
Abstract
In the past decade, there has been a resurgence of interest in elucidating how metabolism is altered in cancer cells and how such dependencies can be targeted for therapeutic gain. At the core of this research is the concept that metabolic pathways are reprogrammed in cancer cells to divert nutrients toward anabolic processes to facilitate enhanced growth and proliferation. Importantly, physiological cellular signaling mechanisms normally tightly regulate the ability of cells to gain access to and utilize nutrients, posing a fundamental barrier to transformation. This barrier is often overcome by aberrations in cellular signaling that drive tumor pathogenesis by enabling cancer cells to make critical cellular decisions in a cell-autonomous manner. One of the most frequently altered pathways in human cancer is the PI3K-Akt-mTOR signaling pathway. Here, we describe mechanisms by which this signaling network is responsible for controlling cellular metabolism. Through both the post-translational regulation and the induction of transcriptional programs, the PI3K-Akt-mTOR pathway coordinates the uptake and utilization of multiple nutrients, including glucose, glutamine, nucleotides, and lipids, in a manner best suited for supporting the enhanced growth and proliferation of cancer cells. These regulatory mechanisms illustrate how metabolic changes in cancer are closely intertwined with oncogenic signaling pathways that drive tumor initiation and progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
A fundamental property of cells in multicellular organisms is the ability to process and integrate a variety of extracellular signals, such as growth factors, cytokines, and nutrients, in order to make decisions about cell fate, including proliferation, growth, survival, differentiation, motility, and metabolism. To accomplish this, cells have developed complex signaling networks that allow them to transduce extracellular signals into cellular decisions. Over the past few decades, much research has been dedicated toward elucidating how cellular signaling pathways are wired, and it has become clear that aberrations in cellular signaling underlie a wide variety of human diseases. In particular, our increasing understanding of the genomic landscape of human cancers over the past few years has revealed that cancer cells are frequently selected for mutations that occur in key nodes of various signaling pathways. These genetic alterations enable cancer cells to escape regulation by extrinsic signals and instead make critical cellular decisions in a cell-autonomous manner to promote uncontrollable cell survival, growth, and proliferation.
In the past decade, there has been a resurgence of interest in elucidating how metabolism is altered in cancer cells, based on observations that components of signal transduction pathways frequently regulate nutrient metabolism. At the core of this research is the concept that metabolic pathways are reprogrammed in cancer cells to divert nutrients toward anabolic processes to facilitate enhanced growth and proliferation. Importantly, physiological cellular signaling mechanisms normally tightly regulate the ability of cells to gain access to and utilize nutrients, posing a fundamental barrier to transformation. Therefore, it is becoming increasingly clear that cancer cells frequently select for mutations that enhance signal transduction through pathways that converge upon a common set of metabolic processes to promote nutrient uptake and utilization in cell building processes. It is the hope that an integrated understanding of oncogenic signaling and its modulation of cancer metabolism will reveal metabolic dependencies that are unique to cancer cells, thereby opening a therapeutic window that can be exploited to selectively target tumor cells.
The phosphoinositide 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) signaling pathway is one of the most frequently altered pathways in human cancer. The importance of the pathway in cancer first became apparent in the 1980s, when it was discovered that a PI kinase activity was associated with transformation mediated by viral oncogenes such as the SRC tyrosine kinase and polyomavirus middle T antigen (Whitman et al. 1985) and that this enzyme carried out a previously unknown reaction, the phosphorylation of PI at the 3 position of the inositol ring (Whitman et al. 1988). Subsequent work placed PI3K at the head of a signaling network in which PI3K transduces upstream signals from receptor tyrosine kinases (RTKs) to generate critical lipid second messengers that serve to further activate downstream signaling effectors such as Akt and mTOR (Toker and Cantley 1997; Toker 2012). Multiple components of the pathway have since been demonstrated to be bona fide oncogenes and tumor suppressors, and activation of this pathway has a critical role in controlling most of the hallmarks of cancer, including proliferation, cell survival, motility, genomic instability, angiogenesis, and metabolism (Hanahan and Weinberg 2011). Studies in cancer genomics over the past decade have reinforced the importance of the PI3K-Akt-mTOR signaling pathway in human cancer, as genetic lesions that drive tumorigenesis are frequently found within components of the signaling network (see Table 1). As a result, numerous drugs have been developed in the past decade that target various nodes of the pathway, many of which are currently in clinical trials (Engelman 2009; Fruman and Rommel 2014; Fruman and Cantley 2014).
Given the critical role of PI3K-Akt-mTOR signaling in driving tumorigenesis, the importance of the pathway in modulating metabolism in cancer cells has also become apparent in recent years. Here, we will focus on mechanisms by which the PI3K-Akt-mTOR signaling pathway is responsible for coordinating nutrient uptake and utilization, and how cancer cells exploit these metabolic processes to support enhanced growth and proliferation. In doing so, we hope to illustrate how metabolic changes in cancer are closely intertwined with oncogenic signaling pathways that drive tumor initiation and progression.
2 Signaling Through the PI3K-Akt-mTOR Pathway
The PI3K-Akt-mTOR pathway coordinates insulin signaling during organismal growth and is a critical mediator of a variety of physiological processes, including glucose homeostasis, protein synthesis, cell proliferation, cell growth, survival, and angiogenesis. In fact, its central role in cellular physiology is underscored by the conservation of the pathway back to Drosophila melanogaster and Caenorhabditis elegans; furthermore, mTOR, but not PI3K/Akt, is conserved as far back as yeast (Vivanco and Sawyers 2002). Given the importance of PI3K-Akt-mTOR signaling in controlling organismal growth, it is not unexpected that this pathway is one of the most frequently dysregulated signaling pathways in human cancers (Engelman 2009).
2.1 PI3K-Akt Pathway Activation
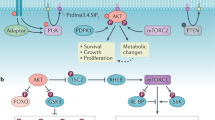
Activation of RTKs and G-protein-coupled receptors (GPCRs) by extracellular stimuli, including growth factors, leads to PI3K activation (Fig. 1). As indicated above, PI3K is a lipid kinase that phosphorylates the 3′-hydroxyl group of the inositol ring of phosphatidylinositol to generate 3′-phosphoinositides (Whitman et al. 1988; Auger et al. 1989; Cantley 2002). Accumulation of these lipid second messengers subsequently activates pathways that control fundamental cellular processes. One key 3′-phosphoinositide is phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3; PIP3), which is produced from phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2; PIP2) by Class I PI3Ks. Negative regulation of the pathway is conferred by the lipid phosphatase, phosphatase and tensin homolog (PTEN), which converts PIP3 back into PIP2 (Li et al. 1997; Maehama and Dixon 1998; Cantley and Neel 1999). PIP3 recruits inactive cytosolic molecules containing pleckstrin homology (PH) domains to the plasma membrane to promote their activation (Vanhaesebroeck and Waterfield 1999). While these PIP3-regulated effectors include multiple proteins such as PREX1, PREX2, and GRP1 that may all have important roles in cancer metabolism, here we focus on the serine/threonine AGC-family protein kinase Akt. PIP3 binds to the PH domain of Akt, recruiting it to the plasma membrane where it is phosphorylated at Thr308 by another PIP3-recruited molecule, phosphoinositide-dependent protein kinase 1 (PDPK1) (Franke et al. 1997; Alessi et al. 1997; Stephens et al. 1998; Storz and Toker 2002). For complete activation, Akt is also phosphorylated on Ser473 by the mammalian target of rapamycin complex 2 (mTORC2) (Bellacosa et al. 2005; Saci et al. 2011). In recent years, further mechanisms that account for full Akt activation have also been described, including Akt ubiquitination, sumoylation, and regulation by the cell cycle (Yang et al. 2009b, 2010, 2013; Chan et al. 2012; Fan et al. 2013a; Li et al. 2013; Risso et al. 2013; de la Cruz-Herrera et al. 2015; Liu et al. 2014). As more details behind these additional layers of regulation are clarified, it is becoming clear that the regulation of signaling downstream of PI3K is highly complex and multifaceted.

PI3K-Akt-mTOR signaling pathway
2.2 Downstream Effectors
PI3K-Akt pathway activation controls a wide variety of cellular processes. Broadly, the majority of the downstream effectors of Akt are regulated by two general mechanisms. First, Akt can acutely regulate various cellular processes in a post-translational manner by phosphorylating and controlling the activity, localization, or stability of its protein substrates. Second, Akt can establish longer-term changes in cellular behavior by phosphorylating and thereby influencing the activity of various transcription factors. It will become evident as we progress through our discussion of metabolism that in many cases, changes in metabolic processes are initiated by post-translational responses and then further reinforced by the induction of transcriptional programs.
2.2.1 Post-translational Modulation
Upon full activation by dual phosphorylation at Ser473 and Thr308, Akt translocates to distinct subcellular compartments and phosphorylates various downstream substrates. Importantly, Akt preferentially phosphorylates substrate proteins in a sequence-specific context, with a minimally required amino acid motif of R-X-R-X-X-S/T-Ψ (where X tends to be a hydrophilic amino acid and Ψ is hydrophobic; Fig. 1) (Alessi et al. 1996; Obata et al. 2000; Manning and Cantley 2007). Currently, close to 200 proposed substrates of Akt have been identified (Manning and Cantley 2007), and these substrates control a wide range of key cellular processes, including proliferation, growth, survival, motility, and metabolism. With regard to metabolism, Akt has been shown to directly phosphorylate several metabolic enzymes such as hexokinase (HK) and phosphofructokinase 2 (PFK2), which we will describe in greater detail in the following sections.
