
Abstract
Microorganisms hold key positions in ecosystem functioning, and thus in biogeochemical cycles. Among these cycles, some, such as chlorine (Cl), are still poorly understood. Recent works have revealed that natural chlorination and dechlorination of organic matter (OM) in most of the ecosystems were much more extensive and ubiquitous than previously suggested. Currently, there are clear evidences that natural chlorination is tightly linked to different defence mechanisms and antagonistic reactions among microorganisms. Likewise, it has been clearly demonstrated that organochlorine (Clorg) formation is also linked to OM degradation, possibly affecting carbon cycle. The chlorination rate of OM depends on several parameters including OM content and quality, microbial activity, chloride (Cl−) input and pH. Once produced, Clorg undergoes oxidative or reductive degradation in the environment depending on the surrounding physico-chemical conditions. Among all enzyme-mediated processes described, the organohalide respiration (an anaerobic bacterial respiratory process) is the only known mechanism leading to the removal of halogens from highly chlorinated compounds, transforming them into biodegradable metabolites. However, despite a significant growth in the literature since the early 1990s, the biogeochemistry of Cl in natural environment is still poorly documented. For instance, the Cl cycling in aquatic environments including Cl− and Clorg pools in sediment and water, are largely missing. The present chapter seeks to review the literature on the natural Cl cycling in environment, with a focus on a freshwater ecosystem, the Lake Pavin.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Freshwater
- Chlorine
- Biogeochemical cycle
- Chlorination
- Dechlorination
- Organohalide respiration
- Abiotic
- Biotic
1 Introduction
Chlorine (Cl) is one of the twenty most abundant elements on earth. Together with fluorine (F), bromine (Br), and iodine (I), Cl belongs to the halogen group in the periodic table. Cl is ubiquitous and naturally present in our environment as well as carbon (C), hydrogen (H), oxygen (O), nitrogen (N) phosphorus (P), and sulphur (S). Cl is essential to most forms of life for diverse reasons (Winterton 2000). Chloride (Cl−) is the only stable ionic form of Cl. Cl− is the main anion in blood, and is present at ~100 mmol . L−1 in plasma and interstitial fluid (Yunos et al. 2010). Cl− takes part to the osmoregulation of cells (White and Broadley 2001), and is an important electrolyte for regulation of muscle function and synaptic transmission in the neural system of animals. Cl− also functions as an essential co-factor in enzymes involved in photosynthesis related to the oxidation of water by the PSII photosystem (Dismukes 1986; Winterton 2000). Thereby, Cl is a critical nutrient and a suggested minimum requirement of Cl for crops is 1 g . kg−1 dry mass (d.m.) (White and Broadley 2001).
Some decades ago, the general opinion was that chlorinated organic compounds (Clorg) detected in ecosystems should have anthropogenic sources and toxic effects. Indeed, many of the most debated organic pollutants are chlorinated (Godduhn and Duffy 2003). But since then, several surveys showed that organic-bound chlorine is more widespread than previously identified and that not all Clorg can be explained by pollution. This assumption was confirmed by a lot of studies. Currently nearly 5000 naturally produced Clorg have been identified and chemically characterized. The production of Clorg has been associated with bacteria, fungi, lichen, plants, marine organisms of all types, insects, and higher animals including humans (Öberg 2002; Gribble 2003; Wagner et al. 2009; Gribble 2010). Some of these have well known physiological functions, including several important antibiotics such as vancomycin and chloramphenicol. Others, such as short-chain volatile Clorg (VOCls), have important effects in the environments because they can, for instance, enhance atmospheric ozone destruction (Winterton 2000). In addition, Cl represents the sixth or seventh most common element of soil OM (SOM) (0.01–0.5 %), at the same level as for P (0.03–0.2 %) and only slightly lower than N (1–5 %) and S (0.1–1.5 %) (Hjelm et al. 1995; Öberg 2002). However, the ecological functions of most Clorg in nature, and the reasons for its production, are largely unknown.
On the earth’s surface, the largest Cl reservoirs are the crust and the ocean (Graedel and Keene 1996). Inorganic Cl by far dominates these reservoirs. Soil (pedosphere), freshwater (rivers, lakes and groundwater), oceans, cryosphere and atmosphere (troposphere and stratosphere) (Graedel and Keene 1996; Öberg 2003; Svensson et al. 2007) are the other important reservoirs. Estimates for these reservoirs are also largely based on Cl− concentration measurements. This assumption of a general dominance of Cl− is problematic for the pedosphere because Clorg levels have been shown to range from 11 to near 100 % of the total Cl pool in a large range of soil types (Johansson et al. 2003; Redon et al. 2011, 2013; Gustavsson et al. 2012).
