
Abstract
Methods for adipose-derived stromal cell (ASC) isolation, characterization and the respective data generated differ between different research groups. Laboratories have developed in-house methods to isolate, expand and differentiate ASCs, which often makes data comparison difficult.
Methods for both research laboratory isolation and clinical applications are explored in this chapter. To use ASCs in the clinical setting, stem cell therapy products need to be prepared using good manufacturing practices (GMPs). This is achieved by eliminating animal-derived products from the manufacturing process. As a result, several human and chemically defined alternatives have been explored, especially during the isolation and expansion process.
ASC characterization criteria remain open ended, but should be compliant within the parameters of (1) adherence to plastic, (2) expression of a defined set of surface antigens and (3) the capacity to differentiate into various cell types.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Adipose stem cells
- Mesenchymal stem cells
- Good manufacturing practices
- Stromal vascular fraction
- Stem cell culture
7.1 Introduction
A stem cell is defined by its ability to self-renew and to differentiate along multiple lineage pathways. Stem cells have the potential to develop into different cell types in the body during their lifespan. Even after long periods of quiescence, stem cells retain the ability to divide, and the two daughter cells have the potential to either remain a stem cell within the stem cell niche or to develop into a more specialized cell with specific functions. Stem cells can broadly be classified into two categories, namely adult stem cells and pluripotent stem cells. Pluripotent stem cells include embryonic and induced pluripotent stem cells and will not be discussed further in this chapter. Adult stem cells are further subdivided into two categories, namely, haematopoietic stem cells (HSCs) and mesenchymal stromal cells (MSCs). The latter contains a small population of stem cells. MSCs are present in most tissues in the body and are required to restore normal tissue function via repair and regeneration mechanisms (Jones et al. 2002). The bone marrow, Wharton’s jelly present in the umbilical cord and adipose tissue are the most common sources used to isolate MSCs.
The clinical translation of MSC research remains a major challenge. Research groups around the world are working on various strategies to apply MSCs in a variety of settings. There is currently a need for consensus in standardization regarding isolation and expansion procedures, characterization of the cells, evaluation of multipotency, including tissue lineage induction, the best route of administration and monitoring of engraftment success.
Stem cells should meet various criteria before being considered for regenerative medicine purposes. The cells should (1) be present in abundant quantities in the tissue of interest, (2) require minimally invasive harvesting procedures, (3) differentiate along multiple cell lineage pathways that are reproducible and can be regulated/controlled, (4) be safe and effective when transplanted in an autologous or allogeneic manner and (5) be manufactured according to GMP guidelines (Gimble et al. 2007).
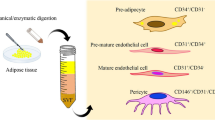
In 2013, the International Society for Cellular Therapy (ISCT) reviewed the nomenclature and characterization guidelines for adipose-derived stromal cells (ASCs) and in a joint statement with the International Society for Cellular Therapy (ISCT ) deemed it important to distinguish between the adipose-derived stromal vascular fraction (AD-SVF) and culture expanded ASCs (Bourin et al. 2013). SVF refers to the cellular component present in adipose tissue after mature adipocytes and peripheral blood cells have been removed during the isolation procedure. AD-SVF consists of a heterogeneous cell population which includes preadipocytes, MSCs, endothelial cells, leukocytes, adipose tissue-associated macrophages and other cell types. ASCs refer to the adherent multipotent cell population within the AD-SVF. It remains to be established whether AD-SVF cells or culture expanded ASCs are better suited for clinical applications.
This chapter will discuss AD-SVF isolation procedures, ASC expansion conditions, phenotypic characterization and qualitative and quantitative techniques used to assess the differentiation capacities of ASCs into the adipogenic, osteogenic, chondrogenic and myogenic lineages.
7.2 Isolation Methods
MSCs have been successfully isolated from nearly all postnatal organs and tissues. To date, bone marrow-derived MSCs (BM-MSCs) are the best described, particularly in the clinical setting. These cells serve as the gold standard to which other sources of MSCs are compared (Kern et al. 2006). Adipose tissue, like the bone marrow, derives from the embryonic mesodermal layer (Zuk et al. 2001). Interestingly, adipose tissue is one of the tissues in the human body with the highest number of stem cells, containing between 100- and 1000-fold more multipotent cells per volume unit when compared to the bone marrow. It has been shown that about 3.5 × 104 preadipocytes can be isolated from 1 g of adipose tissue (Fournier and Otteni 1983; Ersek and Salisbury 1995; von Heimburg et al. 2004; Strem et al. 2005). Fraser and colleagues (2006) also demonstrated that adipose tissue yields a 500-fold greater number of ASCs when compared to the MSC yield obtained from the bone marrow. Liposuction procedures can yield anywhere from 100 ml to >3 l of lipoaspirate, which is routinely discarded as biological waste. A recent review mentioned that 20 mg of lipoaspirate should yield >1 × 107 SVF cells (Banyard et al. 2015). This translates to an average expected yield of 1 × 106 ASCs, as it is estimated that 15–30 % of the AD-SVF consists of ASCs (Bourin et al. 2013). The ease with which these cells can be harvested and abundant cell yields render adipose tissue a preferred stromal/stem cell source (Fraser et al. 2006).
The isolation of ASCs from adipose tissue was first pioneered in the 1960s. Minced rat fat pads were extensively washed to remove contaminating HSCs , then incubated with collagenase and centrifuged to obtain an AD-SVF pellet. The selection for plastic-adherent fibroblast-like cells from the AD-SVF concluded this isolation process (Rodbell 1966; Rodbell and Jones 1966). ASCs, at that time referred to as resident MSCs in human adipose tissue, were first described by Zuk and colleagues in 2001 (Zuk et al. 2001). The initial procedure of mincing human adipose tissue by hand was simplified by the development of liposuction surgery.
7.2.1 Manual Isolation Procedure
In 2006, Dominici and colleagues (Dominici et al. 2006) suggested that the standard isolation protocol developed by Zuk and colleagues (2001, 2002) should be accepted as an established methodology to obtain the AD-SVF from lipoaspirate. In 2010, Estes and colleagues published a detailed method, adapted from the method described by Zuk and colleagues (Zuk et al. 2001), to isolate ASCs (Estes et al. 2010). This commonly accepted approach involves five basic steps, namely, (1) the removal of contaminating peripheral blood through washing steps, (2) enzymatic digestion of the adipose tissue, (3) separation of SVF from mature adipocytes, (4) lysis of red blood cells present in SVF and (5) selective isolation of the adherent cell component present in AD-SVF. Isolation procedures may differ concerning the enzymatic digestion step, type and composition of culture medium, approaches used to lyse red blood cells and cell seeding density. Data comparisons between research groups become problematic due to these different approaches. Other factors that may influence the quality of a cell therapy product include the donor’s age, the location (subcutaneous vs. visceral) of the adipose tissue and the surgical procedures used for tissue harvesting (Gimble et al. 2007; Mizuno 2009; Fossett et al. 2012). These factors should be considered when developing an isolation protocol in order to obtain a reliable source of cells that are safe, free from contamination and are of a high quality for application in the clinic. Please refer to the supplementary material for a brief description of the manual isolation procedure.
7.2.1.1 Seeding
Seeding refers to the cells being placed in a culture flask to allow proliferation (expansion). Seeding density is the number of cells seeded onto a culture surface to ensure a standardized concentration of cells per culture and is reported as the number of cells per cm2. The initial seeding density of the AD-SVF is higher than the subsequent seeding densities that will be used during the expansion phase. This ensures that a sufficient number (15–30 % of AD-SVF) of stromal cells are introduced into the culture flasks. Approximately 5 × 105 cells/cm2 of the AD-SVF is usually used and decreased to 5 × 103 cells/cm2 during the ASC expansion phase. Accurate cell quantification is important to ensure that the correct number of cells have been seeded in the culture flask.
Cell counts may be obtained by using either a manual method such as the trypan blue (0.4 %) dye exclusion method or automated cell counting devices, including benchtop flow cytometers. Benchtop flow cytometers are usually not volumetric and provide an absolute count (cells/μl) when a known concentration of counting beads is added to the sample. The Accuri benchtop flow cytometer (BD Biosciences, San Jose, USA) is an exception; it is a volumetric system that allows for direct determination of an abso lute count without the addition of counting beads.
Various flow cytometry counting beads are available commercially, including Flow Count™ counting beads (Beckman Coulter, Miami, USA) and CountBright™ absolute counting beads (Invitrogen/Molecular Probes, Life Technologies, Eugene, USA). Counting beads are commonly referred to as fluorospheres with a broad excitation/emission range. Certain flow cytometers, such as the FC500 and Navios flow cytometers (Beckman Coulter, Miami, USA), have algorithms built into the instrument software that will perform the absolute count calculations in the background and report the absolute cell counts as the number of cells/μl. It is crucial in these flow cytometric systems that the volume of beads to the volume of cell suspension is the same (1:1; vol/vol) to ensure the reporting of an accurate absolute count. The absolute count can also be calculated manually by using Eqs. (7.1) or (7.2) below.
