
Abstract
Barrett’s esophagus is the condition in which metaplastic columnar epithelium that predisposes to cancer development replaces stratified squamous epithelium in the distal esophagus. Potential sources for the cell or tissue of origin for metaplastic Barrett’s epithelium are reviewed including native esophageal differentiated squamous cells, progenitor cells native to the esophagus located within the squamous epithelium or in the submucosal glands or ducts, circulating bone marrow-derived stem cells, and columnar progenitor cells from the squamocolumnar junction or the gastric cardia that proximally shift into the esophagus to fill voids left by damaged squamous epithelium. Wherever its source the original cell must undergo molecular reprogramming (i.e., either transdifferentiation or transcommitment) to give rise to specialized intestinal metaplasia. Transcription factors that specify squamous, columnar, intestinal, and mucus-secreting epithelial differentiation are discussed. An improved understanding of how esophageal columnar metaplasia forms could lead to development of effective treatment or prevention strategies for Barrett’s esophagus. It could also more broadly inform upon normal tissue development and differentiation, wound healing, and stem cell biology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Barrett’s esophagus
- Metaplasia
- Cell of origin
- Transcommitment
- Transdifferentiation
- Multilayered epithelium
- Submucosal gland
- Gastric cardia
- Squamocolumnar junction
- Limiting ridge
Barrett’s esophagus is the metaplastic change of the distal esophageal epithelium from stratified squamous to columnar that can arise as a complication of gastroesophageal reflux disease (GERD) [1]. Barrett’s esophagus is considered by many as an adaptive response to chronic exposure to gastric acid and intestinal bile salts since a columnar-lined esophagus (CLE) should be more protective against these insults. The metaplastic esophageal columnar epithelium can be one of three types: specialized intestinal metaplasia , characterized by (intestinal mucin) MUC2-expressing goblet cells and other intestinal cell types; cardia-type epithelium, characterized by mucus containing cells; and gastric fundic-type epithelium, characterized by mucus secreting, parietal, and chief cells [2]. While all three types of CLE are classified as Barrett’s metaplasia outside of the United States, the American Gastroenterological Association (AGA) more strictly defines Barrett’s esophagus as “the condition in which any extent of metaplastic columnar epithelium that predisposes to cancer development replaces the stratified squamous epithelium that normally lines the distal esophagus” [2]. Since only specialized intestinal metaplasia is clearly at increased risk of transforming into esophageal adenocarcinoma, the more prevalent form of esophageal cancer in Western countries, the presence of goblet cells is required for the histopathologic definition of Barrett’s esophagus in the United States [2].
Metaplasia is the replacement of one fully differentiated tissue by another [3]. In Barrett’s esophagus , the columnar epithelium that replaces the squamous epithelium originates either from a native esophageal cell or from a cell external to the esophagus that relocates to the esophagus and undergoes molecular reprogramming (i.e., transdifferentiation or transcommitment ). Wherever the source of the Barrett’s esophagus cell or tissue of origin, the original cell is unlikely intestinal and therefore must undergo molecular reprogramming to convert into epithelium classified as specialized intestinal metaplasia . Furthermore, the columnar cell would need to possess or acquire the ability to form three-dimensional (3D) structures such as the glands observed in Barrett’s esophagus.
There are four general possibilities for the source of the Barrett’s esophagus cell or tissue of origin (Table 10.1). First, a native esophageal differentiated squamous cell could give rise to an intestinal columnar cell through irreversible direct phenotypic conversion, also known as transdifferentiation . Second, a native esophageal progenitor cell, which could be located within the squamous epithelium or in a submucosal gland or its duct, differentiates to become an intestinal columnar cell (esophageal progenitor cell transcommitment ). Third, an external circulating bone marrow-derived stem cell migrates to the esophagus and undergoes intestinal columnar epithelial differentiation (circulating stem cell transcommitment ). Fourth, an external columnar progenitor cell from the squamocolumnar junction (SCJ ) or gastric cardia proximally shifts to fill a void left by damaged stratified squamous epithelium and then undergoes intestinal differentiation (columnar progenitor cell transcommitment ).
Identifying the cell or tissue origin of Barrett’s esophagus could lead to development of effective treatment or prevention strategies for this condition. Prevention is especially relevant in current clinical practice where Barrett’s esophagus is often treated with radiofrequency ablation (RFA) and frequently recurs postablation [4]. Understanding the process of how Barrett’s esophagus forms could also provide new insights into normal tissue development and differentiation, wound healing, and stem cell biology. Data supporting various possible sources of the Barrett’s esophagus cell or tissue of origin have arisen from patient biopsies or human esophagectomy specimens; in vivo animal models (i.e., surgically induced reflux esophagitis or genetically engineered mice); cell culture experiments utilizing human esophageal squamous and Barrett’s epithelial cell lines; and novel experimental systems such as organ explant culture, 3D organotypic culture, or in vivo transplant culture.
Transdifferentiation
Support for transdifferentiation is derived from the description of multilayered epithelium (MLE) , demonstrating the existence of biphenotypic squamous and columnar cells in adult human patients and experimental animals. By scanning electron microscopy (SEM) , a distinctive superficial “transition zone cell” was identified in mucosal biopsies from patients with Barrett’s esophagus where squamous and Barrett’s epithelium were apposed [5]. Squamous epithelium was characterized by SEM as having prominent intercellular ridges and distinct microridges while every Barrett’s esophagus cell exhibited microvilli and had neither intercellular ridges nor microridges. In contrast to both squamous and Barrett’s epithelium, transition zone cells displayed both intercellular ridges and short, stubby microvilli with some transition zone cells also demonstrating bulging mucus. Thus, these distinctive transition zone cells possessed SEM features of both squamous and columnar epithelium. In addition, SEM could further differentiate gastric surface-like cells from intestinal absorptive-like cells from goblet cells, which contained a central mound of protruding mucus, by surface topology. The investigators next examined biopsies taken from the normal gastroesophageal junction (GEJ) from several patients and did not find cells resembling transition zone cells, suggesting that transition zone cells were unique to Barrett’s esophagus in the gastrointestinal (GI) tract. Later investigations found these distinctive transition zone cells overlying squamous epithelial cells in areas of squamo-Barrett’s transition [6]. Now termed “multilayered epithelium ,” cytokeratin (CK) expression analysis by immunohistochemistry (IHC) demonstrated that individual basal cells found in MLE simultaneously expressed both squamous CK4 and columnar CK19 [7]. Mucin expression profiling of both basal and superficial cells showed that MLE more closely resembled Barrett’s epithelium and esophageal submucosal gland duct epithelium than gastric cardia epithelium, arguing against proximal shifting of gastric cardia cells [8]. The observation of MLE in association with Barrett’s esophagus in rats that have undergone esophagogastroduodenal anastomosis to surgically induce bile reflux suggested that MLE could be a precursor lesion to Barrett’s esophagus [9, 10]. Ultrastructural analysis of mouse esophageal basal epithelial cells in which the intestinal transcription factor Cdx2 is overexpressed using the CK14 promoter revealed that the cells acquired some characteristics of MLE with both squamous and secretory features [11].
A common objection to transdifferentiation as a mechanism of Barrett’s metaplasia is that direct phenotypic conversion of a squamous esophageal cell into an intestinal goblet cell has not yet been achieved in vitro. Overexpression of various transcription factors in differentiated human esophageal squamous cell lines such as HET-1A (SV-40 immortalized) [12, 13], EPC2 (hTERT immortalized, proximal esophagus) [14, 15], NES-B3T, and NES-B10T (hTERT immortalized, distal esophagus) [16, 17] has led to expression of columnar, intestinal, or mucin associated genes that are found in Barrett’s esophagus but have not led to full phenotypic conversion of a squamous cell into a specialized intestinal metaplastic cell. It is likely that a less differentiated cell that still maintains multipotent potential (i.e., a progenitor or stem cell) is required to form an intestinal goblet cell under these conditions .
Native Esophageal Squamous Progenitor Cells
Evidence for transcommitment of esophageal epithelial progenitor cells comes from observations made during esophageal embryogenesis. The entire GI tract, including the esophagus, embryologically develops from a columnar epithelium-lined gut tube that then regionalizes and undergoes organ-specific differentiation. In humans, the epithelium lining the esophagus begins as a pseudostratified columnar epithelium, seen as early as 7 weeks gestational age in autopsy specimens [18]. By 8–10 weeks gestational age, the esophageal epithelium has become ciliated and simple columnar. Early in the 5th month of gestation, the epithelium changes from columnar to squamous near the mid-esophagus [19]. The entire esophageal epithelium then bidirectionally transitions into stratified squamous from this starting point, concluding at both the proximal and distal ends of the esophagus. IHC staining revealed expression of squamous CK5 and CK13 in the 14-week-old embryo; however, columnar CK20 was still observed in esophageal epithelium in the 17-week-old embryo [18]. Based on these observations, either a mixed phenotype of squamous and columnar epithelium may line the esophagus prior to its final differentiation into stratified squamous beginning at 5 months of gestation or a subpopulation of epithelial cells may express both columnar and squamous CK’s simultaneously.
This phenotypic switch was subsequently more carefully studied in mouse embryos . In mice, normal esophageal epithelial development occurs between E (embryonic day) 11.5, when the esophagus separates from the trachea, and 1 month after birth when the esophageal epithelium fully keratinizes. The timing of this change varies based on the mouse strain [20–22]. It was initially thought that the mechanism causing epithelial phenotype switching from columnar to stratified squamous was luminal sloughing of columnar cells followed by replacement by underlying squamous cells. However, an elegant study using outbred CD1 mice demonstrated otherwise [22]. Investigators from the Tosh lab found that during normal esophageal embryogenesis some epithelial cells simultaneously expressed columnar and squamous CKs. This was demonstrated using columnar CK8 immunostaining in conjunction with a squamous CK14-nuclear green fluorescent protein (GFP) reporter or with CK8 and CK14 coimmunostaining. The acquisition of expression of a squamous CK by the CK8 positive columnar cells occurred in the absence of cell division and ended with CK8 being silenced through de novo promoter methylation .
Multiple studies have characterized adult mouse esophageal epithelial progenitor cells and identified several cell surface markers and populations. Epperly and colleagues harvested esophageal epithelium from male GFP positive C57BL/6 mice and isolated esophageal epithelial progenitor cells using serial preplating or sorting for side population cells after staining with Hoechst dye [23]. Side population cells are characterized by their ability to exclude the DNA dye Hoechst 33342, and this technique has been used successfully to identify stem cells [24]. Identified esophageal epithelial progenitor cells had high expression of the cell surface markers Sca-1 and Thy-1, homed to the esophagus when injected into the tail vein of female GFP negative mice that had been irradiated to induce esophagitis, and gave rise to colonies of cells within the esophagus. These GFP positive cells also could be serially passaged in later generations of female GFP negative mice. Though it was likely that the serially passaged GFP positive cells were epithelial, no costaining experiments were performed to confirm this.