All these compartments are linked by the hydrological cycle. Although the greatest quantity and variety of naturally-produced chlorinated compounds are found in the marine environment, it is now well accepted that terrestrial and non-marine aquatic ecosystems receive significant fluxes of Cl from the deposition of sea-salt aerosol, through wet and dry precipitations, from leaching processes or from biological processes linked to the formation or the degradation of the OM. However, the terrestrial biogeochemistry of Cl and its cycle between soil, water, biota and dead OM is still ill-understood. Most of previous studies have primarily dealt with Cl− and Clorg separately because they were considered not connected to each other. However, currently, there is irrefutable evidence that Cl undergoes a more complex biogeochemical cycling than expected in terrestrial environments where Cl− can be biotically or abiotically transformed into Clorg in nature, and vice versa.
2 Chlorine Compounds Origin and Formation in Aquatic Ecosystems
2.1 Chloride Ions
2.1.1 Chloride Ions in the Environment
Cl− has long been believed to take part in geochemical processes only, i.e. carried from oceans via soil back to the oceans again, being only negligibly affected by biological processes or interactions with OM. Cl− in the environment can originate from some common chloride salts such as sodium chloride (NaCl), potassium chloride (KCl) and magnesium chloride (MgCl2) (used for de-icing of roads and walkways), calcium chloride (CaCl2) (used as a dust suppressant on roads), aluminium chloride (AlCl2) (used in municipal drinking water and wastewater treatment facilities) as well as ferric chloride (FeCl3) (used at municipal wastewater treatment plants). Cl− containing compounds are highly soluble in water and hence they easily dissociate and tend to remain in their ionic forms once dissolved in water (e.g. Na+ and Cl−). Cl− is the dominating chlorine pool globally and has a high enrichment factor when comparing oceanic and riverine concentrations (i.e. sea water concentrations are in the order of 2500 times larger than freshwater concentrations; Winterton 2000). At the first glance this indicates that Cl− is unreactive in ecosystems and this has been a prevailing view for a long time (e.g. White and Broadley 2001). Accordingly, Cl− has been seen as an inexpensive and suitable tracer of soil and ground water movements (Herczeg and Leaney 2011; Hruška et al. 2012) and studies using Cl− as a water tracer has been a foundation for contaminant transport models (e.g. Kirshner et al. 2000). However, this view has gradually been revised during the last few decades and there is now clear evidence that Cl− is highly reactive in some ecosystems (Öberg 2002; Lovett et al. 2005). For instance, in soil, some type of ‘sorption’ process is present and the retention of Cl− in this compartment has been reported (Bastviken et al. 2007). Hence, the concentrations of Clorg in surface soil layers are in most cases higher than Cl− levels (Johansson et al. 2003; Redon et al. 2011, 2013; Gustavsson et al. 2012). Thus, a large proportion of Cl− deposited in terrestrial ecosystems can be transformed into Clorg in soil, but also certainly in other compartments such as aquatic ones, although the underlying processes are not yet completely elucidated (Öberg 2002; Öberg et al. 2005).
In contrast to soils, Cl− concentrations generally exceed Clorg concentrations in water. For instance, the Cl− concentration in diverse waters is measured in mg . L−1, while Clorg is typically measured in μg . L−1 and VOCls are in the range of ng . L−1 (Eriksson 1960; Asplund and Grimvall 1991; Enell and Wennberg 1991; McCulloch 2003; Svensson et al. 2007). This means that the atmospheric deposition of Clorg is in the order of 1000-fold lower than deposition of Cl− and thereby often assumed to be negligible from a bulk chlorine perspective. While ground water has the highest Cl− concentrations in comparison with rain water and surface waters, Clorg and VOCl concentrations can be highest in surface waters. The environmental quality criteria with regard to Cl− levels in groundwater published by Swedish food agency use a Cl− threshold level of 100 mg. L−1. Regarding the lakes, the ambient Cl− concentrations in the Atlantic region of Canada are normally <10 mg . L−1 in inland lakes, with concentrations as high as 20–40 mg . L−1 in lakes located closer to coastal areas (Mayer et al. 1999). Unimpacted lakes on Canadian shield of Canada’s central region have measured Cl− concentrations of <1–7 mg . L−1, with higher concentrations (20–40 mg . L−1) measured in the lower Great Lakes and St Lawrence River. However, Cl− concentrations above background are commonly detected in densely populated areas, and result usually from human activities. Indeed, anthropogenic Cl− input from irrigation and fertilization can represent substantial inputs to terrestrial ecosystems. Other anthropogenic sources include application of chloride brine solutions for dust in summer, water softeners, industrial effluent, domestic sewage, or yet landfill leachate. Moreover, since the start of de-icing of roads in mid-twentieth century, studies have shown increased Cl− concentrations in both surface water and groundwater in the vicinity of roads. In Canada, elevated concentrations of Cl− associated with de-icing have been documented in groundwater, wetlands, streams and ponds adjacent to snow dumps and salt-storage areas, and also those draining major roadways and urban areas. In the Laxemar-Simpevarp area in South East Sweden, 35–56 % of the total Cl− input was estimated to come from road salt (Tröjbom et al. 2008).