Equation (7.1) is used to manually calculate the absolute count when the ratio of beads to cells is 1:1 (vol/vol):
Equation (7.2) is used in situations where the ratio of sam ple volume to bead suspension volume is not equal [Eq. (7.2)]:
Please refer to the supplementary material for a brief descri ption of the trypan blue (0.4 %) dye exclusion assay as well as an example of how an absolute count is obtained using a benchtop flow cytometer.
7.2.1.2 Expansion of ASCs In Vitro
Several investigators suggest that seeding density may affect cell proliferation. There is once again no consensus among investigators regarding the seeding densities used, although most investigators use a seeding density of 5 × 103 cells/cm2 during the ASC expansion phase. The effect of seeding density on MSC proliferation was demonstrated with BM-MSCs that were seeded at 1 × 102 and 5 × 103 cells/cm2 (Both et al. 2007). The cells seeded at the lower density reached the target of 2 × 108 cells in total in a shorter time period compared to cells seeded at 5 × 103 cells/cm2. The cells seeded at the higher density took approximately 4 days longer to achieve the same target. Similar results were observed by Lode and colleagues in 2008 using synovial fat pad-derived MSCs seeded on three-dimensional scaffolds (Lode et al. 2008) and by Fossett and colleagues (2012) who showed that low seeding densities increase the proliferation capacity in vitro.
7.2.1.2.1 Cell Expansion for Research Purposes
The in vitro expansion of primary cells such as ASCs to reach therapeutic numbers requires that growth conditions mimic the physiological environment in vivo. Such an environment is achieved in vitro through the use of a synthetic basal culture medium enriched with growth factors, hormones and other necessary nutrients (Freshney 2006; Freshney 2010). In addition, ASC proliferation may be stimulated by several exogenous supplements including fibroblast growth factor 2 (FGF-2), sphingosylphosphorylcholine, platelet-derived growth factors and others (Song et al. 2005; Kang et al. 2005; Chiou et al. 2006; Jeon et al. 2006; Mizuno 2009).
Traditionally, ASCs are expanded and maintained in culture medium (DMEM or α-MEM) containing serum. The use of serum during ASC expansion plays a vital role in ASC attachment, longevity and proliferation as it contains essential components such as amino acids, growth factors, hormones, lipids, vitamins, adhesion factors, binding proteins, spreading factors and other trace elements (Lennon et al. 1995; Lennon et al. 1996; Van Der Valk et al. 2004, 2010). Foetal bovine serum (FBS) is widely accepted as the stand ard serum supplement for in vitro studies in the research and experimental setting (Sotiropoulou et al. 2006; Chen et al. 2009). Many disadvantages such as batch-to-batch variation and xeno-immunization are however associated with the use of FBS. Another associated risk is the transmission of zoonotic disease through contamination with Mycoplasma, viruses and prions (Lennon et al. 1995, 1996; Van Der Valk et al. 2004; Kocaoemer et al. 2007; Van Der Valk et al. 2010; Chieregato et al. 2011; Kølle et al. 2013; Kyllonen et al. 2013).
Preparation of cellular therapy products under GMP conditions requires the minimal use of animal-derived products in the manufacturing process (Müller et al. 2006; Crespo-Diaz et al. 2011). Consequently, several human and chemical alternatives are being explored in GMP manufacturing of ASC cell therapy products. Human-derived alternatives such as human serum (Stute et al. 2004), platelet lysate (Schallmoser et al. 2007) and platelet-rich plasma (Doucet et al. 2005) (Table 7.1) may potentially result in an enhanced culture environment that more accurately mimics the in vivo environment (Azouna et al. 2012). However, their relative efficacy is still largely debated, and there is currently no consensus on which of the alternatives is the best to replace FBS (Baer et al. 2010; Bieback et al. 2012; Patrikoski et al. 2013; Koellensperger et al. 2014).
7.2.1.2.2 Xeno- and Serum-Free Cell Expansion for Clinical Application
Synthetic, chemically defined serum-free medium has been recommended as a potential replacement to animal-derived ser um as it contains the necessary growth factors required to sustain ASC expansion (Van Der Valk et al. 2004, 2010). Nonetheless, many studies have revealed that when using serum-free medium, ASCs are incapable of adhering to plastic without the addition of coating agents. In addition, ready-to-use serum-free medium is expensive and might therefore be a less cost-effective alternative for the replacement of FBS in the clinical setting (Lennon et al. 1995; Lund et al. 2009; Baer et al. 2010; Rajala et al. 2010; Yang et al. 2012; Kyllonen et al. 2013; Patrikoski et al. 2013).
7.2.2 Isolation Using Automated Closed Systems
An automated closed system allows for minimal handling of a sample by the operator, resulting in a potentially safer, less labour-intensive isolation procedure. Contamination is a highly undesirable event in the clinical setting and is limited through strict control of standard operating procedures (SOPs) and processing according to GMP (Gimble et al. 2011). Although the manual isolation procedure is relatively easy to reproduce and is carried out in a sterile environment, there is a higher contamination risk when compared to a closed system. The introduction of contaminating particles into the culture system is significantly reduced using closed systems. Hicok and colleagues suggest that in order to comply with the regulations involved in a clinical trial, an automated system for cell isolation should be used (Hicok and Hedrick 2011). Commercial companies such as Cytori Therapeutics Inc (San Diego, USA) and Biosafe Ltd (Lake Geneva, Switzerland) have designed state-of-the art cell-processing devices that allow for the isolation of the AD-SVF from lipoaspirate in automated closed systems. It has been suggested that automated systems may contribute to the advancement of clinical research and the translation thereof into clinical trials.
The Sepax® cell-processing system (Biosafe Ltd, Eysins, Switzerland) was developed to isolate and concentrate nucleated cells from the umbilical cord blood, peripheral blood or bone marrow (Zingsem et al. 2003; Aktas et al. 2008; Zinno et al. 2011). The successful application of the system in the HSC field resulted in the system also being validated for the isolation of AD-SVF cells from human adipose tissue (Guven et al. 2012). The Sepax® system automates the cell washing and concentration steps during adipose tissue processing. The device is comprised of the main Sepax® sample processing unit, single-use sterile disposable sample processing kits and ready-to-use application-specific software protocols.
In Asia, Australia and Europe, but not in the USA, the Cytori’s Celution® sample processing system has been granted regulatory approval for the use in adipose tissue processing and AD-SVF cell isolation. Multiple clinical trials are ongoing in several countries using the Celution® system (Akita et al. 2012; Houtgraaf et al. 2012). This automated system was specifically designed for fat processing and to mimic the modified manual isolation steps from the method de scribed by Zuk and colleagues (Zuk et al. 2001, 2002). The Celution® system is a fully closed automated system, which uses a single-use sterile disposable kit comprised of a processing canister, connection tubes and waste bags. The Celase® enzyme is used for adipose tissue digestion instead of collagenase. One of the advantages of the system is that it performs several quality control processes before the automated sample processing is initiated. For example, upon insertion of the sample processing kit into the device, the Celution® system performs a wet test to validate the integrity of the kit by checking for any leakages in the system and consequently ensures that the sample will be processed in a fully closed manner. After the lipoaspirate suspension is introduced into the processing canister, the system automatically weighs the sample and performs rinsing steps with Ringer’s lactate solution to remove blood cells and other contaminants. The Celution® system automatically calculates the amount of Celase® enzyme required for proper digestion of the volume of adipose tissue processed. The instrument will indicate the volume of Celase® enzyme which needs to be added manually. Enzymatic digestion takes place automatically through constant agitation. Following digestion and separation, the fraction of cells released from the adipose tissue matrix is pumped into the centrifuge chamber where it is washed and concentrated into the output chambers on either end of the centrifuge chamber. The final AD-SVF cell product is then aspirated from the output chambers (~5 ml).
Evaluation of the different automated systems needs to take into account factors such as the ability to process a large variety of lipoaspirate volumes and the degree of automation it provides. Practical considerations include the cost of the device and the consumables required. Sample processing time (which is influenced by the volume of lipoaspirate introduced) may also be a consideration. It should be noted that the Sepax® system is not fully automated as adipose tissue digestion needs to be carried out manually. However, an automated adipose tissue protocol has been developed for the isolation of the AD-SVF following digestion. In comparison, the Celution® system i s a fully automated system validated for the processing of adipose tissue and requires minimal intervention by the operator.
7.3 In Vitro Characterization of ASCs
In 2006, the ISCT released a position statement defining the minimal criteria required to identify ASCs (Dominici et al. 2006). According to these guidelines, ASCs and BM-MSCs share the same characteristics, namely, that they are multipotent, plastic-adherent cells that express specific surface antigens and have the capacity to differentiate into adipocytes, chondrocytes and osteoblasts. These criteria became the gold standard for all in vitro studies involving ASCs. However, in 2013 a new set of criteria was suggested by the IFATS and the ISCT. The overall criteria remained essentially unchanged, except that the report suggested a repertoire of protein surface markers that will allow investigators to distinguish between AD-SVF, ASCs and BM-MSCs (Bourin et al. 2013).