Kalabis and colleagues identified a population of label retaining cells (LRC) , using BrdU or tritiated thymidine incorporation, comprising approximately 1 % of all CK14 positive basal epithelial cells in the mouse esophagus [25]. Since either type of DNA label is diluted by successive cell divisions, this technique can be used to identify the slowest cycling cells which many consider to be quiescent progenitor or stem cells. These LRC were proven to be epithelial by staining with pan-CK and could be sorted among side population cells following staining with Hoechst dye. Side population cells contained a higher percentage of CD34 positive cells compared to the total esophageal epithelial cell population (34 % versus 0.4 %), formed colonies, and made a fully differentiated epithelium with keratinization in 3D organotypic culture. The 3D organotypic cultures made with side population cells also had more robust immunostaining for CK4 and CK13 which are normally expressed by suprabasal layers of squamous epithelium, and cells from these cultures could produce second-generation 3D organotypic cultures with a similar mature CK differentiation pattern. Finally, in mice in which the esophageal epithelium was mechanically wounded GFP positive, CD34 positive cells were shown to participate in repair within 48 h while GFP positive, CD34 negative cells did not. Based on these results, it was concluded that CD34 positive LRC represented esophageal epithelial progenitor cells.
Croagh and colleagues divided esophageal basal epithelial cells into four groups based on their expression of α6 integrin and CD71 [26]. Cells that were α6brightCD71dim, α6brightCD71bright, or α6dim had similar clonogenic potential compared to cells that were CD71 very bright (CD71+++) which had a much lower clonogenic potential. In an in vivo transplant culture system in which mouse esophageal epithelial cells were injected into a denuded rat trachea which was then placed subcutaneously into NOD/SCID mice, α6brightCD71dim and α6brightCD71bright cells generated a differentiated stratified squamous epithelium while α6dim cells did not. Finally, α6brightCD71dim cells were shown to have the highest number of LRC for the longest period of time, consistent with being the least differentiated progenitor cells. DeWard and colleagues also identified α6 integrin as a marker of cells that have stem cell-like features [27]. Single cells isolated from mouse esophageal epithelium gave rise to organoids in growth media supplemented with exogenous stem cell factors. This ability to form organoids in culture is a property retained by stem cells [28]. Further analysis found that this organoid-forming ability was dependent on positive Sox2 expression and limited to cells located in the basal layer. Sox2 positive basal cells were further sorted based on expression of α6 integrin, β4 integrin, and CD73. α6/β4 integrin high, CD73 positive cells possessed the highest organoid forming activity. Treatment with the differentiating agent all-trans retinoic acid (ATRA) led to a decrease in α6/β4 integrin high, CD73 positive or α6/β4 integrin high, CD73 negative cells and an increase in α6/β4 integrin low cells. These studies support a hierarchy of cells within the basal epithelial cell layer, with α6/β4 integrin high, CD73 positive cells being the least differentiated.
Further work on identifying progenitor cells in the mouse esophagus was performed in the Jones lab [29]. They identified 0.4 % of esophageal basal epithelial cells as LRC, but these LRC did not stain for CK14 or CD34 as Kalabis and colleagues had reported. Instead these LRC stained for CD45, consistent with a hematopoietic origin. Using a tamoxifen-inducible Ahcre mouse line [30] to activate a yellow fluorescent protein (YFP) reporter , these investigators tracked individual cell clones in the mouse esophagus over 1 year. Unexpectedly, the number of YFP positive clones decreased while the size of each clone increased linearly over time. Based on these findings, Doupé and colleagues hypothesized that all esophageal progenitor cells were functionally equivalent, stochastically dividing to equally give rise to proliferating and differentiating daughter cells, and that the esophageal epithelium did not have a slow cycling stem cell population. Mice were then treated with ATRA at a dose selected to induce hyperproliferation and lineage tracing was performed. ATRA doubled both basal cell proliferation and differentiation with no observed difference in the proportion of symmetric (proliferating daughter cells) versus asymmetric (progenitor cells) cell division, thus establishing a new homeostatic state. The mouse esophageal epithelium was next injured by performing a microendoscopic biopsy to investigate the role of esophageal progenitors in wound repair. Through the use of two separate reporter mouse strains and EdU incorporation experiments, these investigators demonstrated that injury caused esophageal progenitors to favor proliferation over differentiation for a period of 5 days after which they returned to equal states of proliferation and differentiation .
Unlike mouse esophageal epithelium which is typically divided into basal and suprabasal compartments, the human esophageal epithelium is divided into two basal cell regions: one overlying the stromal papillae and the other overlying interpapillary regions [31] (Fig. 10.1). Seery and Watt described cells in the interpapillary basal cell regions as undergoing asymmetric division, while proliferating cells overlying papillae underwent symmetric division. Proliferating Ki-67 positive cells were found to be four times more common in cells overlying papillae, which had higher levels of β1 integrin. In contrast, studies from the Fitzgerald lab demonstrated that in human esophageal epithelium the most proliferative basal cells are found within the interpapillary region with no mitoses seen in the region overlying the tips of the papillae [32]. Cells at the tip of the papillae were quiescent and costained for CD34 and β1 integrin. Barbera and colleagues then sorted human esophageal cells into four groups using a CD34 antibody and an antibody against EpCAM, a marker for cells in the suprabasal layer. They found no difference in clonogenic capacity between the four groups and concluded that esophageal epithelial progenitor cells were widespread, not restricted to either basal cell region, and included cells that were committed to epithelial differentiation. In another study, Pan and colleagues injected four patients with adenocarcinoma who were scheduled to undergo esophagectomy with IdU to label proliferative cells and collected esophageal squamous tissue at the time of surgical resection 7, 11, 29, and 67 days postinjection [33]. Using IHC for Ki-67, they found that proliferating cells were located within the basal epithelium of both interpapillary and papillary regions and should have incorporated IdU during the infusion. After 7 days, IdU positive squamous cells were observed within the basal layer as well as in differentiating cells transiting toward the lumen. By 29 and 67 days postinjection, LRC were rare and limited to basal epithelium. In agreement with studies by Barbera and colleagues [32], LRC were concentrated in the basal cell region overlying papillae and costained with pan-CK confirming an epithelial phenotype. In addition, these LRC were located in close proximity to Ki-67 positive proliferating cells suggesting that perhaps these LRC gave rise to proliferative Ki-67 positive cells .

Human esophagus (20×). Stromal papillae are indicated by the arrows. Basal cells found between two papillae are within the interpapillary basal cell layer or region (IBL). Basal cells overlying papillae are within the papillary basal cell layer or region (PBL) which is indicated by the curly brace
A conclusion drawn from these studies is that esophageal squamous cells likely have a hierarchy in terms of differentiation status but may not contain true stem cells. If squamous cells in the basal layer of the esophagus are the precursor cell pool then these cells could undergo transcommitment to give rise to Barrett’s esophagus . If, on the other hand, cells throughout the stratified cell layers as well as cells in the basal layer equally function as progenitor cells, then transdifferentiation would be functionally synonymous with transcommitment . Direct evidence of a squamous progenitor cell undergoing a full phenotypic conversion into a specialized intestinal metaplastic cell remains lacking. Lineage tracing experiments using reporter mice may resolve this in the future.
Native Esophageal Submucosal Gland or Duct Progenitor Cells
Submucosal glands are found in the human esophagus and are distributed throughout its entirety [34, 35]. A single submucosal gland consists of acini lined by simple columnar cells surrounded by myoepithelial cells and a single duct lined by cuboidal cells that transition into squamous cells as the duct reaches the esophageal lumen [35]. Acinar secretions include acidic and neutral mucins, bicarbonate, epidermal growth factor, and prostaglandins. Submucosal gland ducts have been observed in direct continuity with overlying squamous epithelium, MLE, Barrett’s epithelium, and neosquamous islands found within areas of Barrett’s esophagus [8, 34, 36] supporting the hypothesis that these glands harbor epithelial progenitor cells. The existence of progenitor cells within submucosal glands or their ducts was further supported by clonal studies tracking DNA mutations [37]. Using laser capture microdissection and DNA sequencing, Leedham and colleagues located a squamous island arising from a submucosal gland duct. While a surrounding area of Barrett’s metaplasia was P53 mutant, both the squamous island and the submucosal gland duct were P53 wild type. In another case, a silent point mutation in the second exon of the P16 gene was discovered in an area of Barrett’s metaplasia and the same P16 mutation was identified in an adjoining submucosal gland duct. These experiments strongly support the contention that cells within submucosal gland ducts can give rise to both squamous and Barrett’s epithelium .
In 1988, Gillen and colleagues reported the results of an insightful experiment [38]. These investigators assigned 25 mongrel dogs either to a control group or to one of four experimental groups. Animals in the four experimental groups underwent circumferential mucosal stripping of two separate 2 cm wide sections of squamous epithelium lining the distal esophagus separated by a 2 cm section of intact squamous epithelium. Animals in three of the four experimental groups then underwent creation of a hiatal hernia while animals in the fourth group did not. Of the animals which underwent hiatal hernia creation, one group was given pentagastrin daily to induce acid secretion, a second group underwent an additional biliary diversion and ligation of the common bile duct to the lesser curvature of the stomach to induce acid secretion and bile reflux, and a third group underwent biliary diversion and ligation of the common bile duct to the lesser curvature of the stomach followed by treatment with cimetidine, an H2 blocker which suppresses acid, twice daily postoperatively to induce exposure to bile salts only. After 3 months, the GEJ and distal esophagus were evaluated in all animals. Animals with a hiatal hernia which received pentagastrin (acid group) or those with a hiatal hernia and biliary diversion (acid and bile salt group) had columnar epithelialization in the lower stripped ring. Regenerating columnar epithelium contained both goblet and parietal cells. Animals with a hiatal hernia and biliary diversion treated with cimetidine (bile salt group) or those without a hiatal hernia (no reflux) reepithelialized only with squamous epithelium. Of the animals with a hiatal hernia treated with pentagastrin, two also developed columnar epithelium in the upper stripped ring. This led to the conclusion that the presence of columnar epithelium in the upper ring beyond an intact squamous barrier ruled out “creeping substitution” from the gastric cardia and that the source of columnar cells was submucosal gland ducts since ducts contiguous with esophageal surface ulcerations were observed .
Another study reported several years later also supported submucosal glands as the source of esophageal epithelial progenitor cells. Li and colleagues performed rectangular-shaped mucosal stripping of the distal esophagus in 12 mongrel dogs, leaving 1 cm of intact squamous epithelium entirely around the stripped area [39]. Sutures were placed at the four corners of the stripped area to demarcate it. The animals underwent creation of a hiatal hernia and then were treated with pentogastrin daily for 3 months. After 3 months, the regenerated epithelium within the boundaries of the sutures was stripped from ten surviving animals and analyzed. Seven animals were found to have columnar epithelium without evidence of squamous islands. No goblet cells were observed, but in most animals the columnar epithelium was contiguous with ducts of deep submucosal glands. Six of the ten animals then underwent surgical repair of the hiatal hernia followed by treatment with omeprazole for acid suppression. Three months later, the animals were sacrificed and the restripped epithelium within the four suture boundaries was analyzed. All six animals again had columnar metaplasia but now multiple squamous islands were found throughout the metaplastic epithelium. These squamous islands were contiguous with submucosal gland ducts.
