
Abstract
LC-MS/MS, particularly when linked with immunoaffinity enrichment, has emerged as a highly capable bioanalytical technique for the quantitative measurement of protein biomarkers and therapeutic proteins, thus impacting translational pharmacology. A key advantage of a protein LC-MS/MS assay over other bioanalytical techniques is the high measurement specificity that can be achieved. Immunoaffinity enrichment techniques using anti-protein or anti-peptide antibodies, or both in a sequential manner, extend LC-MS/MS assay sensitivity for protein biomarkers into the pg/mL range. Assay translation between species can be facilitated by selecting proteotypic peptides that are conserved in the same protein across species, if available, to allow the same MS detection method, the same SIL standard peptide and the same anti-peptide antibody can be used. Practical challenges to routine implementation in clinical assays are being overcome by the use of standardized workflows, liquid handling robotics, and robust LC-MS/MS configurations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Liquid chromatography tandem mass spectrometry
- Translational pharmacology
- Proteotypic peptide
- Selected reaction monitoring
- Protein and peptide immunoaffinity
Protein Biomarkers and the Need for Their Selective Bioanalysis
A major aim of translational pharmaceutical and biomedical research is to transition drug candidates and their targets and pathways from preclinical discovery to clinical development. Biomarkers can facilitate decisions in translational pharmacology , safety as well as precision medicine for patient stratification [1]. While many classes of endogenous molecules can constitute biomarkers, the need to quantitatively measure endogenous proteins as biomarkers is undeniable. In addition, the bioanalysis of the therapeutic targets themselves, which are proteins in many cases, can be a key biomarker measurement. Target analysis in normal and disease, or following treatment, both in systemic circulation and in tissues can support translational pharmacology by assisting with the construction of the pharmacokinetic/pharmacodynamic relationship. An important extension is the analysis of target engagement by the therapeutic, which is frequently important for rationalizing the selection of the dosing regimen. Downstream pharmacodynamic biomarkers are mechanistically linked to the therapeutic target and are a direct measure of pathway modulation by the therapeutic.
Localization, abundance, internalization, or turnover rate, to name a few, are diverse attributes of the proteins investigated as biomarkers. Most commonly, the concentration of a soluble protein biomarker in a biological fluid is determined. However, depending on the question, biomarker assays are also needed to determine the amount of membrane-associated protein in solid tissues, such as in clinical biopsies. Moreover, the occurrence and abundance of posttranslational modifications such as phosphorylation, glycosylation or ubiquitination, and splice variants or the rate of protein synthesis or degradation can also be a biomarker. A constant evolution of technologies and assay strategies is required to be able to address these, often times, challenging bioanalytical questions for protein biomarkers. There are many competent technologies available for biomarker measurements, many of these are described in this book. This section will focus on quantitative protein mass spectrometry, which is still a comparatively young application area, but has matured substantially in recent years and is increasingly becoming a major player in the quantification of protein biomarkers in translational and clinical research. One key advantage quantitative protein mass spectrometry has over other bioanalytical tools is that one can achieve high measurement specificity.
From Mass Spectrometry-Based Proteomics to Quantitative LC-MS/MS Protein Biomarker Assays
Since 1990s, qualitative proteomics has focused on the identification of proteins from biological samples resulting in the creation of valuable catalogs of detectable proteins [2]. Detection is typically facilitated via digestion of proteins into peptides using specific proteases such as trypsin. Over the years, mass spectrometry has become the dominant proteomics detection tool, employing mostly quadrupole time-of-flight (QTOF) and orbitrap mass analyzers. With evolving instrumentation and advancing workflows for sample preparation, it has been possible to increase the number of proteins identified per sample from hundreds to thousands in each individual experiment including the detection of posttranslational modifications. In the last decade, many global unbiased proteomics studies have been enabled by semiquantitative workflows, allowing comparison of relative protein abundance in different samples and conditions. This facilitated a proteomics study design that could link identification of putative protein biomarkers and their relative abundance to a functional biological endpoint. To this end, data-dependent acquisition (DDA) methods are employed that are either based on label-free comparison of mass spectrometry signals or based on chemical or metabolic labeling with 13C or 15N, which can be distinguished by the mass spectrometer from isotopes that occur naturally in high abundance [2]. Major examples include labeling techniques such as iTRAQ, TMT, or SILAC [3]. Recent advances in data-independent acquisition (DIA) methods demonstrate not only deep proteome coverage [4], but also the ability to semiquantitate all measured proteins, and require no a priori knowledge of anticipated protein biomarker changes. Proteomics researchers also perform hypothesis-driven studies, where the experiment focuses on the detection of a known set of putative protein biomarkers. The mass spectrometer is programmed to only analyze proteins of interest, providing improved sensitivity compared to a global proteomics survey experiment where as many proteins as possible are detected. Hypothesis-driven proteomics experiments are typically done in a semiquantitative manner by comparing relative signal intensities. Both, unbiased and hypothesis-driven experimentation can discover new protein biomarkers or signatures. These can be followed up with fully quantitative assays capable of analyzing larger sample sets with higher analytical rigor to achieve biological validation of the putative biomarker.