One of the major effectors that has emerged as a critical signaling component downstream of PI3K-Akt activation is the mammalian target of rapamycin complex 1 (mTORC1) (Fig. 1). mTORC1 activation occurs at the lysosome, where it is activated by the GTP-bound form of the small G-protein Rheb. An important negative regulator of mTORC1 is the tuberous sclerosis (TSC) complex, which consists of the tumor suppressors tuberous sclerosis 1 (TSC1), tuberous sclerosis 2 (TSC2), and a recently described third protein, TBC1D7 (Tee et al. 2002; Dibble et al. 2012). The TSC complex suppresses mTORC1 by acting as a GTPase-activating protein (GAP) for Rheb (Manning and Cantley 2003; Tee et al. 2003). Akt stimulates mTORC1 activity by inhibiting the TSC complex through phosphorylation of multiple sites on TSC2 (Manning et al. 2002). TSC2 phosphorylation leads to dissociation of the TSC complex from the lysosome, thus relieving its inhibition of Rheb and resulting in mTORC1 activation (Menon et al. 2014). Akt can also directly phosphorylate mTOR and an mTOR-associated protein, PRAS40 (AKT1S1), and these events may also promote activation of mTORC1 (Nave et al. 1999; Sekulić et al. 2000; Vander Haar et al. 2007). Importantly, mTORC1 activation requires both active PI3K-Akt signaling and sufficient intracellular pools of amino acids. Hence, mTORC1 is considered to be a key integrator of systemic signals (such as growth factors) and local signals (nutrient availability), with its activation culminating in anabolic processes that promote growth such as protein synthesis and metabolism (Dibble and Manning 2013; Bar-Peled and Sabatini 2014). Indeed, the most well-studied substrates of mTORC1 are the ribosomal S6 kinases, S6K1 and S6K2, and the eIF4E (eukaryotic initiation factor 4E)-binding proteins, 4E-BP1 and 4E-BP2, which all have critical roles in regulating mRNA translation to enhance cellular growth and proliferation (Ma and Blenis 2009). In addition, recent emerging evidence has also demonstrated that signaling downstream of mTORC1 regulates anabolic metabolic processes, through translational and even direct post-translational modulation of metabolic enzymes.
2.2.2 Transcriptional Output
In addition to acutely regulating various cellular processes through post-translational modifications, PI3K-Akt-mTOR signaling can also establish longer-term changes in cellular behavior by influencing the activity of various transcription factors (Fig. 1). Here, we will briefly describe how PI3K-Akt-mTOR signaling regulates a few of these transcription factors, with particular emphasis on those that have a major role in modulating cellular metabolism.
2.2.2.1 FoxO
The forkhead box O (FoxO) transcription factor family, which consists of FoxO1, FoxO3, FoxO4, and FoxO6, functions primarily downstream of the insulin and insulin-like growth factor receptors to regulate diverse cellular processes, including proliferation, apoptosis, antioxidant responses, longevity, and metabolism (Tzivion et al. 2011). As a major effector of insulin signaling, Akt was identified in early genetic and biochemical studies as a key regulator of FoxO function (Paradis and Ruvkun 1998). FoxO1/3/4 each contains three Akt phosphorylation sites, while FoxO6 contains two. These phosphorylation events primarily serve as docking points for interactions with 14-3-3 proteins (Tzivion et al. 2011). Upon binding, 14-3-3 proteins regulate the cellular localization of FoxO, both by increasing nuclear export of FoxO and by sequestering FoxO in the cytoplasm (Brunet et al. 2002). 14-3-3 also masks the DNA-binding domain of FoxO, thus preventing it from binding to the promoters of its target genes (Obsil et al. 2003; Silhan et al. 2009). Together, the functional consequence of phosphorylation by Akt and subsequent 14-3-3 binding is the inhibition of FoxO-mediated transcriptional activity.
2.2.2.2 c-Myc
c-Myc is a key transcription factor encoded by the MYC oncogene that controls a number of physiological cellular processes, including cell growth, proliferation, apoptosis, and energy metabolism (Eilers and Eisenman 2008). c-Myc can also cooperate with PI3K to promote cell proliferation and transformation (Zhao et al. 2003). Given the potential for this adverse consequence, multiple PI3K-mediated regulatory mechanisms exist to modulate and fine-tune c-Myc activity. For example, activation of Akt relieves FoxO-mediated inhibition of c-Myc target genes and enables the transformative capacity of c-Myc (Bouchard et al. 2004). PI3K-Akt signaling also controls c-Myc protein stability through glycogen synthase kinase-3β (GSK-3β), which is directly phosphorylated and inhibited by Akt. Inhibition of GSK-3β prevents c-Myc phosphorylation at Thr58 and its subsequent degradation, since phosphorylation at this site promotes its ubiquitination by the SCFFbw7 ubiquitin ligase followed by proteasomal degradation (Gregory et al. 2003; Welcker et al. 2004). Finally, S6K phosphorylates Ser145 on Mad1, which is an antagonist of c-Myc. Mad1 phosphorylation promotes its ubiquitination and degradation, leading to the upregulation of Myc-mediated transcription (Zhu et al. 2008).
2.2.2.3 HIFs
The hypoxia-inducible factor (HIF) family of transcription factors, which consists of three members, HIF1, HIF2, and HIF3, are activated in response to hypoxia in order to induce genes that facilitate adaptation to the hypoxic environment. These gene programs enable adaptation by directing glucose away from oxidative phosphorylation toward glycolysis and lactate production and by the stimulation of angiogenesis to restore oxygen homeostasis. Each HIF consists of an oxygen-dependent α subunit and a constitutively expressed β subunit, and the active heterodimer is stabilized under hypoxic conditions (reviewed in Brahimi-Horn et al. 2007). PI3K-Akt-mTOR signaling can also regulate HIF1 independently of oxygen concentration to stimulate its increased accumulation and activity in tumor cells (Zundel et al. 2000; Zhong et al. 2000; Blancher et al. 2001; Chan et al. 2002; Bardos and Ashcroft 2004). This regulation occurs primarily at the level of both transcription and translation by mTORC1. Transcriptionally, both HIF1α and HIF2α message levels are elevated in an mTORC1-dependent manner in TSC2-null cells (Düvel et al. 2010). mTORC1 also selectively increases the translation of HIF1α through the 5′-untranslated region of the HIF1α mRNA by inhibiting 4E-BP1 to activate cap-dependent translation (Laughner et al. 2001; Thomas et al. 2006; Düvel et al. 2010).
2.2.2.4 SREBPs
The sterol regulatory element-binding proteins (SREBPs), which consist of three isoforms SREBP-1a, SREBP-1c, and SREBP2, are transcription factors that function as central regulators of lipid homeostasis. SREBPs reside in the endoplasmic reticulum membrane in their inactive, full-length forms. In response to sterol depletion, the SREBPs are trafficked to the Golgi, where they are proteolytically activated. The cleaved N-terminal domain of SREBPs subsequently translocates to the nucleus and binds to sterol regulatory elements (SRE) to upregulate genes involved in the uptake and biosynthesis of cholesterol, fatty acids, triglycerides, and phospholipids (reviewed in Espenshade and Hughes 2007). PI3K-Akt-mTOR signaling has been implicated in SREBP activation, since signaling through S6K1 promotes the accumulation of the active form of SREBP1 (Porstmann et al. 2008; Düvel et al. 2010). Furthermore, inhibition of GSK-3β by Akt also enhances the stability of SREBP, which can be phosphorylated by GSK-3β and directed for ubiquitination by the SCFFbw7 ubiquitin ligase and subsequent proteasomal degradation (Kim et al. 2004b; Sundqvist et al. 2005).
3 The PI3K-Akt-mTOR Pathway Regulates Glucose Metabolism and the Warburg Effect
The altered metabolism of cancer cells was first noted in 1924, when Otto Warburg observed that tumors in rats take up more than ten times as much glucose as compared to corresponding normal tissue, and that this glucose is primarily converted to lactate (Warburg 1956). This led to the hypothesis that through a process known as aerobic glycolysis, or the “Warburg effect,” cancer cells promote the glycolytic conversion of glucose into lactate even in the presence of sufficient oxygen to support mitochondrial oxidative phosphorylation. Recently, it has become evident that the switch to aerobic glycolysis provides tumor cells with a proliferative advantage (Vander Heiden et al. 2009). Hence, there has been intense investigation into the mechanisms by which this process is activated and regulated in an effort to exploit this pathway for therapeutic gain.
One major mechanism by which this occurs is through the oncogenic activation of PI3K-Akt-mTOR signaling. For example, immortalized hematopoietic cells transformed by a constitutively active mutant of Akt display aerobic glycolysis—that is, higher rates of glycolysis without effects on the rate of oxidative phosphorylation (Elstrom et al. 2004). Similar results were found in human glioblastoma cells possessing constitutive Akt activity. Importantly, these cells are dependent on aerobic glycolysis for growth and survival, since they are more susceptible to cell death after glucose withdrawal. Together, these findings indicate that PI3K-Akt-mTOR signaling is sufficient to stimulate the switch to aerobic glycolysis. Since the studies described above, it has become clear that this switch is achieved through a number of mechanisms that converge on the regulation of glycolysis, which we describe below.