More recently, rodents have become the preferred model system for studying Barrett’s pathogenesis through surgically induced reflux procedures such as esophagojejunostomy or esophagoduodenostomy. Investigators at Mayo Clinic have even developed a method to use magnets to induce fistula formation between the distal esophagus and small bowel [40]. Rat and mouse esophagi do not contain esophageal submucosal glands [35, 41], and thus in these animal models the source of metaplastic epithelium must be elsewhere .
Circulating Bone Marrow-Derived Stem Cells
In esophageal injury models, bone marrow-derived stem cells can migrate to the esophagus and reconstitute esophageal epithelium. Epperly and colleagues irradiated female C57BL/6 mice with 30 Gray (Gy) to the upper torso, inducing lethal radiation esophagitis, and then injected the female mice with GFP positive male whole bone marrow cells [23]. The female GFP negative mice were sacrificed 14–21 days later and the esophagi evaluated. After 14 days, GFP and Y chromosome positive foci were seen in esophageal squamous epithelium. These GFP positive foci further increased in size after 21 days. The cells were confirmed to be epithelial by hematoxylin and eosin (H&E) staining and pan-CK IHC.
Since the reflux of acid and bile salts injures the esophageal squamous epithelium in patients with GERD, bone marrow-derived cells might also migrate to the esophagus to repair GERD-induced injury. To model this, Sarosi and colleagues irradiated female Sprague-Dawley rats at 6 weeks of age with 900cGy followed by tail vein injection of bone marrow cells obtained from age-matched male rats [42]. Ten days later, the female rats were randomized to either esophagojejunostomy or a sham operation. The esophagi were then harvested and analyzed 8 weeks postoperatively. Four of ten animals who underwent esophagojejunostomy and nine of ten sham-operated animals survived until 8 weeks. Animals that underwent esophagojejunostomy were found to have loss of keratinization, papillary lengthening, basal cell hyperplasia, and mucosal ulceration in addition to intestinal metaplasia while sham-operated animals exhibited none of these features. Fluorescence in situ hybridization (FISH) demonstrated that Y chromosome containing cells gave rise to both squamous and metaplastic columnar epithelium as differentiated by squamous CK14 expression. Further, in sham-operated animals Y chromosome containing cells were also found in female esophageal epithelium, demonstrating that bone marrow-derived cells can contribute to esophageal epithelium even in the absence of injury.
In another study, Hutchinson and colleagues evaluated bile salt injury in 12 lethally irradiated C57BL/6 mice [43]. These mice subsequently received bone marrow transplants from Gt(ROSA)LacZ mice [44] and underwent esophagojejunostomy. Twelve mice survived until the end of the study and were evaluated after 20 weeks. One-third of the mice were found to have intestinal metaplasia with half of the metaplastic glands expressing β-galactosidase. The β-galactosidase expressing cells were found in groups of 3–4 and never comprised an entire gland. They were shown to be epithelial by costaining with E-cadherin. Interestingly, Kalabis and colleagues reported a similar experiment in FVB/N mice but did not perform esophagojejunostomy. Recipient FVB/N mice were irradiated with 12Gy and given the bone marrow of Gt(ROSA)-enhanced GFP mice. Though there was 95 % engraftment in the bone marrow, no GFP positive cells were found in the esophagus [25]. Esophageal injury, similar to that caused by reflux of bile salts, may be required to cause migration of bone marrow-derived stem cells to the mouse esophagus .
Hutchinson and colleagues also described the development of an esophageal carcinoma in a 45-year-old male who had received a bone marrow transplant at age 35 from his sister for M2 acute myeloid leukemia. He presented with dysphagia and an esophagogastroduodenoscopy (EGD) identified an 8 cm distal esophageal tumor. Biopsy revealed adenosquamous carcinoma, and X/Y chromosome FISH demonstrated that the tumor contained at least 6 % XX, 0Y cells. XX carcinoma cells were intermingled with Y containing cells and colocalized with E-cadherin. It was unclear whether the patient had gastroesophageal reflux or whether his tumor was due to a chronically immunosuppressed state as the patient was reported to have severe graft versus host disease.
These studies raise the possibility that human Barrett’s esophagus might originate from a circulating, multipotent bone marrow-derived stem cell. What is important to note is that bone marrow-derived cells did not give rise to complete glands in either rodents on in the case report of the patient with adenosquamous carcinoma. Though bone marrow-derived stem cells can contribute to esophageal epithelium, they may not be the sole source of metaplastic epithelium .
Proximally Shifting Columnar Progenitor Cells
As described earlier, experiments in mongrel dogs supported submucosal gland ducts as the source of Barrett’s epithelium. However, prior to those reports Bremner and colleagues came to a different conclusion in 1970 [45]. These investigators divided 35 mongrel dogs into three groups. All animals underwent stripping of the mucosa from the distal 6–10 cm of the esophagus and ending at the GEJ. Two groups of animals then underwent surgical destruction of the lower esophageal sphincter and creation of a hiatal hernia to induce reflux, with one of these groups treated between two and 12 weeks with histamine to enhance acid secretion. In animals that only underwent mucosal stripping, reepithelialization occurred quickly and was predominantly squamous, though in nine of ten animals columnar epithelium was also found in the distal esophagus. This would be consistent with a wound healing-like mechanism. In animals exposed to reflux, reepithelialization was much slower. Out of 14 animals who did not receive histamine, 12 animals had presence of columnar epithelium and squamous epithelium, with one animal having no squamous epithelium at all. In 11 animals who received histamine, one did not reepithelialize. In half of the remaining ten animals, only columnar epithelium was present. These investigators concluded that reepithelialization can occur with columnar epithelium, that columnar epithelium is favored in the setting of reflux, and this epithelium comes from gastric or junctional epithelium through “creeping substitution.” The finding of columnar metaplasia in dogs in the setting of gastroesophageal reflux was confirmed by others several years later [46].
More recent studies in support of the “creeping substitution” hypothesis have utilized mouse genetic models. As discussed before, the mouse esophagus differs from the human esophagus in that it lacks submucosal glands and stromal papillae and its epithelium is keratinized at the luminal surface (Fig. 10.2). Unlike in humans where the normal squamous epithelium-lined esophagus directly meets the columnar epithelium-lined gastric cardia at the GEJ, in mice the squamous epithelium-lined esophagus ends in a pouch at the junction of the squamous epithelium-lined forestomach and columnar-lined glandular stomach. The forestomach is separated from the glandular stomach through the rest of its circumference by a structure known as the limiting ridge (Fig. 10.3). The entire limiting ridge is covered by squamous epithelium, which transitions into columnar epithelium as the limiting ridge meets the glandular stomach. Thus, in humans the SCJ occurs at the GEJ while in mice the SCJ occurs at the distal end of the limiting ridge at the junction of the forestomach and glandular stomach.

Mouse esophagus (40×). Unlike the human esophagus, the mouse esophagus lacks submucosal glands and stromal papillae. The epithelium is keratinized at the luminal surface (marked with *). Basal cells are found attached to the basement membrane and are indicated by the arrows. Suprabasal cells, which are more differentiated, are indicated by the curly brace

The mouse squamocolumnar junction (20×). The squamous epithelium-lined forestomach (right) and the columnar epithelium-lined glandular stomach (left) are separated by the squamous epithelium-lined limiting ridge (LR). The transition from stratified squamous to columnar epithelium occurs where the limiting ridge meets the glandular stomach. The first gland of the gastric fundus is indicated by the box. The top of the gland reaches the luminal surface in close proximity to the CK7 positive, Dclk1 positive, Lgr5 positive cells (**) as described in Refs. [47, 52, 58]
Recent interest has focused on the distal limiting ridge as the putative source of Barrett’s epithelium. The first of several high profile studies found similarities between forestomach epithelium from p63−/− mouse embryos and Barrett’s epithelium as p63 is required for stem cell maintenance in stratified epithelium and is absent in human Barrett’s esophagus [47]. At E18, wild-type embryos had forestomachs covered by a stratified squamous epithelium while p63−/− embryos had forestomachs lined with columnar epithelium that secreted mucus. In addition, p63−/− epithelium had higher expression levels of Villin and Agr2, intestinal and mucin associated genes, respectively, that are expressed in human Barrett’s esophagus. Using gene expression microarrays (GEM) , 17 of the top 50 upregulated genes in the mutant p63 forestomach were found to be upregulated in two separate Barrett’s esophagus GEM datasets. Interestingly, the p63−/− forestomach did not express Cdx2, an intestinal transcription factor frequently found in Barrett’s epithelium. A time course examining epithelial development of the wild-type forestomach between E13 and E19 demonstrated that the forestomach was initially lined by a Carbonic anhydrase 4 (Car4) expressing monolayered epithelium. As embryogenesis progressed, these Car4 expressing epithelial cells were then pushed toward the lumen by a p63 positive epithelial cell population beginning at E13–14. This led to decreased proliferative capacity (measured by Ki-67 IHC) of the Car4 positive cells as they lost contact with the basement membrane. These investigators from the McKeon lab postulated that in p63−/− embryos, Car4 positive cells remained proliferative and in contact with the basement membrane and gave rise to columnar mucin secreting cells by E18. As embryogenesis continued in the wild-type mouse, Car4 cells expressed CK7 though not all CK7 positive cells expressed Car4. They then tracked CK7 cells during esophageal and forestomach development in wild-type embryos and discovered that this CK7 expressing epithelium was completely sloughed off except for approximately 30 cells which remained at the luminal surface at the SCJ at birth. In association with these remaining CK7 positive cells was an occasional Car4 expressing cell attached to the basement membrane. The remaining CK7 positive cells also expressed Muc4 and survived into adulthood as “residual embryonic cells.” Similar CK7, MUC4 positive cells were also found at the luminal surface of the human embryonic and adult SCJ. Induction of injury in CK14 positive squamous epithelium overlying the distal limiting ridge in 3-week-old wild-type mice using Diptheria toxin A (DTA), in an attempt to simulate acid-induced injury, caused CK7 positive epithelium at the SCJ to proximally shift and repopulate the distal limiting ridge . The theory that Barrett’s esophagus arises by proximal shifting of residual embryonic cells into voids left by damaged squamous epithelium was put forth. Over 50 years ago, Barrett’s epithelium was postulated to represent a persistence of embryonic esophageal epithelium in the distal esophagus that was not replaced during the transition to stratified squamous [48]. Thus, the renewed interest in this concept is clearly warranted .
