sabanay, H., et al. (2008). DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death and Differentiation, 15(12), 1875–1886. doi:10.1038/cdd.2008.121.
Guertin, D. A., & Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell, 12(1), 9–22. doi:10.1016/j.ccr.2007.05.008.
Hara, T., Takamura, A., Kishi, C., Iemura, S., Natsume, T., Guan, J. L., et al. (2008). FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. The Journal of Cell Biology, 181(3), 497–510. doi:10.1083/jcb.200712064.
Jin, S. (2006). Autophagy, mitochondrial quality control, and oncogenesis. Autophagy, 2(2), 80–84.
Jin, S., & White, E. (2008). Tumor suppression by autophagy through the management of metabolic stress. Autophagy, 4(5), 563–566.
Kang, R., Zeh, H. J., Lotze, M. T., & Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death and Differentiation, 18(4), 571–580. doi:10.1038/cdd.2010.191.
Kimmelman, A. C. (2011). The dynamic nature of autophagy in cancer. Genes & Development, 25(19), 1999–2010. doi:10.1101/gad.17558811.
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biology, 12(3), 213–223. doi:10.1038/ncb2021.
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell, 131(6), 1149–1163. doi:10.1016/j.cell.2007.10.035.
Kono, H., & Rock, K. L. (2008). How dying cells alert the immune system to danger. Nature Reviews Immunology, 8(4), 279–289. doi:10.1038/nri2215.
Kosta, A., Roisin-Bouffay, C., Luciani, M. F., Otto, G. P., Kessin, R. H., & Golstein, P. (2004). Autophagy gene disruption reveals a non-vacuolar cell death pathway in Dictyostelium. The Journal of Biological Chemistry, 279(46), 48404–48409. doi:10.1074/jbc.M408924200.
Kroemer, G., & Levine, B. (2008). Autophagic cell death: The story of a misnomer. Nature Reviews. Molecular Cell Biology, 9(12), 1004–1010. doi:10.1038/nrm2529.
Lau, A., Wang, X. J., Zhao, F., Villeneuve, N. F., Wu, T., Jiang, T., et al. (2010). A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Molecular and Cellular Biology, 30(13), 3275–3285. doi:10.1128/MCB.00248-10.
Levine, B., Mizushima, N., & Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature, 469(7330), 323–335. doi:10.1038/nature09782.
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402(6762), 672–676. doi:10.1038/45257.
Maiuri, M. C., Criollo, A., Tasdemir, E., Vicencio, J. M., Tajeddine, N., Hickman, J. A., et al. (2007). BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy, 3(4), 374–376.
Mathew, R., Karantza-Wadsworth, V., & White, E. (2007). Role of autophagy in cancer. Nature Reviews Cancer, 7(12), 961–967. doi:10.1038/nrc2254.
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell, 137(6), 1062–1075. doi:10.1016/j.cell.2009.03.048.
Mathew, R., & White, E. (2007). Why sick cells produce tumors: The protective role of autophagy. Autophagy, 3(5), 502–505.
Mathew, R., & White, E. (2011). Autophagy, stress, and cancer metabolism: What doesn’t kill you makes you stronger. Cold Spring Harbor Symposia on Quantitative Biology, 76, 389–396. doi:10.1101/sqb.2012.76.011015.
Mizushima, N. (2007). Autophagy: Process and function. Genes & Development, 21(22), 2861–2873. doi:10.1101/gad.1599207.
Mizushima, N., Levine, B., Cuervo, A. M., & Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature, 451(7182), 1069–1075. doi:10.1038/nature06639.
Munoz-Gamez, J. A., Rodriguez-Vargas, J. M., Quiles-Perez, R., Aguilar-Quesada, R., Martin-Oliva, D., de Murcia, G., et al. (2009). PARP-1 is involved in autophagy induced by DNA damage. Autophagy, 5(1), 61–74.
Munster, T., Gibbs, J. P., Shen, D., Baethge, B. A., Botstein, G. R., Caldwell, J., et al. (2002). Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis and Rheumatism, 46(6), 1460–1469. doi:10.1002/art.10307.