The proteomics field has developed numerous sample preparation techniques including digestion and enrichment approaches as well as mass spectrometry detection methods that are fundamental to today’s quantitative protein LC-MS/MS assays. Methodologies used in contemporary LC-MS/MS quantification of protein biomarkers also draw on the vast experience that exists with LC-MS/MS quantification of small molecule biomarkers. This includes basic quantification concepts such as the use of stable isotope labeled standards, determination of quantitative LC-MS/MS assay performance, and how to handle endogenously detectable analyte during assay development. This has led to an ongoing discussion of how bioanalytical performance and acceptance criteria can be adapted for the quantification of protein biomarkers.
Preparing the Sample for Mass Spectrometry
In most cases, the sample containing the protein to be quantified is digested into measurable, proteotypic peptides prior to LC-MS/MS using a specific protease. Most commonly, trypsin is employed which cleaves C-terminal to arginine and lysine amino acids, except if adjacent to a proline. Other enzymes can be used as well, such chymotrypsin or endoproteases Asp-N, Lys-C, Arg-C, or Glu-C. Chemical digestion with acid [5] or cyanogen bromide [6, 7] has also been demonstrated for protein quantification. The advantage of digestion is that higher mass spectrometric sensitivity can be achieved when measuring peptides compared to proteins as instrumental sensitivity declines with increasing mass. Amino acid sequence homology of the selected peptide with relevant database entries needs to be confirmed in silico. Furthermore, when measuring peptides as quantitative surrogates of the proteins they originate from, it is important to experimentally confirm that peptide abundance is truly representative of protein abundance. This is central to quantitative protein LC-MS/MS, particularly for the analysis of protein biomarkers, where premature forms, posttranslational modifications or splice variants, or other forms resulting from biological processing can correlate with biological effect. If possible and depending on the application, additional peptides should be measured simultaneously, to ensure that results are consistent. In fact, it is a key advantage of LC-MS/MS that several peptides from the same protein can be measured simultaneously. This allows obtaining more complete amino acid sequence coverage of the protein biomarker to span multiple domains of relevance to biological function. Multiplexed quantification of several peptides originating from one or more proteins can then be incorporated into one protein biomarker assay.
Sample types for protein biomarker quantification can range from cell cultures, plasma or serum and other fluids to solid tissues. The dynamic range of protein abundance in these samples can be large, spanning >10 orders of magnitude in human serum [8]. A sample preparation workflow consisting only of protease digestion prior to conventional LC-MS/MS is typically not very sensitive which limits the application of the assay to only highly abundant proteins. This is due to a number of factors including the limited loading capacity on liquid chromatography columns and the dynamic detection range of the mass spectrometer. Therefore, in order to measure biomarker proteins of lower abundance, sample fractionation or enrichment needs to be incorporated in an assay workflow.
Fractionation
There are well-established fractionation techniques, both at protein or peptide level, which can be used in quantitative protein biomarker LC-MS/MS assays. These include fractionation based on differential solubility or hydrophobicity, molecular weight, charge, and pI. Many proteomics fractionation techniques have been used in the quantitative analysis of proteins biomarkers; however, prominent examples of methods that can be easily implemented in protein biomarker assays are solid-phase extraction (SPE), protein precipitation or ion exchange chromatography. SPE can be used as a positive or negative selection tool depending on the stationary phase and analyte characteristics [9]. Fractionation based on ion exchange can also be incorporated into a protein quantification workflow, for example at digest level using a weak cation exchange monolithic trap in an online configuration prior to LC-MS/MS [7].