This intriguing study by Wang and colleagues raises several important questions. First, can the human SCJ population of CK7, MUC4 expressing cells give rise to Barrett’s esophagus ? The expression of CK7 has been detected in both superficial and deep metaplastic glands in patients with Barrett’s esophagus [49]. If so, then the CK7 cells would need to proximally shift into the esophagus and undergo phenotypic and proliferative change (i.e., molecular reprogramming) to form a glandular tissue. Second, do CK7 positive cells and Car4 positive cells share similar characteristics and have the capacity to phenotypically change into intestinal cells (i.e., express Cdx2)? Though the mouse SCJ population of CK7 positive cells was able to proximally shift following injury to adjacent squamous epithelial cells, Car4 cells did not move. These long-term questions remained unanswered because mice could only be followed out to 10 days post-DTA induction due to collateral damage of nearby squamous epithelium. Third, if residual embryonic cells reside superficially at the SCJ, wouldn’t they have maximum exposure to acid and bile salts, the physiological components of gastric refluxate? Conceivably as a resident stem cell, CK7 positive cells located at the luminal surface might be more resistant to reflux-induced injury. As the investigators noted in a later review, their work did not directly address how Barrett’s metaplasia occurs in surgical rodent models of bile reflux (esophagojejunostomy or esophagoduodenostomy) in which the SCJ region is bypassed [50]. They did suggest that residual embryonic cells in other parts of the GI tract could potentially give rise to Barrett’s metaplasia in these surgical models and that the validity of their theory depends on identification of these other populations of residual embryonic cells .
The second study examined the phenotype of mice overexpressing human IL-1β in stratified squamous epithelial cells of the oral cavity, esophagus, and forestomach in which the ED-L2 (an Epstein–Barr virus) promoter is activated [51]. These animals developed a systemic inflammatory reaction as exhibited by splenomegaly; increased serum levels of IL-1β, TNFα, and IL-6; and acute and chronic inflammatory infiltrates in the esophagus and forestomach [52]. At 6 months of age, these mice developed epithelial hyperplasia at the SCJ . By 12–15 months of age, these mice developed columnar metaplasia without goblet cells at the SCJ with Muc5ac, Cdx2, and Tff2 (a marker for spasmolytic polypeptide-expressing metaplasia or SPEM) expressing cells. Between 20 and 22 months, 20 % of the animals developed high-grade dysplasia or intramucosal adenocarcinoma at the SCJ. In adjacent stroma, an increase in myofibroblasts and global hypomethylation was seen, consistent with a stromal role in tumorigenesis. Molecularly, there was increased Wnt (indicated by nuclear β-catenin), Notch, Sonic hedgehog (Shh), Bone morphogenetic protein 4 (Bmp4), and Akt signaling with occasional loss of p16. While acidified water (pH 2.0) did not accelerate the metaplasia time course, adding the unconjugated bile acid deoxycholate to drinking water led to increased inflammatory infiltrates and development of SCJ metaplasia earlier at 9 months and higher degrees of dysplasia at 12 and 15 months with some mice developing tumors at 15 months. Adding the carcinogen N-methyl-N-nitrosurea to deoxycholate led to SCJ tumor development at 12 months. GEM analysis of the mouse SCJ metaplastic lesions in 15-month-old EDL2-IL-1β and 9-month-old EDL2-IL-1β + bile acid-treated mice revealed overlap of 609 genes with human Barrett’s esophagus . Finally, Quante and colleagues demonstrated that the metaplasia induced by IL-1β overexpression led to an expansion of Lgr5 and Dclk1 positive stem cells. This is relevant as LGR5 and DCLK1 positive cells have been detected in human Barrett’s metaplasia [53, 54]. The location of the Lgr5 cells appeared to overlap with the CK7 positive cell population reported by Wang and colleagues [47].
The third study examined the phenotype of mice overexpressing Bmp4 in CK14 expressing basal squamous epithelium in the esophagus and forestomach. Epithelial Bmp4 overexpression upregulated stromal expression of the Bmp4 inhibitor Noggin, with higher levels of Noggin noted in the proximal esophagus as compared to the forestomach. At 20 weeks, mice were found to have metaplastic glands at the SCJ which expressed phosphorylated Smads 1/5/8, transcription factors that mediate downstream Bmp signaling. A subset of cells in these glands stained for squamous markers such as CK5, CK14, and the squamous stem cell marker p63, whereas other cells stained with columnar markers CK8, CK19, and the intestinal and gastric cardia stem cell marker Lgr5. The glands also contained mucus cells and Tff2 positive cells but did not express Cdx2, Muc2, or Muc5ac, thus resembling cardia-type metaplasia [55]. Twelve weeks after an esophagojejunostomy established with a microsurgical technique using magnets in wild type mice, metaplasia developed at the neo-SCJ in the setting of inflammation. The metaplasia resembled human cardia epithelium with a few Cdx2 expressing cells, but no cells exhibited expression of Muc2. Later at 16 weeks, the metaplastic glands expressed both Cdx2 and Muc2. In vitro experiments further demonstrated that coexpression of Bmp4 and Cdx2 was required to induce Muc2 expression and that a Smad/Cdx2 transcriptional complex was necessary to transactivate Muc2. These findings suggested that the nonintestinal, cardia type of metaplasia evolved over time to an intestinal type of metaplasia in the setting of reflux induced injury. In fact, indirect evidence supports this in human patients [55–57].
Additional insight into the mouse gastric cardia was provided by a fourth study in which mice null for Smad3, a mediator of TGF-β signaling, developed tumors at the SCJ [58]. At 6 months, these mice had grossly exophytic growths on the lesser curvature of their stomachs. Histologically, metaplastic glands with cells expressing Tff2 were observed just distal to the limiting ridge . At 10 months, these metaplastic gastric glands exhibited dysplastic changes with increased expression of Ki-67 and phosphorylated Stat3, a key mediator of inflammation and cancer. In addition, some cystic structures lined by metaplastic epithelium invaded into submucosal and muscle tissue. In the normal fundus of wild-type mice, these investigators found Dclk1 positive cells in the first gland of the gastric fundus just distal to the limiting ridge , at the junction of squamous and columnar epithelium. In the Smad3−/− mice, Dclk1 positive cells were expanded in their normal location and were seen in invasive and noninvasive metaplastic glands. Thus, the first gland of the gastric fundus may be the origin of gastric metaplasia as well as tumorigenesis in this model. Interestingly, the location of the Dclk1 positive cells appeared to be similar to the Lgr5 cells reported by Quante and colleagues [52].
While Wang and colleagues demonstrated a proximal shift of CK7 positive columnar cells into an area previously inhabited by squamous epithelium, the other studies did not demonstrate movement of metaplastic glandular tissue into the esophagus. This may be because the immediately adjacent proximal squamous epithelium was not injured in the latter studies. It remains to be seen if the metaplastic glands reported in EDL2-IL-1β, CK14-Bmp4, or Smad3−/− mice would proximally shift in the setting of injury to adjacent squamous epithelium .
Transcommitment
Transcommitment , which refers to molecular reprogramming of stem or progenitor cells, is almost certainly required for the development of Barrett’s epithelium because some type of phenotypic change is required to generate specialized intestinal metaplasia regardless of the Barrett’s esophagus cell or tissue of origin. For example, Barrett’s epithelium can contain Paneth cells, enteroendocrine cells, cells that resemble jejunal absorptive cells, and mature goblet cells [59–61], cell types that are found in both gastric and intestinal tissues. Further, proximal shifting of either residual embryonic cells at the SCJ or gastric cardia does not explain how patients who have undergone partial esophagectomy with esophagogastric anastomosis can develop columnar metaplasia in the residual esophagus [62, 63]. In these patients the anastomosis of oxyntic stomach to squamous cervical esophagus removes both the SCJ and gastric cardia [62]. Finally, following ablation of Barrett’s esophagus, reepithelialization by squamous cells is favored in the setting of acid suppression whereas recurrent Barrett’s epithelium is favored when acid suppression is inadequate suggesting that the progenitor cell has the capacity to differentiate into either a squamous or columnar cell phenotype depending on the local esophageal environment [64, 65].
Transcommitment could explain how progenitor cells found in esophageal submucosal glands or their ducts give rise to multiple phenotypes. Using endoscopic mucosal resection (EMR) or esophagectomy specimens and mitochondrial DNA mutation analysis in cytochrome c oxidase deficient cells, Nicholson and colleagues demonstrated that esophageal submucosal glands are made up of clonal units [66]. More importantly, they went on to show in a specimen of Barrett’s metaplasia from a patient who had undergone ablative therapy that the metaplastic glands and overlying neosquamous epithelium shared the same mitochondrial DNA mutation, suggesting they arose from the same progenitor cell.
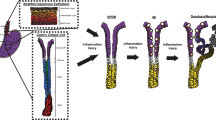
Conceptually, an epithelial progenitor cell that gives rise to Barrett’s esophagus , regardless of origin, would need to acquire or maintain a columnar phenotype, undergo intestinalization, and secrete mucus as goblet cells are the sine qua non of specialized intestinal metaplasia (Fig. 10.4). Switching from a squamous to a columnar phenotype would require the ability to activate transcription factors that characterize columnar cells (i.e., Sox9) and/or downregulate transcription factors that characterize squamous cells (i.e., Sox2 and p63) [13, 67]. This would need to be followed by activation of intestinal (i.e., Cdx1 and Cdx2) and mucin associated transcription factors (i.e., Foxa2) [16, 68].

Transcommitment model for Barrett’s Esophagus. Following acid and bile injury and/or resultant inflammation, a squamous progenitor cell could upregulate the columnar transcription factor SOX9 and downregulate squamous transcription factors SOX2 and P63 to convert into a columnar cell. This metaplastic columnar cell could then upregulate intestinal transcription factors CDX1 and CDX2 to become an intestinal cell or upregulate the mucin associated transcription factor FOXA2 to become a mucin secreting goblet cell. To become a specialized intestinal metaplastic cell, the intestinalized columnar cell may become an intestinalized goblet cell or the goblet cell could become intestinalized
Sox9, a member of the SOX gene family, was first identified in the GI tract within proliferative cells of intestinal crypts as well as Paneth cells [69]. Sox9 is expressed in the CLE during embryogenesis along with CK8 and CK18, but Sox9 expression is lost when the epithelium becomes squamous [13]. To determine whether SOX9 may have functional relevance to the development of Barrett’s esophagus , Wang and colleagues performed SOX9 IHC on esophageal tissue microarrays representing 96 esophagectomy cases containing Barrett’s esophagus and/or esophageal adenocarcinoma. Nuclear SOX9 was expressed in 100 % of patients with Barrett’s epithelium and 85 % of patients with esophageal adenocarcinoma but not in adjacent squamous epithelium [13]. SOX9 was activated in Barrett’s epithelium through acid and bile-induced Hedgehog ligand secretion by epithelial cells that in turn activated BMP4 secretion by adjacent stromal cells. This stromal BMP4 then acted back on the epithelium to induce SOX9 expression . Though Sox9 is a Hedgehog target gene in chondrocytes and the skin [70, 71] and it has a distant enhancer region containing a Gli1 binding site [72], SOX9 expression could not be directly induced by Hedgehog pathway activation in human esophageal epithelial cells. Instead, treatment of esophageal squamous HET-1A cells with human recombinant BMP4 or transfection with a constitutively active form of the BMP type I receptor BMPRIA led to increased mRNA expression of SOX9 by quantitative real-time PCR (qPCR). Overexpression of either SOX9 or constitutively active BMPRIA in HET-1A cells led to expression of columnar CK8/18 [13]. In an in vivo transplant culture system, esophageal epithelial cells from a Shh transgenic mouse induced stromal Bmp4 and epithelial Sox9 expression, establishing that SOX9 is a target of Hedgehog-BMP4 signaling in Barrett’s esophagus [13]. Using wild type C57BL/6 mouse esophageal epithelium in the same in vivo transplant culture system, Clemons and colleagues found that retroviral transduction of Sox9 induced expression of columnar CK8 and of the intestinal glycoprotein A33 and changed the stratified squamous epithelium into one to two layers of cuboidal or columnar shaped epithelial cells [73]. In contrast, retroviral transduction of Cdx2 did not alter squamous differentiation or induce either columnar or intestinal gene expression. These data demonstrated that Sox9 expression in esophageal squamous epithelial cells induced markers and morphological changes characteristic of a columnar phenotype.
