
Abstract
Nutrition and lifestyle factors play an important role in human health as dietary imbalances are major determinants of several diseases including cancer. Emerging studies suggest that diet and nutrition can impact gene expression through epigenetic mechanisms. Epigenetic modifications are heritable and cause potentially reversible changes in gene expression that do not require alteration in DNA sequence. Epigenetic marks include changes in DNA methylation, histone modifications, and small noncoding miRNA. Aberrant epigenetic modifications probably occur at an early stage in neoplastic development and are widely described as essential players in cancer progression. Epigenetic modifications also mediate environmental signals and provide links between susceptibility genes and environmental factors in the etiology of cancer. The present chapter initially highlights the role of various epigenetic mechanisms in the regulation and maintenance of mammalian genome. Focusing on the effect of various endogenous factors that include environmental, lifestyle, nutritional, and social-economic/racial aspects; this chapter discusses their impact on the process of carcinogenesis through various epigenetic modifications. Elucidating the impact of nutrition and lifestyle factors on epigenetic mechanisms may serve as a personalized prediction tool assessing cancer susceptibility and in providing recommendation and guide for prevention and therapeutic options against cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- DNA methylation
- Histone modification
- Noncoding RNA
- Dietary agents
- Gene−environment interaction
- Carcinogens
- Chemoprevention
Introduction
Cancer is widely recognized as a heterogeneous disease resulting from genetic and epigenetic alterations inherited by a series of transformations in clonally selected cells exhibiting selective growth advantage, sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis. Although genetic lesions drive tumor progression, however it is becoming clear that epigenetic perturbations are equally important in cancer development. Epigenetics is referred to as the study of stable inheritance of gene expression that occurs without changes in the DNA sequence. A majority of cancers result from changes that accumulate throughout the lifespan as a result of exposure to various endogenous factors that include environmental, lifestyle, nutritional, and social-economic/racial aspects. Epigenetic disruption of gene expression by these endogenous factors plays a critical role in cancer progression. A number of epigenetic mechanisms have now been identified in mammals. There are three major epigenetic mechanisms which are known to regulate gene expression. These include DNA methylation, modulation of chromatin structure by posttranslational modification of histone or nonhistone proteins, and small noncoding microRNAs (miRNAs) that alter gene expression by either inhibiting translation or causing targeted degradation of specific mRNAs. These mechanisms are critical components in the normal development and growth of cells and their modifications contribute to neoplastic phenotype.
Mechanisms Underlying Epigenetics
DNA Methylation
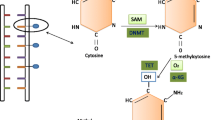
Methylation of cytosine residues within the dinucleotide sequence-CpG is one of the most widely studied epigenetic modifications in mammals [1]. Forming an essential component of the cellular epigenetic machinery, DNA methylation in collaboration with histone modification regulates gene expression by modulating DNA packaging and chromatin architecture [2]. DNA methylation is a chemical modification that involves transfer of a methyl (CH3) moiety from the donor S-adenosyl methionine (SAM) to the 5′ position of cytosine residue that precedes guanine in the CpG dinucleotide sequence, forming 5-methyl cytosine and S-adenosyl-l-homocysteine (SAH) [1, 3–6]. The mammalian genome has been reported to harbor 3 × 107 methylated cytosine residues mostly within CpG dinucleotide sequences [4]. Although CpG sequences are unevenly distributed throughout the human genome, they are frequently enriched in gene promoters (often referred as CpG islands) and large repetitive sequences such as Long interspersed nuclear element (LINE) and ALU retrotransposon elements [7]. DNA methylation is catalyzed by a group of enzymes known as DNA methyltransferases (DNMTs) [1, 4]. There are three major DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b) identified in mammals. Evidence from phenotypic analyses of mice with mutant DNMT genes have provided useful mechanistic insights into the role and establishment of DNA methylation patterns during development [4, 8]. Dnmt1 enzyme has been demonstrated to have a 5–30-fold more preference for hemimethylated substrates and therefore popularly designated as maintenance methyltransferase. It preserves the existing methylation patterns in the daughter DNA strands by adding methyl groups to hemimethylated CpG sequences following replication. However Dnmt1 has also been demonstrated to be involved in de novo methylation activity in embryo lysates and its sequence specificity was shown to be confined to 5′-CpG-3′ dinucleotide sequence with little dependence on sequence context or density [9]. Dnmt3a and Dnmt3b enzymes are essential for global de novo methylation as they preferentially target unmethylated CpG sequences [10]. They have been shown to be highly expressed in developing mouse embryos and establish methylation patterns postimplantation [10]. Although Dnmt3L, the fourth family member, lacks intrinsic DNMT activity by itself, it colocalizes with Dnmt3a and Dnmt3b to establish genomic imprints in maternal germ line [11] and facilitate methylation of retroposons. Dnmt2, another member of DNMT family, was found to lack biochemical detectable DNMT activity and its deletion in mice had no obvious phenotypic effects on genomic methylation pattern or methylation of retroviral DNA [10].
Hypermethylation of CpG islands is usually associated with gene silencing. There are multiple routes through which DNA methylation can suppress transcription. A general mechanism is to exclude binding of proteins that modulate transcription through their DNA binding domains [12]. For example, binding of chromatin boundary element binding protein CTCF to DNA is blocked by CpG methylation, which allows the enhancer to activate transcription [13, 14]. This mechanism has been demonstrated to be essential for imprinting of Igf2 gene [15]. Beside this, CpG methylation has been shown to block the binding of several other transcription factors; however, their biological consequences remain unknown [16]. Another mechanism for DNA methylation mediated gene repression involves binding of specialized DNA binding proteins to the methylated CpG stretches, which form repressor complexes with histone deacetylases (HDACs) and cause chromatin compaction [17–19]. In mammals six methyl-CpG-binding proteins have been characterized to date, which include MeCp2, MBD1-4, and Kaiso. Studies demonstrate that all (except mammalian MBD3) possess a domain that specifically targets them to methylated CpG regions in vitro and in vivo [20, 21].
Histone Modifications
In addition to DNA methylation, posttranslational modification of N-terminal histone tails play a significant role in epigenetic regulation of gene expression [22, 23]. A typical nucleosome unit consists of ~146 bp of DNA wrapped around an octamer of histones (H2A, H2B, H3, and H4) representing the fundamental building unit of eukaryotic chromatin. A diverse array of covalent chemical modification of less structured, protruding N-terminal tails of core histones by methylation, acetylation, ubiquitination, phosphorylation, sumoylation, and ADP-ribosylation dictate the dynamics of chromatin state [24]. Euchromatin is lightly packed form of chromatin where DNA is accessible for transcription, whereas heterochromatin represents tightly packed chromatin state inaccessible to cellular transcriptional machinery. Most of the chemical modifications occur at Lysine (K), Arginine (R), and Serine (S) residues within the histone tails. These distinct histone modifications on one or more histone tails (often referred to as ‘Histone code’) which may act sequentially or in combination are recognized by other proteins that signal further downstream events. A number of enzymes have been implicated in catalyzing (addition or removal) various histone modifications. Examples include histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), histone demethylases (HDMs), histone kinases, etc. In brief, HATs catalyze the addition of acetyl group on the ε-amino group of lysine residues in the N-terminal tail of histones, which neutralize the positive charge, relax the chromatin and facilitate the binding of transcriptional machinery to the DNA [25]. Till date 25 HATs have been characterized which are divided into four families. Examples include GNAT (hGCN5, PCAF), MYST (MYST, Tip60), p300/CBP (p300/CBP, SRC (SRC-1), and TAFII250 families (TAFII250) [3, 6, 26]. In contrast, HDACs catalyze the removal of acetyl groups from lysine residues resulting in the compaction of chromatin configuration which repress transcription [27]. HDACs are classified into four groups. HDAC-1, -2, -3, and -8 are members of Class I HDAC family while HDAC-4, -5, -6, -7, -9, and -10 belong to class II HDAC family. HDAC-11 belongs to Class IV HDAC group. Sirtuins, which require NAD+ as cofactor for their activity, are structurally unrelated to other HDAC classes, constitute Class III HDAC family [28, 29]. HMTs catalyze the addition of methyl groups to lysine or arginine residues while HDMs act to remove them [30–32]. Examples of histone lysine methyltransferase include EZH2 (Enhancer of zeste homolog 2) and that of histone lysine demethylase include LSD1 (Lysine specific demethylase 1) [33, 34]. Depending on the site of lysine methylation (K4, K9, K27, etc. in Histone H3) and methylation status (mono, di, or tri methylation), histone methylation may have activating or repressive effect on gene expression [26, 34]. H3K4, H3K36, and H3K79 methylation have activating effects on gene transcription, whereas methylation of H3K9, H3K27, and H4K20 is generally associated with gene silencing or transcriptional repression [26, 32, 35]. A plethora of literature is available on each group of histone modifying enzymes, their mechanism of action and various histone modifications, which is beyond the scope of this chapter.