2.1.2 Chloride ions in Lake Pavin
Still to date, the water balance of Lake Pavin is not really well constrained except for the water inputs by direct precipitation (Qp) onto the lake surface which are estimated to be 18 L . s−1 (Fig. 17.1). Those from the main surface streams feeding the lake (Qr) are estimated to be about 20 L . s−1 (Aeschbach-Hertig et al. 2002). The water outflow (Qout) via the surface outlet is estimated to a mean value of 50 L . s−1 and the water outputs by evaporation (Qev), to be 8 L . s−1. The difference between water inputs and outputs leads to a deficit of about 20 L . s−1, which is assumed to be balanced by two sub-surface springs, one located in the mixilimnion (Q45) and the second in the monimolimnion (Aeschbach-Hertig et al. 2002; Assayag et al. 2008). Concerning Cl content in Lake Pavin, only Cl− concentrations were measured regularly in water column over the past decades because Cl− was used as a tracer of water circulation. Its concentrations vary from 1.7 to 2.1 mg . L−1 along the water column. Cl− content is almost 30 % higher in the deep anoxic waters than in surface waters (Assayag et al. 2008; Jézéquel et al. 2012). The main streams and springs feeding the lake display similar Cl− contents (~1–4 mg . L−1). No significant increase of Cl− concentrations was detected between 1992 (~1.6 mg . L−1 in the mixolimnion and ~2.1 mg . L−1 in the monimolimnion) (Michard et al. 1994) and 2006 (~1.7 mg . L−1 in the mixolimnion and ~2.1 mg . L−1 in the monimolimnion) (Jézéquel et al. 2012) which may suggest a relatively good preservation of this ecosystem from human activities.

Summary figure recapitulating the hydrological budget of lake Pavin, with the water inputs: precipitation (Qp), surface streams (Qr), sub-surface springs located in the mixolimnion (Q45) and in the monimolimnion (Q90) and water outputs: evaporation (Qev), output (Qout), and their respective water flow rates. AKz/e is the turbulent water exchange term between the monimolimnion and the mixolimnion (from Assayag et al. 2008)
2.2 Processes of OM Chlorination
Oceans, with a water volume of about 1.36 × 108 km3 and a Cl content of approximately 26 × 1015 tons (t), are the primary reservoir for this element in Earth’s hydrosphere. In comparison, surface waters (including lakes and rivers) have a volume estimated at about 1 × 105 km3 and contain approximately 58 × 107 t of Cl (Graedel and Keene 1996; Bastviken et al. 2013). As ecosystems are open systems, Cl can be supplied and be taken away from the system by various ways. In surface waters, Clorg, like Cl−, originate from atmospheric precipitations but it is not certainly the main source. Other inter-reservoir transfer fluxes have been identified (Table 17.1, Graedel and Keene 1996; Winterton 2000). Rock-water interactions (dissolution and desorption), thermal and mineral springs in volcanic areas but also, to a lesser extent, watershed runoff or leaching can further increase Clorg concentrations in surface waters (Svensson et al. 2007). In addition, higher molecular weight OM found in soil, groundwater and sediment (from rivers, reservoirs and lakes) has a significant Clorg content believed to be of natural and sometimes ancient origin. These Clorg range from peptides, polyketides, indoles, terpenes, acetogenins and phenols to volatile VOCls (for example chloroform) that are produced on a very large scale (Edwards et al. 2004; Gribble 2010). The majority, not biologically active, can be easily transformed into smaller chlorinated by-products or completely degraded by various organisms (Öberg 2002). Finally, a multitude of complex biotic and abiotic transformation processes takes place directly in surface waters and can also lead to the production of a wide variety of Clorg.