Sox2, another member of the SOX family of transcription factors, is expressed in the embryonic esophagus where its presumed role is to regulate endoderm differentiation into stratified squamous epithelium [74]. In Sox2 hypomorphic mice, the esophageal epithelium was observed to be thinner, characterized by multilayered columnar cells, and expressed mucin. There was also decreased expression of both p63 and CK14. Sox2 overexpression in the mouse intestine using a conditional Villin promoter led to loss of villi, appearance of p63 expressing basal cells (characteristic of the forestomach and esophagus), and decreased binding of Cdx2 to the promoters of its target genes [75]. A role in squamous differentiation was further supported by the findings of increased SOX2 expression in squamous cancers of the GI tract and lung and that SOX2 and P63 colocalized on genes required for squamous cell carcinoma growth [67, 76, 77]. In addition to its role as a squamous differentiation factor, Sox2 also plays a role in stem cell maintenance. In mice with thymidine kinase (TK) inserted into the endogenous Sox2 locus, treatment with ganciclovir over 2 weeks led to loss of Sox2 expressing esophageal basal cells but maintenance of suprabasal epithelium; these epithelial changes were reversible with a shorter duration of ganciclovir treatment , suggesting that this shorter exposure allowed some Sox2 positive basal cells to survive [78]. Overexpression of Sox2 in the basal epithelium of the mouse esophagus using a conditional CK5 promoter caused basal cell hyperplasia [79]. Combining Sox2 overexpression in esophageal basal epithelial cells with constitutive activation of Stat3 using a Lentiviral construct, led to formation of squamous cell carcinomas. These findings demonstrated that Sox2 is an essential factor for squamous cell differentiation and tissue maintenance in the esophagus. In the normal adult esophagus of both rodents and humans, Sox2 is expressed in the basal cells of the stratified squamous epithelium. In contrast, Sox2 is not expressed in MLE or intestinal metaplasia of the esophagus. Thus, downregulation of Sox2 may be required to reprogram esophageal progenitor cells into Barrett’s epithelium [9].
P63 is a member of the P53 family of transcription factors and has six isoforms [80]. Three full length proteins, known as TAp63, contain an amino terminal transactivation domain. Three proteins transcribed from an alternate promoter in the third intron, known as ΔNp63, do not contain the transactivation domain but retain the carboxyl terminal DNA binding domain. Because of alternative splicing at the C-terminus, there are three forms each of TAp63 and ΔNp63 designated as α, β, and γ. Given that mice null for p63 completely lacked stratified squamous epithelium and have esophagi lined by simple columnar epithelium [81], it is likely that downregulation of p63 also plays a role in the formation of Barrett’s metaplasia . As described earlier, p63−/− mice developed a Barrett’s like metaplasia in the forestomach [47]. Multiple studies have examined P63 expression in esophageal squamous epithelium, esophageal squamous cell carcinoma, Barrett’s esophagus , and esophageal adenocarcinoma. Using an antibody against the carboxyl terminus of P63, which recognized all 6 isoforms, one group of investigators reported moderate to strong P63 expression in esophageal squamous epithelium, absent to moderate expression in Barrett’s esophagus, and high expression in Barrett’s esophagus with high-grade dysplasia and esophageal adenocarcinoma [82]. Other investigators performed IHC with the commonly used 4A4 antibody followed by RT-PCR using primers specific for TAp63 and ΔNp63 [83]. They found that esophageal squamous epithelium and esophageal squamous cell carcinoma strongly expressed P63, while Barrett’s esophagus and esophageal adenocarcinoma did not. ΔNp63 was the predominant isoform expressed by esophageal epithelium. Finally, a third group used the 4A4 antibody as well as an antibody that specifically recognized ΔNp63 [84]. They found that ΔNp63 was strongly expressed by esophageal squamous epithelium and esophageal squamous carcinomas while Barrett’s epithelium and esophageal adenocarcinoma rarely expressed P63. Adenocarcinomas that did express P63 expressed the TA isoforms in 63 % of cases. Although these studies had somewhat conflicting results, it appeared that ΔNp63 is the predominant form of p63 in the normal esophagus and that it is required for squamous differentiation. In addition, Barrett’s esophagus without dysplasia is not likely to express P63 while adenocarcinomas may weakly express P63, favoring TAp63 isoforms. Another study examined the effect of bile acids and acidified media on P63 expression in primary esophageal epithelial cells and in esophageal squamous cell carcinoma cell lines [85]. It was found that the predominant P63 isoform expressed was ΔNp63α and that ΔNp63α expression could be synergistically repressed with deoxycholic acid and acidic media (pH 5) in a squamous carcinoma cell line. In primary esophageal epithelium, ΔNp63α repression was mostly mediated by the bile salt with minimal additive effect from acidified media. Taken together, these data suggested that bile salts in patients with GERD can suppress expression of ΔNp63 in the squamous-lined esophagus leading to reprogramming of esophageal progenitor cells and the development of Barrett’s esophagus.
Following the acquisition and maintenance of a columnar phenotype, the progenitor cell still must undergo intestinalization to generate specialized intestinal metaplasia characterizing Barrett’s esophagus . Cdx1 and Cdx2 are members of the caudal related homeobox gene family which are expressed in the intestine [86]. Cdx1 is expressed in the proliferative crypt compartment, while Cdx2 is expressed in the differentiated villus compartment [87]. Cdx1 is expressed in two waves during embryogenesis, initially between E7.5 and 12.5 in the ectoderm and mesoderm. The second wave begins at E12.5 in the gut endoderm and continues through adulthood [87]. Cdx1 knockout mice had anterior homeotic shift of the axial skeleton but no known gut phenotype [88]. Targeting Cdx1 expression to gastric parietal cells using a rat H/K-ATPase promoter, investigators found Cdx1 transgenic mice developed intestinal metaplasia of the gastric epithelium with all four cell types of the adult colon represented including enterocytes, Paneth cells, goblet cells, and enteroendocrine cells [89]. This is consistent with Cdx1 reprogramming columnar progenitors into intestinal columnar cells. CDX1 mRNA has been found in Barrett’s metaplastic tissue, but not in normal esophageal squamous tissue [90]. By bisulfite sequencing, the CDX1 promoter was found to be methylated and silenced in squamous epithelium but demethylated and active in Barrett’s epithelium. Treatment of the esophageal squamous cell carcinoma cell line OE21 with 5-azacitadine, a demethylating agent, led to CDX1 expression. NFk-B, TNF-α, and IL-1β (proinflammatory cytokines elevated in reflux esophagitis) were all able to induce CDX1 expression in the colorectal cell line C32. Further, exposure to bile salts or acidified bile salts led to CDX1 expression in C32 cells while exposure to acid alone did not. Investigators in the Kinoshita lab performed esophagojejunostomy in Wistar rats using the Levrat procedure [68]. Seven weeks postoperatively, classic features of reflux esophagitis such as basal cell hyperplasia and papillary lengthening were observed. Cdx1 nuclear staining was seen in squamous epithelium above the anastomosis. At 6 months, columnar metaplasia with goblet cells had arisen and expressed Cdx1. This colocalized with Cdx2 expression within metaplastic Barrett’s epithelium. In vitro, bile salts activated CDX1 promoter luciferase activity in HET-1A and esophageal adenocarcinoma OE33 cells and induced CDX1 protein expression in primary cultured esophageal keratinocytes. Overexpression of Cdx1 in HET-1A cells caused expression of MUC2. Finally, these investigators demonstrated that Cdx1 and Cdx2 can autoregulate their own expression and the expression of each other, establishing a positive feed forward intestinalization loop .
By IHC, CDX2 expression has been found in 100 % of biopsy samples from nondysplastic and dysplastic Barrett’s metaplasia and esophageal adenocarcinoma [91, 92]. CDX2 expression has been found in inflamed esophageal squamous epithelium of GERD patients, but not in normal noninflamed esophageal epithelium [93]. In another study, CDX2 expression was found in esophageal squamous epithelium in up to one third of GERD patients with Barrett’s esophagus , but not in any samples of esophageal squamous epithelium from GERD patients without Barrett’s esophagus [94]. We showed that human esophageal squamous epithelial cells from GERD patients with Barrett’s esophagus differentially respond to acid and bile salt exposure by upregulating CDX2 as compared to human esophageal squamous epithelial cells from GERD patients without Barrett’s esophagus [17]. These findings suggest that CDX2 may be involved in reprogramming esophageal progenitor cells in those patients with GERD that develop Barrett’s esophagus. Like Sox9, Cdx2 is expressed early in the embryonic gut [87]. Unlike Sox9, which is expressed in the embryonic esophagus, Cdx2 is expressed from the duodenum through the gut distally beginning as early as E12.5 [95]. Some insight into its function can be obtained from knockout or overexpression studies in mice. Cdx2 homozygote knockout mice died at E3.5 because of an implantation defect in the trophectoderm [96]. Cdx2 heterozygote mice have been found to develop multiple intestinal adenomatous polyps. Interestingly, keratinized squamous epithelial metaplasia , resembling esophagus or forestomach, was found within these adenomas [96]. Conditional knockout of Cdx2 using the Villin intestinal promoter led to loss of microvilli and intestinal epithelium expressing squamous genes such as p63 and Sox2 [97]. While Cdx2 overexpression in the murine stomach led to intestinal metaplasia [98], Cdx2 overexpression in the murine esophagus using the squamous CK14 promoter did not lead to macroscopic changes of intestinal metaplasia [11]. In vitro, acid induced Cdx2 expression in cultured murine keratinocytes [99]. Additional insight was obtained from CDX2 expression studies in human cells. CDX2 overexpression in vitro in human HET-1A cells led to gland formation [12]. Cdx2’s transcriptional targets include intestinal genes such as sucrase-isomaltase [100], MUC2 [101], CK20 [12], Villin [12], and CDX1 [97]. In esophageal EPC2 cells, treatment with the demethylating agent DAC was required for CDX2 to transactivate its target genes. Collectively these data support a simplistic view that CDX2 is insufficient to induce an intestinal phenotype in squamous cells (unless changes in methylation states or other epigenetic changes are induced), while CDX2 can induce intestinal metaplasia in columnar cells. Within the intestine Cdx2 is a major transcriptional activator and thus, Cdx2 loss and resultant loss of its downstream target genes seem to cause a reprogramming of intestinal progenitor cells into squamous cells .