Driving Sensitivity with Immunoaffinity Enrichment
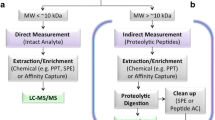
Using antibodies to enrich the protein biomarker, the enzymatically released peptide or both in a sequential manner can provide tremendous gains in LC-MS/MS assay performance [10], particularly with respect to sensitivity, dynamic range and throughput. Immunoaffinity (IA) at protein level using an antibody (Fig. 1a), which is considered equivalent to a protein capture step in a ligand binding assay, allows LC-MS/MS analysis of protein biomarkers that are in the low to mid pg/mL concentration range or above in plasma or serum. Sensitivity is typically scalable with sample volume if sufficient capture antibody is used. A preferred practical implementation, which has been tested both in preclinical and routine clinical protein biomarker assays, is the use of biotinylated antibodies paired with streptavidin-coated paramagnetic beads. This workflow is well suited to operation on liquid handling robotics [7, 11–15] providing a technical solution that can be easily standardized and validated for clinical implementation. One key feature of the protein IA technique is that further analyte selection can be performed, similar to a ligand binding assay. For example, selecting a capture antibody to a specific epitope on the protein biomarker or therapeutic target, allows either intentionally competing or not competing with an endogenous binding partner, or a binding biotherapeutic, such as a monoclonal antibody. These types of measurements can be critical to developing an understanding for example of mechanistic pharmacology in preclinical studies and early clinical drug trials.

Schematic of immunoaffinity workflows for LC-MS/MS quantification of protein biomarkers. a Immunoaffinity extraction of the protein biomarker from the sample using an anti-protein antibody prior to digestion and LC-MS/MS; b digestion of the sample into peptides followed by immunoaffinity extraction of the targeted peptide using an anti-peptide antibody prior to LC-MS/MS; c sequential protein immunoaffinity extraction followed by digestion and immunoaffinity extraction of the targeted peptide using an anti-peptide antibody
Another frequently employed IA strategy is the use an of anti-peptide antibody for enrichment at the level of the peptide that has been enzymatically released from the protein biomarker of interest as part of the assay procedure (Fig. 1b). This approach is termed stable isotope standards and capture by anti-peptide antibodies (SISCAPA) [16]. Polyclonal and monoclonal anti-peptide antibodies can be used. Furthermore, bead-based [17] and column-based online flow formats [16, 18] are successfully used. Achievable sensitivity is in the pg/mL to low ng/mL range and above depending on the quality of the capture antibody and the sample volume used. One of the unique advantages of this workflow is the compatibility with harsh, denaturing conditions during sample preparation, for example as might be needed for extraction of protein biomarkers from tissues. To this end, a successful workflow might include tissue homogenization and extraction, protein precipitation followed by enzymatic digestion of the pellet prior to anti-peptide antibody-based enrichment. Harsh conditions during samples handling, for example using strong detergents, can be incompatible with protein IA methods irrespective whether the end-point detection is based on ligand binding or mass spectrometry.
Finally, protein and peptide IA methods can be combined (Fig. 1c) in a protein biomarker assay [11, 12]. Although the sequential protein and peptide IA approach is a more complex assay format, this configuration can deliver ultimate assay performance with respect to sensitivity and throughput. This assay format has been successfully employed for the routine analysis of thousands of clinical samples [14]. Which IA technique is selected depends on the availability of capture reagents, assay feasibility, available sample volume, required sensitivity, and other bioanalytical goals.
Liquid Chromatography Options
With the exception of some chromatography-free workflow developments, such as iMALDI [19], most quantitative protein biomarker MS assays require liquid chromatography (LC)-based separation of the analytes. Assays based on quantification of enzymatically released peptides mostly employ C18 reverse phase chromatography. Most LC configurations reported in the literature for quantitative protein assays use conventional, high flow rates typically at or above the mid-microlitre per minute range, with or without analyte trapping prior to analytical separation. These LC configurations are well tested and robust, mostly characterized by a short total cycle time and are easy to implement using standard equipment. In contrast, the proteomics community has been using mostly nanoflow rates for reverse phase chromatography of complex digests, typically in the mid-nanoliter per minute range. This improves mass spectrometric sensitivity, which is inversely correlated with flow rate. Chromatography cycle times are typically longer (>1 h) in order to maximize separation for improved peptide identification or quantification. Advances in protein biomarker quantification workflows have illustrated the symbiosis between IA enrichment and nanoflow LC. Antibody directed analyte enrichment and therefore complexity reduction of the sample makes it possible to run short nanoflow gradients routinely for larger sample sets. Biomarker assays that employ such a workflow with total LC cycle times between 10 and 15 min have been published recently using either online anti-peptide antibody enrichment, offline anti-protein antibody enrichment, or both [11, 12, 14, 15, 18, 20]. Finally, capillary flow rates (low microlitre per minute range) are being used to bridge the sensitivity gap between methods that use high or nanoflow LC [21, 22].