2.2.1 Abiotic Chlorination
Although most of the natural chlorinated compounds identified so far are without doubt the results of reactions mediated by enzymes, there is also some evidence of the existence of abiotic formation processes leading to a large number of Clorg (Keppler et al. 2000; Hamilton et al. 2003). These compounds include the majority of chlorinated hydrocarbons and their derivatives formed as a result of various geothermal (e.g. volcano, burning, lightning, erosion) and geochemical (e.g. Fenton reaction) processes.
2.2.1.1 Sources from Geothermal Processes
Several geothermal processes can lead to the production of Clorg. Among these ones, volcanism may be a significant natural source of an extraordinarily large array of Clorg. Indeed, few hundreds of organic substances were detected in fumarolic and lava gases of several volcanoes, of which at least 100 were chlorinated (Jordan et al. 2000). The chloromethanes, chloroethenes, and chlorobenzenes represent the most concentrated molecule families. For instance, the chloroform concentrations (CHCl3) detected in volcanic gases (up to 40 nmol . L−1) are between 1.5 and 2 orders of magnitude higher than those observed above the oceans (Isidorov 1990; Laturnus et al. 2002). Chlorofluorocarbons (CFCs) were also identified but in negligible concentrations as compared to the anthropogenic CFC burden (Jordan et al. 2000).
Another source of Clorg is rocks where they are present in gas pockets or are components of some minerals (apatite, biotite, hornblende among others). Thus when rocks are crushed or as a result of wheathering processes, small quantities of Clorg such as methyl chloride, dichloromethane (DCA) or also carbon tetrachloride can be released (Gribble 2004). Some human activities can also accelerate these natural processes. For instance, we estimate that potassium-salt mining alone liberates thousands of tons of CHCl3 per year.
Finally, significant amount of abiotic Clorg originates from biomass burning, though a minor fraction of these events is considered to be entirely natural, caused for example by volcanic eruptions or lightning. Forest fires are known to produce chloromethane, polychlorinated dibenzodioxins (PCDDs) and polychlorinated dibenzofurans (PCDFs) (Kim et al. 2003).
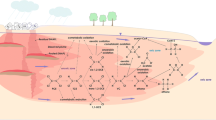
Regarding Lake Pavin, water inputs by direct precipitation onto the lake surface are currently reasonably well constrained (Aeschbach-Hertig et al. 2002) and could supply the ecosystem in a wide variety of Clorg of abiotic origin emanating both from natural sources and anthropogenic activities (Fig. 17.2). Furthermore, because the lake is set in a maar crater and thus has volcanic structures and because it is fed by several sub-surface mineral springs in the monimolimnion and in the mixolimnion (Aeschbach-Hertig et al. 2002; Assayag et al. 2008), it is reasonable to consider erosion or weathering processes as possible sources of Clorg, but certainly in negligible amount.

Chlorine cycle in lake Pavin area with transformation processes of chloride (Cl−) depicted in orange and organically bound chlorine (Clorg) in red
2.2.1.2 OM Chlorination Through Chemical Processes
In forest soil, spontaneous chlorination of OM can occur during degradation by an oxidant (e.g FeIII) in presence of Cl− but without microbial mediation (Keppler et al. 2000). The thermodynamically labile OM is oxidized and the redox partner (for example iron) is reduced. During this process, halides such as Cl− are methylated and the resulting methyl chlorides represent degradation products of oxidized OM. One other way of chlorination can be explained by the Fenton reaction. In the presence of hydrogen peroxide (H2O2) and iron, Cl− can react with OM to form chloroacetic acids. There is evidence for the formation of four classes of Clorg through these processes: volatyl alkyl chlorides, chloroacetates, PCDDs and chlorinated humic substances (Fahimi et al. 2003).
Lake Pavin being continuously fed by OM, essentially plant material (leaves and plant debris) coming from the watershed densely covered by mixed deciduous-coniferous forest, several processes may support the presence of Clorg in the water column (Fig. 17.2). First, OM decomposition in the water column could lead to the release of Clorg in this zone. Furthermore, chemical processes described above, i.e. spontaneous chlorination during oxidative degradation of OM and Fenton reaction, could also take place, though this has never been demonstrated. Indeed, labile OM might also react with Cl− and iron hydroxides, both present in the water column (Viollier et al. 1995; Bura-Nakic et al. 2009), to produce Clorg.