7.3.1 Immunophenotypic Characterization of ASCs
The challenge in phenotyping ASCs is that none of the surface expression markers used is specific to ASCs (Table 7.2). Therefore, to be more confident in the phenotypic assessment of ASCs, it is advisable to follow a multicolour flow cytometric approach, where multiple surface protein markers are simultaneously stained with various fluorochrome-conjugated antibodies to provide a more accurate co-expression profile of the cells (Zimmerlin et al. 2013; Bourin et al. 2013; Baer 2014; Donnenberg et al. 2015). According to the IFATS criteria, more than 80 % of the cells should co-express the following markers: CD13, CD29, CD36, CD44, CD73, CD90 and CD105 (Bourin et al. 2013). There should be less than 2 % haematopoietic cells as well as endothelial cells present in an ASC isolate. Consequently less than 2 % of the cells should stain positive for CD31 (endothelial cells), CD45 (leukocytes) and CD235a (erythrocytes) (Bourin et al. 2013). Simultaneous staining of cell surface markers allows for better discrimination of ASCs from other cells that may be present in the isolation, but do not adhere to the above-mentioned co-expression profile.
7.3.2 In Vitro Differentiation Assessments and Limitations
It is well established that ASCs or subpopulations thereof have the ability to differentiate into adipocytes, osteoblasts, chondrocytes and myocytes. The differentiation capacity of ASCs suggests a role in the turnover and maintenance of adult mesenchymal tissues (Caplan 2009). In vitro differentiation of ASCs into these lineages (adipocytes, chondrocytes, osteoblasts and myocytes) is therefore traditionally used to confirm the multipotent characteristics of the isolated ASCs. However, true stem cell plasticity entails the ability of these cells to form tissues from different germ layers. Investigators have shown that under appropriate culture conditions, ASC subpopulations display stem cell plasticity by differentiating into non-mesodermal tissues such as neurons (Ning et al. 2006) and epithelial cells (Brzoska et al. 2005). However, a significant amount of research is still needed to confirm the purported plasticity of ASCs. Effective clinical application of ASCs will rely on understanding the biological effectors that are responsible for maintaining a specific differentiation state. In addition to the criteria set out by the IFATS and ISCT above, it has been suggested that histochemical staining techniques used to establish differentiation are qualitative and that a quantitative approach, such as quantitative reverse transcription polymerase chain reaction (RT-qPCR), is required. The minimum number of genes required to confirm the differentiation of ASCs into adipocytes, osteoblasts, chondrocytes and myocytes, respectively, is summarized in Table 7.3.
7.3.2.1 Adipogenesis
High-calorie intake without energy expenditure promotes adipocyte hyperplasia. Signalling factors are responsible for this increase in adipocytes by inducing the conversion of ASCs to preadipocytes and finally differentiation into mature adipocytes (Tang and Lane 2012). ASCs become committed to preadipocytes by bone morphogenetic protein (BMP) -2 and -4 (Huang et al. 2009) and Wnt signalling (Bowers and Lane 2008), causing them to go into growth arrest (Fig. 7.1). Differentiation inducers such as hormones and mitogens (insulin, glucocorticoids and cAMP) then initiate mitotic clonal expansion where the cells’ fibroblastic morphology is lost and the accumulation of cytoplasmic triglycerides occurs, eventually forming mature adipocytes (Tang et al. 2004). Peroxisome proliferator-activated receptor-γ (PPAR-γ) and CCAAT/enhancer-binding proteins (C/EBP) are key regulators of adipogenesis (Kang et al. 2007).

Regulation of ASC differ entiation. Fat accumulation, cartilage formation, bone formation and bone resorption are closely related. Adipocytes, osteoblasts and chondrocytes have a common origin, the mesenchymal stem cell, while osteoclasts originate from the haematopoietic stem cell. Osteoblasts regulate bone resorption through the RANKL/RANK/OPG pathway. Osteoblast differentiation is regulated by RUNX2. Key regulators of adipogenesis are PPAR-γ and C/EBP. Adipocytes secrete inflammatory cytokines and hormones which are capable of modulating bone formation. Chondrogenesis is mainly regulated by BMPs, TGF-β and Sox9. Modified from Fig. 7.1 in Cao (2011) and Rosen and Bouxsein (2006). RUNX2 runt-related transcription factor 2, RANK/RANKL receptor activator of nuclear factor-kB ligand, OPG osteoprotegerin, PPAR-γ peroxisome proliferator-activated receptor-γ, C/EBP CCAAT/enhancer-binding proteins, BMP bone morphogenetic protein, TGF transforming growth factor, Sox sex-determining region Y-type high mobility group box (Cao 2011; Rosen and Bouxsein 2006)
In the laboratory, in vitro adipogenic differentiation takes 2–3 weeks after initiating differentiation following exposure of the cells to the appropriate induction media . The composition of the adipogenic induction medium is provided in the supplementary material. Adipogenesis is associated with the accumulation of lipid droplets in the cytoplasm of the adipocytes. The number and size of lipid droplets increase as the ASCs differentiate from preadipocytes to mature adipocytes. The lipid droplets are highly organized organelles, consisting of an outer layer of amphipathic lipids such as phospholipids and a core of neutral lipids including triacylglyceride and sterol esters (Guo et al. 2009; Krahmer et al. 2009). Studies using immortalized preadipocytes, such as the mouse 3 T3-L1 cell line, revealed that combinations of 3-isobutyl-methylxanthine with dexamethasone or insulin induce intracellular lipid accumulation. Therefore, to promote the effective differentiation of ASCs into adipocytes, the induction medium is supplemented with IBMX, insulin and dexamethasone. 3-Isobutyl-methylxanthine (IBMX), a cAMP-phosphodiesterase inhibitor, has been shown to inhibit soluble cyclic nucleotide phosphodiesterase activity causing an increase in intracellular cAMP levels by stimulating the cAMP-dependent protein kinase pathway. This increase in cAMP levels is subsequently responsible for the synthesis of lipogenic enzymes during adipogenesis (Spiegelman and Green 1981; Tang and Lane 2012). Ali and colleagues have shown that the removal of IBMX from the induction medium almost completely blocks the normally observed increase in intracellular lipid accumulation (Ali et al. 2006). Insulin acts as a potent adipogenic hormone by triggering the induction of a series of transcription factor, which facilitate differentiation of preadipocytes into mature adipocytes via the insulin-like growth factor 1 (IGF-1) receptor (Ntambi and Young-Cheul 2000). Dexamethasone is a synthetic glucocorticoid agonist used to stimulate the glucocorticoid receptor pathway. Although each agent is able to initiate adipogenesis, an additive effect is observed when the factors are combined (Russell and Ho 1976; Rubin et al. 1978). Please refer to the supplementary material for a brief description of the method used to induce adipogenesis in vitro.
7.3.2.1.1 Assessment of Adipogenic Differentiation
Adipogenic differentiation may be measured using both quantitative and qualitative (qPCR) assays. The minimum number of genes required to confirm the differentiation of ASCs into adipocytes is summarized in Table 7.3. Most of the histological, qualitative assays make use of lipophilic dyes which mainly stain the triacylglycerides present in the lipid droplet core. IFATS and ISCT jointly recommend using the lipid-specific stains Oil Red O and Nile Red (9-diethylamino-5H-benzo[α]phenoxazine-5-one) as histological determinants of adipogenic differentiation (Bourin et al. 2013).
7.3.2.1.1.1 Quantitative Adipogenesis Assessment Using Oil Red O
Oil Red O is a nonfluorescent hydrophobic stain with a high affinity for neutral lipids, such as triacylglycerides, present in lipid droplets (Fig. 7.2). The stain can then be extracted from the cells using isopropanol and quantified by measuring the absorbance values spectrophotometrically, but this method of quantification lacks specificity due to non-specific Oil Red O staining (Ramírez-Zacarías et al. 1992). Please refer to the supplementary material for a brief description of the qualitative method used to evaluate adipogenesis differentiation usi ng Oil Red O in vitro.

Adipogenic lineage microscopy analysis. (a) Oil Red O-stained and 1 % Toluidine Blue-counter stained non-induced culture. The red arrow indicates Oil Red O residue overlaying the culture. The cells are fibroblast-like, small, slender and elongated as indicated by the blue arrow and conform to describe ASC morphology. (b) Oil Red O-stained and 1 % Toluidine Blue-counterstained adipogenic induced culture. Visible Oil Red O droplets confirm differentiation into the adipogenic linage. (c) A magnification snapshot of (b). The red arrow indicates the presence Oil Red O residue overlaying the cell. Yellow circles demonstrate adipocytes (cytoplasm completely filled with lipid droplets and also fusion of the droplets with associated lipid vacuole formation). Green rectangle demonstrates a more mature preadipocyte with incomplete lipid droplet formation. Blue triangle demonstrates a preadipocyte with an enlarged triangular shaped cell that conforms to the morphology of adipocytes but does not contain any visual Oil Red O-stained lipid vacuoles
7.3.2.1.1.2 Quantitative Adipogenesis Assessment Using Nile Red
Lipid droplet formation during adipocyte differentiation may also be visualized using fluorescent microscopy techniques (Fig. 7.3). Nile Red and Bodipy 493/503 are currently the most common lipid-specific dyes used during fluorescence microscopy assessment of lipid droplet formation (McNeil et al. 1991; Smyth and Wharton 1992; Brasaemle et al. 2000; Lo Surdo et al. 2013; Aldridge et al. 2013).