Noncoding RNAs
Recent evidence indicates that noncoding RNA (ncRNA) transcripts play a fundamental role in epigenetic regulation of gene expression and have been implicated in various epigenetic mechanisms such as transposon silencing, X-chromosome inactivation, DNA imprinting, and paramutation [36–38]. In humans, ncRNAs include microRNA (miRNA), small interfering RNA (siRNA), and piwi-interacting RNA (piRNA) which account for majority of transcripts, representing approximately 98 % of all human transcriptional output [39, 40]. Based on the size, ncRNA can be classified into small ncRNA which are generally less than 200 nucleotides in length and long ncRNA (lncRNA) transcripts that are more than 200 nucleotides in length. They can be divided into further subtypes based on their genomic origin and biogenic processes [37]. Both types of ncRNAs have been shown to be essential ‘epigenetic modifiers’ constituting a hidden layer of complex internal signals controlling multiple levels of gene expression associated with development and physiology of an organism [38, 41–43]. lncRNAs have been demonstrated to be involved in gene silencing via mechanisms involving both histone modifications and DNA methylation. For example, the antisense lncRNA located in the p14/p15/INK4 locus, ANRIL, was reported to cause gene silencing via recruitment of polycomb proteins (PcG) [44, 45]. Another well studied example includes the involvement of a 17 kb lncRNA, XIST, in X-chromosome inactivation which ensures X-linked gene dosage compensation in mammalian females [46–49]. This process involves the recruitment of mammalian PRC2 complex containing the histone methyltransferase EZH2 to the locus by a short repeat RNA (RepA) within XIST and deposition and spreading of repressive H3K27me3 marks throughout the X-chromosome. In addition to histone modifications, lncRNAs were also reported to mediate gene silencing through DNA methylation. One such example includes Kcnq1ot1, which in addition to interacting with PRC2 complex and G9a, has been implicated in the recruitment of Dnmt1 through a critical 890 bp region to the CpG island of the imprinted genes [50].
Small ncRNAs particularly miRNAs regulate key epigenetic mechanisms. Short RNAs (50–200 nucleotides) were reported to be transcribed from H3K27me3-enriched PRC2 target genes and cause cell-type specific gene silencing in cis by stabilizing the PRC2 complex near the transcription site through interactions via formation of stem-loop structures [51]. MiRNAs are known to regulate various components of cellular epigenetic machinery particularly polycomb complexes and thus affect multiple downstream effects [33, 52–54]. One such example include miR-214 which downregulates Ezh2 expression by targeting its 3′-UTR region and accelerates skeletal muscle differentiation and transcription of developmental regulators in embryonic stem cells [55]. There are other miRNAs which have been implicated in the repression of Bmi1, a component of PRC1 complex [56–58]. DNA methylation has also shown to be modulated by miRNAs. Dnmt1 and 3 have been reported to be targeted by the miR-29 family in lung cancer and leukemia cells [59, 60]. In addition to the role of small ncRNAs as regulators of various epigenetic mechanisms, in many instances they are themselves targets of the same epigenetic processes which may lead to further downstream alterations. For example, in human breast tumorigenesis and metastasis decreased expression of a set of miRNAs was attributed to gene hypermethylation [61–63]. In summary, recent evidences suggest that ncRNAs have emerged has key regulators of epigenetic mechanisms and also, that the modulation of these RNA transcripts by the same epigenetic processes may lead to major consequences.
Factors Affecting the Epigenome
Effect of Environmental Factors on the Epigenome
Environmental factors including chemical carcinogens, environmental pollutants, dietary contaminants, and physical carcinogens play important role in the etiology of human cancer. In general, the degree to which environmental factors influence carcinogenesis depends on the presence of specific hazardous entity and duration of exposure. However, the degree to which hazardous exposures affect cancer largely reflects variation in susceptibility to a given environmental exposure. Generally environmental factors that are capable of initiating tumor development by altering the epigenome include agents which are capable of inducing changes either directly or indirectly in the genomic DNA, and agents that affect critical cellular regulatory processes of gene transcription such as DNA damage and repair, cell cycle control, and cell death process.
Studies demonstrate that the mismatch repair gene MHL1 is frequently hypermethylated in sporadic tumors exhibiting microsatellite instability [64]. Similarly, silencing of MGMT, the DNA repair gene encoding the protein responsible for the removal of carcinogen-induced O6-methylguanine adducts from DNA (which if left unrepaired results in G to A transition mutation), appears to increase the mutation rate in critical cellular regulators, including tumor suppressors and oncogenes [65]. These studies provide cues that environmental exposures alter either the expression or the activity of enzymes involved in de novo DNA methylation (Dnmt3A and Dnmt3B) and/or the maintenance of DNA methylation (Dnmt1) may predispose to mutational events [64, 65]. Additionally, different agents in the environment may also induce mutational events through preferential binding to hypermethylated DNA. Studies on benzo(a)pyrene diol epoxide (BPDE), a carcinogen from tobacco smoke that exhibits preference for methylated CpG sites, resulting in formation of DNA adducts and G to T transfersions, often found in cancers of the aero-digestive tract in tobacco smokers. It has been shown that certain infectious agents such as human papillomavirus (HPV) induce gene silencing via DNA hypermethylation of the promoters of host genes including CDH1, RB1, INK4a/p16, CDNK2A, MTHFR, PEG3, and others listed in Table 4.1.
The agents in the second group may alter the pattern of chromatin modifications (histone code) in a transient manner and are likely to induce changes in key cellular processes including gene transcription, DNA damage response, and DNA repair. Primary epigenetic targets for environmental factors in this group may be the proteins and protein complexes responsible for histone modifications such as HATs and HDACs, whose activities are often found deregulated in cancer. Recent studies showed that HATs are involved in the process of DNA repair, suggesting that even moderate and transient inhibition of HAT activity induced by environmental exposures may compromise DNA repair, leading to mutation fixation and genomic instability [110]. Similarly, HDAC was shown to be required for efficient DNA repair, suggesting that the removal of histone acetylation is required for restoration of normal (default) chromatin structure following the completion of DNA repair. A tight regulation of HAT and HDAC activity is thus essential for proper regulation of gene transcription and DNA repair. Reduced levels of histone acetylation or enhanced histone deacetylation may result in the compaction of chromatin, blocking access of transcription factors to DNA and/or impeded progression of RNA polymerase. Therefore, different environmental factors may transiently alter chromatin-modifying/remodeling activities and alter patterns of histone modifications impeding DNA repair and other chromatin-based processes.
Another possible epigenetic ‘target’ of adverse environmental exposure may be general methyl-C-binding proteins, a group of proteins (including MBD1, MBD2, MBD3, MeCP2, and KAISO) that bind to methylated CpG sites [111]. Some members of this family, exemplified by MeCP2, were found to bind and recruit HDAC to chromatin. Changes in MeCP2 protein stability and function elicited by the hazardous agents in diet and environment may thus affect normal gene transcription, leading to aberrant cell proliferation and cancer [112]. Given that histone modifications and DNA methylation appear to work together to establish a permissive or repressive chromatin state, agents in the environment and diet that affect one of these intimately linked and self-reinforcing mechanisms would inevitably affect the other. Although poorly understood, the molecular mechanisms by which epigenetic carcinogens in environment and diet may exhibit adverse effects on histone modifications are beginning to emerge. Several recent studies have examined the effect of specific environmental carcinogens on histone modifications and suggest that these agents may affect the pattern of histone modifications through different mechanisms (Table 4.1).
Another prospective mechanism by which environmental exposure including ingestion affect the epigenome involve transposable elements. Transposons when activated may cause genetic mutations and transcriptional noise [112]. For example, the Alu family alone consists of several hundred thousand elements and is shown to be heavily methylated and transcriptionally silent in somatic cells. It is well documented that the activation of transposable element-derived promoters may be a consequence of perturbed DNA methylation, transposable elements were shown to be activated by various kinds of cellular stress. Therefore, stress induced by environmental agents may activate transposable elements, leading to altered establishment and maintenance of epigenetic states.
Epigenetic Modifications by Nutritional Factors
Studies have demonstrated that maternal nutrition imbalance and metabolic disturbances during embryonic development have a persistent effect on the health of the offspring and may be passed down to the next generation [113]. The potential effect of nutritional factors on phenotype has best demonstrated by studies on the risk of cancer for pregnant women and fetuses. When mother is exposed to adverse conditions, the fetal nutrition may cause alterations in structure, physiology, and metabolism that predispose individuals to several diseases including cancer. Selected dietary components consumed during early pregnancy may influence postnatal risk of cancer development, although all dietary components are not harmful. In those cases where adverse effects on fetal development were observed, a proposed mechanism includes methylation of genes due to dietary food components in the mother’s diet. Both hypermethylation and hypomethylation of selected genes were observed. Genes that were overexpressed included Klf6, Klf9, Nid2, Ntn4, Per1, and Txnip, and genes that were repressed included Bcar3, Cldn12, Csf1, Jag1, Lgals3, Lypd3, Nme1, Ptges2, Ptgs1, and Smarcb1 [113, 114]. In animal models, deficiencies of macronutrients during placental growth have been shown to affect fetal growth. Most of the genes that contribute to reduced fetal growth are regulated by imprinting, and the maternal allele is affected in these cases. Functionally, the nutrient transport from mother to fetus via the placenta is affected dramatically by the hypomethylation of genes in the embryonic trophectoderm [115].