2.2.2 Biochlorination
To date, evidence suggests that the chlorination rate is largely controlled by biosynthetic processes which are mainly conducted by prokaryotes and single cell eukaryotes (Bastviken et al. 2007). They use this strategy to increase the biological activity of secondary metabolites, compounds that are often effective like drugs (Bengtson et al. 2009, 2013). The Clorg could thus be used by the organism (i) to depolimerize the lignocellulose in order to access carbon, a limited resource in some environments, (ii) to dissolve cell wall for cell penetration, (iii) as a chemical defence (e.g. antibiotics), (iv) to compete with other organisms for limited resources by means of antagonistic interactions or (v) yet as a defence against oxygen stress.
Although literature is most impressive on marine algae and terrestrial fungi, bacteria and lichens, reports on halogenating organisms living in fresh water also exist. Aquatic organisms can produce Clorg by enzymatic-mediated processes associated with the transformation and degradation of OM, particularly fulvic and humic acids (Asplund and Grimvall 1991; Öberg 2002). However, Cl− is not particularly reactive unless it is activated, typically by oxidation. Chlorinating enzymes have been discovered from a broad range of organisms and mainly can be grouped into two classes, i.e. the less specific chloroperoxidases (CPOs) utilizing hydrogen peroxide (H2O2) and the highly substrate-specific halogenases requiring O2 for enzymatic activity. In O2-dependent halogenases, either flavin (FADH2-dependent halogenases) or α-ketoglutarate (αKG) (non-heme iron (FeNH)/αKG/O2-dependent halogenases) (Vaillancourt et al. 2005a, b) are found to function as co-substrates. Furthermore, other enzymes requiring S-adenosyl-L-methionine (SAM) as catalyst have been identified to be involved in chlorination (Eustaquio et al. 2008).
2.2.2.1 OM Chlorination Through Chloroperoxidases (CPOs)
Haloperoxidases have traditionally been classified on the basis of the most electrophilic halide that is readily oxidized. Thus, CPOs oxidize chloride, bromide and iodide by H2O2, whereas iodoperoxidases oxidize only iodide in this way. Hydrogen peroxide lacks the thermodynamic potential to oxidize fluoride; thus, enzymes catalysing fluorination are not peroxidases. The overall stoichiometry of the haloperoxidase reaction is consumption of one equivalent of H2O2 per halogenated compound produced. The reaction is:
However, CPO can also catalyse a number of oxidation reactions in the absence of Cl−. These oxidation reactions are generally carried out at neutral pH, whereas chlorination occurs only at low pH. Indeed, the structural features shared with both heme peroxidases and cytochromes P450 make CPOs the most versatile of the known heme enzymes. In addition to catalyzing chlorination, CPOs also catalyse characteristic reactions of heme peroxidases (dehydrogenation), catalases (H2O2 dismutation) and cytochromes P450 (monooxygenation) (Grisham 1991; Rai et al. 2001). Most importantly, CPOs are especially adept in catalyzing the stereoselective epoxidation of alkenes (Allain et al. 1993), benzylic hydroxylation (Zaks and Dodds 1995), propargylic oxidation of 2-alkyenes to chiral alcohol (Hu and Hager 1999) and oxidation of organic sulfides to chiral sulfoxides (Trevisan et al. 2004).
Among the current known CPOs, the heme-depedent and non-heme vanadium-dependent ones require the transition metals heme and vanadium as cofactors, while the few cofactor-free CPOs, also named perhydrolase enzymes, do not require any metal cofactor.
Heme-iron CPOs The first halogenating enzyme to be discovered, in the 1960s, was the heme (iron-containing porphyrin) CPO from the terrestrial fungus Caldariomyces fumago, which produces the chlorinated natural product caldariomycin (Shaw and Hager 1959). The optimal pH for chlorination turnover by heme CPO is pH 2.7 (Hager et al. 1966). In this reaction, the heme iron centre functions as a redox catalyst (Fig. 17.3). Hydrogen peroxide oxidizes the heme Fe(III) centre to compound I, the Fe(IV)-oxo π-cation radical species (O = Fe(IV)-heme + •), via the short-lived compound-0 state, characterized as a peroxo-anion complex, HOO–Fe(III)-heme (Wagenknecht and Woggon 1997). Glutamate residue 183 is proposed to assist in formation of both compound 0 and compound I. Compound I oxidizes Cl− by two electrons, reforming the heme Fe(III) centre and generating an oxidized chlorine intermediate that is formally at the oxidation level of the hypochlorite anion (OCl−). The oxidized chlorine intermediate can then chlorinate the organic substrate or react with a second equivalent of H2O2, producing O2 (in the singlet excited state).