3.1 Glucose Uptake
Enhanced glucose uptake is a requisite event for aerobic glycolysis. Under normal physiological conditions, the PI3K-Akt-mTOR signaling pathway is a major regulator of glucose uptake, through both post-translational and transcriptional effects. At the post-translational level, glucose uptake is stimulated primarily through the regulation of glucose transporter trafficking (Fig. 2). For example, acute insulin treatment in adipocytes and muscle cells leads to the translocation of the glucose transporter GLUT4 to the plasma membrane (Huang and Czech 2007), in a manner that is dependent on PI3K and Akt (Okada et al. 1994; Kotani et al. 1995; Kohn et al. 1996; Cong et al. 1997). Interestingly, insulin-stimulated GLUT4 translocation is specifically dependent on the Akt2 isoform, since translocation is impaired in adipocytes upon Akt2 knockout or knockdown and cannot be rescued by Akt1 expression (Katome et al. 2003; Bae et al. 2003). Following insulin stimulation of adipocytes, Akt2 is preferentially localized to the plasma membrane, where it phosphorylates and inhibits the RabGAP AS160 (Gonzalez and McGraw 2009). Inhibition of AS160 is required for GLUT4 vesicle docking and fusion with the plasma membrane (Sano et al. 2003; Larance et al. 2005). As a consequence, insulin stimulation leads to GLUT4 localization at the plasma membrane, which enables glucose uptake. Another contributing mechanism is the activating phosphorylation of the phosphatidylinositol 3-phosphate 5-kinase PIKfyve by Akt (Berwick et al. 2004). PIKfyve participates in endosomal trafficking through the generation of PI(3,5)P2 from PI(3)P, and these lipid species appear to positively regulate GLUT4 trafficking to the plasma membrane through a poorly understood mechanism (Ikonomov et al. 2002; Sbrissa et al. 2004; Berwick et al. 2004; Ikonomov et al. 2007; Shisheva 2008; Ikonomov et al. 2009).

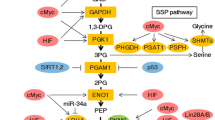
PI3K-Akt-mTOR signaling regulates glucose uptake and glycolysis. Filled green square target genes for c-Myc; filled green star target genes for HIF; filled red circle genes that are repressed by FoxO activation. HK hexokinase; G6P glucose 6-phosphate; GPI glucose 6-phosphate isomerase; F6P fructose 6-phosphate; PFK1/2 phosphofructokinase 1/2; FBP fructose 1,6-bisphosphate; DHAP dihydroxyacetone phosphate; Ga3P glyceraldehyde 3-phosphate; TPI triose phosphate isomerase; GAPDH glyceraldehyde 3-phosphate dehydrogenase; 1,3-BPG 1,3-bisphosphoglycerate; PGK phosphoglycerate kinase; 3-PG 3-phosphoglycerate; PGM phosphoglycerate mutase; 2-PG 2-phosphoglycerate; PEP phosphoenolpyruvate; PK pyruvate kinase; LDH lactate dehydrogenase; PDH pyruvate dehydrogenase; PDK1 pyruvate dehydrogenase kinase 1; ATP adenosine triphosphate; ADP adenosine diphosphate; NADH nicotinamide adenine dinucleotide
Although regulation of GLUT4 by Akt has been relatively well studied, it is important to note that GLUT4 is a muscle- and fat-cell-specific glucose transporter. In other words, most cancer cells do not express GLUT4, and rather express the embryonic glucose transporter isoform, GLUT1. Notably, GLUT1 has a high affinity for glucose and may be selected for by cancer cells to increase glucose transport efficiency (Ganapathy et al. 2009). PI3K-Akt signaling has also been proposed to regulate GLUT1 trafficking to the plasma membrane (Rathmell et al. 2003; Bentley et al. 2003). However, the specific mechanisms by which such regulation occurs remains an important open question. Of interest, thioredoxin-interacting protein (TXNIP) was recently shown to negatively regulate glucose uptake by binding to GLUT1 and inducing its internalization through clathrin-coated pits. Upon energy stress, TXNIP is phosphorylated by AMP-dependent protein kinase (AMPK), which leads to its rapid degradation and subsequent induction of GLUT1 activity (Parikh et al. 2007; Stoltzman et al. 2008; Wu et al. 2013). Given this regulation of GLUT1 trafficking by AMPK signaling, it would be interesting to evaluate whether TXNIP plays a role in GLUT1 regulation downstream of PI3K-Akt-mTOR signaling.
The glucose transporters are also regulated by PI3K-Akt-mTOR signaling at the transcriptional level (Fig. 2). Constitutive Akt activation results in elevated GLUT1 mRNA and protein levels, but not GLUT4 (Kohn et al. 1996; Barthel et al. 1999). Increased expression of GLUT1 is dependent on mTORC1 and its subsequent upregulation of HIF1α levels and activity (Wieman et al. 2007; Zhou et al. 2008; Düvel et al. 2010). GLUT1 can also be transcriptionally upregulated by c-Myc (Osthus et al. 2000; Ying et al. 2012), which lies downstream of PI3K.
Taken together, oncogenic activation of the PI3K-Akt-mTOR signaling network promotes post-translational regulation of glucose transporter trafficking to the plasma membrane, which is reinforced by transcriptional upregulation of the glucose transporter genes. These mechanisms combine to robustly stimulate glucose uptake in cancer cells, thus providing them with sufficient amounts of the substrate required for aerobic glycolysis.
3.1.1 Glucose Uptake in the Clinic
Well before the mechanistic details concerning growth factor- and/or oncogene-mediated glucose uptake were understood, an appreciation for the glucose avidity of cancer cells was exploited clinically. Indeed, for more than 30 years now, uptake of the radioactive glucose analog 18-fluoro-deoxyglucose (18FDG) has been used to diagnose and stage tumors and to monitor response to treatment. This is done using a technique that allows tumors to be imaged by positron emission tomography (PET), where cellular uptake of 18FDG is directly proportional to PET response (Ben-Haim and Ell 2009). Importantly, given the relationship between PI3K-Akt-mTOR signaling and glucose uptake, 18FDG-PET has been used as a method to monitor tumor responses to drugs that target the PI3K-Akt-mTOR pathway. For example, in a mouse model of lung cancer driven by the oncogenic PIK3CA(H1047R) mutation, 18FDG-PET imaging of lung tumors following administration of a dual PI3K-mTOR inhibitor for 4 days revealed that the 18FDG signal was significantly reduced, consistent with a measurable decrease in tumor size (Engelman et al. 2008). 18FDG-PET was similarly used in a triple-negative breast cancer mouse model to demonstrate that tumors show a decrease in 18FDG uptake in response to PI3K inhibition (Juvekar et al. 2012). Significantly, these results suggest that inhibition of 18FDG uptake by tumors may be an early and predictive pharmacodynamic marker for the efficacy of PI3K pathway inhibitors in the clinic. Indeed, a recent phase I study showed that the subset of patients who obtained a clinical benefit (partial tumor shrinkage) from PI3K inhibitor therapy exhibited a decline in 18FDG-PET signal within two weeks of initiating drug treatment, while those whose tumors did not show this early decline in 18FDG-PET had no tumor shrinkage (Mayer et al. 2014). Thus, early changes in 18FDG-PET might be more effective than mutational analysis in predicting which patients will respond to PI3K inhibitors (see Rodon et al. 2013 for review).
3.2 Regulation of Glycolytic Enzymes
PI3K-Akt-mTOR signaling also promotes aerobic glycolysis by directly regulating multiple nodes in glycolysis. For example, an early study demonstrated that both Akt and S6K can phosphorylate the bifunctional enzyme PFK-2/FBPase-2 (PFK2) in vitro, resulting in increased PFK-2 activity (Deprez et al. 1997). PFK2 regulates glycolysis by generating fructose 2,6-bisphosphate, which is the most potent known allosteric activator of PFK1, a rate-limiting enzyme for glycolysis (Fig. 2). Hence, PI3K-Akt signaling can indirectly activate PFK1 and stimulate glycolysis through PFK2 phosphorylation. It should be noted that PFK2 phosphorylation and glycolytic flux studies for PFK2 activity in the context of PI3K-Akt-mTOR signaling have not been carefully done in vivo. Furthermore, although one study has shown in prostate cancer cells that androgen treatment stimulates glycolysis by increasing PFK2 activity in a PI3K-dependent manner (Moon et al. 2011), a more comprehensive understanding of the broader applicability of Akt- or S6K-mediated phosphorylation of PFK2 in the reprogrammed metabolism of cancer cells remains to be determined.
Akt activation has also been implicated in increasing the activity of hexokinase (HK), which is overexpressed in multiple cancers and has been shown to be a key mediator of aerobic glycolysis, increased cell proliferation, and therapeutic resistance (Pastorino et al. 2005; Mathupala et al. 2006; Ahn et al. 2009; Wolf et al. 2011). HK catalyzes the first, and first rate-limiting, step of glycolysis by phosphorylating glucose to form glucose 6-phosphate (G6P) (Rathmell et al. 2003) (Fig. 2). Akt promotes the association of hexokinase I (HK1) and hexokinase II (HK2) with mitochondria through interactions with voltage-dependent anion channels (VDAC) and the outer mitochondrial membrane (OMM) (Majewski et al. 2004a; Robey and Hay 2009). The specific mechanisms by which this occurs are not yet entirely clear, but, at least for HK2, mitochondrial association is promoted in part by Akt-mediated phosphorylation of HK2 at Thr473 (Roberts et al. 2013). Akt also indirectly promotes mitochondrial HK2 association by inhibiting GSK-3β, which when active phosphorylates VDAC and inhibits its ability to bind HK2 (Pastorino et al. 2005).
Mitochondrial HK association serves several functions. First, it has been suggested to increase HK activity and to direct G6P toward glycolysis, as opposed to glycogen synthesis (Gottlob et al. 2001; John et al. 2011; Yeo et al. 2013). Mitochondrial HKs also directly couple mitochondrial ATP synthesis to glycolysis by preferentially using intramitochondrial ATP to phosphorylate glucose, thus efficiently regenerating ADP that can be directed back to support oxidative phosphorylation (Gottlob et al. 2001; Robey and Hay 2009). Finally, mitochondrial HKs are required for Akt-mediated inhibition of apoptosis in response to a variety of apoptotic stimuli, since they maintain mitochondrial integrity and prevent cytochrome c release by competing with the binding of Bax and Bak on the OMM (Pastorino et al. 2002, Majewski et al. 2004a, b; Miyamoto et al. 2008). In this way, the anti-apoptotic role of Akt is closely linked with its regulation of glucose metabolism, which provides a mechanism by which metabolism and cell survival are coupled.