Direct effects of nutritional factors on epigenetic changes are most studied and among the best understood is the relationship between dietary methionine and DNA methylation [115]. Methionine, an essential amino acid, plays a central role in the epigenetic regulation by serving as methyl donor for methylation reactions. In the process of cytosine methylation, DNMT enzyme converts SAM to S-adenosylhomocysteine (SAH); therefore, an optimal supply of SAM or removal of SAH is essential for the normal establishment of genome-wide DNA methylation patterns [116]. CpG methylation patterns are largely erased in the early embryos and then re-established in a tissue-specific manner. Therefore, early embryonic development may represent a sensitive stage, and dietary and environmental factors that affect DNA methylation reaction and the activity of DNMTs may result in permanent fixation of aberrant methylation patterns [110, 116]. In postnatal development and adulthood, established patterns of DNA methylation and histone modifications must be maintained through multiple mitotic divisions; therefore, inappropriate quantities of methionine, other food components, and environmental agents may affect normal patterns of DNA methylation and histone modifications. In this respect, it is interesting to note that in adult men with hyperhomocysteinemia, a disorder occurring in several genetically determined and acquired diseases with uremia, treatment with high doses of folate increases methylation levels at specific genes and restores normal expression [110]. In addition to methylation of DNA, methylation of histones, a distinct epigenetic mechanism dependent on 1-carbon groups, may be affected by consuming excessive levels of specific nutritional factors. Therefore, nutrition factors are likely to directly or indirectly (through changes in DNA methylation) affect histone modifications such as histone methylation.
Lifestyle Factors Affecting the Epigenome
Lifestyle factors including exercise and diet plays an important role in regulating the epigenome and altering gene expression. Exercise can modify the epigenome in order to preserve and prolong life. Exercise has been shown to induce positive changes in DNA methylation within adipose tissue and regulate metabolism in both healthy and diseased individuals [117]. Increased DNA methylation of genes Hdac4 and Ncor2 has also shown to increase lipogenesis following exercise [117]. Exercise also leads to beneficial changes in DNA methylation patterns in skeletal muscle [118]. Not only is obesity an indicator for diseases such as type 2 diabetes and cardiovascular disease, but also puts additional stress on the system which can itself negatively impact health [119]. Acute exercise is associated with DNA hypomethylation of the entire genome in skeletal muscle cells of sedentary individuals and high intensity exercise tends to cause reduction in promoter methylation of certain genes [120]. Exercise is also known to positively influence the expression patterns of miRNAs in leukocyte cells [121]. The health benefits of physical exercise, especially on a long-term and strenuous basis, have a positive effect on epigenetic mechanisms and ultimately may reduce incidence and severity of cancer [122].
Studies in genomic imprinting have revealed how DNA methylation patterns are influenced by diet, and how epigenomic sensitivity to specific diet influences cancer susceptibility. Dietary fat comprises a large part of the Westernized diet, which results in increased adipose tissue via adipocyte hypertrophy and hyperplasia [123]. Dietary fat influences adipokine release through their influence on the epigenome affecting DNA methylation and posttranslation modification of the histone proteins. This represents one of the methods by which dietary fat may influence cancer progression. Overconsumption of well-done meats or saturated fats causes increase in somatic GSTP1 inactivation by CpG island methylation in the promoter region increasing susceptibility to prostate cancer [124].
Several studies have provided evidence that alcohol consumption is associated with different epigenetic changes in human cancer [125]. In a large epidemiological study (the Netherlands Cohort Study on diet and cancer), analysis of DNA methylation showed that the prevalence of promoter hypermethylation of several genes including APC-1A, CDKN2D, CDKN2A, hMLH1, MGMT, and RASSF1A was higher in colorectal cancer patients with high alcohol (and low folate) intake than among colorectal cancer patients with high folate/low alcohol intake. In addition, the study of human head and neck squamous cell carcinoma showed that the promoter hypermethylation of MGMT gene and the genes known to regulate the WNT pathway occurs more frequently in both heavy and light drinkers compared to nondrinkers. The mechanism underlying the epigenetic changes caused by alcohol abuse may also involve SAM. This small metabolite is regenerated from demethylated SAM via the methionine cycle, which involves folate. Therefore, imbalance of this cycle through alcohol consumption may result in depletion of SAM and aberrant epigenetic patterns. In addition, it was shown that the human class I alcohol dehydrogenase (ADH) genes may be regulated by epigenetic mechanism. The class I ADH genes were found to be repressed in human hepatoma through epigenetic modification suggests that changes associated with alcohol-metabolizing genes may also enhance other toxic effects of alcohol on different organs, most notably the liver, including hepatic tumorigenesis [110].
Tobacco smoke is a complex aerosol that contains polycyclic aromatic hydrocarbons (PAHs), mostly benzo[a]pyrene, which is considered the most carcinogenic. Epigenetic targets of the PAHs from tobacco smoke induce DNA damage through preferential binding to methylated CpG sites, a phenomenon already demonstrated for BPDE, a carcinogen found in tobacco smoke. Several studies have demonstrated hypermethylation and silencing of several genes in lung cancer associated with smoking [126]. The genes frequently altered by promoter hypermethylation in lung cancers of smokers are p53, p16 and MGMT. In addition, different components in tobacco smoke induce histone changes and alter histone code. Some potentially novel histone marks, including acetylation, monomethylation, and dimethylation, in specific lysine and arginine residues of histones H3 and H4 in mouse lungs.
Nutrients extracted from the diet enter metabolic pathways and are transformed into useful molecules. These nutrients are known to have epigenetic targets in cells such that they can be used to modify the epigenome in order to correct abnormally activated or silenced genes and can be combined into an “epigenetic diet” useful as a therapeutic and/or chemopreventive measure. During this transitory phase methyl groups are formed from key nutrients including folic acid, B vitamins and s-adenosyl methionine (SAMe), and these methyl groups comprise important epigenetic marks for gene silencing. Diets high in such methyl rich nutrients may significantly alter gene expression and offer protective health benefits [123]. Deficiencies in folate and methionine, both of which are involved in cellular processes that supply methyl groups needed for DNA methylation, can change the expression (imprinting) of growth factor genes such as (IGF1) influencing cancer progression [127]. In addition, several natural nutrients products have interesting biological properties and structural diversity. These include polyphenols present in fruits, vegetables, and other dietary botanicals. Phenolic acids, flavonoids, stilbenes, and lignans are the most abundantly occurring polyphenols that are also an integral part of everyday nutrition in populations worldwide. Certain food components epigenetically increase the levels of DNA repair enzymes such as MGMT and MLH1, others such as blueberry anthocyanins actively decrease DNA damage. Anthocyanin is an effective antioxidant for humans that is found in plants and are easily identified by its potent red or purple pigment. It is found in plants such as eggplant, plums, pomegranate, red onion, cranberries, blueberries, kidney beans, and cherries which all possess anthocyanins. This flavonoid serves as a powerful antioxidant that contributes to scavenging of DNA-damaging free radicals. While the direct fate of anthocyanins in vivo following digestion may be less than 5 % (the majority being rapidly excreted), the potent residual antioxidant property remains in blood following consumption of anthocyanin-rich foods due to metabolic breakdown of the flavonoids and resultant increase in uric acid levels. Some of the common examples of the most studied and promising cancer preventive polyphenols include EGCG (from green tea), curcumin (from curry plant), genistein (from soy), resveratrol (from grapes and berries), and sulforaphane (from broccoli). A large number of dietary agents on DNA methylation, histone modifications, and regulation of expression of noncoding miRNAs in various human cancers are shown in Tables 4.2 and 4.3. Significant gains have been made in understanding the molecular mechanisms underpinning the chemopreventive effects of polyphenols, and consequently, a wide range of mechanisms and gene targets have been identified for individual compounds.
Several studies have demonstrated that green tea polyphenol (GTP) constituent, EGCG is a potent demethylating agent which inhibits enzymes involved in DNA methylation as well as an effective histone modifying agent [33, 173, 205, 206]. It is well known that CpG island hypermethylation at the promoter region leads to epigenetic repression of several critical tumor suppressor genes during tumorigenesis. A study suggests that EGCG acts as a competitive inhibitor of DNMT (Ki = 6.89 μM), which binds to the catalytic pocket and inhibit DNMT activity in a dose-dependent manner [168]. Furthermore, EGCG treatment (5–50 μM for 12–144 h) was found to effectively reactivate methylation-silenced genes—p16 INK4a, retinoic acid receptor beta RARbeta, O(6)-methylguanine methyltransferase MGMT, and human mutL homolog 1, hMLH1 in human esophageal cancer KYSE 510 cells. EGCG was also reported to inhibit HDACs and increase permissive or active histone modifications such as histone acetylation at the target gene promoters. Studies from our laboratory showed that exposure of prostate cancer cells to GTP caused re-expression of epigenetically silenced glutathione S-transferase pi, GSTP1 gene which correlated with the promoter demethylation due to DNMT1 inhibition and histone modifications at the promoter region [173]. However, GTP treatment did not show any global hypomethylation effect which could result in genomic instability as the methylation status of LINE-1 promoter remained unaffected as demonstrated by methylation-specific PCR. GTP treatment decreased mRNA and protein levels of MBD1, MBD4, MeCP2, and HDAC 1-3, whereas acetylated histone H3 (LysH9/18) and H4 were found to be elevated. In another study, we demonstrated that GTP treatment caused cell cycle arrest and apoptosis by inducing proteasomal degradation of class I HDACs in human prostate cancer cells [207]. Studies by Li et al. [206] demonstrated that EGCG in combination with trichostatin A (TSA) could synergistically reactivate ERα expression in ERα negative MDA-MB-231 breast cancer cells by modulating histone methylation and acetylation patter at the gene promoter. In addition, they also reported that treatment with EGCG and/or TSA contributes to transcriptional activation of estrogen receptor (ER)-α by causing a decreased binding of transcription repressor complex, Rb/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 to the regulatory region of the gene.