Proposal catalytic cycle for heme CPOs (from Butler and Sandy 2009). The Fe(III)-heme resting state is oxidized by H2O2, forming compound I. Compound I oxidizes Cl− by two electrons, reforming the heme Fe(III) centre and generating an oxidized chlorine intermediate that is formally at the oxidation level of OCl−. This oxidized chlorine intermediate could chlorinate the organic substrate (shown in red) or oxidize a second equivalent of H2O2 (shown in blue), depending on the reaction conditions. L, cysteine
Heme CPOs are produced by organisms generally associated with dead plant material (wood and litter decomposers) such as bacteria belonging to Actinomycetes and various fungi such as ascomycetes and most of decomposing basidiomycetes (Verhagen et al. 1996). This process is also largely used by the bacteria to synthesize antibiotics.
Vanadium chloroperoxidases The second sub-class of CPOs, called vanadium chloroperoxidases (V-CPOs), requires the transition metal vanadium as the necessary cofactor, instead of a heme-iron group in its reactive center. V-CPOs have been isolated from fungi (van Schijndel et al. 1993) and are predicted to be present in marine bacteria (Winter et al. 2007). Although chlorinated natural products have not been isolated from the fungi containing V-CPO, these enzymes may be important in fungus-mediated chlorination and degradation of lignin (Ortiz-Bermúdez et al. 2007). In the terrestrial environment, V-CPOs are widespread among plant-associated microorganisms (living plants or decomposing plant material) such as Streptomyces and Pseudomonas, and symbiotic or parasitic Bradyrhizobium, Burkholderia, Cupriavidus, Frankia and Rhizobium spp. They may catalyze the synthesis of chlorinated antibiotics in bacteria (Bernhardt et al. 2011).
V-CPOs seem to be exclusively exo-enzymes. In contrast to the heme CPOs that function as redox catalysts, V-CPOs function as Lewis acid catalysts of Cl− oxidation by H2O2. The catalytic reaction is initiated by coordination of one equivalent of H2O2 to the resting V(v) state of the enzyme (Fig. 17.4) (Butler and Sandy 2009). The X-ray structure of the peroxo adduct of V-CPO reveals that a Lysine side chain is hydrogen bonded to the coordinated peroxide. This is probably an essential feature of the catalytic reaction because it would increase the potential of the oxoperoxo-V(v) centre for Cl− oxidation. The oxo-peroxo-V(v) species can then oxidize Cl− by two electrons, forming an oxidized halogen that is formally at the OCl− oxidation state. Electrophilic halogenation results from reaction of OCl− either with the organic substrate or, in the absence of a good organic substrate, with a second equivalent of H2O2, forming O2 and Cl−.

Proposal catalytic cycle for Vanadium-CPOs (from Butler and Sandy 2009). The catalytic reaction is initiated by coordination of one equivalent of H2O2 to the resting V(v) state of the enzyme. In red: chlorination of an organic substrate (RH); in blue: oxidation of a second equivalent of H2O2, depending on the reaction conditions. L, ligand
Cofactor-free CPOs (or perhydrolases) The perhydrolases had been previously known as prosthetic group-free bacterial haloperoxidases, particularly non-metal and non-heme haloperoxidases (Song et al. 2006). They have the same catalytic triad as serine hydrolases (Ser-Asp-His) and belong to the large and diverse α/β hydrolase fold enzyme class. These enzymes catalyze the reversible formation of peroxyacids from carboxylic acids and H2O2 in aqueous media (Fig. 17.5) (Kirk and Conrad 1999). The peroxyacids oxidize Cl− to OCl−, which then halogenates organic substrates non-specifically. Only few bacterial perhydrolase genes have been sequenced and characterized from Pseudomonas (Wiesner et al. 1988; Kirner et al. 1996), Serratia (Burd et al. 1995), Burkholderia (Song et al 2006) and Streptomyces genera (Bantleon et al. 1994; Pelletier et al. 1994).