Visualization of adipocyt e formation following adipocyte differentiation using fluorescence microscopy. (a) ASCs were stained with Nile Red (2 μg/ml) and DAPI. (b) ASCs were stained with Bodipy 493/503 (20 μg/ml) and DAPI. Both images were captured at Day 21 after adipocyte differentiation was induced
7.3.2.1.1.3 Quantitative Adipogenesis Assessment Using Flow Cytometry
Flow cytometry provides a more quantitative measurement of both the proportion of cells containing intracellular lipid droplets and the degree of lipid accumulation within each cell (Fig. 7.4) (Fink et al. 2004; Schaedlich et al. 2010; Chazenbalk et al. 2011; Aldridge et al. 2013; Ceppo et al. 2014). Most investigators make use of the hydrophobic dye, Nile Red (Fink et al. 2004; Lu et al. 2010; Menssen et al. 2011; Lo Surdo et al. 2013; Aldridge et al. 2013). Nile Red is a solvatochromatic dye, meaning that it can change fluorescent colour in different polar environments (Fowler and Greenspan 1985; Greenspan et al. 1985). Nile Red emits yellow-gold fluorescence (emission >528 nm) when dissolved in neutral lipids such as triglycerides. The fluorescence emission spectrum of Nile Red shifts to the deep-red spectrum (>590 nm) when it dissolves in an amphipathic lipid environment such as phospholipids (Greenspan et al. 1985). However, when dissolved in lipid droplets, the yellow-gold fluorescence is more easily visualized than the deep-red fluorescence. This may be contributed to by the ratio of neutral lipids to phospholipids present in a lipid droplet.

Flow cytometric analysis of a dipocyte differentiation of ASCs after staining with Nile Red (20 ng/ml final concentration). Adipocyte differentiation is associated with the intracellular accumulation with lipid droplets, which consequently results in an increase in cellular complexity. Green-coloured events indicate early stages of intracellular lipid accumulation. Orange-coloured events are indicative of cells with higher levels of intracellular neutral lipid content due to the accumulation of intracellular lipid droplets (increase in cellular complexity). Flow cytometric analysis was performed on a Beckman Coulter 3-laser, 10-colour Gallios flow cytometer, after 21 days of incubation of ASCs in adipogenic medium
There are also several other hydrophobic fluorescent dyes available that may be useful in the quantitative assessment of adipogenesis, such as Bodipy 493/503. There are reports that Bodipy 493/503 is more specific than Nile Red, but further studies are needed to validate this statement (Gocze and Freeman 1994). A definite advantage of Bodipy 493/503 is that it has a more specific fluorescent emission spectrum range. This allows for easier combination of cell surface marker s with the lipid-specific dyes to study the phenotypic characteristics of adipocytes in more detail.
7.3.2.2 Osteogenesis
There appears to be a complex relationship between adipogenesis and osteogenesis . Both adipocytes and osteoblasts are derived from a common multipotent MSC, thus leading one to believe that the two pathways are interconnected (Fig. 7.1). Obesity increases adipocyte differentiation and fat accumulation and is believed to decrease osteoblast differentiation and bone formation. Several recent investigators have reviewed the complex relationship between adipogenesis and osteogenesis (Cao 2011; Liao 2014; Nuttall et al. 2014; Romagnoli and Brandi 2014; Atashi et al. 2015).
The osteogenic maturation pathway involves cell proliferation, differentiation and matrix deposition followed by mineralization. Differentiation of ASCs into osteoblasts in vitro involves incubating a monolayer of ASCs with ascorbic acid, β-glycerophosphate and dexamethasone for 3 weeks. The composition of the osteogenic induction medium is provided in the supplementary material. Ascorbic acid acts as a cofactor for collagen synthesis through the hydroxylation of proline and lysine residues in collagen and induces extracellular matrix (ECM) production through the synthesis of non-collagenous bone matrix proteins. β-Glycerophosphate is needed for calcification and mineralization of the ECM. Dexamethasone regulates osteoblastic gene expression (Fiorentini et al. 2011). Dexamethasone treatment has been shown to increase alkaline phosphatase activity in vitro which is required for matrix mineralization and morphological transformation to cuboidal-shaped cells (Cheng et al. 1994). Alizarin Red S staining is commonly used for identifying calcific deposition during matrix mineralization in osteogenic differentiation cultures which is an early marker for differentiation (Fig. 7.5). Please refer to the supplementary material for a brief description of the qualitative method used to evaluate osteogenic induction in vitro.

Light microscopy analysis of osteo genesis. (a) Alizarin Red S-stained non-induced culture (osteogenesis control) showing no visible calcium depositions. (b) Alizarin Red S-stained osteogenic induced culture showing the red calcium deposits within the bone matrix characteristic of bone formation
7.3.2.3 Chondrogenesis
Cartilage is a specialized connective tissue which consists of chondrocytes surrounded by an ECM. Chondrocytes produce and maintain an ECM which is rich in glycosaminoglycans and proteoglycans and interact with collagen and elastic fibres. The cartilage matrix is composed mainly of type II collagen, providing tensile strength, and aggrecan, which provides compressive strength. Cartilage is classified according to its composition. Hyaline cartilage consists mainly of type II collagen fibres, elastic cartilage of type II collagen and elastic fibres and fibrocartilage of type I collagen fibres (Naumann et al. 2002). Chondrogenesis involves the recruitment and condensation of MSCs followed by differentiation into chondroblasts, chondrocyte maturation, and ECM production.
In vitro chondrogenesis is induced by artificially condensing the cells by centrifugation followed by addition of TGF-β3, dexamethasone and BMP-2 to the medium. The composition of the chondrogenic induction medium is provided in the supplementary material. Johnstone and colleagues (1998) developed a culture system to facilitate chondrogenic differentiation for BM-MSCs (Johnstone et al. 1998). This pellet culture system allows for cell-cell interaction similar to those that occur during embryonic development (Fell 1925). It has been shown that a defined medium, to which dexamethasone and TGF-β1 have been added, is required (Johnstone et al. 1998). Dexamethasone induces a metachromatic staining pattern in aggregated cells. This indicates the synthesis of a cartilaginous matrix which was confirmed by collagen type II immunohistochemistry. The use of TGF-β1 either alone or in combination with dexamethasone induces chondrogenesis in aggregated cells. It was later found that the addition of BMP-6 further increased chondrogenesis (Sekiya et al. 2001). Critical roles are played by BMPs in compaction of MSCs and for the shaping of the condensations (Zuscik et al. 2008). Comparisons between BMP-2, -4, and -6 with regard to their ability to enhance in vitro formation of cartilage demonstrated that BMP-2 was the most effective, resulting in increased pellet weight along with more proteoglycan and collagen type II production (Sekiya et al. 2005). However, the addition of BMPs to cartilage induction medium is not necessarily required. Please refer to the supplementary material for a brief description of the method used to induce chondrogenesis in vitro.
More detailed strategies for the assessment of chondrogenic differentiation in vitro include techniques such as light microscopy, transmission electron microscopy and immunohistochemistry (Ichinose et al. 2005). Please refer to the supplementary material for a brief description of the qualitative method used to evaluate the induction of chondrogenesis in vitro. Other studies have shown that after a week in induction medium, cell pellets consist of three layers: the superficial zone, containing fibroblast-like cells; the middle zone, containing apoptotic cells; and the deep zone, containing matrix-producing chondrocyte-like cells. After 14 days, the middle zone disappears, and the deep zone dominates after an induction period of 21 days (Ichinose et al. 2005). Biochemical analysis of chondrogene sis includes methods that quantify the total glycosaminoglycan content in pellets (Naumann et al. 2002; Estes et al. 2010).
7.3.2.4 Myogenesis
The in vivo myogenic differentiation capacity of stem cells is highly sought after in regenerative medicine, as this property brings hope that these cells may restore regenerative capacity in muscular and neuromuscular disorders, myoskeletal tissue trauma, sport injuries and urologic incontinence. Benefits associated with this type of intervention include faster recovery time and a reduced risk of graft rejection, and preclinical evidence supports the application of MSCs across allogeneic barriers (Joo et al. 2014). Several lines of evidence exist for successful in vitro myogenic differentiation of MSCs from various origins (Gang et al. 2004). However, successful clinical translation remains to be convincingly demonstrated.
ASCs display superior myogenic differentiation potential (Stern-Straeter et al. 2014; Zych et al. 2013) and might be an ideal candidate for application in muscle tissue engineering of fibrotic muscle (Choi et al. 2012). ASCs can also be induced to differentiate into cardiomyocytes following induction by 5-azacytidine (Cao et al. 2004; Carvalho et al. 2013). Co-culturing of ASCs with primary myoblasts, in addition to stimulation with dexamethasone and FGF-2, promotes differentiation up to multinucleated myotubes (Beier et al. 2011; Bitto et al. 2013). Although no minimal criteria have been established for the myogenic differentiation potential of ASCs, the molecular markers in Table 7.3 have been used for quantitative assessmen t of myogenic differentiation.