EGCG has been reported to modulate polycomb proteins such as Bmi-1 and EZH2 [33, 172, 208]. EGCG alone or in combination with DZNep was shown to decrease PcG proteins including EZH2, EED, SUZ12, MEL18, and BMI-1 via a mechanism involving proteasome-associated degradation. The reduction in PcG protein levels correlated with a decrease in repressive chromatin marks—H3K27me3 and H2AK119ub and HDAC-1 levels, whereas accumulation of acetylated H3 levels was found to be elevated. In a recent study, we reported that in breast cancer cells, EGCG or GTP treatment induced expression of epigenetically repressed TIMP-3 gene is mediated by modulating epigenetic mechanisms involving EZH2 and class I HDACs independent of the promoter DNA methylation [33]. After EGCG or GTP treatment, the protein levels of class I HDACs and EZH2 were significantly reduced. Interestingly, transcriptional activation of TIMP-3 was associated with decreased EZH2 localization and H3K27me3 at the promoter with a concomitant elevation in H3K9/18 acetylation levels.
Numerous epidemiological and experimental studies have demonstrated the chemopreventive effects of genistein and other isoflavones on various cancer types [209]. The role of genistein and other soy isoflavones as epigenetic modulators regulating gene expression has been widely reported by several studies. Genistein has been shown to be more potent DNMT inhibitor as compared to biochanin A or daidzein. A study reported that genistein (2–20 μM/L) could reactivate methylation-silenced genes such as RARbeta, p16INK4a, and MGMT in esophageal squamous carcinoma cells KYSE 510 and prostate cancer LNCaP and PC3 cells [129, 136]. Another study demonstrated that genistein treatment in breast MCF10AT benign cells and MCF-7 cancer cells depletes telomerase (hTERT) activity through epigenetic modulation which involves genistein mediated decrease in Dnmt1, Dnmt3a, and Dnmt3b levels [130, 135]. Furthermore, genistein was shown to repress hTERT promoter by chromatin remodeling which involved increase in trimethyl-H3K9 enrichment with a concomitant decrease in dimetyl-H3K4 chromatin marks. A study by King-Batoon et al. [187] showed that a low, nontoxic dose of genistein (3.125 μM, re-supplemented every 48 h for 1 week) could partially demethylate GSTP1, a tumor suppressor gene, in MCF-7, MDA-MB-468, and MCF10A breast cells. Similar in vitro studies in other cancer types provide evidence that genistein is a potent demethylating as well as histone modifying agent, which could reverse the silenced state of critical tumor suppressor genes [131, 132, 210, 211]. A study by Basak et al. [212] demonstrated that AR downregulation in prostate cancer cell line LNCaP by genistein was attributed to the inhibition of HDAC6-Hsp90 co-chaperone function, which is required for AR protein stabilization. Genistein and other soy isoflavones are known to modulate miRNAs as well [135, 197, 198, 213, 214]. Parker et al. [197, 198] performed miRNA profiling of genistein treated and untreated UL-3A and UL-3B cell lines and found 53 miRNAs which were differentially expressed. Upregulation of miR-200 and let-7 by isoflavones was shown to downregulate ZEB1, slug, and vimentin and therefore cause reversal of epithelial to mesenchymal transition (EMT) in gemcitabine resistant pancreatic cancer cells [135]. In human uveal melanoma cells, genistein treatment was demonstrated to cause significant growth inhibition by targeting miR-27a and its target ZBTB10 [214]. However, in vivo clinical studies were inconclusive and did not fall in line with the studies performed in cell line models.
Curcumin has been shown to modulate multiple intracellular pathways associated with proliferation, survival, invasion, apoptosis, and inflammation [215]. In the context of epigenetic pathways, several studies have reported curcumin to be a potent modulator of DNMTs, histone modifying enzymes such as HDACs and HATs as well as miRNAs [216]. In silico molecular docking studies of curcumin with Dnmt1 revealed that it can block or inhibit the catalytic thiol group of C1226 binding site in the enzyme resulting in decreased DNMT activity [216, 217]. This study was further validated by in vitro experimental studies which showed curcumin to be a potent DNA hypomethylating agent [165]. Curcumin was reported to be an effective HDAC inhibitor. Docking studies performed for curcumin binding to HDAC-8 revealed curcumin to be a more potent HDAC inhibitor than known pharmacological inhibitors such as sodium butyrate and valproic acid [164]. Another study reported that curcumin treatment of B-NHL cell line, Raji cells could reduce HDAC-1,-3. and -8 protein levels in a dose-dependent manner and increase H4 acetylation levels [162]. In agreement with earlier findings, studies by Chen et al. [163] reported significant reduction in p300/CREB binding protein (CBP), HDAC-1, and HDAC-3 levels after exposure of Raji cells to curcumin. Studies revealed curcumin to be a specific inhibitor of p300/CBP HAT, which has emerged a novel target for cancer treatment [149, 154, 218]. Curcumin treatment caused proteasomal degradation of p300 and other closely related CBP proteins with no such effect on HATs such as GCN5 and PCAF [154]. Curcumin has also been closely linked to its ability to modulate miRNAs in cancer cells. A microarray based study of the effect of curcumin (10 μM) on the miRNA profile in pancreatic cancer cells PxBC-3 showed significant changes in the expression of 29 miRNAs (11 upregulated and 18 downregulated) after 72 h treatment [195]. Further studies confirmed that MiRNA-22, which has tumor suppressive function, was upregulated after exposure to curcumin and its downstream target genes SP1 and ESR1 were suppressed in these pancreatic cells. Ali et al. [219] demonstrated that treatment of pancreatic cancer cells with curcumin and its analog CDF could induce gemcitabine sensitivity via induction of miR-200 and inactivation of miR-21 expression.
Epigenetic studies on resveratrol have been previously focused on SIRT1 and acetyl transferase p300 [138, 139, 177, 220]. Resveratrol was identified as a potent dietary activator of SIRT1, which lowers the K m (Michaelis constant) for both acetylated substrate and NAD+. It was reported to stimulate SIRT1-dependent p53 deacetylation which ultimately contributes to increased cell survival [138]. In another study by Wood et al. [139], resveratrol was shown to activate sirtuins from metazoans—Caenorhabditis elegans and Drosophila melanogaster and delay aging without any effect on fecundity. The antitumor effect of resveratrol was reported to be mediated partly by SIRT1 [221]. In addition, resveratrol was shown to have a negative effect on Survivin gene expression through histone deacetylation at the gene promoter and display a more profound inhibitory effect on BRCA-1 mutant cells both in vitro and in vivo [178]. In prostate cancer cells, resveratrol was reported to cause downregulation of MTA1 (metastasis associated protein) and destabilize the NuRD (Nucleosome remodeling deacetylase) complex thus allowing p53 acetylation. Furthermore, activation of p53 was shown to induce proapoptotic pathways causing apoptosis in prostate cancer cells [222].
Sulforaphane (SFN) at physiological concentrations has been shown to downregulate Dnmt1 gene expression in human colon Caco-2 cells [223]. Studies by Meeran et al. [224] demonstrated that in MCF-7 and MDA-MB-231 breast cancer cells, SFN treatment cause dose and time-dependent inhibition of hTERT (Human telomerase reverse transcriptase) via an epigenetic mechanism involving DNA methylation and histone modifications. SFN treatment was shown to cause downregulation of Dnmt1 and Dnmt3a, which induced site-specific demethylation at hTERT gene first exon facilitating the binding of CTCF associated with hTERT repression. Furthermore, ChIP analysis of hTERT promoter revealed that active histone chromatin marks such as acetyl-H3, acetyl-H3K9, and acetyl-H4 were increased, whereas repressive chromatin marks which include trimethyl-H3K9 and trimethyl-H3K27 were reduced after SFN treatment in a dose-dependent manner. The SFN-induced hyperacetylation was reported to promote the binding of repressor proteins such as MAD1 and CTCF to the hTERT regulatory region. In another study, Myzak et al. [183] reported that SFN metabolites—SFN–cysteine and SFN-N-acetylcysteine—were more potent HDAC inhibitors in vitro as compared to SFN or its glutathione conjugate. Furthermore, SFN treatment in HCT116 human colorectal cancer cells increased β-catenin-responsive reporter (TOPflash) activity in a dose-dependent manner and inhibited HDAC activity. Consequently, there was an induction in acetylated histone levels bound to p21 (Cip1/Waf1) promoter. In human prostate epithelial cells BPH-1, LNCaP, and PC3, SFN treatment was shown to inhibit HDAC activity, which was accompanied by an increase in acetylated histone levels by 50–100 % and a corresponding induction of p21 and Bax expression which lead to downstream events such as cell cycle arrest and apoptosis [225]. SFN treatment was shown to inhibit HDAC activity in breast cancer cells, but no change in H3 or H4 acetylation was observed [226]. Studies by Myzak et al. [225] provided first evidence for inhibition of in vivo HDAC activity and suppression of tumorigenesis in APC-min mice.