Nieto, M. A. (2013). Epithelial plasticity: A common theme in embryonic and cancer cells. Science, 342(6159), 1234850. doi:10.1126/science.1234850.
Noman, M. Z., Buart, S., Van Pelt, J., Richon, C., Hasmim, M., Leleu, N., et al. (2009). The cooperative induction of hypoxia-inducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. Journal of Immunology, 182(6), 3510–3521. doi:10.4049/jimmunol.0800854.
Noman, M. Z., Janji, B., Berchem, G., Mami-Chouaib, F., & Chouaib, S. (2012). Hypoxia-induced autophagy: A new player in cancer immunotherapy? Autophagy, 8(4), 704–706. doi:10.4161/auto.19572.
Noman, M. Z., Janji, B., Kaminska, B., Van Moer, K., Pierson, S., Przanowski, P., et al. (2011). Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Research, 71(18), 5976–5986. doi:10.1158/0008-5472.CAN-11-1094.
Pierdominici, M., Vomero, M., Barbati, C., Colasanti, T., Maselli, A., Vacirca, D., et al. (2012). Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB Journal, 26(4), 1400–1412. doi:10.1096/fj.11-194175.
Qu, X., Yu, J., Bhagat, G., Furuya, N., Hibshoosh, H., Troxel, A., et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of Clinical Investigation, 112(12), 1809–1820. doi:10.1172/JCI20039.
Rosenfeldt, M. T., & Ryan, K. M. (2009). The role of autophagy in tumour development and cancer therapy. Expert Reviews in Molecular Medicine, 11, e36. doi:10.1017/S1462399409001306.
Rosenfeldt, M. T., & Ryan, K. M. (2011). The multiple roles of autophagy in cancer. Carcinogenesis, 32(7), 955–963. doi:10.1093/carcin/bgr031.
Schweichel, J. U., & Merker, H. J. (1973). The morphology of various types of cell death in prenatal tissues. Teratology, 7(3), 253–266. doi:10.1002/tera.1420070306.
Shen, H. M., & Codogno, P. (2012). Autophagy is a survival force via suppression of necrotic cell death. Experimental Cell Research, 318(11), 1304–1308. doi:10.1016/j.yexcr.2012.02.006.
Siemens, H., Jackstadt, R., Hunten, S., Kaller, M., Menssen, A., Gotz, U., et al. (2011). miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle, 10(24), 4256–4271. doi:10.4161/cc.10.24.18552.
Sinha, S., & Levine, B. (2008). The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene, 27(Suppl 1), S137–S148. doi:10.1038/onc.2009.51.
Smale, S. T. (2011). Hierarchies of NF-kappaB target-gene regulation. Nature Immunology, 12(8), 689–694. doi:10.1038/ni.2070.
Stein, M., Lin, H., Jeyamohan, C., Dvorzhinski, D., Gounder, M., Bray, K., et al. (2010). Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. The Prostate, 70(13), 1388–1394. doi:10.1002/pros.21172.
Strowig, T., Henao-Mejia, J., Elinav, E., & Flavell, R. (2012). Inflammasomes in health and disease. Nature, 481(7381), 278–286. doi:10.1038/nature10759.
Takamura, A., Komatsu, M., Hara, T., Sakamoto, A., Kishi, C., Waguri, S., et al. (2011). Autophagy-deficient mice develop multiple liver tumors. Genes & Development, 25(8), 795–800. doi:10.1101/gad.2016211.
Tasdemir, E., Maiuri, M. C., Galluzzi, L., Vitale, I., Djavaheri-Mergny, M., D’Amelio, M., et al. (2008). Regulation of autophagy by cytoplasmic p53. Nature Cell Biology, 10(6), 676–687. doi:10.1038/ncb1730.
Tett, S. E., Day, R. O., & Cutler, D. J. (1993). Concentration-effect relationship of hydroxychloroquine in rheumatoid arthritis—A cross sectional study. The Journal of Rheumatology, 20(11), 1874–1879.