Scheme of the halogenation reaction catalyzed by perhydrolases (Song et al. 2006). The enzymatic formation of peroxyacids in the presence of H2O2 is followed by the non-enzymatic oxidation of substrates, for example that of halide ions (Cl−) to hypohalite ions (OCl−). The subsequent chlorination reaction is finished by the incorporation of OCl− into organic compounds (R-H). This perhydrolase reaction that can provide the peroxyacid for the oxidation is not regio-, chemo- or stereoselective
2.2.2.2 OM Chlorination Through FADH2-Dependent Halogenases (FDHs)
The second class of enzymes generating group of halogenating enzyme using oxidative mechanisms is FDHs. These enzymes used O2 as the oxidant, flavin as the redox active co-factor and the presence of a nicotinamide adenine dinucleotide (NADH)-dependent reductase to reduce flavin adenine dinucleotide (FAD) (Keller et al. 2000). The reaction is (X: halogen atom, R, R’: alkyl group):

In contrast to CPOs, these enzymes have a high degree of substrate specificity and are able of regioselective halogen incorporation. The first FDH found was the tryptophan 7-halogenase PrnA which catalyzes the chlorination of tryptophan to 7-chlorotryptophan, the first step in pyrrolnitrin biosynthesis (Keller et al. 2000). In this reaction, the flavin reductase produces FADH2 from FAD and reduced NADH. FADH2 is bound by PrnA where it reacts with O2 to form a flavin hydroperoxide. A single Cl− is bound close to the isoalloxazine ring of the FAD and attacks the flavin hydroperoxide leading to the formation of hypochlorous acid (HOCl) (Dong et al. 2005; Lang et al. 2011). However, since the substrate tryptophan is bound about 10 Å away from the isoalloxazine ring, HOCl is guided through a “tunnel” towards the substrate. The lysine residue, located close to the substrate and conserved in all flavin-dependent halogenase, is suggested to react with HOCl to form a chloramine as the halogenating intermediate (Yeh et al. 2007; Lang et al. 2011).
This class of enzymes is responsible for the halogenation of many bacterial secondary metabolites including many antibiotics, growth hormones as well as antitumor and antifungal compounds. Nearly all known FDHs are involved in the halogenation of aromatic or heteroaromatic ring molecules. Two distinct subgroups of enzymes exist: one uses phenol or pyrol as substrates and the other tryptophan (Murphy 2006). Homologues of FDHs have been found in various bacterial phyla including the Actinobacteria, Cyanobacteria, Planctomycetes and Proteobacteria (Murphy 2006; Bayer et al. 2013).
2.2.2.3 Additional Chlorination Processes
Dramatic advances in deciphering the logic of halogenation enzymes have occurred in the recent past through bacterial genomic and bioinformatics analyses which allow identification of new classes of enzymes, i.e. halogenases of the mononuclear non-heme iron family and chlorinases.
The mononuclear non-heme iron-containing enzymes are a new class of αKG-dependent FeNH halogenase enzymes. The first example was the enzyme SyrB2, which generates a 4-chloro-L-threonine residue incorporated into the framework of the nonribosomal lipopeptidolactone syringomycin produced by Pseudomonas syringae (Vaillancourt et al. 2005a). Detailed evaluation of cofactor requirement established that Fe2+, O2, αKG, and Cl− are required for enzymatic activity. The requirement for Fe2+, O2 and αKG is the hallmark of the two-His, one-carboxylate non-FeNH αKG-dependent oxygenase reactions, and SyrB2 falls into that protein superfamily. However, this enzyme effects chlorination rather than hydroxylation of unactivated methyl groups in substrates (Fujimori and Walsh 2007; Neumann et al. 2008; Butler and Sandy 2009). The reaction mechanism involves the binding of Cl− directly to iron before decarboxylation of a αKG to produce succinate, CO2 and a high-energy ferryl-oxo intermediate that is very reactive and acts as a hydrogen-abstracting species (Blasiak et al. 2006). The reaction is:

Another class of halogenases is that of non-metallo-halogenating enzymes which use SAM as a co-substrate. The first of the SAM-dependent halogenases to be discovered was the methyl chloride transferase enzyme in seaweeds and other plants, which is responsible for the production of profuse levels of methyl chloride (Ni and Hager 1998), raising questions about its biological function. Other SAM-dependent halogenases, including chlorinase (Eustaquio et al. 2008), were recently discovered. The reaction is:

These enzymes catalyse nucleophilic halogenation reactions, rather than electrophilic or radical halogenation reactions. The SAM-dependent chlorinase SalL, from the marine bacterium Salinospora tropica, chlorinates SAM by nucleophilic Cl− addition generating 5′-chloro-5′-deoxyadenosine (5′-ClDA) as a precursor to chloroethylmalonyl-CoA, which is ultimately incorporated into salinosporamide A (Dong et al. 2004).