To identify additional proteins that may participate in the pathogenesis of Barrett’s esophagus , we performed GEM analysis of RNA isolated from whole esophagus of C57BL/6 embryos at E12.5 versus postnatal day (P)1 pups [16]. These timepoints were chosen to compare gene expression in simple columnar epithelium in the former and stratified squamous epithelium in the latter. From this microarray analysis, we identified another Hedgehog target gene, the transcription factor Foxa2 (Hnf3β), as having a similar esophageal developmental expression pattern as Sox9 [102, 103]. Foxa2 is expressed within the embryonic CLE but not in the adult squamous-lined esophagus [104]. On esophageal tissue microarrays, we found nuclear expression of FOXA2 in Barrett’s epithelium but not in normal esophageal squamous epithelium. Further, we found increased expression of FOXA2 mRNA by qPCR in tissue samples from six cases of Barrett’s esophagus. We also found FOXA2 expression by Western blot in telomerase-immortalized Barrett’s epithelial cell lines (BAR-T cells) but not in telomerase-immortalized squamous epithelial cell lines from patients with GERD with (NES-BT) or without (NES-GT) Barrett’s esophagus [17, 105]. These telomerase immortalized cells showed no signs of tumorigenesis, altered differentiation, or dysregulation of cell proliferation [105–108]. Since NES-BT cells did not express FOXA2, we electroporated them with plasmids containing Shh, Gli1 (a Hedgehog pathway transcriptional activator), and constitutively active BMPRIA. Consistent with prior reports [103, 109], we found that Shh and Gli1 induced FOXA2 expression. We also found that Hedgehog signaling activated FOXA2 via a Gli dependent enhancer found 3′ to its coding region and that FOXA2 expression in BAR-T cells was decreased following GLI1 siRNA mediated knockdown. In an in vivo transplant culture system, Foxa2 expression was only seen in cultures made from esophageal epithelium from activated Shh transgenic mice. No Foxa2 expression was seen in cultures made from wild type esophageal epithelium. Thus, expression of Shh in mouse esophageal epithelium led to stromal expression of Bmp4 and epithelial expression of Sox9, Foxa2, and columnar CK 8/18 in our in vivo transplant culture system [13, 16]. Foxa2 has been demonstrated to transcriptionally regulate expression of MUC2, the mucin specifically expressed by intestinal epithelium and found in Barrett’s esophagus [110, 111]. Proper processing of mature MUC2 protein is regulated by AGR2, a protein disulfide isomerase localized to the endoplasmic reticulum [112]. Analysis of Agr2 knockout mice revealed that they have decreased intestinal mucus and lack morphologically normal goblet cells [113]. As expected, these mice lacked mature Muc2 protein but express Muc2 transcript within their intestine. We examined expression of MUC2 mRNA and protein in NES-B3T and NES-B10T cells and found these cells expressed little to no MUC2 mRNA and no protein. Overexpression of FOXA2 in both squamous cell lines led to expression of both MUC2 mRNA and protein. Further, we found that FOXA2 expression in NES-B3T and NES-B10T cells led to expression of AGR2 mRNA and protein. siRNA mediated knockdown of FOXA2 in both BAR-T and BAR-10T cells led to decreased expression of AGR2 mRNA and protein. Together, these data suggested that FOXA2 induced production of intestinal mucus. It does this through presumed transcriptional regulation of MUC2 itself and of AGR2, which is required for proper processing of the MUC2 protein. Though FOXA2 expression led to MUC2 protein expression, the cells did not acquire a full goblet cell phenotype. It is likely that other factors may be required in addition to FOXA2 to induce a goblet cell phenotype. Based on data from esophageal development, these other factors could include downregulation of SOX2 and P63. Similar to Noggin null mice, in which Bmp4 signaling is unopposed, Sox2 null or p63 null mouse embryos have esophagi with columnar epithelium containing goblet-like cells [47, 74]. Notch pathway modulation may also be required as treatment with gamma secretase inhibitors and the resultant loss of Notch signaling in a surgical model of reflux esophagitis and Barrett’s metaplasia led to almost a complete conversion of metaplastic epithelial cells to differentiated goblet cells [114]. Furthermore, the loss of Notch signaling would increase expression of ATOH1 , a Notch pathway component which can also regulate MUC2 [115].
Conclusions
Based on available data generated from human patients, dog and rodent models of surgically induced reflux esophagitis, and various cell culture systems, it can be concluded that cells that give rise to Barrett’s esophagus can come from multiple tissue sources and most likely undergo transcommitment . Transdifferentiation is unlikely because a nonproliferating differentiated squamous cell could not sustain Barrett’s esophagus tissue indefinitely and full phenotypic conversion of a cultured squamous cell has not yet been demonstrated. Esophageal squamous epithelial progenitor cells that retain the embryonic capacity to switch between squamous and columnar phenotype must still undergo molecular reprogramming to generate specialized intestinal metaplasia . Submucosal glands or their ducts have been shown to be contiguous with normal squamous, Barrett’s, MLE, and regenerating neosquamous epithelium in human patients and in dogs which have undergone reflux surgery. More convincingly, mutational analysis of P53 and P16 has shown that the same mutation present in a submucosal gland duct is also present in either overlying Barrett’s or squamous epithelium. Progenitor cells in the submucosal glands or their ducts would be native to the esophagus, but would have to move out of the glands and ducts into the esophageal epithelium, and undergo molecular reprogramming to give rise to specialized intestinal metaplasia . Circulating bone marrow-derived stem cells have been shown to migrate to the esophagus and regenerate epithelium following injury induced by radiation, surgically induced reflux, or bone marrow transplant preparative regimens in mice, rats, and humans. Residual embryonic cells at the SCJ in mice have been shown to proximally shift to repair immediately adjacent squamous epithelium injured with DTA. A similar mechanism is supported by proximal shifting of gastric cardia cells giving rise to columnar epithelium in dogs as reported by Bremner and colleagues and suggested by the EDL2-IL-1β mouse model. Circulating bone marrow-derived cells, residual embryonic cells at the SCJ, and gastric cardia cells would all have to undergo molecular reprogramming to give rise to specialized intestinal metaplasia . In human patients, data exists to support each of the currently proposed hypotheses and as of yet none can be completely excluded in an individual patient.
Objections to some of these possible sources of Barrett’s esophagus include lack of submucosal glands in rodents that develop Barrett’s metaplasia from surgically induced reflux, the belief that bone marrow-derived cells do not migrate to the esophagus in the absence of major injury or the finding that they only partially contribute to glands, and the absence of definitive evidence in humans of either transdifferentiation of squamous epithelium or proximal shifting of SCJ or gastric cardia cells. Moreover, these data suggest that sources for the cell or tissue of origin of Barrett’s esophagus may vary depending on the species. For example, there is great interest in recently reported transgenic and knockout mouse models and the resulting metaplasia found at the SCJ in many of these mice. Further studies will likely show that the first gland of the gastric fundus, found at the junction of the forestomach and glandular stomach, serves as a reservoir of multipotent progenitors in mice similar to esophageal submucosal glands or ducts in humans that can give rise to both squamous and columnar epithelium. This notion is supported by the finding of MLE, characterized by both squamous and columnar-like cells in CK14-Bmp4 mice.
We speculate that human Barrett’s metaplasia can arise from progenitor cells that originate in the esophagus, circulate in the bloodstream, or proximally shift from the SCJ or gastric cardia to fill in voids left by injured squamous epithelium. Regardless of their origin, these progenitors must then undergo transcommitment through molecular reprogramming of the expression levels of different combinations of transcription factors to give rise to the specialized intestinal metaplasia characteristic of Barrett’s esophagus . Most likely, progenitor cells give rise initially to epithelial cells with biphenotypic potential (such as seen in MLE), followed by columnar differentiation, intestinalization, and for some mucus differentiation. Sequential activation or knockdown of a logical sequence of transcription factors in human cells in novel cell culture systems or in the appropriate animal model in the future may shed further insight into how Barrett’s metaplasia develops in patients with GERD.
References
Spechler SJ, Souza RF. Barrett’s esophagus. N Engl J Med. 2014;371(9):836–45.
Spechler SJ, Sharma P, Souza RF, Inadomi JM, Shaheen NJ. American Gastroenterological Association technical review on the management of Barrett’s esophagus. Gastroenterology. 2011;140(3):e18–52. quiz e13.
Li WC, Yu WY, Quinlan JM, Burke ZD, Tosh D. The molecular basis of transdifferentiation. J Cell Mol Med. 2005;9(3):569–82.
Cotton CC, Wolf WA, Pasricha S, Li N, Madanick RD, Spacek MB, Ferrell K, Dellon ES, Shaheen NJ. Recurrent intestinal metaplasia after radiofrequency ablation for Barrett’s esophagus: endoscopic findings and anatomic location. Gastrointest Endosc. 2015;81(6):1362–9.
Shields HM, Zwas F, Antonioli DA, Doos WG, Kim S, Spechler SJ. Detection by scanning electron microscopy of a distinctive esophageal surface cell at the junction of squamous and Barrett’s epithelium. Dig Dis Sci. 1993;38(1):97–108.
Sawhney RA, Shields HM, Allan CH, Boch JA, Trier JS, Antonioli DA. Morphological characterization of the squamocolumnar junction of the esophagus in patients with and without Barrett’s epithelium. Dig Dis Sci. 1996;41(6):1088–98.
Boch JA, Shields HM, Antonioli DA, Zwas F, Sawhney RA, Trier JS. Distribution of cytokeratin markers in Barrett’s specialized columnar epithelium. Gastroenterology. 1997;112(3):760–5.
Glickman JN, Chen YY, Wang HH, Antonioli DA, Odze RD. Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett’s esophagus. Am J Surg Pathol. 2001;25(5):569–78.
Chen X, Qin R, Liu B, Ma Y, Su Y, Yang CS, Glickman JN, Odze RD, Shaheen NJ. Multilayered epithelium in a rat model and human Barrett’s esophagus: similar expression patterns of transcription factors and differentiation markers. BMC Gastroenterol. 2008;8:1.
Su Y, Chen X, Klein M, Fang M, Wang S, Yang CS, Goyal RK. Phenotype of columnar-lined esophagus in rats with esophagogastroduodenal anastomosis: similarity to human Barrett’s esophagus. Lab Invest. 2004;84(6):753–65.
Kong J, Crissey MA, Funakoshi S, Kreindler JL, Lynch JP. Ectopic Cdx2 expression in murine esophagus models an intermediate stage in the emergence of Barrett’s esophagus. PLoS One. 2011;6(4):e18280.
Liu T, Zhang X, So CK, Wang S, Wang P, Yan L, Myers R, Chen Z, Patterson AP, Yang CS, et al. Regulation of Cdx2 expression by promoter methylation, and effects of Cdx2 transfection on morphology and gene expression of human esophageal epithelial cells. Carcinogenesis. 2007;28(2):488–96.