Thiery, J., Keefe, D., Boulant, S., Boucrot, E., Walch, M., Martinvalet, D., et al. (2011). Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nature Immunology, 12(8), 770–777. doi:10.1038/ni.2050.
Thiery, J., Keefe, D., Saffarian, S., Martinvalet, D., Walch, M., Boucrot, E., et al. (2010). Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood, 115(8), 1582–1593. doi:10.1182/blood-2009-10-246116.
Trocoli, A., & Djavaheri-Mergny, M. (2011). The complex interplay between autophagy and NF-kappaB signaling pathways in cancer cells. American Journal of Cancer Research, 1(5), 629–649.
Viry, E., Baginska, J., Berchem, G., Noman, M. Z., Medves, S., Chouaib, S., et al. (2014). Autophagic degradation of GZMB/granzyme B: A new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy, 10(1), 173–175. doi:10.4161/auto.26924.
White, E., Karp, C., Strohecker, A. M., Guo, Y., & Mathew, R. (2010). Role of autophagy in suppression of inflammation and cancer. Current Opinion in Cell Biology, 22(2), 212–217. doi:10.1016/j.ceb.2009.12.008.
Wong, K. K., Engelman, J. A., & Cantley, L. C. (2010). Targeting the PI3K signaling pathway in cancer. Current Opinion in Genetics & Development, 20(1), 87–90. doi:10.1016/j.gde.2009.11.002.
Wu, Y. T., Tan, H. L., Huang, Q., Ong, C. N., & Shen, H. M. (2009). Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy, 5(6), 824–834.
Xu, C., Liu, J., Hsu, L. C., Luo, Y., Xiang, R., & Chuang, T. H. (2011). Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB Journal, 25(8), 2700–2710. doi:10.1096/fj.10-167676.
Yang, Z., & Klionsky, D. J. (2009). An overview of the molecular mechanism of autophagy. Current Topics in Microbiology and Immunology, 335, 1–32. doi:10.1007/978-3-642-00302-8_1.
Yang, Z., & Klionsky, D. J. (2010). Eaten alive: A history of macroautophagy. Nature Cell Biology, 12(9), 814–822. doi:10.1038/ncb0910-814.
Young, A. R., Narita, M., Ferreira, M., Kirschner, K., Sadaie, M., Darot, J. F., et al. (2009). Autophagy mediates the mitotic senescence transition. Genes & Development, 23(7), 798–803. doi:10.1101/gad.519709.
Yu, S. W., Baek, S. H., Brennan, R. T., Bradley, C. J., Park, S. K., Lee, Y. S., et al. (2008). Autophagic death of adult hippocampal neural stem cells following insulin withdrawal. Stem Cells, 26(10), 2602–2610. doi:10.1634/stemcells.2008-0153.
Yu, Y., Yang, L., Zhao, M., Zhu, S., Kang, R., Vernon, P., et al. (2012). Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia, 26(8), 1752–1760. doi:10.1038/leu.2012.65.
Yue, Z., Jin, S., Yang, C., Levine, A. J., & Heintz, N. (2003). Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proceedings of the National Academy of Sciences of the United States of America, 100(25), 15077–15082. doi:10.1073/pnas.2436255100.
Acknowledgments
A part of research results presented in this chapter was generated at Luxembourg Institute of Health and Gustave Roussy Cancer Center. Research projects related to these results were funded by grants from Luxembourg Ministry of Culture, Higher Education and Research (Grant LHCE 2013 11 05); Fondation Cancer Luxembourg; FNRS Televie (7.4517.14; 7.4571.15 and 7.4664.15) and “Equipe labélisée la ligue contre le cancer”.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Janji, B., Chouaib, S. (2016). Role of Autophagy in Tumor Progression and Regression. In: Yang, JM. (eds) Targeting Autophagy in Cancer Therapy. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-319-42740-9_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-42740-9_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42738-6
Online ISBN: 978-3-319-42740-9
eBook Packages: MedicineMedicine (R0)