7.3.3 Experimental Animal Models
In order to reach the long-term aim of clinical translation of cell-based therapy, preclinical safety and efficacy need to be demonstrated in a nimal models. Transplantation of in vitro expanded ASCs in the appropriate experimental animal model is therefore an important step in the development of cell therapy products. Sensitive cell-tracking techniques are needed in order to determine the most optimal route of administration as well as the homing ability of the transplanted ASCs. Transduced ASCs containing GFP lentiviral vectors could offer a feasible in vivo tracking system as it has been shown that transduction of ASCs with a GFP-expressing lentiviral vector does not affect their phenotypic expression or their differentiation potential (van Vollenstee et al. 2016).
7.4 Conclusion
Although manual isolation procedures are less costly than closed automated systems, the risk of contamination associated with manual procedures makes them a less attractive option for clinical applications. GMP isolation and expansion procedures require the minimal use of xenogeneic components, and only cell therapy products that are free of animal-derived products will lead to successful translation into the clinic. ASCs are regarded as non-minimally manipulated cells and are classified as a drug by the Food and Drug Administration (FDA). However, it is currently unclear whether in vitro expanded ASCs hold any significant benefit over AD-SVF, particularly in the clinical setting.
One of the major pitfalls in the rapidly expanding field of ASCs is that no inclusive panel of either cellular or molecular markers exists that specifically characterize these cells. The ISCT and IFATS regularly publish updated guidelines for the classification of ASCs. However, the current guidelines are still not specific and are based on the ability of the cells to adhere to plastic, to express a set of non-specific cellular markers and to differentiate into adipogenic, osteogenic, chondrogenic and myogenic lineages.
For example, although classical stains (Oil Red O, Alizarin Red S and Toluidine Blue) confirm lineage differentiation in vitro, it is unclear if all induced ASCs have differentiated into the respective lineage. The translation of in vitro assays to the patient is also unclear, and well-characterized in vivo experimental models are needed to validate the engrafting, homing and differentiation potential of the cell therapy in question. The availability of reliable and reproducible in vivo experimental models will therefore contribute to a more confident and potentially more rapid translation of ASC research to the clinical setting.
References
Akita S, Yoshimoto H, Ohtsuru A et al (2012) Autologous adipose-derived regenerative cells are effective for chronic intractable radiation injuries. Radiat Prot Dosimetry 151:656–660
Aktas M, Radke TF, Strauer BE et al (2008) Separation of adult bone marrow mononuclear cells using the automated closed separation system Sepax. Cytotherapy 10:203–211
Aldridge A, Kouroupis D, Churchman S et al (2013) Assay validation for the assessment of adipogenesis of multipotential stromal cells-a direct comparison of four different methods. Cytotherapy 15:89–101
Ali AT, Penny CB, Paiker JE et al (2006) The relationship between alkaline phosphatase activity and intracellular lipid accumulation in murine 3T3-L1 cells and human preadipocytes. Anal Biochem 354:247–254
Atashi F, Modarressi A, Pepper MS (2015) The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev 4:1150–1163
Azouna NB, Jenhani F, Regaya Z et al (2012) Phenotypical and functional characteristics of mesenchymal stem cells from bone marrow: comparison of culture using different media supplemented with human platelet lysate or fetal bovine serum. Stem Cell Res Ther 3:6
Baer PC, Griesche N, Luttmann W et al (2010) Human adipose-derived mesenchymal stem cells in vitro: evaluation of an optimal expansion medium preserving stemness. Cytotherapy 12:96–106
Baer PC (2014) Adipose derived mesenchymal stromal/stem cell: An update on their phenotype in vivo and in vitro. World J Stem Cells 6(3):256–265
Banyard DA, Salibian AA, Widgerow AD et al (2015) Implications for human adiposederived stem cells in plastic surgery. J Cell Mol Med 19(1):21–30
Beier JP, Bitto FF, Lange C et al (2011) Myogenic differentiation of mesenchymal stem cells co-cultured with primary myoblasts. Cell Biol Int 35:397–406
Bernardo ME, Avanzini MA, Perotti C et al (2006) Optimization of in vitro expansion of human multipotent mesenchymal stromal cells for cell-therapy approaches: Further insights in the search for a fetal calf serum substitute. J Cell Physiol 211(1):121–130
Bernardo ME, Cometa AM, Pagliara D et al (2011) Ex vivo expansion of mesenchymal stromal cells. Best Pract Res Clin Haematol 24(1):73–81
Bieback K, Hecker A, Kocaömer A et al (2009) Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells 27(9):2331–2341
Bieback K, Hecker A, Schlechter T et al (2012) Replicative aging and differentiation potential of human adipose tissue-derived mesenchymal stromal cells expanded in pooled human or fetal bovine serum. Cytotherapy 14:570–583
Bitto FF, Klumpp D, Lange C et al (2013) Myogenic differentiation of mesenchymal stem cells in a newly developed neurotised av-loop model. BioMed Res Int 2013:935046
Both SK, van der Muijsenberg AJ, van Bitterswijk CA (2007) A rapid and efficient method for expansion of human mesenchymal stem cells. Tissue Eng 13(1):3–9
Bourin P, Bunnell BA, Casteilla L et al (2013) Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: a joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International So. Cytotherapy 15:641–648
Bowers RR, Lane MD (2008) Wnt signaling and adipocyte lineage commitment. Cell Cycle 7:1191–1196
Brasaemle DL, Rubin B, Harten IA et al (2000) Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J Biol Chem 275(49):38486–38493
Brzoska M, Geiger H, Gauer S et al (2005) Epithelial differentiation of human adipose tissue-derived adult stem cells. Biochem Biophys Res Commun 330(1):142–150
Bunnell BA, Flaat M, Gagliardi C et al (2008) Adipose-derived stem cells: isolation, expansion and differentiation. Methods 45:115–120
Cao J (2011) Effects of obesity on bone metabolism. J Orthop Surg Res 6:30
Cao F, Niu LL, Meng L et al (2004) Cardiomyocyte-like differentiation of human bone marrow mesenchymal stem cells after exposure of 5-azacytidine in vitro. Shi Yan Sheng Wu Xue Bao 37:118–124
Caplan AI (2009) Why are MSCs therapeutic? New data: new insight. J Pathol 217:318–324
Carvalho PH, Daibert APF, Monteiro BS et al (2013) Diferenciação de células-tronco mesenquimais derivadas do tecido adiposo em cardiomiócitos. Arq Bras Cardiol 100:82–89
Ceppo F, Berthou F, Jager J et al (2014) Implication of the Tpl2 kinase in inflammatory changes and insulin resistance induced by the interaction between adipocytes and macrophages. Endocrinology 155:951–964
Chazenbalk G, Bertolotto C, Heneidi S et al (2011) Novel pathway of adipogenesis through cross-talk between adipose tissue macrophages, adipose stem cells and adipocytes: evidence of cell plasticity. PLoS One 6(3), e17834. doi:10.1371/journal.pone.0017834
Chen HH, Decot V, Ouyang JP et al (2009) In vitro initial expansion of mesenchymal stem cells is influenced by the culture parameters used in the isolation process. Biomed Mater Eng 19:301–309
Cheng SL, Yang JW, Rifas L et al (1994) Differentiation of human bone marrow osteogenic stromal cells in vitro: induction of the osteoblast phenotype by dexamethasone. Endocrinology 134:277–286
Chieregato K, Castegnaro S, Madeo D et al (2011) Epidermal growth factor, basic fibroblast growth factor and platelet-derived growth factor-bb can substitute for fetal bovine serum and compete with human platelet-rich plasma in the ex vivo expansion of mesenchymal stromal cells derived from adipose tissue. Cytotherapy 13(8):933–943
Chiou M, Xu Y, Longaker MT (2006) Mitogenic and chondrogenic effects of fibroblast growth factor-2 in adipose-derived mesenchymal cell. Biochem Biophys Res Commun 343:644–652
Choi YS, Vincent LG, Lee AR et al (2012) Mechanical derivation of functional myotubes from adipose-derived stem cells. Biomaterials 33:2482–2491
Crespo-Diaz R, Behfar A, Butler GW et al (2011) Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplant 20:797–811
Dominici M, Le Blanc K, Mueller I et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Donnenberg AD, Meyer EM, Rubin JP et al (2015) The cell-surface proteome of cultured adipose stromal cells. Cytometry 87(7):665–674
Doucet C, Ernou I, Zhang Y et al (2005) Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell-based therapy applications. J Cell Physiol 205:228–236. doi:10.1002/jcp.20391
Ersek RA, Salisbury AV (1995) Circumferential liposuction of knees, calves and ankles. Aesthetic Plast Surg 19:321–333
Estes BT, Diekman BO, Gimble JM, Guilak F (2010) Isolation of adipose-derived stem cells and their induction to a chondrogenic phenotype. Nat Protoc 5:1294–1311