Mass Spectrometry Techniques
Triple quadrupole mass spectrometers (Fig. 2) are most commonly used for quantification of surrogate, proteotypic peptides using the selected reaction monitoring (SRM) data acquisition mode. Selected fragment ions obtained from predefined precursor ions via collision with gas in a collision cell are monitored by the mass spectrometer [23]. SRM has been used for several decades for the quantitative bioanalysis of small molecules, which provides the foundation for the recent advances made in LC-MS/MS quantification of proteins. In peptide SRM assays, typically 3–5 fragment ions per precursor are recorded, which are frequently those that are most abundant and free from interferences. These ion transitions are typically the most sequence informative, but that selection is not always needed particularly when paired with an immunoaffinity workflow that provides additional selectivity. A stable isotope labeled (SIL) standard is typically employed to coelute with the analyte of interest. This increases confidence in assay selectivity via correct assignment and quantification of the signal, especially from complex biological samples [24], including those from samples that have been enriched using immunoaffinity. Importantly, SIL peptide standards also mirror the expected intensity ratio of SRM transitions of the analyte which can be used to confirm correct signal assignment. Recommendations relating to the quantification, storage, and handling of peptide standards for mass spectrometry-based workflows have recently been published [25]. Finally, in addition to using recombinant protein calibrators where possible as well as endogenous and recombinant quality control samples, SIL peptide standards are utilized for MS response normalization as well as for normalizing parts of the sample preparation workflow (Table 1).

Selected reaction monitoring (SRM) of peptide ions on a triple quadrupole mass spectrometer
High-resolution (HR) MS instruments are increasingly explored for quantification of protein biomarkers via their surrogate peptides. Product ion scans on a QTOF mass spectrometer provide high measurement specificity. Akin to this approach, contemporary quantification methods on orbitrap MS instruments can use targeted higher energy collisional dissociation (tHCD) methods. Only precursor masses are preset and high-resolution and high mass accuracy allows simultaneous identification and quantification of multiple fragment ions from complex tandem mass spectra. Other HRMS quantification techniques rely on high measurement resolution and accuracy of the precursor peptide ions without fragmentation using selected ion monitoring (SIM) on both QTOF and orbitrap mass spectrometers [20]. A promising feature of HRMS is that in addition to the targeted quantification of the peptides of interest, the mass spectrometer can simultaneously acquire qualitative information from other components of the sample. Finally, quantification workflows that include protein immunoaffinity enrichment, but do not rely on digestion and instead analyze the intact protein by MS are beginning to be explored. This approach holds great potential, as workflows are simpler due to the absence of the digestion step. Furthermore, no structural or sequence information about the biomarker protein is lost when only one or a few peptides are monitored as surrogates. However, the currently achievable sensitivity is limited mostly due to multiple charging of the protein precursor during ionization, which makes this approach mostly suitable to quantification of biomarker proteins that are in higher abundance.
Building Translatable Assays
As candidate drug compounds and their targets and pathways progress through drug discovery stage gates toward investigations in clinical trials, the need for developing clinical biomarkers increases. Oftentimes, this necessitates de novo development of assays. Ideally, the same or similar bioanalytical method is used during the research and development continuum to facilitate better interpretation and translation of results between species and investigations. High measurement specificity and good sensitivity of protein biomarker LC-MS/MS are key drivers to implement the technology in translational research. However, additional opportunities exist to realize synergies for clinical assay development, for example when preclinical assays are also developed, optimizing the investment in bioanalytical resources. Specifically, a number of factors should be considered when developing protein biomarker LC-MS/MS assays that can be used across different species with no or only minor modifications. Assay translation between species can be facilitated by selecting proteotypic peptides that are conserved in the same protein across species, if available. This allows the use of the same MS detection method and the same SIL standard peptide. Furthermore, other reagents specific for the targeted peptides can then also be used in a cross-species assay, such as anti-peptide antibodies. In some cases, where the protein itself is highly homologous or conserved between the species of interest, cross-species binding of the antibody used for protein IA enrichment can be investigated as a desired reagent property. In a protein biomarker IA-LC-MS/MS assay, such a capture antibody may be preferentially chosen over other antibodies that do not cross-react. Examples of sequential protein and peptide IA-LC-MS/MS assays have been reported recently where all antibody reagents and SIL peptides were successfully employed across matrices from different species [11, 12, 15].