In addition to post-translational regulation of glycolytic enzymes, PI3K-Akt-mTOR signaling also controls the expression of glycolytic genes (Fig. 2). One of the primary transcription factors through which this is regulated is HIF1. mTORC1-mediated upregulation of HIF1α increases the expression of GLUT1 and GLUT3, as well as almost all genes involved in glycolysis (Semenza et al. 1994, 1996; Denko 2008; Düvel et al. 2010). Interestingly, in addition to upregulating glycolysis, HIF1 also inhibits mitochondrial respiration, primarily by modulating the fate of pyruvate. HIF1 induces the expression of pyruvate dehydrogenase kinase 1 (PDK1), which phosphorylates and inhibits pyruvate dehydrogenase (PDH) (Kim et al. 2006; Papandreou et al. 2006). PDH is responsible for the oxidation of pyruvate by converting it into acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle and condenses with oxaloacetate to form citrate (Fig. 2). Therefore, PDH inhibition by PDK1 shunts pyruvate away from mitochondrial metabolism and oxidative phosphorylation. Another consequence of this, however, is that the reduced nicotinamide adenine dinucleotide (NADH) produced from glycolysis is no longer recycled by oxidative phosphorylation to regenerate NAD+, which is a critical cofactor for glycolytic activity. This is resolved by the upregulation of lactate dehydrogenase A (LDHA) by HIF1 (Semenza et al. 1996). LDHA regenerates NAD+ from NADH in the process of converting pyruvate into lactate, thus explaining the increased lactate production that is a hallmark of aerobic glycolysis.
Other transcription factors in the PI3K-Akt-mTOR signaling network, such as c-Myc and FoxO, also contribute to glycolytic gene expression. Like HIF1, c-Myc can directly regulate almost all the genes involved in glycolysis, including GLUT1, glucose phosphate isomerase (GPI), PFK1, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), triosephosphate isomerase (TPI), phosphoglycerate kinase 1 (PGK1), and enolase (ENO1) (Osthus et al. 2000; Kim et al. 2004a). It is important to note that such regulation can be highly fine-tuned and context dependent, as c-Myc activation does not necessarily always upregulate all glycolytic genes in a given tumor. c-Myc can also increase the expression of LDHA and PDK1, which shifts glycolysis toward lactate production as opposed to mitochondrial metabolism, as described above (Shim et al. 1997; Dang 2007). Finally, activation of the FoxO transcription factors results in the decreased expression of several glycolytic genes, including glucokinase (GCK), GPI, aldolase B (ALDOB), ENO1, and pyruvate kinase 1 (PK1) (Zhang et al. 2006). Hence, inhibition of FoxO by Akt relieves the suppression of glycolytic gene expression. Interestingly, an Akt-independent mechanism of FoxO regulation that impinges on glycolysis has also been demonstrated in glioblastoma (Masui et al. 2013). Specifically, mTORC2 promotes the inactivating phosphorylation of several histone deacetylases, which results in the acetylation and inactivation of FoxO. The regulation of glycolysis by FoxO occurs, at least in part, through transcriptional upregulation of TSC1 by FoxO, which attenuates mTORC1 activation and its effects on glycolytic gene expression (Khatri et al. 2010). FoxO also antagonizes c-Myc, in part through the increased expression of miR-145 and miR-34c, which limits c-Myc mRNA stability and translation (Gan et al. 2010; Kress et al. 2011; Peck et al. 2013).
Taken together, activation of the PI3K-Akt-mTOR network initiates a variety of mechanisms at both the post-translational and the transcriptional levels that converge to strongly stimulate the metabolism of glucose in glycolysis and to divert glucose carbon from being oxidized in the mitochondria. Several of the alternative biosynthetic fates of this carbon are described below.
4 Beyond Aerobic Glycolysis: Coordination with Anabolic Metabolism
Despite clear evidence that cancer cells reprogram glucose metabolism to favor aerobic glycolysis, the precise molecular reasons for this shift remain to be determined. Initial explanations proposed that aerobic glycolysis constituted a shift to glycolytic ATP production and energy dependence, as opposed to generation of ATP through mitochondrial oxidative phosphorylation. In fact, Warburg initially hypothesized that most cancer cells likely have mitochondrial defects that impair oxidative phosphorylation, thus forcing them to rely on glycolysis for energy production (Warburg 1956). However, several studies have shown that mitochondrial function is intact in most cancer cells (Weinhouse 1976; Fantin et al. 2006; Moreno-Sanchez et al. 2007).
The predominant logic in the field now posits that aerobic glycolysis is activated to provide cells with unrestricted access to basic cellular building blocks, in the form of glucose carbon, from which to draw for biosynthetic purposes (Vander Heiden et al. 2009). In particular, cancer cells must promote an anabolic state to produce the nucleotides, amino acids, and lipids needed to grow and divide. In other words, the enhanced glycolytic flux in cancer cells functions primarily to catabolize glucose into biosynthetic intermediates that are used for anabolic processes to meet the increased demands of enhanced growth and proliferation.
This concept is particularly evident when considering the integration of oncogenic PI3K-Akt-mTOR signaling with metabolic reprogramming in cancer. Activation of the signaling pathway does not merely stimulate aerobic glycolysis, but also tightly coordinates it with downstream anabolic processes to synthesize metabolites required for malignant cell growth and proliferation. The specific mechanism by which the PI3K-Akt-mTOR network regulates the metabolism of cellular building blocks such as nucleotides, lipids, and amino acids is an active area of investigation, and much remains to be delineated. Here, we describe a few mechanisms by which PI3K-Akt-mTOR signaling controls these processes.
4.1 Pentose Phosphate Pathway
The pentose phosphate pathway (PPP) branches off from glycolysis and uses glycolytic intermediates to generate reducing power in the form of nicotinamide adenine dinucleotide phosphate (NADPH) and 5-carbon sugars. There are two distinct but related phases of the pathway: the oxidative branch and the nonoxidative branch. In the oxidative PPP, glucose is oxidized to facilitate the production of NADPH, a reducing equivalent that contributes both to maintaining redox homeostasis and to providing sufficient reducing power for various anabolic processes. The reduction of NADP+ in the oxidative arm of the PPP occurs in two steps, ultimately converting glucose 6-phosphate into ribulose 5-phosphate (Ru5P), which is then directed into ribose 5-phosphate (R5P) (Fig. 3). This R5P can be used as a precursor in RNA and DNA biosyntheses, or it can be recycled back into glycolytic intermediates through the nonoxidative arm of the PPP. In certain contexts, the nonoxidative arm can be used to generate R5P from the glycolytic intermediates fructose 6-phosphate (F6P) and glyceraldehyde 3-phosphate (Ga3P), thereby bypassing the oxidative arm. This pathway has been described to be operative when the oxidative arm is inhibited and/or when alternative sources of NADPH generation dominate (Ying et al. 2012; Stanton 2012; Son et al. 2013).

PI3K-Akt-mTOR signaling coordinates the pentose phosphate pathway with lipid and nucleotide metabolism. Filled Green square genes that are upregulated by mTORC1 activation; filled green star target genes for SREBP1; filled red circle genes that are repressed by FoxO activation. For the purposes of representing these pathways schematically, information about the subcellular location of enzymes or signaling proteins is not conveyed. PPP pentose phosphate pathway; G6PD glucose 6-phosphate dehydrogenase; GδL6P 6-phosphogluconolactone; PGLS 6-phosphogluconolactonase; 6PG 6-phosphogluconate; PGD 6-phosphogluconate dehydrogenase; NADPH nicotinamide adenine dinucleotide phosphate; Ru5P ribulose 5-phosphate; RPE ribulose 5-phosphate epimerase; RPIA ribulose 5-phosphate isomerase; X5P xylulose 5-phosphate; R5P ribose 5-phosphate; TKT transketolase; S7P sedoheptulose 7-phosphate; E4P erythrose 4-phosphate; TALDO1 transaldolase 1; SBP sedoheptulose-1,7-bisphosphate; CPT1A carnitine palmitoyltransferase 1A; ACL ATP citrate lyase; Gln glutamine; CAD carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase; PRPP phosphoribosyl pyrophosphate. Double asterisk this reaction (SBP synthesis) has not been demonstrated in mammalian cells and has thus far only been observed in yeast
Both branches of the PPP have been shown to be important in human cancers. For example, activity of the rate-limiting enzyme glucose 6-phosphate dehydrogenase (G6PD) in the oxidative arm and expression of transketolase (TKT) in the nonoxidative arm are frequently elevated in a variety of cancers (Zampella et al. 1982; Bokun et al. 1987; Dessì et al. 1988; Langbein et al. 2006; Krockenberger et al. 2007; Langbein et al. 2008). Indeed, PI3K-Akt-mTOR signaling has been implicated in the regulation of the pentose phosphate pathway (Fig. 3). For example, activation of mTORC1 is sufficient to upregulate the expression of genes involved in the pathway, including G6PD, 6-phosphogluconate dehydrogenase (PGD), ribulose 5-phosphate epimerase (RPE), ribulose 5-phosphate isomerase (RPIA), and transaldolase 1 (TALDO1). G6PD expression in particular is mediated by SREBP1 activation downstream of S6K1, while how mTORC1 induces the expression of the other PPP genes is unknown (Düvel et al. 2010). PI3K-Akt-mTOR signaling also stimulates the production of R5P, although the relative contribution from the oxidative branch versus the nonoxidative branch varies in a cell-type and context-dependent manner. For example, constitutive mTORC1 activity stimulates R5P generation predominantly by increased flux through the oxidative PPP (Düvel et al. 2010). In contrast, several studies have suggested that the nonoxidative branch of the pathway supplies the majority of the R5P used in nucleotide synthesis (Raivio et al. 1981; Boss and Pilz 1985; Boros et al. 1997, 2001; Ying et al. 2012), and PI3K inhibition can selectively inhibit the nonoxidative branch of the PPP (Wang et al. 2009). From these studies, it is clear that the PI3K-Akt-mTOR signaling pathway has a critical role in dictating PPP activity; however, the exact mechanisms explaining tissue-specific and context-dependent effects remain to be fully characterized.