Fei W, Du X, Yang H (2011) Seipin, adipogenesis and lipid droplets. Trends Endocrinol Metab 22:204–210. doi:10.1016/j.tem.2011.02.004
Fell HB (1925) The histogenesis of cartilage and bone in the long bones of the embryonic fowl. J Morphol 40:417–459
Fink T, Abildtrup L, Fogd K et al (2004) Induction of adipocyte-like phenotype in human mesenchymal stem cells by hypoxia. Stem Cells 22:1346–1355
Fiorentini E, Granchi D, Leonardi E et al (2011) Effects of osteogenic differentiation inducers on in vitro expanded adult mesenchymal stromal cells. Int J Artif Organs 34:998–1011
Fossett E, Khan WS, Longo UG et al (2012) Effect of age and gender on cell proliferation and cell surface characterization of synovial fat pad derived mesenchymal stem cells. J Orthop Res 30:1013–1018
Fournier PF, Otteni FM (1983) Lipodissection in body sculpturing: the dry procedure. Plast Reconstr Surg 72:598–609
Fowler SD, Greenspan P (1985) Application of Nile red, a fluorescent hydrophobic probe, for the detection of neutral lipid deposits in tissue sections: comparison with oil red O. J Histochem Cytochem 33:833–836
Fraser JK, Wulur I, Alfonso Z et al (2006) Fat tissue: an underappreciated source of stem cells for biotechnology. Trends Biotechnol 24:150–154
Freshney RI (2006) Basic principles of cell culture. Culture of cells for tissue engineering. Wiley, New Jersey. doi:10.1002/0471741817.ch1
Freshney RI (2010) Culture of animal cells: a manual of basic technique and specialized applications, 6th edn. Wiley, New York
Gang EJ, Jeong JA, Hong SH et al (2004) Skeletal myogenic differentiation of mesenchymal stem cells isolated from human umbilical cord blood. Stem Cells 22:617–624
Gimble JM, Adam JK, Bruce AB (2007) Adipose-derived stem cells for regenerative medicine. Circ Res 100:1249–1260
Gimble JM, Bunnell BA, Chiu ES et al (2011) Concise review: adipose-derived stromal vascular fraction cells and stem cells: let’s not get lost in translation. Stem Cells 29:749–754
Gocze PM, Freeman D (1994) Factors underlying the variability of lipid droplet fluorescence in MA-10 Leydig tumor cells. Cytometry 17:151–158
Greenspan P, Mayer EP, Fowler SD (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 100:965–973
Guo Y, Cordes KR, Farese RV et al (2009) Lipid droplets at a glance. J Cell Sci 122:749–752
Guven S, Karagianni M, Schwalbe M et al (2012) Validation of an automated procedure to isolate human adipose tissue-derived cells by using the Sepax(R) technology. Tissue Eng Part C Methods 18:575–582
Hicok KC, Hedrick MH (2011) Automated isolation and processing of adipose-derived stem and regenerative cells. Methods Mol Biol 702:87–105
Houtgraaf JH, den Dekker WK, van Dalen BM et al (2012) First experience in humans using adipose tissue-derived regenerative cells in the treatment of patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol 59:539–540
Huang H, Song TJ, Li X et al (2009) BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A 106:12670–12675
Ichinose S, Tagami M, Muneta T et al (2005) Morphological examination during in vitro cartilage formation by human mesenchymal stem cells. Cell Tissue Res 322:217–226
Jeon ES, Song HY, Kim MR et al (2006) Sphingosylphosphorylcholine induces proliferation of human adipose tissue-derived mesenchymal stem cells via activation of JNK. J Lipid Res 47:653–664
Johnstone B, Hering TM, Caplan AI et al (1998) In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp Cell Res 238:265–272
Jones A, Kinsey SE, English A et al (2002) Isolation and characterisation of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum 46:3349–3360
Joo S, Lim HJ, Jackson JD et al (2014) Myogenic-induced mesenchymal stem cells are capable of modulating the immune response by regulatory T cells. J Tissue Eng 5:2041731414524758
Kang YJ, Jeon ES, Song HY et al (2005) Role of c-Jun N-terminal kinase in the PDGF-induced proliferation and migration of human adipose tissue-derived mesenchymal stem cells. J Cell Biochem 95:1135–1145
Kang S, Bennett CN, Gerin I et al (2007) Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor γ. J Biol Chem 282:14515–14524
Kern S, Eichler H, Stoeve J et al (2006) Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 24:1294–1301
Kocaoemer A, Kern S, Klüter H et al (2007) Human AB serum and thrombin-activated platelet-rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells 25:1270–1278
Koellensperger E, Bollinger N, Dexheimer V et al (2014) Choosing the right type of serum for different applications of human adipose tissue-derived stem cells: influence on proliferation and differentiation abilities. Cytotherapy 16:789–799
Kølle ST, Oliveri RS, Glovinski PV et al (2013) Pooled human lysate versus fetal bovine serum—Investigating the proliferation rate, chromosome stability and angiogenic potential of human adipose tissueederived stem cells intended for clinical use. Cytotherapy 15(9):1086–1097
Krahmer N, Guo Y, Farese RV et al (2009) SnapShot: lipid droplets. Cell 139:10–11
Kyllonen L, Haimi S, Mannerstrom B et al (2013) Effects of different serum conditions on osteogenic differentiation of human adipose stem cells in vitro. Stem Cell Res Ther 4:1–17
Lennon DP, Haynesworth SE, Young RG et al (1995) A chemically defined medium supports in vitro proliferation and maintains the osteochondral potential of rat marrow-derived mesenchymal stem cells. Exp Cell Res 219:211–222
Lennon DP, Haynesworth SE, Bruder SP et al (1996) Human and animal mesenchymal progenitor cells from bone marrow: identification of serum for optimal selection and proliferation. Vitro Cell Dev Biol Animal 32(10):602–611
Lo Surdo JL, Millis B, Bauer SR (2013) Automated microscopy as a quantitative method to measure differences in adipogenic differentiation in preparations of human mesenchymal stromal cells. Cytotherapy 15:1527–1540
Lode A, Bernhardt A, Gelinsky M (2008) Cultivation of human bone marrow stromal cells on three-dimensional scaffolds of mineralized collagen: influence of seeding density on colonization, proliferation and osteogenic differentiation. J Tissue Eng Regen Med 2:400–407
Lu X, Alshemali S, de Wynter EA et al (2010) Mesenchymal stem cells from CD34(-) human umbilical cord blood. Transfus Med 20:178–184
Lund P, Pilgaard L, Duroux M et al (2009) Effect of growth media and serum replacements on the proliferation and differentiation of adipose-derived stem cells. Cytotherapy 11:189–197
McNeil M, Daffe M, Brennan PJ (1991) Location of the mycolyl ester substituents in the cell walls of mycobacteria. J Biol Chem 266:13217–13223
Menssen A, Häupl T, Sittinger M et al (2011) Differential gene expression profiling of human bone marrowderived mesenchymal stem cells during adipogenic development. BMC Genomics 12:461–477
Mizuno H (2009) Adipose-derived stem cells for tissue repair and regeneration: ten years of research and a literature review. J Nippon Med Sch 76:56–66
Müller I, Kordowich S, Holzwarth C et al (2006) Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy 8:437–444
Naumann A, Dennis JE, Awadallah A et al (2002) Immunochemical and mechanical characterization of cartilage subtypes in rabbit. J Histochem Cytochem 50:1049–1058
Ning H, Lin G, Lue TF et al (2006) Neuron-like differentiation of adipose tissue-derived stromal cells and vascular smooth muscle cells. Differentiation 74:510–518
Ntambi JM, Young-Cheul K (2000) Adipocyte differentiation and gene expression. J Nutr 130:3122S–3126S
Nuttall ME, Shah F, Singh V et al (2014) Adipocytes and the regulation of bone remodeling: A balancing act. Calcif Tissue Int 94(1):78–87
Patrikoski M, Juntunen M, Boucher S et al (2013) Development of fully defined xeno-free culture system for the preparation and propagation of cell therapy compliant human adipose stem cells. Stem Cell Res Ther 4:1–15
Rajala K, Lindroos B, Hussein SM et al (2010) A defined and xeno-free culture method enabling the establishment of clinical-grade human embryonic, induced pluripotent and adipose stem cells. PLoS One 5:1–14
Ramírez-Zacarías JL, Castro-Muñozledo F, Kuri-Harcuch W (1992) Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with oil red O. Histochem Cell Biol 97:493–497
Rodbell M (1966) Metabolism of isolated fat cells II. The similar effects of phospholipase c (clostridium perfringens alpha toxin) and of insulin on glucose and amino acid metabolism. J Biol Chem 241:130–139
Rodbell M, Jones AB (1966) Metabolism of isolated fat cells. III. The similar inhibitory action of phospholipase c (clostridium perfringens alpha toxin) and of insulin on lipolysis stimulated by lipolytic hormones and theophylline. J Biol Chem 241:140–142
Romagnoli C, Brandi ML (2014) Adipose mesenchymal stem cells in the field of bone tissue engineering. World J Stem Cells 6(2):144–152
Rosen CJ, Bouxsein ML (2006) Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol 2(1):35–43
Rubin CS, Hirsch A, Fung C et al (1978) Development of hormone receptors and hormonal responsiveness in vitro. Insulin receptors and insulin sensitivity in the preadipocyte and adipocyte forms of 3T3-L1 cells. J Biol Chem 253:7570–7578