The effect of apigenin on epigenetic related enzymes and their mechanisms was not recognized until recently. Apigenin treatment has been shown to cause a marked decrease in DNMT activity in vitro [137]. Studies from our laboratory demonstrated that apigenin mediated growth arrest and apoptosis in prostate cancer cells was due to the inhibition of class I HDACs [227]. In vivo studies using PC-3 xenografts in athymic nude mice further confirmed that oral intake of apigenin (20 and 50 μg/mouse/d over an 8-week period) reduces tumor burden, HDAC activity, and HDAC -1/-3 protein levels. HDAC-1 and HDAC-3 mRNA and protein levels were found to be significantly decreased in apigenin treated (20–40 μM) PC-3 and 22Rv1 prostate cancer cell lines, which resulted in a global decline in histone H3 and H4 acetylation levels. A corresponding elevation in p21/waf1 and bax levels was observed in both in vitro and in vivo studies, which resulted in the induction of downstream events, that is, apoptosis and cell cycle arrest. In a recent study by Paredes-Gonzalez et al. [228], apigenin was shown to reactive Nrf2 gene which encodes a key transcription factor known for regulating antioxidative defense system and skin homeostasis, in mouse skin epidermal JB6 P+ cells via epigenetic mechanisms. Hypermethylation of 15 CpG sites in Nrf2 promoter was demonstrated to be reversed by apigenin treatment in a dose-dependent manner. Furthermore, apigenin treatment resulted in decreased expression of Dnmt1, Dnmt3A, Dnmt3B, and HDAC (1–8) levels. However, the nuclear localization of Nrf2 was shown to be enhanced and there was increased expression of Nrf2 as well as its target gene NQO1 after apigenin treatment.
Social-Economic and Racial Factors Affecting the Epigenome
Few studies have reported significant epigenetic differences in socio-economic/racial status that account for the differences in cancer and their outcomes [229]. Certain populations are prone to specific types of cancer such as African Americans (AA) who have 14 % higher incidence and 34 % higher death rates than Caucasian Americans (CA) men. Although access to quality healthcare, socioeconomic status, and genetic make-up is implicated in this disparity, the fundamental causes of such cancer disparity seem to be a complex phenomenon. Many investigators are trying to address various sociocultural determinants as a major cause of cancer disparity and in understanding and underpinning mechanisms for designing better community specific interventions for different populations. For example, AA have been found to have statistically significant lower plasma concentrations of certain antioxidants such as vitamin E, alpha-carotene, beta-carotene, lutein, and zeaxanthin than CA [230]. This report indicates that low levels of antioxidants may affect the epigenome and gene expression leading to higher susceptibility and differential cancer outcomes. More research is needed to fully understand how these epigenetic modifications occur and subsequently affecting cancer outcome in diverse population.
Summary and Conclusions
From the studies described herein, it is clear that nutritional and lifestyle factors hold great promise in cancer prevention and in therapy by causing epigenetic modifications. As the importance of epigenetic modifications in cancer is well recognized, precise contribution of epigenetic mechanisms and cellular targets of epigenetic alterations by various endogenous factors in human cancer needs further investigation. Although recent advances in the field of cancer epigenetics has enhanced our understanding of epigenetic changes in normal cellular processes and abnormal events leading to tumorigenesis, however deeper understanding of the global patterns of epigenetic modifications by dietary compounds and lifestyle factors in cancer will lead to the design of better strategies to prevent and cure cancer. Moreover, sufficient preclinical and clinical data is required on the epigenetic changes induced by dietary phytochemicals which will lead to better understanding of the epigenetic targets and pathways altered by these agents to elicit their efficacy in cancer. Additional preclinical and clinical studies are required to analyze the safety profile of doses, route of administration, organ bioavailability alone, and in combination in order to obtain maximum beneficial effects. At last, systematic well-designed randomized placebo-controlled trials with adequate power and relevant clinical epigenetic endpoints are needed. Despite these challenges, research on diet and nutrition continues to emerge and will offer new epigenetic targets and promising agents with more opportunities for prevention, and perhaps therapy of cancer in the near future.
Abbreviations
- AA:
-
African American
- BPDE:
-
Benzo(a)pyrene diol epoxide
- CA:
-
Caucasian American
- DNA:
-
Deoxyribonucleic acid
- DNMT:
-
DNA methyltransferases
- EGCG:
-
Epigallocatechin-3-gallate
- ER:
-
Estrogen receptor
- EZH2:
-
Enhancer of zeste homolog 2
- GSTP1:
-
Glutathione S-transferase pi
- GTP:
-
Green tea polyphenols
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylases
- HDM:
-
Histone demethylases
- hMLH1:
-
Human mutL homolog 1
- HMT:
-
Histone methyltransferases
- HPV:
-
Human papillomavirus
- IGF:
-
Insulin-like growth factor
- LINE:
-
Long interspersed nuclear element
- lncRNA:
-
Long noncoding RNA
- LSD1:
-
Lysine specific demethylase 1
- MBD:
-
Methyl-binding domain proteins
- MGMT:
-
O(6)-methylguanine methyltransferase
- miRNA:
-
MicroRNA
- ncRNA:
-
Noncoding RNA
- PAH:
-
Polycyclic aromatic hydrocarbons
- PcG:
-
Polycomb-group proteins
- piRNA:
-
Piwi-interacting RNA
- RARbeta:
-
Retinoic acid receptor beta
- RepA:
-
Short repeat RNA
- SAH:
-
S-adenosyl-l-homocysteine
- SAM:
-
S-adenosyl methionine
- SFN:
-
Sulforaphane
- siRNA:
-
Small interfering RNA
- TIMP-3:
-
Tissue inhibitor of metalloproteinases-3
References
Issa JP, Kantarjian HM (2009) Targeting DNA methylation. Clin Cancer Res 15(12):3938–3946
Dehan P, Kustermans G, Guenin S et al (2009) DNA methylation and cancer diagnosis: new methods and applications. Expert Rev Mol Diagn 9(7):651–657
Gerhauser C (2013) Epigenetic impact of dietary isothiocyanates in cancer chemoprevention. Curr Opin Clin Nutr Metab Care 16(4):405–410
Bestor TH (2000) The DNA methyltransferases of mammals. Hum Mol Genet 9(16):2395–2402
Santi DV, Garrett CE, Barr PJ (1983) On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell 33(1):9–10
Gerhauser C (2013) Cancer chemoprevention and nutriepigenetics: state of the art and future challenges. Top Curr Chem 329:73–132
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16(1):6–21
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254
Yoder JA, Walsh CP, Bestor TH (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13(8):335–340
Okano M, Bell DW, Haber DA et al (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99(3):247–257
Bourc’his D, Xu GL, Lin CS et al (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294(5551):2536–2539
Watt F, Molloy PL (1988) Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev 2(9):1136–1143
Bell D, Chomarat P, Broyles D et al (1999) In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med 190(10):1417–1426
Ohlsson R, Renkawitz R, Lobanenkov V (2001) CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17(9):520–527
Hark AT, Schoenherr CJ, Katz DJ et al (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405(6785):486–489
Tate PH, Bird AP (1993) Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev 3(2):226–231
Jones PL, Veenstra GJ, Wade PA et al (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19(2):187–191
Nan X, Ng HH, Johnson CA (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393(6683):386–389
Feng Q, Zhang Y (2001) The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev 15:827–832
Hendrich B, Bird A (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 18(11):6538–6547
Nan X, Meehan RR, Bird A (1993) Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res 21:4886–4892
Luger K, Richmond TJ (1998) DNA binding within the nucleosome core. Curr Opin Struct Biol 8(1):33–40
Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98(3):285–294
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403(6765):41–45
Struhl K (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12(5):599–606
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395
Kuo MH, Allis CD (1998) Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20(8):615–626
Sauve AA, Wolberger C, Schramm VL et al (2006) The biochemistry of sirtuins. Annu Rev Biochem 75:435–465
Mottet D, Castronovo V (2008) Histone deacetylases: target enzymes for cancer therapy. Clin Exp Metastasis 25(2):183–189
Shi X, Kachirskaia I, Yamaguchi H et al (2007) Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 27(4):636–646
Rice JC, Allis CD (2001) Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol 13(3):263–273
Upadhyay AK, Cheng X (2011) Dynamics of histone lysine methylation: structures of methyl writers and erasers. Prog Drug Res 67:107–124
Deb G, Singh AK, Gupta S (2014) EZH2: not EZHY (easy) to deal. Mol Cancer Res 12(5):639–653
Kouzarides T (2007) SnapShot: histone-modifying enzymes. Cell 128(4):802
Suzuki T, Miyata N (2006) Rational design of non-hydroxamate histone deacetylase inhibitors. Mini Rev Med Chem 6(5):515–526
Costa FF (2008) Non-coding RNAs, epigenetics and complexity. Gene 410(1):9–17