4.2 Nucleotide Biosynthesis
By stimulating the production of R5P through the PPP, the PI3K-Akt-mTOR network provides the building block for de novo nucleotide biosynthesis (Fig. 3). As such, it would seem logical that there would be coordination between PI3K-Akt-mTOR signaling-mediated increases in R5P levels and enhanced nucleotide synthesis. Indeed, mTORC1 acutely stimulates flux through the de novo pyrimidine biosynthetic pathway through S6K1. S6K1 directly phosphorylates S1859 on CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase), the rate-limiting multifunctional enzyme that mediates the first three steps of de novo pyrimidine synthesis. This phosphorylation event drives the activity of CAD to increase pyrimidine biosynthetic flux (Ben-Sahra et al. 2013; Robitaille et al. 2013). Whether CAD phosphorylation by S6K1 is necessary to sustain the enhanced growth and proliferation of cancer cells in the context of oncogenic signaling is an important question that will need to be addressed.
There is also evidence that PI3K-Akt signaling may regulate purine metabolism, although the mechanisms are not as clear. In C2C12 cells, inhibition of PI3K or Akt, but not mTORC1, decreased flux through both the de novo purine biosynthetic pathway and the purine salvage pathway, in part through reducing the availability of phosphoribosyl pyrophosphate (PRPP), which is required for the first step of both purine biosynthesis and purine salvage. Reduced PRPP levels are likely due to inhibition of R5P production through the PPP, since R5P is incorporated into PRPP (Fig. 3). Interestingly, it was also found in cell extracts that the activity of ATIC, which catalyzes the last two steps of de novo purine synthesis, is reduced by PI3K inhibition (Wang et al. 2009). ATIC does not have a consensus Akt substrate motif, however, suggesting that the potential regulation of ATIC enzymatic activity by Akt is indirect. Furthermore, the expression of the genes involved in purine synthesis and salvage was not assessed upon PI3K inhibition. It is clear that much remains to be understood regarding how the PI3K-Akt-mTOR network regulates purine metabolism, and especially in the context of oncogenic signaling in cancer cells.
4.3 Lipid Synthesis
Most normal cells have low rates of lipid synthesis, and only a few specialized cell types, such as hepatocytes and adipocytes, are designed to produce lipids. On the other hand, nearly all tumor cells have an increased rate of de novo lipid synthesis (Menendez and Lupu 2007). This metabolic feature allows cancer cells to autonomously generate lipids to form new membranes and to support other features associated with cell division and proliferation.
PI3K-Akt-mTOR signaling also has an important role in initiating both post-translational and transcriptional programs to stimulate de novo lipid synthesis. At the post-translational level, Akt has been proposed to phosphorylate Ser454 on ATP citrate lyase (ACL), which converts citrate to cytosolic acetyl-CoA that is required for lipid biosynthesis (Fig. 3) (Berwick et al. 2002; Sale et al. 2006; Migita et al. 2008). ACL phosphorylation on Ser454 increases its enzymatic activity in vitro (Potapova et al. 2000), although careful in vivo studies evaluating the contribution of phosphorylation at this site still need to be performed. It is also important to note that the proposed Akt phosphorylation site lacks the critical Arg at the −5 position, suggesting that there may be an intermediate kinase between Akt and ACL. Nevertheless, there is clear genetic evidence linking ACL phosphorylation to PI3K-Akt signaling. For instance, in nonsmall cell lung cancer, ACL phosphorylation is associated with PI3K-Akt activation, since inhibition of the pathway blocks ACL phosphorylation and activity. Direct knockdown of ACL induces cell-cycle arrest in vitro and inhibits tumor growth in vivo (Migita et al. 2008). ACL knockdown also impairs Akt-mediated leukemogenesis (Bauer et al. 2005). Taken together, ACL is a critical downstream effector of PI3K-Akt signaling that is post-translationally regulated to enhance de novo lipid synthesis.
The PI3K-Akt-mTOR network also establishes multiple transcriptional programs that converge to stimulate fatty acid and lipid synthesis. First, as previously described, mTORC1-mediated upregulation of the oxidative PPP generates NADPH. NADPH is required for lipid synthesis, as it provides the reducing power necessary for extending the carbon units on fatty acid chains (Fig. 3). mTORC1 also upregulates genes involved in the cholesterol and fatty acid biosynthetic pathways, primarily through activation of SREBP (Porstmann et al. 2005, 2008; Düvel et al. 2010). Indeed, one key target of SREBP, fatty acid synthase (FASN), is highly expressed in many types of human cancer (Van de Sande et al. 2002, 2005; Menendez and Lupu 2007). Moreover, at least in the liver, FoxO transcription factors inhibit lipogenesis by suppressing the expression of lipogenic genes, in part through their suppression of SREBP-1c (Zhang et al. 2006; Deng et al. 2012). Therefore, Akt-mediated inhibition of FoxO relieves this suppression of lipid synthesis. Finally, in addition to stimulating lipid synthesis, PI3K-Akt signaling also suppresses β-oxidation, the process by which lipids are catabolized, by decreasing the expression of the β-oxidation enzyme carnitine palmitoyltransferase 1A (CPT1A) (Deberardinis et al. 2006). The PI3K-regulated transcription factor responsible for this effect, however, has not yet been identified. Taken together, these transcriptional programs combine with the post-translational levels of regulation described above to robustly enhance lipogenesis in cancer cells driven by oncogenic PI3K-Akt-mTOR signaling.
4.4 Glutamine Metabolism
Due to a metabolic shift to aerobic glycolysis in cancer cells, many of the central glucose-derived intermediates are directed away from the mitochondria, where the TCA cycle and oxidative phosphorylation occur. Biosynthetic activity in the TCA cycle, however, is essential to proliferating cells as it provides intermediates for amino acid, nucleotide, and lipid biosynthesis. As a result, cancer cells must coordinate a shift to aerobic glycolysis with mechanisms to compensate for the depletion of TCA cycle intermediates, in a process known as anaplerosis. One major pathway by which anaplerosis occurs is through the utilization of glutamine, which can be converted into α-ketoglutarate (αKG) and fed into the TCA cycle (Fig. 4) (Hensley et al. 2013). In fact, some cancer cells generate more than half of their ATP from glutamine-derived αKG (Reitzer et al. 1979; Fan et al. 2013b).

PI3K-Akt-mTOR signaling regulates glutamine uptake and utilization. Filled green square target genes for CREB2; filled green star target genes for c-Myc; filled red circle target genes for FoxO. Gln glutamine; GLUL glutamine synthetase; GLS glutaminase; GDH glutamate dehydrogenase; NEAA nonessential amino acid
Since PI3K-Akt-mTOR signaling stimulates aerobic glycolysis, it must, in some way, also initiate mechanisms to facilitate anaplerosis. Early studies indicated that insulin can acutely stimulate glutamine transport, at least in primary cultures of rat skeletal muscle cells and hepatocytes (Gebhardt and Kleemann 1987; Tadros et al. 1993; Low et al. 1996). IGF-1 has also been shown to stimulate glutamine uptake in a human neuroblastoma cell line when extracellular glutamine concentrations are limited (Wasa et al. 2001). In addition, mTORC1 was recently shown to increase glutamine uptake in TSC2-null MEFs and in various human epithelial tumor cell lines (Csibi et al. 2013). Although one study in Xenopus oocytes suggested that the stability of the glutamine transporter SN1 may be promoted by Akt and the related serum- and glucocorticoid-dependent kinases SGK1 and SGK3 (Boehmer et al. 2003), the detailed mechanisms behind the acute stimulation of glutamine transport by insulin are not known. At the transcriptional level, glutamine uptake may be stimulated in part by upregulation of the glutamine transporters ASCT2 and SN2 by c-Myc (Wise et al. 2008; Li and Simon 2013), which can be activated by the PI3K-Akt-mTOR network.
The first and rate-limiting step for anaplerotic utilization of glutamine carbon within the mitochondria is its deamination to glutamate (Fig. 4). This reaction is mediated by the enzyme glutaminase (GLS). The reverse reaction is catalyzed by glutamine synthetase (GLUL), which condenses glutamate and ammonia in an ATP-dependent reaction to form glutamine. Both of these enzymes can be regulated transcriptionally by PI3K-Akt-mTOR signaling. GLS is a c-Myc target gene (Wise et al. 2008; Gao et al. 2009; Li and Simon 2013), while GLUL expression is increased by FoxO activation. Hence, upon PI3K-Akt-mTOR pathway activation, c-Myc activation and FoxO inhibition result in the upregulation of GLS concurrent with the downregulation of GLUL (van der Vos et al. 2012). Together, this promotes increased glutaminolysis and subsequent utilization of glutamate.