Wang DH, Clemons NJ, Miyashita T, Dupuy AJ, Zhang W, Szczepny A, Corcoran-Schwartz IM, Wilburn DL, Montgomery EA, Wang JS, et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett’s metaplasia. Gastroenterology. 2010;138(5):1810–22.
Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, Lynch JP, Rustgi AK. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett’s esophagus. PLoS One. 2008;3(10):e3534.
Kong J, Nakagawa H, Isariyawongse BK, Funakoshi S, Silberg DG, Rustgi AK, Lynch JP. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis. 2009;30(1):122–30.
Wang DH, Tiwari A, Kim ME, Clemons NJ, Regmi NL, Hodges WA, Berman DM, Montgomery EA, Watkins DN, Zhang X, et al. Hedgehog signaling regulates FOXA2 in esophageal embryogenesis and Barrett’s metaplasia. J Clin Invest. 2014;124(9):3767–80.
Huo X, Zhang HY, Zhang XI, Lynch JP, Strauch ED, Wang JY, Melton SD, Genta RM, Wang DH, Spechler SJ, et al. Acid and bile salt-induced CDX2 expression differs in esophageal squamous cells from patients with and without Barrett’s esophagus. Gastroenterology. 2010;139(1):194–203. e1.
De Hertogh G, Van Eyken P, Ectors N, Geboes K. On the origin of cardiac mucosa: a histological and immunohistochemical study of cytokeratin expression patterns in the developing esophagogastric junction region and stomach. World J Gastroenterol. 2005;11(29):4490–6.
DeNardi FG, Riddell RH. The normal esophagus. Am J Surg Pathol. 1991;15(3):296–309.
Raymond C, Anne V, Millane G. Development of esophageal epithelium in the fetal and neonatal mouse. Anat Rec. 1991;230(2):225–34.
Duan H, Gao F, Li S, Nagata T. Postnatal development and aging of esophageal epithelium in mouse: a light and electron microscopic radioautographic study. Cell Mol Biol (Noisy-le-Grand). 1993;39(3):309–16.
Yu WY, Slack JM, Tosh D. Conversion of columnar to stratified squamous epithelium in the developing mouse oesophagus. Dev Biol. 2005;284(1):157–70.
Epperly MW, Guo H, Shen H, Niu Y, Zhang X, Jefferson M, Sikora CA, Greenberger JS. Bone marrow origin of cells with capacity for homing and differentiation to esophageal squamous epithelium. Radiat Res. 2004;162(3):233–40.
Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183(4):1797–806.
Kalabis J, Oyama K, Okawa T, Nakagawa H, Michaylira CZ, Stairs DB, Figueiredo JL, Mahmood U, Diehl JA, Herlyn M, et al. A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. J Clin Invest. 2008;118(12):3860–9.
Croagh D, Phillips WA, Redvers R, Thomas RJ, Kaur P. Identification of candidate murine esophageal stem cells using a combination of cell kinetic studies and cell surface markers. Stem Cells. 2007;25(2):313–8.
DeWard AD, Cramer J, Lagasse E. Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep. 2014;9(2):701–11.
Little MH, Takasato M. Generating a self-organizing kidney from pluripotent cells. Curr Opin Organ Transplant. 2015;20(2):178–86.
Doupe DP, Alcolea MP, Roshan A, Zhang G, Klein AM, Simons BD, Jones PH. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science. 2012;337(6098):1091–3.
Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126(5):1236–46.
Seery JP, Watt FM. Asymmetric stem-cell divisions define the architecture of human oesophageal epithelium. Curr Biol. 2000;10(22):1447–50.
Barbera M, di Pietro M, Walker E, Brierley C, MacRae S, Simons BD, Jones PH, Stingl J, Fitzgerald RC. The human squamous oesophagus has widespread capacity for clonal expansion from cells at diverse stages of differentiation. Gut. 2015;64(1):11–9.
Pan Q, Nicholson AM, Barr H, Harrison LA, Wilson GD, Burkert J, Jeffery R, Alison MR, Looijenga L, Lin WR, et al. Identification of lineage-uncommitted, long-lived, label-retaining cells in healthy human esophagus and stomach, and in metaplastic esophagus. Gastroenterology. 2013;144(4):761–70.
Lorinc E, Oberg S. Submucosal glands in the columnar-lined oesophagus: evidence of an association with metaplasia and neosquamous epithelium. Histopathology. 2012;61(1):53–8.
Long JD, Orlando RC. Esophageal submucosal glands: structure and function. Am J Gastroenterol. 1999;94(10):2818–24.
Coad RA, Woodman AC, Warner PJ, Barr H, Wright NA, Shepherd NA. On the histogenesis of Barrett’s oesophagus and its associated squamous islands: a three-dimensional study of their morphological relationship with native oesophageal gland ducts. J Pathol. 2005;206(4):388–94.
Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, Harrison R, Novelli MR, Jankowski JA, Wright NA. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008;57(8):1041–8.
Gillen P, Keeling P, Byrne PJ, West AB, Hennessy TP. Experimental columnar metaplasia in the canine oesophagus. Br J Surg. 1988;75(2):113–5.
Li H, Walsh TN, O’Dowd G, Gillen P, Byrne PJ, Hennessy TP. Mechanisms of columnar metaplasia and squamous regeneration in experimental Barrett’s esophagus. Surgery. 1994;115(2):176–81.
Mari L, Milano F, Parikh K, Straub D, Everts V, Hoeben KK, Fockens P, Buttar NS, Krishnadath KK. A pSMAD/CDX2 complex is essential for the intestinalization of epithelial metaplasia. Cell Rep. 2014;7(4):1197–210.
Rishniw M, Rodriguez P, Que J, Burke ZD, Tosh D, Chen H, Chen X. Molecular aspects of esophageal development. Ann N Y Acad Sci. 2011;1232:309–15.
Sarosi G, Brown G, Jaiswal K, Feagins LA, Lee E, Crook TW, Souza RF, Zou YS, Shay JW, Spechler SJ. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus. 2008;21(1):43–50.
Hutchinson L, Stenstrom B, Chen D, Piperdi B, Levey S, Lyle S, Wang TC, Houghton J. Human Barrett’s adenocarcinoma of the esophagus, associated myofibroblasts, and endothelium can arise from bone marrow-derived cells after allogeneic stem cell transplant. Stem Cells Dev. 2011;20(1):11–7.
Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1.
Bremner CG, Lynch VP, Ellis Jr FH. Barrett’s esophagus: congenital or acquired? An experimental study of esophageal mucosal regeneration in the dog. Surgery. 1970;68(1):209–16.
Pollara WM, Cecconello I, Zilberstein B, Iria K, Pinotti HW. Regeneration of esophageal epithelium in the presence of gastroesophageal reflux. Arq Gastroenterol. 1983;20(2):53–9.
Wang X, Ouyang H, Yamamoto Y, Kumar PA, Wei TS, Dagher R, Vincent M, Lu X, Bellizzi AM, Ho KY, et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell. 2011;145(7):1023–35.
Goldman MC, Beckman RC. Barrett syndrome. Case report with discussion about concepts of pathogenesis. Gastroenterology. 1960;39:104–10.
Ormsby AH, Goldblum JR, Rice TW, Richter JE, Falk GW, Vaezi MF, Gramlich TL. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30(3):288–94.
Xian W, Ho KY, Crum CP, McKeon F. Cellular origin of Barrett’s esophagus: controversy and therapeutic implications. Gastroenterology. 2012;142(7):1424–30.
Nakagawa H, Wang TC, Zukerberg L, Odze R, Togawa K, May GH, Wilson J, Rustgi AK. The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus and forestomach. Oncogene. 1997;14(10):1185–90.
Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, Lee Y, Friedman R, Asfaha S, Dubeykovskaya Z, et al. Bile Acid and inflammation activate gastric cardia stem cells in a mouse model of barrett-like metaplasia. Cancer Cell. 2012;21(1):36–51.
Becker L, Huang Q, Mashimo H. Lgr5, an intestinal stem cell marker, is abnormally expressed in Barrett’s esophagus and esophageal adenocarcinoma. Dis Esophagus. 2010;23(2):168–74.
Vega KJ, May R, Sureban SM, Lightfoot SA, Qu D, Reed A, Weygant N, Ramanujam R, Souza R, Madhoun M, et al. Identification of the putative intestinal stem cell marker doublecortin and CaM kinase-like-1 in Barrett’s esophagus and esophageal adenocarcinoma. J Gastroenterol Hepatol. 2012;27(4):773–80.
Hahn HP, Blount PL, Ayub K, Das KM, Souza R, Spechler S, Odze RD. Intestinal differentiation in metaplastic, nongoblet columnar epithelium in the esophagus. Am J Surg Pathol. 2009;33(7):1006–15.
Chaves P, Pereira AD, Cruz C, Suspiro A, Mendes de Almeida JC, Leitao CN, Soares J. Recurrent columnar-lined esophageal segments—study of the phenotypic characteristics using intestinal markers. Dis Esophagus. 2002;15(4):282–6.
Dias Pereira A, Chaves P. Columnar-lined oesophagus without intestinal metaplasia: results from a cohort with a mean follow-up of 7 years. Aliment Pharmacol Ther. 2012;36(3):282–9.
Nam KT, O’Neal R, Lee YS, Lee YC, Coffey RJ, Goldenring JR. Gastric tumor development in Smad3-deficient mice initiates from forestomach/glandular transition zone along the lesser curvature. Lab Invest. 2012;92(6):883–95.
Schreiber DS, Apstein M, Hermos JA. Paneth cells in Barrett’s esophagus. Gastroenterology. 1978;74(6):1302–4.
Griffin M, Sweeney EC. The relationship of endocrine cells, dysplasia and carcinoembryonic antigen in Barrett’s mucosa to adenocarcinoma of the oesophagus. Histopathology. 1987;11(1):53–62.
Levine DS, Rubin CE, Reid BJ, Haggitt RC. Specialized metaplastic columnar epithelium in Barrett’s esophagus. A comparative transmission electron microscopic study. Lab Invest. 1989;60(3):418–32.
Lord RV, Wickramasinghe K, Johansson JJ, Demeester SR, Brabender J, Demeester TR. Cardiac mucosa in the remnant esophagus after esophagectomy is an acquired epithelium with Barrett’s-like features. Surgery. 2004;136(3):633–40.
Oberg S, Johansson J, Wenner J, Walther B. Metaplastic columnar mucosa in the cervical esophagus after esophagectomy. Ann Surg. 2002;235(3):338–45.
Berenson MM, Johnson TD, Markowitz NR, Buchi KN, Samowitz WS. Restoration of squamous mucosa after ablation of Barrett’s esophageal epithelium. Gastroenterology. 1993;104(6):1686–91.
Brandt LJ, Blansky RL, Kauvar DR. Repeat laser therapy of recurrent Barrett’s epithelium: success with anacidity. Gastrointest Endosc. 1995;41(3):267.