Peschansky VJ, Wahlestedt C (2014) Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 9(1):3–12
Zhou H, Hu H, Lai M (2010) Non-coding RNAs and their epigenetic regulatory mechanisms. Biol Cell 102(12):645–655
Mattick JS (2005) The functional genomics of noncoding RNA. Science 309(5740):1527–1528
Szymanski M, Barciszewska MZ, Erdmann VA et al (2005) A new frontier for molecular medicine: noncoding RNAs. Biochim Biophys Acta 1756(1):65–75
Ulitsky I, Shkumatava A, Jan CH et al (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147(7):1537–1550
Magistri M, Faghihi MA, St Laurent G 3rd et al (2012) Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet 28(8):389–396
De Lucia F, Dean C (2011) Long non-coding RNAs and chromatin regulation. Curr Opin Plant Biol 14(2):168–173
Pasmant E, Laurendeau I, Héron D et al (2007) Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 67(8):3963–3969
Kotake Y, Nakagawa T, Kitagawa K et al (2011) Longnon-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30(16):1956–1962
Mak W, Baxter J, Silva J et al (2002) Mitotically stable association of polycomb group proteins eed and enx1 with the inactive x chromosome in trophoblast stem cells. Curr Biol 12(12):1016–1020
Plath K, Fang J, Mlynarczyk-Evans SK et al (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300(5616):131–135
Silva J, Mak W, Zvetkova I et al (2003) Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4(4):481–495
Zhao J, Sun BK, Erwin JA et al (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322(5902):750–756
Mohammad F, Mondal T, Guseva N et al (2010) Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137(15):2493–2499
Kanhere A, Viiri K, Araújo CC et al (2010) Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol Cell 38(5):675–688
Deb G, Thakur VS, Gupta S (2013) Multifaceted role of EZH2 in breast and prostate tumorigenesis: epigenetics and beyond. Epigenetics 8(5):464–476
Sander S, Bullinger L, Klapproth K et al (2008) MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 112(10):4202–4212
Varambally S, Cao Q, Mani RS et al (2008) Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322(5908):1695–1699
Juan AH, Kumar RM, Marx JG et al (2009) Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell 36(1):61–74
Liu R, Chen X, Du Y et al (2012) Serum microRNA expression profile as a biomarker in the diagnosis and prognosis of pancreatic cancer. Clin Chem 58(3):610–618
Godlewski J, Nowicki MO, Bronisz A et al (2008) Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res 68(22):9125–9130
Wellner U, Schubert J, Burk UC et al (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 11(12):1487–1495
Fabbri M, Ivan M, Cimmino A et al (2007) Regulatory mechanisms of microRNAs involvement in cancer. Expert Opin Biol Ther 7(7):1009–1019
Garzon R, Calin GA, Croce CM (2009) MicroRNAs in cancer. Annu Rev Med 60:167–179
Lehmann U, Hasemeier B, Römermann D et al (2007) Epigenetic inactivation of microRNA genes in mammary carcinoma. Verh Dtsch Ges Pathol 91:214–220
Hsu RJ, Yang HJ, Tsai HJ (2009) Labeled microRNA pull-down assay system: an experimental approach for high-throughput identification of microRNA-target mRNAs. Nucleic Acids Res 37(10):77
Lujambio A, Calin GA, Villanueva A et al (2008) A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A 105(36):13556–13561
de Leeuw WJ, Dierssen J, Vasen HF, Wijnen JT et al (2000) Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J Pathol 192(3):328–335
Khin SS, Kitazawa R, Kondo T et al (2011) Epigenetic alteration by DNA promoter hypermethylation of genes related to transforming growth factor-β (TGF-β) signaling in cancer. Cancers (Basel) 3(1):982–993
Chen JX, Zheng Y, West M et al (1998) Carcinogens preferentially bind at methylated CpG in the p53 mutational hot spots. Cancer Res 58:2070–2075
Yoon JH, Smith LE, Feng Z et al (2001) Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res 61:7110–7117
Osada H, Takahashi T (2002) Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene 21:7421–7434
Belinsky SA, Klinge DM, Liechty KC et al (2004) Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis 25:1063–1067
Cameron EE, Bachman KE, Myohanen S (1999) Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 21:103–107
Sutherland JE, Costa M (2003) Epigenetics and the environment. Ann N Y Acad Sci 983:151–160
Broday L, Cai J, Costa M (1999) Nickel enhances telomeric silencing in Saccharomyces cerevisiae. Mutat Res 440:121–130
Karaczyn AA, Golebiowski F, Kasprzak KS (2005) Truncation, deamidation, and oxidation of histone H2B in cells cultured with nickel(II). Chem Res Toxicol 18:1934–1942
Zhang YJ, Ahsan H, Chen Y et al (2002) High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol Carcinog 35:85–92
Zhang YJ, Chen Y, Ahsan H et al (2003) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation and its relationship to aflatoxin B1-DNA adducts and p53 mutation in hepatocellular carcinoma. Int J Cancer 103:440–444
Zhang YJ, Rossner P Jr, Chen Y et al (2006) Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int J Cancer 119:985–991
Takiguchi M, Achanzar WE, Qu W et al (2003) Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res 286:355–365
Zhao CQ, Young MR, Diwan BA et al (1997) Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci U S A 94:10907–10912
Chen H, Liu J, Zhao CQ et al (2001) Association of c-myc overexpression and hyperproliferation with arsenite-induced malignant transformation. Toxicol Appl Pharmacol 175:260–268
Van Doorn R, Gruis NA, Willemze R et al (2005) Aberrant DNA methylation in cutaneous malignancies. Semin Oncol 32:479–487
Mittal A, Piyathilake C, Hara Y et al (2003) Exceptionally high protection of photocarcinogenesis by topical application of (–)-epigallocatechin-3-gallate in hydrophilic cream in SKH-1 hairless mouse model: relationship to inhibition of UVB-induced global DNA hypomethylation. Neoplasia 5:555–565
Maekita T, Nakazawa K, Mihara M et al (2006) High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 12:989–995
Sugiyama A, Maruta F, Ikeno T et al (1998) Helicobacter pylori infection enhances N-methyl-N-nitrosourea-induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res 58:2067–2069
Szaleczky E, Pronai L, Molnar B et al (2000) Increased cell proliferation in chronic Helicobacter pylori positive gastritis and gastric carcinoma–correlation between immuno-histochemistry and Tv image cytometry. Anal Cell Pathol 20:131–139
De Capoa A, Musolino A, Della Rosa S et al (2003) DNA demethylation is directly related to tumour progression: evidence in normal, pre-malignant and malignant cells from uterine cervix samples. Oncol Rep 10:545–549
Van Tine BA, Kappes JC, Banerjee NS et al (2004) Clonal selection for transcriptionally active viral oncogenes during progression to cancer. J Virol 78:11172–11186
Zheng ZM, Baker CC (2006) Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci 11:2286–2302
Kalantari M, Calleja-Macias IE, Tewari D et al (2004) Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol 78:12762–12772
Wiley DJ, Huh J, Rao JY et al (2005) Methylation of human papillomavirus genomes in cells of anal epithelia of HIV-infected men. J Acquir Immune Defic Syndr 39:143–151
Zhou L, Jiang W, Ren C et al (2005) Frequent hypermethylation of RASSF1A and TSLC1, and high viral load of Epstein-Barr Virus DNA in nasopharyngeal carcinoma and matched tumor-adjacent tissues. Neoplasia 7:809–815
Zazula M, Ferreira AM, Czopek JP et al (2006) CDH1 gene promoter hypermethylation in gastric cancer: relationship to Goseki grading, microsatellite instability status, and EBV invasion. Diagn Mol Pathol 15:24–29
Jicai Z, Zongtao Y, Jun L et al (2006) Persistent infection of hepatitis B virus is involved in high rate of p16 methylation in hepatocellular carcinoma. Mol Carcinog 45:530–536
Zhang J, Martins CR, Fansler ZB et al (2005) DNA methylation in anal intraepithelial lesions and anal squamous cell carcinoma. Clin Cancer Res 11:6544–6549
Feng Q, Balasubramanian A, Hawes SE et al (2005) Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst 97(4):273–282
Van Engeland M, Weijenberg MP, Roemen GM et al (2003) Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer Res 63:3133–3137
Giovannucci E, Rimm EB, Ascherio A et al (1995) Alcohol, low-methionine–low-folate diets, and risk of colon cancer in men. J Natl Cancer Inst 87:265–273
Puri SK, Si L, Fan CY, Hanna E (2005) Aberrant promoter hypermethylation of multiple genes in head and neck squamous cell carcinoma. Am J Otolaryngol 26:12–17
Marsit CJ, McClean MD, Furniss CS et al (2006) Epigenetic inactivation of the SFRP genes is associated with drinking, smoking and HPV in head and neck squamous cell carcinoma. Int J Cancer 119:1761–1766
Olaharski AJ, Rine J, Marshall BL et al (2005) The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases. PLoS Genet 1:e77
Haigis MC, Guarente LP (2006) Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev 20:2913–2921
Waterland RA (2006) Assessing the effects of high methionine intake on DNA methylation. J Nutr 136:1706S–1710S
Waterland RA, Jirtle RL (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23:5293–5300
Richardson B (2003) Impact of aging on DNA methylation. Ageing Res Rev 2:245–261
Wilson VL, Jones PA (1983) DNA methylation decreases in aging but not in immortal cells. Science 220:1055–1057
Sakatani T, Kaneda A, Iacobuzio-Donahue CA et al (2005) Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science 307:1976–1978