Russell TR, Ho R (1976) Conversion of 3T3 fibroblasts into adipose cells: triggering of differentiation by prostaglandin F2alpha and 1-methyl-3-isobutyl xanthine. Proc Natl Acad Sci U S A 73:4516–4520
Schaedlich K, Knelangen JM, Santos AN et al (2010) A simple method to sort ESC-derived adipocytes. Cytometry 77A:990–995
Schallmoser K, Bartmann C, Rohde E et al (2007) Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion 47:1436–1446
Schallmoser K, Bartmann C, Rohde E et al (2010) Replicative senescence-associated gene expression changes in mesenchymal stromal cells are similar under different culture conditions. Haematologica 95(6):867–874
Sekiya I, Colter DC, Prockop DJ (2001) BMP-6 enhances chondrogenesis in a subpopulation of human marrow stromal cells. Biochem Biophys Res Commun 284:411–418
Sekiya I, Larson BL, Vuoristo JT et al (2005) Comparison of effect of BMP-2, −4, and −6 on in vitro cartilage formation of human adult stem cells from bone marrow stroma. Cell Tissue Res 320(2):269–276
Smyth MJ, Wharton W (1992) Differentiation of A31T6 preadipocytes to adipocytes: a flow cytometric analysis. Exp Cell Res 199:29–38
Song HY, Jeon ES, Jung JS et al (2005) Oncostatin M induces proliferation of human adipose tissue derived mesenchymal stem cells. Int J Biochem Cell Biol 37:2357–2365
Sotiropoulou PA, Perez SA, Salagianni M et al (2006) Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells 24:462–471
Spiegelman BM, Green H (1981) Cyclic AMP-mediated control of lipogenic enzyme synthesis during adipose differentiation of 3T3 cells. Cell 24:503–510
Stedman TL (2006) Stedman’s medical dictionary. Lippincott Williams & Wilkins, Baltimore, MD
Stern-Straeter J, Bonaterra GA, Juritz S et al (2014) Evaluation of the effects of different culture media on the myogenic differentiation potential of adipose tissue- or bone marrow-derived human mesenchymal stem cells. Int J Mol Med 33:160–170
Strem BM, Hicok KC, Zhu M et al (2005) Multipotential differentiation of adipose tissue-derived stem cells. Keio J Med 54:132–141
Stute N, Holtz K, Bubenheim M et al (2004) Autologous serum for isolation and expansion of human mesenchymal stem cells for clinical use. Exp Hematol 32:1212–1225
Tang QQ, Lane MD (2012) Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem 81:715–736
Tang QQ, Otto TC, Lane MD (2004) Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A 101(26):9607–9611
Van Der Valk J, Mellor D, Brands R et al (2004) The humane collection of fetal bovine serum and possibilities for serum-free cell and tissue culture. Toxicol In Vitro 18(1):1–12
Van Der Valk J, Brunner D, De Smet K et al (2010) Optimization of chemically defined cell culture media—replacing fetal bovine serum in mammalian in vitro methods. Toxicol In Vitro 24(4):1053–1063
van Vollenstee FA, Jackson C, Hoffmann D et al (2016) Human adipose derived mesenchymal stromal cells transduced with GFP lentiviral vectors: assessment of immunophenotype and differentiation capacity in vitro. Cytotechnology. doi:10.1007/s10616-016-9945-6
Von Heimburg D, Hemmrich K, Haydarlioglu S et al (2004) Comparison of viable cell yield from excised versus aspirated adipose tissue. Cells Tissues Organs 178:87–92
Yang S, Pilgaard L, Chase LG et al (2012) Defined xenogeneic-free and hypoxic environment provides superior conditions for long-term expansion of human adipose-derived stem cells. Tissue Eng Part C Methods 18:593–602
Zimmerlin L, Donnenberg VS, Rubin JP et al (2013) Mesenchymal markers on human adipose stem/progenitor cells. Cytometry 83(1):134–140
Zingsem J, Strasser E, Weisbach V et al (2003) Cord blood processing with an automated and functionally closed system. Transfusion 43:806–813
Zinno F, Landi F, Scerpa MC et al (2011) Processing of hematopoietic stem cells from peripheral blood before cryopreservation: use of a closed automated system. Transfusion 51:2656–2663
Zuk PA, Zhu M, Mizuno H et al (2001) Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7:211–228
Zuk PA, Zhu M, Ashjian P et al (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279–4295
Zuscik MJ, Hilton MJ, Zhang X et al (2008) Regulation of chondrogenesis and chondrocyte differentiation by stress. J Clin Invest 118:429–438
Zych J, Stimamiglio MA, Senegaglia AC et al (2013) The epigenetic modifiers 5-aza-2’-deoxycytidine and trichostatin A influence adipocyte differentiation in human mesenchymal stem cells. Braz J Med Biol Res 46:405–416
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix: Brief Description of Current Methods
Appendix: Brief Description of Current Methods
7.1.1 Manual Isolation Procedure
The isolation protocol is adapted from the procedures described by Zuk et al. (2001), Bunnell et al. (2008) and Estes et al. (2010) (Fig. A.1).

Outline of the procedure used f or isolating adipose-derived stromal cells. Lipoaspirate samples are processed by enzymatic digestion, and the stromal vascular fraction (SVF) is collected. Adipose-derived stromal cells from the SVF adhere to the plastic culture dish, and non-adherent cells are washed away after 24 h
The method requires the transfer of lipoaspirate material into 50 ml tubes (30 ml lipoaspirate/tube), followed by the addition of 20 ml phosphate buffer saline (PBS), supplemented with antibiotics (Fig. A.1). Depending on the future application of the adipose-derived stromal cell (ASC) isolates, culture medium may contain either FBS or human derivatives and antibiotics, usually penicillin and streptomycin (pen/strep). The adipose tissue is separated from peripheral blood contaminants by centrifugation at 1660 g for 3 min. The top oil layer is aspirated with a suction-assisted glass pipette system, and the compact lipoaspirate is carefully transferred to a sterile 50 ml tube. It is recommended that the washing steps are repeated until the compacted lipoaspirate material is golden yellow in colour without any visible evidence of peripheral blood contamination. The volume of the compacted lipoaspirate material should be recorded once most of the blood contamination has been removed.
To release resident ASCs in adipose tissue from the fibrous network, the lipoaspirate is enzymatically digested in culture plates. The most popular approach makes use of the enzyme, collagenase type I. Other enzymatic alternatives include dispase and trypsin. A filter-sterilized 0.1 % collagenase type I solution is prepared using PBS supplemented with 2 % antibiotics. The volume of collagen solution required is dependent on the volume of the compacted adipose tissue previously recorded. The ratio of compacted adipose tissue volume to the volume of collagen solution should be at least 2:1, meaning that the final volume of collagen digesting solution added to the adipose tissue in the culture plates should be half that of the washed compacted adipose tissue volume. The ratio of collagenase to adipose tissue should be optimized by each laboratory as well as for each isolation technique used. A sterile plastic pipette is used to mix the adipose tissue well, before incubation at 37 °C, 5 % CO2 for 45 min. The sample may either be continuously agitated using an automated rotating system or agitated manually every 15 min with a plastic pipette to aid the mechanical breakdown of the fibrous tissue.
The collagenase-digested sample is then transferred to sterile 50 ml tubes. The tubes are shaken vigorously and centrifuged at 500 g for 5 min, resulting in the SVF pellet settling to the bottom of the tube. The compacted adipose tissue and collagenase solution supernatants are carefully aspirated, and the collagenase activity is neutralized by adding 2 ml of stromal medium to the AD-SVF pellet. Stromal medium may consist of either Dulbecco’s Modified Eagle Medium (DMEM) containing GlutaMax™, 4.5 g/L D-glucose and pyruvate or alpha-Modified Eagle Medium (α-MEM) containing GlutaMax™, supplemented with 10 % serum and 1 % antibiotics.
The AD-SVF pellets are pooled into a single tube (15 or 50 ml) and centrifuged at 265 g for 5 min, followed by aspiration of the supernatant. Red blood cells present in the AD-SVF pellet are lysed either by the addition of an ammonium chloride-based lysing solution or an enzymatic-based lysing solution like VersaLyse™ (Beckman Coulter, Miami, USA). After a 10–15 min incubation period at room temperature, the lysing reaction is stopped by filling the tube with PBS supplemented with antibiotics and centrifuged at 184 g for 5 min. The supernatant is removed and the pellet resuspended in stromal medium before filtering the cellular suspension through a 70 μm cell strainer to remove any larger, undigested fragments.
In order to seed the cells at the correct seeding density, an absolute cell count should be performed. Cell counts may be performed by using either a manual approach in which the cells are counted using a haemocytometer or an automated cell counting device such as a flow cytometer. Details are provided below.