Nicholson AM, Graham TA, Simpson A, Humphries A, Burch N, Rodriguez-Justo M, Novelli M, Harrison R, Wright NA, McDonald SA, et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut. 2012;61(10):1380–9.
Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, Fox C, Francis JM, Pedamallu CS, DeLuca DS, et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest. 2014;124(4):1636–45.
Kazumori H, Ishihara S, Kinoshita Y. Roles of caudal-related homeobox gene Cdx1 in oesophageal epithelial cells in Barrett’s epithelium development. Gut. 2009;58(5):620–8.
Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN, Clevers H, Jay P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol. 2004;166(1):37–47.
Vidal VP, Chaboissier MC, Lutzkendorf S, Cotsarelis G, Mill P, Hui CC, Ortonne N, Ortonne JP, Schedl A. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr Biol. 2005;15(15):1340–51.
Tavella S, Biticchi R, Schito A, Minina E, Di Martino D, Pagano A, Vortkamp A, Horton WA, Cancedda R, Garofalo S. Targeted expression of SHH affects chondrocyte differentiation, growth plate organization, and Sox9 expression. J Bone Miner Res. 2004;19(10):1678–88.
Bien-Willner GA, Stankiewicz P, Lupski JR. SOX9cre1, a cis-acting regulatory element located 1.1 Mb upstream of SOX9, mediates its enhancement through the SHH pathway. Hum Mol Genet. 2007;16(10):1143–56.
Clemons NJ, Wang DH, Croagh D, Tikoo A, Fennell CM, Murone C, Scott AM, Watkins DN, Phillips WA. Sox9 drives columnar differentiation of esophageal squamous epithelium: a possible role in the pathogenesis of Barrett’s esophagus. Am J Physiol Gastrointest Liver Physiol. 2012;303(12):G1335–46.
Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development. 2007;134(13):2521–31.
Raghoebir L, Bakker ER, Mills JC, Swagemakers S, Kempen MB, Munck AB, Driegen S, Meijer D, Grosveld F, Tibboel D, et al. SOX2 redirects the developmental fate of the intestinal epithelium toward a premature gastric phenotype. J Mol Cell Biol. 2012;4(6):377–85.
Long KB, Hornick JL. SOX2 is highly expressed in squamous cell carcinomas of the gastrointestinal tract. Hum Pathol. 2009;40(12):1768–73.
Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41(11):1238–42.
Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, Seandel M, Geijsen N, Hochedlinger K. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell. 2011;9(4):317–29.
Liu K, Jiang M, Lu Y, Chen H, Sun J, Wu S, Ku WY, Nakagawa H, Kita Y, Natsugoe S, et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12(3):304–15.
Barbieri CE, Pietenpol JA. p63 and epithelial biology. Exp Cell Res. 2006;312(6):695–706.
Daniely Y, Liao G, Dixon D, Linnoila RI, Lori A, Randell SH, Oren M, Jetten AM. Critical role of p63 in the development of a normal esophageal and tracheobronchial epithelium. Am J Physiol Cell Physiol. 2004;287(1):C171–81.
Hall PA, Woodman AC, Campbell SJ, Shepherd NA. Expression of the p53 homologue p63alpha and DeltaNp63alpha in the neoplastic sequence of Barrett’s oesophagus: correlation with morphology and p53 protein. Gut. 2001;49(5):618–23.
Glickman JN, Yang A, Shahsafaei A, McKeon F, Odze RD. Expression of p53-related protein p63 in the gastrointestinal tract and in esophageal metaplastic and neoplastic disorders. Hum Pathol. 2001;32(11):1157–65.
Geddert H, Kiel S, Heep HJ, Gabbert HE, Sarbia M. The role of p63 and deltaNp63 (p40) protein expression and gene amplification in esophageal carcinogenesis. Hum Pathol. 2003;34(9):850–6.
Roman S, Petre A, Thepot A, Hautefeuille A, Scoazec JY, Mion F, Hainaut P. Downregulation of p63 upon exposure to bile salts and acid in normal and cancer esophageal cells in culture. Am J Physiol Gastrointest Liver Physiol. 2007;293(1):G45–53.
Guo RJ, Suh ER, Lynch JP. The role of Cdx proteins in intestinal development and cancer. Cancer Biol Ther. 2004;3(7):593–601.
Silberg DG, Swain GP, Suh ER, Traber PG. Cdx1 and cdx2 expression during intestinal development. Gastroenterology. 2000;119(4):961–71.
Subramanian V, Meyer BI, Gruss P. Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell. 1995;83(4):641–53.
Mutoh H, Sakurai S, Satoh K, Osawa H, Hakamata Y, Takeuchi T, Sugano K. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: comparative study with Cdx2 transgenic mice. Gut. 2004;53(10):1416–23.
Wong NA, Wilding J, Bartlett S, Liu Y, Warren BF, Piris J, Maynard N, Marshall R, Bodmer WF. CDX1 is an important molecular mediator of Barrett’s metaplasia. Proc Natl Acad Sci U S A. 2005;102(21):7565–70.
Groisman GM, Amar M, Meir A. Expression of the intestinal marker Cdx2 in the columnar-lined esophagus with and without intestinal (Barrett’s) metaplasia. Mod Pathol. 2004;17(10):1282–8.
Phillips RW, Frierson Jr HF, Moskaluk CA. Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am J Surg Pathol. 2003;27(11):1442–7.
Eda A, Osawa H, Satoh K, Yanaka I, Kihira K, Ishino Y, Mutoh H, Sugano K. Aberrant expression of CDX2 in Barrett’s epithelium and inflammatory esophageal mucosa. J Gastroenterol. 2003;38(1):14–22.
Moons LM, Bax DA, Kuipers EJ, Van Dekken H, Haringsma J, Van Vliet AH, Siersema PD, Kusters JG. The homeodomain protein CDX2 is an early marker of Barrett’s oesophagus. J Clin Pathol. 2004;57(10):1063–8.
Beck F, Erler T, Russell A, James R. Expression of Cdx-2 in the mouse embryo and placenta: possible role in patterning of the extra-embryonic membranes. Dev Dyn. 1995;204(3):219–27.
Chawengsaksophak K, James R, Hammond VE, Kontgen F, Beck F. Homeosis and intestinal tumours in Cdx2 mutant mice. Nature. 1997;386(6620):84–7.
Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell. 2009;16(4):588–99.
Satoh K, Mutoh H, Eda A, Yanaka I, Osawa H, Honda S, Kawata H, Kihira K, Sugano K. Aberrant expression of CDX2 in the gastric mucosa with and without intestinal metaplasia: effect of eradication of Helicobacter pylori. Helicobacter. 2002;7(3):192–8.
Marchetti M, Caliot E, Pringault E. Chronic acid exposure leads to activation of the cdx2 intestinal homeobox gene in a long-term culture of mouse esophageal keratinocytes. J Cell Sci. 2003;116(Pt 8):1429–36.
Boudreau F, Rings EH, van Wering HM, Kim RK, Swain GP, Krasinski SD, Moffett J, Grand RJ, Suh ER, Traber PG. Hepatocyte nuclear factor-1 alpha, GATA-4, and caudal related homeodomain protein Cdx2 interact functionally to modulate intestinal gene transcription. Implication for the developmental regulation of the sucrase-isomaltase gene. J Biol Chem. 2002;277(35):31909–17.
Guo M, House MG, Suzuki H, Ye Y, Brock MV, Lu F, Liu Z, Rustgi AK, Herman JG. Epigenetic silencing of CDX2 is a feature of squamous esophageal cancer. Int J Cancer. 2007;121(6):1219–26.
van den Brink GR, Hardwick JC, Tytgat GN, Brink MA, Ten Kate FJ, Van Deventer SJ, Peppelenbosch MP. Sonic hedgehog regulates gastric gland morphogenesis in man and mouse. Gastroenterology. 2001;121(2):317–28.
Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124(7):1313–22.
Besnard V, Wert SE, Hull WM, Whitsett JA. Immunohistochemical localization of Foxa1 and Foxa2 in mouse embryos and adult tissues. Gene Expr Patterns. 2004;5(2):193–208.
Jaiswal KR, Morales CP, Feagins LA, Gandia KG, Zhang X, Zhang HY, Hormi-Carver K, Shen Y, Elder F, Ramirez RD, et al. Characterization of telomerase-immortalized, non-neoplastic, human Barrett’s cell line (BAR-T). Dis Esophagus. 2007;20(3):256–64.
Harley CB. Telomerase is not an oncogene. Oncogene. 2002;21(4):494–502.
Morales CP, Gandia KG, Ramirez RD, Wright WE, Shay JW, Spechler SJ. Characterisation of telomerase immortalised normal human oesophageal squamous cells. Gut. 2003;52(3):327–33.
Zhang HY, Zhang X, Chen X, Thomas D, Hormi-Carver K, Elder F, Spechler SJ, Souza RF. Differences in activity and phosphorylation of MAPK enzymes in esophageal squamous cells of GERD patients with and without Barrett’s esophagus. Am J Physiol Gastrointest Liver Physiol. 2008;295(3):G470–8.
Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, German MS. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc Natl Acad Sci U S A. 2007;104(25):10500–5.
van der Sluis M, Vincent A, Bouma J, Korteland-Van Male A, van Goudoever JB, Renes IB, Van Seuningen I. Forkhead box transcription factors Foxa1 and Foxa2 are important regulators of Muc2 mucin expression in intestinal epithelial cells. Biochem Biophys Res Commun. 2008;369(4):1108–13.
Ye DZ, Kaestner KH. Foxa1 and Foxa2 control the differentiation of goblet and enteroendocrine L- and D-cells in mice. Gastroenterology. 2009;137(6):2052–62.
Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A. 2009;106(17):6950–5.
Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, Ouellette AJ, Andersen B, Lipkin SM. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2−/− mice. Dev Biol. 2010;338(2):270–9.
Menke V, van Es JH, de Lau W, van den Born M, Kuipers EJ, Siersema PD, de Bruin RW, Kusters JG, Clevers H. Conversion of metaplastic Barrett’s epithelium into post-mitotic goblet cells by gamma-secretase inhibition. Dis Model Mech. 2010;3(1–2):104–10.
Tamagawa Y, Ishimura N, Uno G, Yuki T, Kazumori H, Ishihara S, Amano Y, Kinoshita Y. Notch signaling pathway and Cdx2 expression in the development of Barrett’s esophagus. Lab Invest. 2012;92(6):896–909.
Funding
This work was funded by the US National Institutes of Health (R01-DK097340 to D.H.W. and R01-DK63621 to R.F.S.) and by the Office of Research and Development, US Department of Veterans Affairs (I01-BX001061 and I01-BX002666 to R.F.S.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Wang, D.H., Souza, R.F. (2016). Transcommitment: Paving the Way to Barrett’s Metaplasia. In: Jansen, M., Wright, N. (eds) Stem Cells, Pre-neoplasia, and Early Cancer of the Upper Gastrointestinal Tract. Advances in Experimental Medicine and Biology, vol 908. Springer, Cham. https://doi.org/10.1007/978-3-319-41388-4_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-41388-4_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41386-0
Online ISBN: 978-3-319-41388-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)