Fraga MF, Ballestar E, Paz MF et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102:10604–10609
Rakyan V, Whitelaw E (2003) Transgenerational epigenetic inheritance. Curr Biol 13:R6
Pembrey ME, Bygren LO, Kaati G et al (2006) Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet 14:159–166
Kaati G, Bygren LO, Edvinsson S (2002) Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet 10:682–688
Herceg Z (2007) Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis 22(2):91–103
Mazzio EA, Soliman KF (2012) Basic concepts of epigenetics: impact of environmental signals on gene expression. Epigenetics 7(2):119–130
Liyanage VR, Jarmasz JS, Murugeshan N et al (2014) DNA modifications: function and applications in normal and disease States. Biology (Basel) 3(4):670–723
Gallou-Kabani C, Junien C (2005) Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes 54(7):1899–1906
Szarc vel Szic K, Declerck K, Vidaković M et al (2015) From inflammaging to healthy aging by dietary lifestyle choices: is epigenetics the key to personalized nutrition? Clin Epigenetics 7(1):33
Verma M (2012) Cancer control and prevention by nutrition and epigenetic approaches. Antioxid Redox Signal 17(2):355–364
Kanwal R, Gupta S (2010) Epigenetics and cancer. J Appl Physiol 109(2):598–605
Rönn T, Volkov P, Davegårdh C et al (2013) A six month exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet 9(6):e1003572
Nitert MD, Dayeh T, Volkov P et al (2012) Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 61(12):3322–3332
Ronti T, Lupattelli G, Mannarino E (2006) The endocrine function of adipose tissue: an update. Clin Endocrinol (Oxf) 64(4):355–365
Ntanasis-Stathopoulos J, Tzanninis JG et al (2013) Epigenetic regulation on gene expression induced by physical exercise. J Musculoskelet Neuronal Interact 13(2):133–146
Radom-Aizik S, Zaldivar F Jr, Oliver S et al (2010) Evidence for microRNA involvement in exercise-associated neutrophil gene expression changes. J Appl Physiol 109(1):252–261
Sanchis-Gomar F, Garcia-Gimenez JL, Perez-Quilis C et al (2012) Physical exercise as an epigenetic modulator: Eustress, the “positive stress” as an effector of gene expression. J Strength Cond Res 26(12):3469–3472
Kanherkar RR, Bhatia-Dey N, Csoka AB (2014) Epigenetics across the human lifespan. Front Cell Dev Biol 2:49
Nelson WG, Demarzo AM, Yegnasubramanian S (2014) The diet as a cause of human prostate cancer. Cancer Treat Res 159:51–68
Ponomarev I (2013) Epigenetic control of gene expression in the alcoholic brain. Alcohol Res 35(1):69–76
Steenaard RV, Ligthart S, Stolk L et al (2015) Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin Epigenetics 7(1):54
Ross SA, Milner JA (2007) Epigenetic modulation and cancer: effect of metabolic syndrome? Am J Clin Nutr 86(3):s872–s877
Hong T, Nakagawa T, Pan W et al (2004) Isoflavones stimulate estrogen receptor-mediated core histone acetylation. Biochem Biophys Res Commun 317(1):259–264
Fang MZ, Chen D, Sun Y et al (2005) Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res 11:7033–7041
Li Y, Liu L, Andrews LG et al (2009) Genistein depletes telomerase activity through cross-talk between genetic and epigenetic mechanisms. Int J Cancer 125(2):286–296
Majid S, Dar AA, Ahmad AE et al (2009) BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 30(4):662–670
Majid S, Dar AA, Shahryari V et al (2009) Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer 116(1):66–76
Majid S, Kikuno N, Nelles J et al (2008) Genistein induces the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification. Cancer Res 68(8):2736–2744
Kikuno N, Shiina H, Urakami S et al (2008) Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer 123(3):552–560
Li Y, Vanden Boom TG 2nd, Kong D et al (2009) Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res 69(16):6704–6712
Fang MZ, Jin Z, Wang Y et al (2005) Promoter hypermethylation and inactivation of O (6)-methylguanine-DNA methyltransferase in esophageal squamous cell carcinomas and its reactivation in cell lines. Int J Oncol 26:615–622
Fang M, Chen D, Yang CS (2007) Dietary polyphenols may affect DNA methylation. J Nutr 137(1 Suppl):223S–228S
Howitz KT, Bitterman KJ, Cohen HY et al (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425(6954):191–196
Wood JG, Rogina B, Lavu S et al (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430(7000):686–689
Lee WJ, Shim JY, Zhu BT (2005) Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol 68(4):1018–1030
Lea MA, Randolph VM, Patel M (1999) Increased acetylation of histones induced by diallyl disulfide and structurally related molecules. Int J Oncol 15(2):347–352
Lea MA, Randolph VM, Lee JE et al (2001) Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int J Cancer 92(6):784–789
Druesne N, Pagniez A, Mayeur C et al (2004) Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis 25(7):1227–1236
Nian H, Delage B, Pinto JT (2008) Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis 29(9):1816–1824
Lee JY, Kim HS, Song YS (2012) Genistein as a potential anticancer agent against ovarian cancer. J Tradit Complement Med 2(2):96–104
Lea MA, Randolph VM (2001) Induction of histone acetylation in rat liver and hepatoma by organosulfur compounds including diallyl disulfide. Anticancer Res 21(4A):2841–2845
Sbardella G, Castellano S, Vicidomini C et al (2008) Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorg Med Chem Lett 18(9):2788–2792
Mai A, Rotili D, Tarantino D et al (2006) Small-molecule inhibitors of histone acetyltransferase activity: identification and biological properties. J Med Chem 49(23):6897–6907
Balasubramanyam K, Swaminathan V, Ranganathan A et al (2003) Small molecule modulators of histone acetyltransferase p300. J Biol Chem 278(21):19134–19140
Sun Y, Jiang X, Chen S et al (2006) Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett 580(18):4353–4356
Eliseeva ED, Valkov V, Jung M et al (2007) Characterization of novel inhibitors of histone acetyltransferases. Mol Cancer Ther 6(9):2391–2398
Chandregowda V, Kush A, Reddy GC (2009) Synthesis of benzamide derivatives of anacardic acid and their cytotoxic activity. Eur J Med Chem 44(6):2711–2719
Singh N, Misra K (2009) Computational screening of molecular targets in Plasmodium for novel non-resistant anti-malarial drugs. Bioinformation 3(6):255–262
Marcu MG, Jung YJ, Lee S et al (2006) Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem 2(2):169–174
Kang J, Chen J, Shi Y et al (2005) Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol 69(8):1205–1213
Cui L, Miao J, Furuya T et al (2007) PfGCN5-mediated histone H3 acetylation plays a key role in gene expression in Plasmodium falciparum. Eukaryot Cell 6(7):1219–1227
Sng JC, Taniura H, Yoneda Y (2006) Histone modifications in kainate-induced status epilepticus. Eur J Neurosci 23(5):1269–1282
Chiu J, Khan ZA, Farhangkhoee H et al (2009) Curcumin prevents diabetes-associated abnormalities in the kidneys by inhibiting p300 and nuclear factor-kappaB. Nutrition 25(9):964–972
Tikoo K, Meena RL, Kabra DG et al (2008) Change in post-translational modifications of histone H3, heat-shock protein-27 and MAP kinase p38 expression by curcumin in streptozotocin-induced type I diabetic nephropathy. Br J Pharmacol 153(6):1225–1231
Li HL, Liu C, de CG et al (2008) Curcumin prevents and reverses murine cardiac hypertrophy. J Clin Invest 118(3):879–893
Morimoto T, Sunagawa Y, Kawamura T et al (2008) The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest 118(3):868–878
Liu HL, Chen Y, Cui GH et al (2005) Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol Sin 26(5):603–609
Chen Y, Shu W, Chen W et al (2007) Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin Pharmacol Toxicol 101(6):427–433
Bora-Tatar G, Dayangac-Erden D, Demir AS et al (2009) Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: activity and docking studies. Bioorg Med Chem 17(14):5219–5228
Liu Z, Xie Z, Jones W, Pavlovicz RE et al (2009) Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett 19(3):706–709
Kuck D, Singh N, Lyko F et al (2010) Novel and selective DNA methyltransferase inhibitors: docking-based virtual screening and experimental evaluation. Bioorg Med Chem 18(2):822–829
Choi KC, Jung MG, Lee YH et al (2009) Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res 69(2):583–592
Fang MZ, Wang Y, Ai N et al (2003) Tea polyphenol(-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 63(22):7563–7570
Nair S, Hebbar V, Shen G et al (2008) Synergistic effects of a combination of dietary factors sulforaphane and (-) epigallocatechin-3-gallate in HT-29 AP-1 human colon carcinoma cells. Pharm Res 25(2):387–399
Gao Z, Xu Z, Hung MS et al (2009) Promoter demethylation of WIF-1 by epigallocatechin-3-gallate in lung cancer cells. Anticancer Res 29(6):2025–2030
Kato K, Long NK, Makita H et al (2008) Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br J Cancer 99(4):647–654
Balasubramanian S, Adhikary G, Eckert RL (2010) The Bmi-1 polycomb protein antagonizes the (-)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis 31(3):496–503
Pandey M, Shukla S, Gupta S (2010) Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int J Cancer 126(11):2520–2533
Murugan RS, Uchida K, Hara Y, Nagini S (2008 Oct) Black tea polyphenols modulate xenobiotic-metabolizing enzymes, oxidative stress and adduct formation in a rat hepatocarcinogenesis model. Free Radic Res 42(10):873–84