In the absence of conserved, cross-species sequences, the selection of peptides is guided by the analytical aim and the desired assay workflow. For example, if anti-peptide antibodies will be part of the assay, then peptides from the equivalent sequence region of the protein from different species can be selected that share a similar antigenic sequence as part of a proteotypic peptide . A peptide immunogen sequence can then be carefully designed for generating anti-peptide antibodies, frequently in rabbits [16, 26], that are capable of binding the related proteotypic peptides from different species. The anti-peptide antibody reagent generated in such a way can be used in an IA-LC-MS/MS assay for enriching the relevant peptides from the different species. MS detection methods have to be adjusted accordingly.
Although quantitative protein biomarker LC-MS/MS has substantially evolved in recent years, it is still a fairly nascent technique. When developing a workflow based on this technique, perhaps using research grade instrumentation and methods, the technical implementation in a clinical setting needs to be carefully planned. Until recently, the operational complexity, particularly of immunoaffinity workflows prior to LC-MS/MS, has been perceived as a possible limitation to clinical implementation [27]. However, the technical challenges are being overcome by the use of standardized workflows, the implementation of liquid handling robotics, and robust mass spectrometry configurations [11, 12]. This led to recent examples of large-scale implementation of the quantitative LC-MS/MS technique in clinical protein biomarker studies [14]. Although a protein biomarker LC-MS/MS assay is typically developed under the fit-for-purpose paradigm [28], the assays can meet stringent acceptance criteria [11]. This technology is anticipated to mature further as additional precedence is generated and experience is gained in the field.
Chapter Summary
-
LC-MS/MS, especially nanoflow LC-MS/MS, particularly when linked with immunoaffinity enrichment, has emerged as a viable bioanalytical technique for the quantitative measurement of protein biomarkers and therapeutic target proteins, impacting translational pharmacology .
-
A key advantage of protein LC-MS/MS assay is the high measurement specificity that can be achieved.
-
Immunoaffinity enrichment techniques using anti-protein or anti-peptide antibodies, or both, extend LC-MS/MS assay sensitivity for protein biomarkers into the pg/mL range.
-
Assay translation between species can be facilitated by selecting proteotypic peptides that are conserved in the same protein across species, if available, to allow the same MS detection method, the same SIL standard peptide and the same anti-peptide antibody can be used.
-
Practical challenges to routine implementation in clinical assays are being overcome by the use of standardized workflows, liquid handling robotics, and robust LC-MS/MS configurations.
Abbreviations
- Translational pharmacology :
-
Investigations of drug effects on pathways and disease to establish a mechanistic link between in vitro or ex vivo to in vivo systems as well as within and between species. For example, to identify in vivo pharmacology and biomarkers in preclinical species that can also be measured in humans
- Proteotypic peptide :
-
A peptide, enzymatically released from a protein as part of an LC-MS/MS assay, which serves to unambiguously identify that protein
- Selected reaction monitoring :
-
Selected reaction monitoring (SRM) is a tandem mass spectrometry technique in which an ion of specified mass-to-charge ratio is selected in the first mass spectrometry stage. One or several product ions of a specified mass-to-charge ratio resulting from fragmentation of the precursor are detected in the second mass spectrometry stage
- Sequential protein and peptide immunoaffinity :
-
Dual immunoaffinity sample preparation technique used for measuring low abundance protein biomarkers in biological matrices by mass spectrometry. Protein immunoaffinity enrichment of the protein biomarker using an anti-protein capture reagent is followed by digestion and immunoaffinity enrichment of one or several enzymatically released peptides using anti-peptide antibodies
References
Rifai N, Gillette MA, Carr SA (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol 24(8):971–983
Mallick P, Kuster B (2010) Proteomics: a pragmatic perspective. Nat Biotechnol 28(7):695–709
Coombs KM (2011) Quantitative proteomics of complex mixtures. Expert Rev Proteomics 8(5):659–677
Egertson JD, Kuehn A, Merrihew GE et al (2013) Multiplexed MS/MS for improved data-independent acquisition. Nat Methods 10(8):744–746