The AD-SVF is seeded at an initial seeding density of 5 × 105 cells per cm2. In order to determine the volume of cell suspension required for initial seeding, the following formula should be used:
After the cells are seeded, the cultures flasks are swirled gently to ensure uniform distribution. The culture flasks are maintained in an incubator under standard cell culture conditions (humidity, 5 % CO2 and 37 °C). The cultures are washed twice after 24 h with PBS supplemented with antibiotics to remove non-adherent cells, cellular debris and non-viable cells. Fresh stromal medium is added to the culture flasks and incubated under standard conditions until cells are 80–90 % confluent, implying that cells cover 80–90 % of the culture flask surface area. In order to determine the volume of cell suspension required for reseeding after thawing frozen samples, the following formula should be used:
7.1.2 Methods Used to Obtain Absolute Cell Counts
7.1.2.1 Trypan Blue (0.4 %) Dye Exclusion Method
Trypan blue is a non-membrane permeable vital stain that is used to assess the viability of cells. Trypan blue is not able to cross the cell membrane of intact, viable cells and therefore only stains cells with compromised cell membranes. Dead cells display a distinct blue colour after staining, due to the accumulation of trypan blue in the cytoplasm of cells with compromised cell membranes The recommended procedure is as follows: (1) prepare the Neubauer counting chamber (haemocytometer) by carefully placing the cover slip on the counting grid; (2) carefully mix 80 μl of a 0.4 % trypan blue with 100 μl PBS and 20 μl of the suspension; (3) carefully load 10 μl of the solution onto both sides of the Neubauer counting chamber (haemocytometer); and (4) count the viable (unstained) cells as well as dead cells (stained) using a microscope (10 times objective lens). The following formula is used to determine the absolute cell concentration (Fig. A.1):
Absolute cell concentration (cells/ml) \( =\left(\frac{Q1+Q2+Q3+\dots +Q8}{8}\right)\times \), where Q1–Q8 refer to eight quadrants on the haemocytometer. The factor 10 is to correct for the dilution of the sample with the 0.4 % trypan blue solution.
There are several commercial automated counting devices on the market that make use of trypan blue (0.4 %) dye exclusion assay principles. Examples of such devices are the Vi-Cell XR™ automated cell counter (Beckman Coulter, Miami, USA), Countess™ automated cell counter (Invitrogen, Carlsbad, USA) and TC20™ automated cell counter (Bio-Rad, Hercules, USA).
7.1.2.2 Absolute Cell Count Determination Using a Benchtop Flow Cytometer (Beckman Coulter Flow Cytometers)
An example of the strategy that is followed to obtain an absolute count on a benchtop flow cytometer is illustrated in Fig. A.2. The cell population of interest is identified by using a side scatter linear (SS lin) and a forward scatter linear (FS lin) histogram by placing a region around the cell population of interest only, excluding the counting beads and debris. The counting beads are enumerated in the CAL region. In order to obtain an accurate absolute count, it is important to count a sufficient number of cells as well as counting beads (>1000 events if possible) as well as to perform the count as soon as possible after the counting beads are mixed with the sample.

(a) A side scatter linear and forward scatter linear dot histogram displaying all the events measured by the flow cytometer. The flow beads (pink) and a gate were used to encircle the cell population that was counted until the CAL factor was reached. The gate-labelled cell population displays the cell population count that was expressed as the number of cells per μl cell suspension. (b) A histogram displaying the flow beads with a region of interest placed over the peak of the flow cytometry counting beads labelled as CAL. In this example, the specific calibration factor (assayed bead concentration) was 986
7.1.3 Induction of Adipogenesis In Vitro
ASCs are seeded at a density of 5 × 103 cells/cm2 in a six-well plate and maintained under standard culturing conditions (37 °C and 5 % CO2) with complete stromal medium (α-MEM or DMEM, containing 10 % FBS and 1 % pen/strep). When 70–80 % confluency is achieved, the stromal medium is removed and replaced with adipogenic induction medium (Table A.1) in three of the wells of the 6-well plate. DMEM supplemented with serum and antibiotics is added to the remaining three wells. These wells serve as non-induced controls. The cultures are maintained for 21 days under standard culturing conditions of 37 °C, 5 % CO2. During this period the induction and control media are replaced every second day.
7.1.3.1 Qualitative Assessment of Adipogenesis In Vitro Using Oil Red O
After 21 days of induction, the 6-well plates containing both induced and non-induced cultures are fixed by adding a 4 % formaldehyde solution for 1 h. A 0.5 % Oil Red O stock solution is prepared in isopropanol. An Oil Red O working solution is prepared from the stock solution by mixing three parts of the stock solution with two parts of double-distilled water (ddH2O) (volume/volume). After removal (aspiration) of the fixative solution, the cultures are allowed to dry at room temperature before adding 1 ml of Oil Red O working solution to both the adipogenic induced and non-induced cultures, followed by a 20 min incubation at room temperature. The stain is removed, and the wells are thoroughly washed with ddH2O until no pink discoloration of the freshly added ddH2O is visible to the naked eye.
The cultures are then counterstained with Toluidine Blue O. A 0.01 % Toluidine Blue O counterstain solution is prepared (pH 11) by adding 0.005 g Toluidine Blue O and 0.005 g Na2CO3 to 50 ml ddH2O. The Toluidine Blue O counterstain is added to the Oil Red O-stained cultures for 5 min at room temperature, after which the excess stain is washed away with ddH2O. 1 ml ddH2O is added to each well before microscopy analysis.
7.1.4 Induction of Osteogenesis In Vitro
Immunophenotyped ASCs are seeded at a density of 5 × 103 cells/cm2 in a six-well plate and maintained under standard culture conditions (37 °C and 5 % CO2) with stromal medium (α-MEM, containing 10 % FBS and 1 % pen/strep) until about 60–70 % confluency. Osteogenic induction medium (Table A.1) is introduced to half the wells and DMEM supplemented with serum and antibiotics to the other half to serve as non-induced (control) cultures. The cultures are maintained for 21 days under standard culture conditions of 37 °C, 5 % CO2. The induction and non-induction media are replaced every second day.
7.1.4.1 Qualitative Assessment of Osteogenesis In Vitro
Twenty-one days after induction of osteogenic diffe rentiation, the cells are fixed by addition of a 4 % formaldehyde fixative solution for 1 h. A 2 % Alizarin Red S classical stain is used to detect the calcium in the mineral matrix from mature osteocytes. An alizarin stock solution is prepared by adding 2 g Alizarin Red S powder to 100 ml of ddH2O. The solution is mixed thoroughly using a magnetic stirrer until solutes are dissolved before filtering through filter paper.
The induced and non-induced cultures are pre-washed with 2 ml PBS at pH 4.2 for 5 min before introducing 2 ml of the 2 % Alizarin Red S stain and incubating the cultures for 10 min at room temperature. The cultures are washed thoroughly with ddH2O to remove the excess stain. 1 ml ddH2O is added to each well before microscopy analysis.
7.1.5 Induction of Chondrogenesis In Vitro
A suspension culture technique is usually used for the differentiation of ASCs into chondrocytes. ASCs are seeded in a T25 flask at a density of 5 × 103 cells/cm2 and maintained under standard culture conditions until about 60 % confluence. The cells are enzymatically removed from the flask (0.25 % trypsin-EDTA) followed by the neutralization of the enzymatic action with the addition of stromal medium (α-MEM, containing serum and antibiotics).
The cell suspension is transferred into a 15 ml tube, and the sample is centrifuged for 5 min at 400 g. The substrate is carefully aspirated until only the ASC pellet remains in the tube. The ASC pellet is suspended in chondrogenic induction medium (Table A.1), for the chondrogenic induced cultures or DMEM supplemented with serum only and for the non-induced cultures, and centrifuged at 400 g for 10 min. The tubes are carefully placed into the incubator without disrupting the pellet. The tube caps are slightly loosened to allow for gas exchange to occur. Cultures are incubated under standard conditions of 37 °C, 5 % CO2 for 21 days. The induction and control media (0.5 ml) are replaced every second day. After 24 h, the ASC pellets contract into a sphere. The cells that have not been incorporated into the sphere after 48 h are removed from the suspension cultures during medium replacement.
7.1.5.1 Qualitative Assessment of Chondrogenesis In Vitro
Each induced and non-induced chondrocyte sphere is serially dehydrated in 30, 50, 70 and 90 % ethanol, followed by three changes of absolute ethanol for 15 min per dehydration step. The dehydrated chondrocyte spheres are infiltrated with 50 % LR White Resin in absolute ethanol for 1 h, followed by an infiltration in a 100 % LR White Resin overnight. To embed the tissue spheres, they are transferred into resin capsules with 100 % LR White Resin and polymerized for 24 h at 60 °C.
Ten to 15 serial transverse sections of between 0.5 and 1.0 μm (optimal 0.5 μm) are prepared using a microtome, and the sections are stained with 1 % Toluidine Blue O stain for 30 s. The glass slides containing stained sections are gently rinsed with ddH2O. One drop of ddH2O is added on the glass plate and covered using a cover slip before microscopy analysis is performed.
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
van Vollenstee, F.A. et al. (2016). Isolation and Characterization of Adipose-Derived Stromal Cells. In: Pham, P. (eds) Stem Cell Processing . Stem Cells in Clinical Applications. Springer, Cham. https://doi.org/10.1007/978-3-319-40073-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-40073-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-40071-6
Online ISBN: 978-3-319-40073-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)