Wang LG, Beklemisheva A, Liu XM et al (2007) Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol Carcinog 46(1):24–31
Ma X, Fang Y, Beklemisheva A et al (2006) Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int J Oncol 28(5):1287–1293
Wang LG, Liu XM, Fang Y et al (2008) De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol 33(2):375–380
Wang RH, Zheng Y, Kim HS et al (2008) Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell 32(1):11–20
Wang RH, Sengupta K, Li C et al (2008) Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14(4):312–323
Wilkinson JT, Morse MA, Kresty LA et al (1995) Effect of alkyl chain length on inhibition of N nitrosomethylbenzylamine-induced esophageal tumorigenesis and DNA methylation by isothiocyanates. Carcinogenesis 16(5):1011–1015
Schwab M, Reynders V, Loitsch S et al (2008) The dietary histone deacetylase inhibitor sulforaphane induces human beta-defensin-2 in intestinal epithelial cells. Immunology 125(2):241–251
Paluszczak J, Krajka-Kuźniak V, Baer-Dubowska W (2010) The effect of dietary polyphenols on the epigenetic regulation of gene expression in MCF7 breast cancer cells. Toxicol Lett 192(2):119–125
Myzak MC, Karplus PA, Chung FL et al (2004) A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res 64(16):5767–5774
Yang SR, Wright J, Bauter M et al (2007) Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol 292(2):L567–L576
Yang J, Kong X, Martins-Santos ME et al (2009) Activation of SIRT1 by resveratrol represses transcription of the gene for the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) by deacetylating hepatic nuclear factor 4alpha. J Biol Chem 284(40):27042–27053
Stefanska B, Rudnicka K, Bednarek A et al (2010) Hypomethylation and induction of retinoic acid receptor beta 2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. Eur J Pharmacol 638(1–3):47–53
King-Batoon A, Leszczynska JM, Klein CB (2008) Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ Mol Mutagen 49(1):36–45
Tan S, Wang C, Lu C et al (2009) Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy 55(1):6–10
Chakrabarti M, Khandkar M, Banik NL et al (2012) Alterations in expression of specific microRNAs by combination of 4-HPR and EGCG inhibited growth of human malignant neuroblastoma cells. Brain Res 1454:1–13
Wu H, Huang M, Liu Y et al (2014) Luteolin induces apoptosis by up-regulating miR-34a in human gastric cancer cells. Technol Cancer Res Treat 14(6):747–755, PubMed PMID: 24988056
Gandhy SU, Kim K, Larsen L et al (2012) Curcumin and synthetic analogs induce reactive oxygen species and decreases specificity protein (Sp) transcription factors by targeting microRNAs. BMC Cancer 12:564
Saini S, Arora S, Majid S et al (2011) Curcumin modulates microRNA-203-mediated regulation of the Src-Akt axis in bladder cancer. Cancer Prev Res (Phila) 4:1698–1709
Zhang J, Du Y, Wu C et al (2010) Curcumin promotes apoptosis in human lung adenocarcinoma cells through miR-186n signaling pathway. Oncol Rep 24:1217–1223
Gao SM, Yang JJ, Chen CQ et al (2012) Pure curcumin decreases the expression of WT1 by upregulation of miR-15a and miR-16-1 in leukemic cells. J Exp Clin Cancer Res 31:2
Sun M, Estrov Z, Ji Y et al (2008) Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther 7(3):464–473
Chiyomaru T, Yamamura S, Zaman MS et al (2012) Genistein suppresses prostate cancer growth through inhibition of oncogenic microRNA-151. PLoS One 7:e43812
Parker LP, Taylor DD, Kesterson J et al (2009) Modulation of microRNA associated with ovarian cancer cells by genistein. Eur J Gynaecol Oncol 30:616–621
Parker JS, Parizotto EA, Wang M et al (2009) Enhancement of the seed-target recognition step in RNA silencing by a PIWI/MID domain protein. Mol Cell 33(2):204–214
Dhar S, Hicks C, Levenson AS (2011) Resveratrol and prostate cancer: promising role for microRNAs. Mol Nutr Food Res 55:1219–1229
Sheth S, Jajoo S, Kaur T et al (2012) Resveratrol reduces prostate cancer growth and metastasis by inhibiting the Akt/MicroRNA-21 pathway. PLoS One 7:e51655
Tili E, Michaille JJ, Alder H et al (2010) Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFbeta signaling pathway in SW480 cells. Biochem Pharmacol 80:2057–2065
Jiang L, Huang Q, Chang J et al (2011) MicroRNA HSA-miR-125a-5p induces apoptosis by activating p53 in lung cancer cells. Exp Lung Res 37(7):387–398
Chakrabarti M, Banik NL, Ray SK (2013) miR-138 overexpression is more powerful than hTERT knockdown to potentiate apigenin for apoptosis in neuroblastoma in vitro and in vivo. Exp Cell Res 319(10):1575–1585
Wen XY, Wu SY, Li ZQ et al (2009) Ellagitannin (BJA3121), an anti-proliferative natural polyphenol compound, can regulate the expression of MiRNAs in HepG2 cancer cells. Phytother Res 23(6):778–784
Nandakumar V, Vaid M, Katiyar SK (2011) (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 32(4):537–544
Li Y, Yuan YY, Meeran SM et al (2010) Synergistic epigenetic reactivation of estrogen receptor-α (ERα) by combined green tea polyphenol and histone deacetylase inhibitor in ERα-negative breast cancer cells. Mol Cancer 9:274
Thakur VS, Gupta K, Gupta S (2012) Green tea polyphenols causes cell cycle arrest and apoptosis in prostate cancer cells by suppressing class I histone deacetylases. Carcinogenesis 33(2):377–384
Choudhury SR, Balasubramanian S, Chew YC et al (2011) (-)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis 32(10):1525–1532
Banerjee S, Li Y, Wang Z et al (2008) Multi-targeted therapy of cancer by genistein. Cancer Lett 269(2):226–242
Day JK, Bauer AM, DesBordes C et al (2002) Genistein alters methylation patterns in mice. J Nutr 132(8 Suppl):2419S–2423S
Majid S, Dar AA, Saini S et al (2010) Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res 70(7):2809–2818
Basak S, Pookot D, Noonan EJ et al (2008) Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function. Mol Cancer Ther 7(10):3195–3202
Majid S, Dar AA, Shahryari V et al (2010) Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer 116(1):66–76
Sun Q, Cong R, Yan H et al (2009) Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol Rep 22(3):563–567
Teiten MH, Eifes S, Dicato M et al (2010) Curcumin-the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins (Basel) 2(1):128–162
Fu S, Kurzrock R (2010) Development of curcumin as an epigenetic agent. Cancer 116(20):4670–4676
Medina-Franco JL, Caulfield T (2011) Advances in the computational development of DNA methyltransferase inhibitors. Drug Discov Today 16(9-10):418–425
Dekker FJ, Haisma HJ (2009) Histone acetyl transferases as emerging drug targets. Drug Discov Today 14(19-20):942–948
Ali S, Banerjee S, Logna F et al (2012) Inactivation of Ink4a/Arf leads to deregulated expression of miRNAs in K-Ras transgenic mouse model of pancreatic cancer. J Cell Physiol 227(10):3373–3380
Rajendran P, Ho E, Williams DE et al (2011) Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenetics 3(1):4
Boily G, He XH, Pearce B et al (2009) SirT1-null mice develop tumors at normal rates but are poorly protected by resveratrol. Oncogene 28(32):2882–2893
Kai L, Samuel SK, Levenson AS (2010) Resveratrol enhances p53 acetylation an apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int J Cancer 126(7):1538–1548
Traka M, Gasper AV, Smith JA, Hawkey CJ, Bao Y, Mithen RF (2005 Aug) Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. J Nutr 135(8):1865–72
Meeran SM, Patel SN, Tollefsbol TO (2010) Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One 5(7):e11457
Myzak MC, Dashwood WM, Orner GA et al (2006) Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J 20(3):506–508
Pledgie-Tracy A, Sobolewski MD, Davidson NE (2007) Sulforaphane induces celltype-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther 6(3):1013–1021
Pandey M, Kaur P, Shukla S et al (2012) Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: in vitro and in vivo study. Mol Carcinog 51(12):952–962
Paredes-Gonzalez X, Fuentes F et al (2014) Apigenin reactivates Nrf2 anti-oxidative stress signaling in mouse skin epidermal JB6 P + cells through epigenetics modifications. AAPS J 16(4):727–735
Mohammed SI, Springfield S, Das R (2012) Role of epigenetics in cancer health disparities. Methods Mol Biol 863:395–410
Signorello LB, Schlundt DG, Cohen SS et al (2007) Comparing diabetes prevalence between African Americans and Whites of similar socioeconomic status. Am J Public Health 97(12):2260–2267
Tsang WP, Kwok TT (2010) Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem 21:140–146
Sakurai MA, Ozaki Y, Okuzaki D et al (2014) Gefitinib and luteolin cause growth arrest of human prostate cancer PC-3 cells via inhibition of cyclin G-associated kinase and induction of miR-630. PLoS One 9(6):e100124
Acknowledgment
The original work from author’s laboratory outlined in this chapter was supported by United States Public Health Service Grants R01CA115491, R01CA108512, R21CA193080, and R03CA186179, and Department of Veteran Affairs grant 1I01BX002494 to SG.
Conflict of Interest: The authors have no competing interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Shankar, E., Gupta, S. (2016). Nutritional and Lifestyle Impact on Epigenetics and Cancer. In: Berger, N. (eds) Epigenetics, Energy Balance, and Cancer. Energy Balance and Cancer, vol 11. Springer, Cham. https://doi.org/10.1007/978-3-319-41610-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-41610-6_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41608-3
Online ISBN: 978-3-319-41610-6
eBook Packages: MedicineMedicine (R0)