Fernandez Ocaña M, Neubert H, Przyborowska A et al (2004) BSE control: detection of gelatine-derived peptides in animal feed by mass spectrometry. Analyst 129(2):111–115
Neubert H, Grace C, Rumpel K, James I (2008) Assessing immunogenicity in the presence of excess protein therapeutic using immunoprecipitation and quantitative mass spectrometry. Anal Chem 80(18):6907–6914
Neubert H, James I (2009) Online capillary weak cation exchange enrichment hyphenated to nanospray mass spectrometry for quantitation of a basic pegvisomant derived peptide. J Chromatogr A 1216(33):6151–6154
Anderson NL, Anderson NG (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 1(11):845–867
Jian W, Edom RW, Wang D, Weng N, Zhang S (2013) Relative quantitation of glycoisoforms of intact apolipoprotein C3 in human plasma by liquid chromatography-high-resolution mass spectrometry. Anal Chem 85(5):2867–2874
Ackermann BL, Berna MJ (2007) Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev Proteomics 4(2):175–186
Neubert H, Muirhead D, Kabir M, Grace C, Cleton A, Arends R (2013) Sequential protein and peptide immunoaffinity capture for mass spectrometry-based quantification of total human beta-nerve growth factor. Anal Chem 85(3):1719–1726
Palandra J, Finelli A, Zhu M, Masferrer J, Neubert H (2013) Highly specific and sensitive measurements of human and monkey interleukin 21 using sequential protein and tryptic peptide immunoaffinity LC-MS/MS. Anal Chem 85(11):5522–5529
Fernández Ocaña M, Neubert H (2010) An immunoaffinity liquid chromatography-tandem mass spectrometry assay for the quantitation of matrix metalloproteinase 9 in mouse serum. Anal Biochem 399(2):202–210
Schultz GA, Mccardle K, Neubert H (2016) Large-scale implementation of sequential protein and peptide immunoaffinity enrichment LC/nanoLC–MS/MS for human β-nerve growth factor. Bioanalysis 8(8):753–764
Palandra J, Quazi A, Fitz L, Rong H, Morris C, Neubert H (2016) Quantitative measurements of GDF-8 using Immunoaffinity LC-MS/MS. Proteomics Clin Appl 10(5):597–604. doi:10.1002/prca.201500112n/a-n/a
Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW (2004) Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J Proteome Res 3(2):235–244
Whiteaker JR, Zhao L, Anderson L, Paulovich AG (2010) An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics 9(1):184–196
Neubert H, Gale J, Muirhead D (2010) Online high-flow peptide immunoaffinity enrichment and nanoflow LC-MS/MS: assay development for total salivary pepsin/pepsinogen. Clin Chem 56(9):1413–1423
Shah B, Reid J, Kuzyk M, Parker C, Borchers C (2013) Developing an iMALDI Method. Methods Mol Biol 1023:97–120
Fan Y-Y, Neubert H (2016) Quantitative Analysis of Human Neonatal Fc Receptor (FcRn) tissue expression in transgenic mice by online peptide immuno-affinity LC-HRMS. Anal Chem. doi:10.1021/acs.analchem.5b03900
Wang H, Bennett P (2013) Performance assessment of microflow LC combined with high-resolution MS in bioanalysis. Bioanalysis 5(10):1249–1267
Lee AYH, Chappell DL, Bak MJ et al (2016) Multiplexed quantification of proglucagon-derived peptides by immunoaffinity enrichment and tandem mass spectrometry after a meal tolerance test. Clin Chem 62(1):227–235
Picotti P, Aebersold R (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Meth. 9(6):555–566
Sherman J, Mckay MJ, Ashman K, Molloy MP (2009) How specific is my SRM? The issue of precursor and product ion redundancy. Proteomics 9(5):1120–1123
Hoofnagle AN, Whiteaker JR, Carr SA et al (2016) Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin Chem 62(1):48–69
Pope ME, Soste MV, Eyford BA, Anderson NL, Pearson TW (2009) Anti-peptide antibody screening: selection of high affinity monoclonal reagents by a refined surface plasmon resonance technique. J Immunol Methods 341(1–2):86–96
Ackermann BL (2012) Understanding the role of immunoaffinity-based mass spectrometry methods for clinical applications. Clin Chem 58(12):1620–1622
Lee J, Devanarayan V, Barrett Y et al (2006) Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res 23(2):312–328
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Neubert, H. (2016). Quantification of Protein Biomarkers Using Liquid Chromatography Tandem Mass Spectrometry. In: Weiner, R., Kelley, M. (eds) Translating Molecular Biomarkers into Clinical Assays . AAPS Advances in the Pharmaceutical Sciences Series, vol 21. Springer, Cham. https://doi.org/10.1007/978-3-319-40793-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-40793-7_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-40792-0
Online ISBN: 978-3-319-40793-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)