It is worth noting that glutamine can also be converted to glutamate by a number of other mechanisms, including transaminase-mediated nitrogen transfer. Such transaminases include asparagine synthetase (ASNS) and glutamine-fructose-6-phosphate transaminase 1 (GFPT1), which utilize the amide nitrogen of glutamine for anabolic processes such as nucleotide base synthesis and protein glycosylation, respectively (Ying et al. 2012; Balasubramanian et al. 2013). Whether the PI3K-Akt-mTOR pathway regulates these glutamine transaminases is not known.
Glutamate can then be converted into αKG by glutamate dehydrogenase (GDH) or transaminases, and αKG is used to replenish the TCA cycle (Fig. 4). Consistent with increased utilization of glutamine, mTORC1 activation elevates GDH activity in MEFs and human epithelial tumor cell lines by suppressing SIRT4-mediated inhibition of GDH. SIRT4 is a mitochondrial enzyme that ADP ribosylates GDH to inhibit its activity (Haigis et al. 2006). mTORC1 suppresses SIRT4 mRNA expression by promoting the destabilization of the transcription factor CREB2 (Csibi et al. 2013). In another study, however, the opposite was observed: Inhibition of Akt in glioblastoma cells, which presumably also inhibited mTORC1, stimulated GDH activity, and vice versa. It was proposed that Akt activation indirectly suppresses GDH activity through its effects on glycolysis, since adding exogenous pyruvate to the media could reverse the increased GDH activity in response to Akt inhibition (Yang et al. 2009a). It should be noted that the two studies above focus on different nodes of PI3K-Akt-mTOR signaling, and it would be interesting to evaluate the effect of Akt inhibition in epithelial cancer cell lines, as well as the status of mTORC1, CREB2, and SIRT4 in glioblastoma cells, on GDH activity. It is also likely that depending on the context, PI3K-Akt-mTOR signaling may inhibit GDH activity in favor of transaminases (or vice versa), which can also convert glutamate to αKG (Fig. 4). Dependence on the transamination pathway as opposed to GDH activity has been demonstrated in pancreatic adenocarcinoma (Son et al. 2013); however, regulation of transaminases by PI3K-Akt-mTOR signaling remains to be explored.
Taken together, it is clear that oncogenic activation of the PI3K-Akt-mTOR pathway modulates the uptake and utilization of glutamine. Notably, the observations described above involve transcriptional regulation, and it will be interesting to assess whether PI3K-Akt-mTOR signaling exerts any post-translational effects on metabolic enzymes that mediate the uptake and utilization of glutamine. The precise molecular mechanisms underlying this regulation remain an important avenue of investigation, especially in light of the emerging perspective that glutamine metabolism may be an appealing target for novel clinical strategies to detect, monitor, and treat cancer (Hensley et al. 2013).
5 Concluding Remarks
The reprogramming of metabolic processes in cancer cells by the PI3K-Akt-mTOR signaling network clearly illustrates the close relationship between oncogenic cellular signaling and cancer metabolism. As the mechanisms underlying the regulation of metabolism by PI3K-Akt-mTOR signaling are explored more deeply, an emerging theme is the concept that the metabolic differences observed in cancer cells do not occur in isolation. Rather, there exists a layer of regulation established by oncogenic signaling processes that coordinate uptake and utilization of multiple nutrients in a manner best suited for supporting survival, growth, and proliferation. Furthermore, the mode of regulation is often multifaceted, with robust changes in metabolism being initiated by post-translational responses and then further reinforced by the induction of transcriptional programs.
Many open questions remain in our understanding of how the PI3K-Akt-mTOR signaling pathway governs metabolic reprogramming in cancer. In particular, it is important to note that the complexity of cellular metabolism extends beyond glucose, glutamine, nucleotide, and lipid metabolism. In fact, the Human Metabolome Database (www.hmdb.ca) lists more than 29,000 endogenous human metabolites involved in more than 6000 reactions, many of which have not been explored in cancer. Future studies will no doubt reveal more metabolic pathways that are regulated by PI3K-Akt-mTOR signaling.
Another key challenge is that several of the regulatory mechanisms that have been described in this chapter were studied in the context of physiological metabolism in nontumorigenic cells. It will be valuable to assess whether these same mechanisms are functional and relevant in the context of cancer cells. Even more importantly, it will be critical to gain an understanding of how the mechanisms of metabolic regulation by oncogenic PI3K-Akt-mTOR signaling confer cancer cells with metabolic dependencies and vulnerabilities that can be exploited as a therapeutic intervention. Particularly exciting are the prospects of identifying metabolic pathways that share a synthetic lethal relationship with oncogenic PI3K-Akt-mTOR pathway activation, as well as the identification of metabolic enzymes that may potentially be pharmacologically targeted in combination with PI3K-Akt-mTOR pathway inhibitors currently in development. As we increase our understanding of oncogenic signaling and its modulation of cancer metabolism, it is the hope that cancer-specific metabolic dependences will open novel therapeutic strategies that can be exploited to selectively and effectively target tumor cells.
Abbreviations
- PI3K:
-
Phosphoinositide 3-kinase
- mTOR:
-
Mammalian target of rapamycin
- RTKs:
-
Receptor tyrosine kinases
- GPCRs:
-
G-protein-coupled receptors
- PIP3 :
-
Phosphatidylinositol-3,4,5-trisphosphate
- PIP2 :
-
Phosphatidylinositol-4,5-bisphosphate
- PTEN:
-
Phosphatase and tensin homolog
- PH:
-
Pleckstrin homology
- PDK1:
-
Phosphoinositide-dependent protein kinase 1
- mTORC1/2:
-
Mammalian target of rapamycin complex 1/2
- HK:
-
Hexokinase
- PFK1/2:
-
Phosphofructokinase 1/2
- TSC1/2:
-
Tuberous sclerosis 1/2
- GAP:
-
GTPase-activating protein
- S6K1/2:
-
S6 kinase 1/2
- 4E-BP1/2:
-
eIF4E (eukaryotic initiation factor 4E)-binding protein 1/2
- FoxO:
-
Forkhead box O
- GSK-3β:
-
Glycogen synthase kinase-3β
- HIF:
-
Hypoxia-inducible factor
- SREBP:
-
Sterol regulatory element-binding protein
- SRE:
-
Sterol regulatory elements
- TXNIP:
-
Thioredoxin-interacting protein
- AMPK:
-
AMP-dependent protein kinase
- 18FDG:
-
18-fluoro-deoxyglucose
- PET:
-
Positron emission tomography
- G6P:
-
Glucose 6-phosphate
- VDAC:
-
Voltage-dependent anion channel
- OMM:
-
Outer mitochondrial membrane
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- PDH:
-
Pyruvate dehydrogenase
- TCA:
-
Tricarboxylic acid
- NADH:
-
Nicotinamide adenine dinucleotide
- LDHA:
-
Lactate dehydrogenase A
- GPI:
-
Glucose phosphate isomerase
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- TPI:
-
Triosephosphate isomerase
- PGK1:
-
Phosphoglycerate kinase 1
- ENO1:
-
Enolase
- GCK:
-
Glucokinase
- ALDOB:
-
Aldolase B
- PK1:
-
Pyruvate kinase 1
- PPP:
-
Pentose phosphate pathway
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- Ru5P:
-
Ribulose 5-phosphate
- R5P:
-
Ribose 5-phosphate
- F6P:
-
Fructose 6-phosphate
- Ga3P:
-
Glyceraldehyde 3-phosphate
- G6PD:
-
Glucose 6-phosphate dehydrogenase
- TKT:
-
Transketolase
- PGD:
-
6-phosphogluconate dehydrogenase
- RPE:
-
Ribulose 5-phosphate epimerase
- RPIA:
-
Ribulose 5-phosphate isomerase
- TALDO1:
-
Transaldolase 1
- CAD:
-
Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase
- PRPP:
-
Phosphoribosyl pyrophosphate
- ACL:
-
ATP citrate lyase
- αKG:
-
α-ketoglutarate
- GLS:
-
Glutaminase
- GLUL:
-
Glutamine synthetase
- GDH:
-
Glutamate dehydrogenase
- ASNS:
-
Asparagine synthetase
- GFPT1:
-
Glutamine–fructose 6-phosphate transaminase 1
References
Ahn KJ, Hwang HS, Park JH, Bang SH, Kang WJ, Yun M, Lee JD (2009) Evaluation of the role of hexokinase type II in cellular proliferation and apoptosis using human hepatocellular carcinoma cell lines. J Nucl Med 50:1525–1532
Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P (1996) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399:333–338
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7:261–269
Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC (1989) PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 57:167–175
Bae SS, Cho H, Mu J, Birnbaum MJ (2003) Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem 278:49530–49536
Balasubramanian MN, Butterworth EA, Kilberg MS (2013) Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab 304:E789–E799
Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486:405–409
Bar-Peled L, Sabatini DM (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol 24:400–406
Bardos JI, Ashcroft M (2004) Hypoxia-inducible factor-1 and oncogenic signalling. BioEssays 26:262–269
Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JPJ, Roth RA (1999) Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem 274:20281–20286
Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24:6314–6322
Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Benedetti Panici P, Mancuso S, Neri G, Testa JR (1995) Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer 64:280–285
Bellacosa A, Kumar CC, Di Cristofano A, Testa JR (2005) Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res 94:29–86
Ben-Haim S, Ell P (2009) 18F-FDG PET and PET/CT in the evaluation of cancer treatment response. J Nucl Med 50:88–99
Ben-Sahra I, Howell JJ, Asara JM, Manning BD (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339:1323–1328
Bentley J, Itchayanan D, Barnes K, McIntosh E, Tang X, Downes CP, Holman GD, Whetton AD, Owen-Lynch PJ, Baldwin SA (2003) Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J Biol Chem 278:39337–39348
Berwick DC, Dell GC, Welsh GI, Heesom KJ, Hers I, Fletcher LM, Cooke FT, Tavare JM (2004) Protein kinase B phosphorylation of PIKfyve regulates the trafficking of GLUT4 vesicles. J Cell Sci 117:5985–5993
Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM (2002) The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem 277:33895–33900
Blancher C, Moore JW, Robertson N, Harris AL (2001) Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3′-kinase/Akt signaling pathway. Cancer Res 61:7349–7355
Boehmer C, Okur F, Setiawan I, Broer S, Lang F (2003) Properties and regulation of glutamine transporter SN1 by protein kinases SGK and PKB. Biochem Biophys Res Commun 306:156–162
Bokun R, Bakotin J, Milasinović D (1987) Semiquantitative cytochemical estimation of glucose-6-phosphate dehydrogenase activity in benign diseases and carcinoma of the breast. Acta Cytol 31:249–252
Boormans JL, Korsten H, Ziel-van der Made AC, van Leenders GJ, Verhagen PC, Trapman J (2010) E17K substitution in AKT1 in prostate cancer. Br J Cancer 102:1491–1494
Boros LG, Bassilian S, Lim S, Lee WN (2001) Genistein inhibits nonoxidative ribose synthesis in MIA pancreatic adenocarcinoma cells: a new mechanism of controlling tumor growth. Pancreas 22:1–7
Boros LG, Puigjaner J, Cascante M, Lee WN, Brandes JL, Bassilian S, Yusuf FI, Williams RD, Muscarella P, Melvin WS, Schirmer WJ (1997) Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res 57:4242–4248
Boss GR, Pilz RB (1985) Phosphoribosylpyrophosphate synthesis from glucose decreases during amino acid starvation of human lymphoblasts. J Biol Chem 260:6054–6059
Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M (2004) Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J 23:2830–2840
Brahimi-Horn MC, Chiche J, Pouyssegur J (2007) Hypoxia signalling controls metabolic demand. Curr Opin Cell Biol 19:223–229
Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB (2002) 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol 156:817–828
Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA (2004) Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 64:7678–7681
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–1657
Cantley LC, Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 96:4240–4245
Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448:439–444
Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, Hazle JD, Yu D, Wei W, Sarbassov D, Hung MC, Nakayama KI, Lin HK (2012) The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149:1098–1111
Chan DA, Sutphin PD, Denko NC, Giaccia AJ (2002) Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1alpha. J Biol Chem 277:40112–40117
Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR (1996) Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 93:3636–3641
Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A (2014) Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res 74:964–973
Cong LN, Chen H, Li Y, Zhou L, McGibbon MA, Taylor SI, Quon MJ (1997) Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol 11:1881–1890
Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J (2013) The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153:840–854
Dang CV (2007) The interplay between MYC and HIF in the Warburg effect. In: Ernst Schering Foundation Symposium Proceedings, pp 35–53
Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, Lazar AJ, Gershenwald JE, Mills GB (2008) A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer 99:1265–1268
de la Cruz-Herrera CF, Campagna M, Lang V, del Carmen González-Santamaría J, Marcos-Villar L, Rodríguez MS, Vidal A, Collado M, Rivas C (2015) SUMOylation regulates AKT1 activity. Oncogene 34:1442–1450
Deberardinis RJ, Lum JJ, Thompson CB (2006) Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem 281:37372–37380
Deng X, Zhang W, O-Sullivan I, Williams JB, Dong Q, Park EA, Raghow R, Unterman TG, Elam MB (2012) FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J Biol Chem 287:20132–20143
Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713
Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH (1997) Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 272:17269–17275
Dessì S, Batetta B, Cherchi R, Onnis R, Pisano M, Pani P (1988) Hexose monophosphate shunt enzymes in lung tumors from normal and glucose-6-phosphate-dehydrogenase-deficient subjects. Oncology 45:287–291
Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, Manning BD (2012) TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 47:535–546
Dibble CC, Manning BD (2013) Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15:555–564
Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39:171–183
Eilers M, Eisenman RN (2008) Myc’s broad reach. Genes Dev 22:2755–2766
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64:3892–3899
Engelman JA (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9:550–562
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14:1351–1356
Espenshade PJ, Hughes AL (2007) Regulation of sterol synthesis in eukaryotes. Annu Rev Genet 41:401–427
Fan CD, Lum MA, Xu C, Black JD, Wang X (2013a) Ubiquitin-dependent regulation of phospho-AKT dynamics by the ubiquitin E3 ligase, NEDD4-1, in the insulin-like growth factor-1 response. J Biol Chem 288:1674–1684
Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD (2013b) Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 9:712
Fantin VR, St-Pierre J, Leder P (2006) Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9:425–434
Franke TF, Kaplan DR, Cantley LC, Toker A (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275:665–668
Fruman DA, Cantley LC (2014) Idelalisib–a PI3Kδ inhibitor for B-cell cancers. N Engl J Med 370:1061–1062
Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13:140–156
Gan B, Lim C, Chu G, Hua S, Ding Z, Collins M, Hu J, Jiang S, Fletcher-Sananikone E, Zhuang L, Chang M, Zheng H, Wang YA, Kwiatkowski DJ, Kaelin WGJ, Signoretti S, DePinho RA (2010) FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell 18:472–484
Ganapathy V, Thangaraju M, Prasad PD (2009) Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther 121:29–40
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
García JM, Silva J, Peña C, Garcia V, Rodríguez R, Cruz MA, Cantos B, Provencio M, España P, Bonilla F (2004) Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosom Cancer 41:117–124
Gebhardt R, Kleemann E (1987) Hormonal regulation of amino acid transport system N in primary cultures of rat hepatocytes. Eur J Biochem 166:339–344
Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR (2004) Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 64:3014–3021
Gonzalez E, McGraw TE (2009) Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci USA 106:7004–7009
Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N (2001) Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 15:1406–1418
Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM (2014) A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4:554–563
Gregory MA, Qi Y, Hann SR (2003) Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 278:51606–51612
Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126:941–954
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hensley CT, Wasti AT, DeBerardinis RJ (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123:3678–3684
Huang S, Czech MP (2007) The GLUT4 glucose transporter. Cell Metab 5:237–252
Ikonomov OC, Sbrissa D, Dondapati R, Shisheva A (2007) ArPIKfyve-PIKfyve interaction and role in insulin-regulated GLUT4 translocation and glucose transport in 3T3-L1 adipocytes. Exp Cell Res 313:2404–2416
Ikonomov OC, Sbrissa D, Mlak K, Shisheva A (2002) Requirement for PIKfyve enzymatic activity in acute and long-term insulin cellular effects. Endocrinology 143:4742–4754
Ikonomov OC, Sbrissa D, Shisheva A (2009) YM201636, an inhibitor of retroviral budding and PIKfyve-catalyzed PtdIns(3,5)P2 synthesis, halts glucose entry by insulin in adipocytes. Biochem Biophys Res Commun 382:566–570
John S, Weiss JN, Ribalet B (2011) Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE 6:e17674
Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, Sampson JR, Cheadle JP (1999) Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 64:1305–1315
Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, Nardella C, Pandolfi PP, Baselga J, Scully R, Asara JM, Cantley LC, Wulf GM (2012) Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2:1048–1063
Katome T, Obata T, Matsushima R, Masuyama N, Cantley LC, Gotoh Y, Kishi K, Shiota H, Ebina Y (2003) Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. J Biol Chem 278:28312–28323
Khatri S, Yepiskoposyan H, Gallo CA, Tandon P, Plas DR (2010) FOXO3a regulates glycolysis via transcriptional control of tumor suppressor TSC1. J Biol Chem 285:15960–15965
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV (2004a) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 24:5923–5936
Kim KH, Song MJ, Yoo EJ, Choe SS, Park SD, Kim JB (2004b) Regulatory role of glycogen synthase kinase 3 for transcriptional activity of ADD1/SREBP1c. J Biol Chem 279:51999–52006
Kohn AD, Summers SA, Birnbaum MJ, Roth RA (1996) Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 271:31372–31378
Kotani K, Carozzi AJ, Sakaue H, Hara K, Robinson LJ, Clark SF, Yonezawa K, James DE, Kasuga M (1995) Requirement for phosphoinositide 3-kinase in insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes. Biochem Biophys Res Commun 209:343–348
Kress TR, Cannell IG, Brenkman AB, Samans B, Gaestel M, Roepman P, Burgering BM, Bushell M, Rosenwald A, Eilers M (2011) The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol Cell 41:445–457
Krockenberger M, Honig A, Rieger L, Coy JF, Sutterlin M, Kapp M, Horn E, Dietl J, Kammerer U (2007) Transketolase-like 1 expression correlates with subtypes of ovarian cancer and the presence of distant metastases. Int J Gynecol Cancer 17:101–106
Langbein S, Frederiks WM, zur Hausen A, Popa J, Lehmann J, Weiss C, Alken P, Coy JF (2008) Metastasis is promoted by a bioenergetic switch: new targets for progressive renal cell cancer. Int J Cancer 122:2422–2428
Langbein S, Zerilli M, Zur Hausen A, Staiger W, Rensch-Boschert K, Lukan N, Popa J, Ternullo MP, Steidler A, Weiss C, Grobholz R, Willeke F, Alken P, Stassi G, Schubert P, Coy JF (2006) Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br J Cancer 94:578–585
Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE (2005) Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem 280:37803–37813
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL (2001) HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 21:3995–4004
Li B, Simon MC (2013) Molecular pathways: targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin Cancer Res 19:5835–5841
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275:1943–1947
Li R, Wei J, Jiang C, Liu D, Deng L, Zhang K, Wang P (2013) Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res 73:5742–5753
Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, Wan L, Singh A, Zhai B, Yuan M, Wang Z, Gygi SP, Lee TH, Lu KP, Toker A, Pandolfi PP, Asara JM, Kirschner MW, Sicinski P, Cantley L, Wei W (2014) Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508:541–545
Low SY, Taylor PM, Rennie MJ (1996) Responses of glutamine transport in cultured rat skeletal muscle to osmotically induced changes in cell volume. J Physiol 492:877–885
Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10:307–318
Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY (2000) PIK3CA as an oncogene in cervical cancer. Oncogene 19:2739–2744
Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273:13375–13378
Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N (2004a) Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 16:819–830
Majewski N, Nogueira V, Robey RB, Hay N (2004b) Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol 24:730–740
Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, Malara N, Savino R, Rocco G, Chiappetta G, Franco R, Tirino V, Pirozzi G, Viglietto G (2008) Activating E17 K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle 7:665–669
Manning BD, Cantley LC (2003) United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans 31:573–578
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10:151–162
Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS (2013) mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab 18:726–739
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25:4777–4786
Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, Sanders ME, Yap JT, Van den Abbeele AD, Li Y, Cantley LC, Winer E, Arteaga CL (2014) Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol 32:1202–1209
Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7:763–777
Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156:771–785
Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y (2008) ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res 68:8547–8554
Miyamoto S, Murphy AN, Brown JH (2008) Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ 15:521–529
Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, Kim JW, Park SW, Kim KS (2011) Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J 433:225–233
Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E (2007) Energy metabolism in tumor cells. FEBS J 274:1393–1418
Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344(Pt 2):427–431
Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC (2000) Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem 275:36108–36115
Obsil T, Ghirlando R, Anderson DE, Hickman AB, Dyda F (2003) Two 14-3-3 binding motifs are required for stable association of Forkhead transcription factor FOXO4 with 14-3-3 proteins and inhibition of DNA binding. Biochemistry 42:15264–15272
Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M (1994) Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem 269:3568–3573
Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275:21797–21800
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3:187–197
Paradis S, Ruvkun G (1998) Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 12:2488–2498
Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, Jensen CB, Krook A, Bjornholm M, Tornqvist H, Zierath JR, Ridderstrale M, Altshuler D, Lee RT, Vaag A, Groop LC, Mootha VK (2007) TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4:e158
Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Lengauer C, Velculescu VE (2005) Colorectal cancer: mutations in a signalling pathway. Nature 436:792
Pastorino JG, Hoek JB, Shulga N (2005) Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res 65:10545–10554
Pastorino JG, Shulga N, Hoek JB (2002) Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem 277:7610–7618
Peck B, Ferber EC, Schulze A (2013) Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front Oncol 3:96
Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A (2005) PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24:6465–6481
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8:224–236
Potapova IA, El-Maghrabi MR, Doronin SV, Benjamin WB (2000) Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars. Biochemistry 39:1169–1179
Rácz A, Brass N, Heckel D, Pahl S, Remberger K, Meese E (1999) Expression analysis of genes at 3q26-q27 involved in frequent amplification in squamous cell lung carcinoma. Eur J Cancer 35:641–646
Raivio KO, Lazar CS, Krumholz HR, Becker MA (1981) The phosphogluconate pathway and synthesis of 5-phosphoribosyl-1-pyrophosphate in human fibroblasts. Biochim Biophys Acta 678:51–57
Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB (2003) Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol 23:7315–7328
Reitzer LJ, Wice BM, Kennell D (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 254:2669–2676
Risso G, Pelisch F, Pozzi B, Mammi P, Blaustein M, Colman-Lerner A, Srebrow A (2013) Modification of Akt by SUMO conjugation regulates alternative splicing and cell cycle. Cell Cycle 12:3165–3174
Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S (2013) Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem 288:23798–23806
Robey RB, Hay N (2009) Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 19:25–31
Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN (2013) Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339:1320–1323
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10:143–153
Saci A, Cantley LC, Carpenter CL (2011) Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell 42:50–61
Sale EM, Hodgkinson CP, Jones NP, Sale GJ (2006) A new strategy for studying protein kinase B and its three isoforms. Role of protein kinase B in phosphorylating glycogen synthase kinase-3, tuberin, WNK1, and ATP citrate lyase. Biochemistry 45:213–223
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554
Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278:14599–14602
Sasaki H, Okuda K, Kawano O, Yukiue H, Yano M, Fujii Y (2008) AKT1 and AKT2 mutations in lung cancer in a Japanese population. Mol Med Rep 1:663–666
Sbrissa D, Ikonomov OC, Strakova J, Shisheva A (2004) Role for a novel signaling intermediate, phosphatidylinositol 5-phosphate, in insulin-regulated F-actin stress fiber breakdown and GLUT4 translocation. Endocrinology 145:4853–4865
Sekulić A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT (2000) A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 60:3504–3513
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271:32529–32537
Semenza GL, Roth PH, Fang HM, Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269:23757–23763
Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV (1997) c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A 94:6658–6663
Shisheva A (2008) PIKfyve: partners, significance, debates and paradoxes. Cell Biol Int 32:591–604
Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, Uehara Y, Sone K, Miyamoto Y, Hiraike H, Hiraike-Wada O, Nei T, Kawana K, Kuramoto H, Aburatani H, Yano T, Taketani Y (2009) The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br J Cancer 101:145–148
Silhan J, Vacha P, Strnadova P, Vecer J, Herman P, Sulc M, Teisinger J, Obsilova V, Obsil T (2009) 14-3-3 protein masks the DNA binding interface of forkhead transcription factor FOXO4. J Biol Chem 284:19349–19360
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496:101–105
Staal SP (1987) Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA 84:5034–5037
Stanton RC (2012) Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 64:362–369
Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT (1998) Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279:710–714
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van ‘t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404
Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE (2008) Glucose sensing by MondoA: Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci USA 105:6912–6917
Storz P, Toker A (2002) 3’-phosphoinositide-dependent kinase-1 (PDK-1) in PI 3-kinase signaling. Front Biosci 7:d886–d902
Sun X, Huang J, Homma T, Kita D, Klocker H, Schafer G, Boyle P, Ohgaki H (2009) Genetic alterations in the PI3 K pathway in prostate cancer. Anticancer Res 29:1739–1743
Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J (2005) Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 1:379–391
Tadros LB, Taylor PM, Rennie MJ (1993) Characteristics of glutamine transport in primary tissue culture of rat skeletal muscle. Am J Physiol 265:E135–E144
Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J (2002) Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA 99:13571–13576
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13:1259–1268
Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL (2006) Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 12:122–127
Toker A (2012) Phosphoinositide 3-kinases-a historical perspective. Subcell Biochem 58:95–110
Toker A, Cantley LC (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387:673–676
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 1813:1938–1945
Van de Sande T, De Schrijver E, Heyns W, Verhoeven G, Swinnen JV (2002) Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer Res 62:642–646
Van de Sande T, Roskams T, Lerut E, Joniau S, Van Poppel H, Verhoeven G, Swinnen JV (2005) High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. J Pathol 206:214–219
van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, Verhagen LP, Groot Koerkamp MJ, Braat AK, Dansen TB, Holstege FC, Gebhardt R, Burgering BM, Coffer PJ (2012) Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 14:829–837
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9:316–323
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Vanhaesebroeck B, Waterfield MD (1999) Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res 253:239–254
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2:489–501
Wang W, Fridman A, Blackledge W, Connelly S, Wilson IA, Pilz RB, Boss GR (2009) The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. J Biol Chem 284:3521–3528
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Wasa M, Wang HS, Tazuke Y, Okada A (2001) Insulin-like growth factor-I stimulates amino acid transport in a glutamine-deprived human neuroblastoma cell line. Biochim Biophys Acta 1525:118–124
Weinhouse S (1976) The Warburg hypothesis fifty years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol 87:115–126
Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA 101:9085–9090
Whitman M, Downes CP, Keeler M, Keller T, Cantley L (1988) Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332:644–646
Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM (1985) Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239–242
Wieman HL, Wofford JA, Rathmell JC (2007) Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 18:1437–1446
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105:18782–18787
Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, Guha A (2011) Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med 208:313–326
Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC (2013) AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49:1167–1175
Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ (2009a) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69:7986–7993
Yang WL, Jin G, Li CF, Jeong YS, Moten A, Xu D, Feng Z, Chen W, Cai Z, Darnay B, Gu W, Lin HK (2013) Cycles of ubiquitination and deubiquitination critically regulate growth factor-mediated activation of Akt signaling. Sci Signal 6:ra3
Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, Lin HK (2009b) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325:1134–1138
Yang WL, Wu CY, Wu J, Lin HK (2010) Regulation of Akt signaling activation by ubiquitination. Cell Cycle 9:487–497
Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, Cantley LC, Paik JH (2013) FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J 32:2589–2602
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149:656–670
Zampella EJ, Bradley EL, Pretlow TG (1982) Glucose-6-phosphate dehydrogenase: a possible clinical indicator for prostatic carcinoma. Cancer 49:384–387
Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, Heidenreich KA, Sajan MP, Farese RV, Stolz DB, Tso P, Koo SH, Montminy M, Unterman TG (2006) FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 281:10105–10117
Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC (2003) Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3:483–495
Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL (2000) Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60:1541–1545
Zhou QL, Jiang ZY, Holik J, Chawla A, Hagan GN, Leszyk J, Czech MP (2008) Akt substrate TBC1D1 regulates GLUT1 expression through the mTOR pathway in 3T3-L1 adipocytes. Biochem J 411:647–655
Zhu J, Blenis J, Yuan J (2008) Activation of PI3 K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci USA 105:6584–6589
Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ (2000) Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev 14:391–396
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Lien, E.C., Lyssiotis, C.A., Cantley, L.C. (2016). Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. In: Cramer, T., A. Schmitt, C. (eds) Metabolism in Cancer. Recent Results in Cancer Research, vol 207. Springer, Cham. https://doi.org/10.1007/978-3-319-42118-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-42118-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42116-2
Online ISBN: 978-3-319-42118-6
eBook Packages: MedicineMedicine (R0)