Although not yet described, the presence of all necessary precursors in the water column of Lake Pavin assumes that enzymatic chlorination is plausible in this ecosystem (Fig. 17.6). Over the past decade, numerous studies exploring microbial diversity in pelagic zone of this ecosystem revealed the presence of parasitic and saprophytic fungi belonging to the three major divisions, i.e., Chytridiomycota, Basidiomycota and Ascomycota (Monchy et al. 2011; Jobard et al. 2012), well known to synthetize various CPOs. Regarding bacteria, the main phyla involved in OM chlorination through enzyme-mediated processes (Actinobacteria, Proteobacteria, Cyanobacteria, Planctomycetes) were also identified (Biderre-Petit et al. 2011a, b; Lehours et al. 2007).

Schematic representation of main processes of chlorination and dechlorination mediated by microorganisms potentially occurring in water column of lake Pavin (OM organic matter; V vanadium, e − electrons, CPOs chloroperoxidases, Cl in inorganic chlorine, Cl org organic chlorine)
3 Chlorinated OM Degradation
As stated above, several thousands of chlorinated compounds are naturally produced in the environment, including non-marine aquatic ecosystems whereas tens of thousands are synthetized by human. Some of them are aromatics, i.e., containing one or more aromatic rings with one or more chlorine atoms while others are aliphatics i.e., formed by carbon chains without benzene ring but with one or more hydrogen atoms replaced by a chlorine atom. Because the freshwaters (lakes, rivers, groundwaters among others) display different and complex geochemical and biological patterns, they may exhibit different potential to degrade these chlorinated compounds which can be from both natural and human origin. Like chlorination, dechlorination can occur through both biotic and abiotic processes with abiotic dechlorination usually slower than microbial one. Because bio-mineralization of chlorinated compounds occurs frequently through multiple-step processes producing various intermediate molecules, it usually requires the involvement of microbial consortia (Men et al. 2014).
Most of the processes involved in the biodegradation of chlorinated compounds are catalyzed by enzymes carried by microorganisms. Indeed, microorganisms, due to the immense reservoir of their metabolic capacities, can adapt to almost every environmental conditions found on Earth (Guerrero and Berlanga 2006). Many of them can use Clorg as a main carbon and/or energy source through enzymes with specific dehalogenase activities. Over the past decades, investigation of the microbial degradation capacities allowed the identification of a broad range of this type of enzymes. All are capable to cleave carbon-halogen bonds through totally different reaction mechanisms, under aerobic or anaerobic conditions (Janssen et al. 2001; de Jong and Dijkstra 2003; van Pée and Unversucht 2003). Furthermore, some microorganisms are also able to degrade chlorinated compounds through fortuitous reactions mediated by non-specific enzymes, also called co-metabolism. While relatively lightly chlorinated compounds can generally be degraded by aerobes, heavily chlorinated relatives are often recalcitrant to biodegradation under aerobic conditions. However, overall, the aerobic processes are low for most chlorinated compounds.
3.1 Thermodynamic Feasibility of Dehalogenation Mechanisms
The thermodynamic properties of a large number of biochemical compounds and reactions have been calculated experimentally under different conditions (Thauer et al. 1977; Alberty 2003; Goldberg et al. 2004; Finley et al. 2009) but for most of the known biochemical compounds such information is lacking. Dolfing and Janssen (1994) used group contribution to classify conversion steps in a biodegradation pathway of halogenated compounds as endergonic or exergonic, and determine whether the reactions yield adequate energy to sustain microbial growth. Thermodynamics has also been used to compare biodegradation routes involving reductive dechlorination to those involving oxidative and fermentative degradation reactions (Dolfing 2000, 2003; Smidt and de Vos 2004).
Estimation of Gibbs free energies (∆G0') and redox potentials (E0'0) for a wide range of aliphatic and aromatic Clorg have indicated that chlorinated compounds should be excellent electrons acceptors, yielding ∆G0' ranging between −130 and −180 kJ . mol−1 of Cl removed by hydrogenolytic reductive dehalogenation (Dolfing 1990; Dolfing and Harrisson 1992; Dolfing and Janssen 1994). Corresponding E0' range between +300 and +550 mV which is considerably higher than that of the reduction of sulfate (SO4 2-/H2S, E0'=−220 mV) and comparable to the potential of NO3 −/NO2 − (E0' = +443 mV) (Table 17.2) (Löffler et al. 1999). From these thermodynamic considerations it has been predicted that reductive dehalogenation should also be occurring, though rarely, under aerobic conditions (Dolfing 2003).
While hydrogenolysis is the main mechanism involved in reductive dehalogenation reactions, a second type has also been observed for nonaromatic Clorg, i.e. the dichloroelimination. In this reaction, two rather than one chlorine groups are simultaneously removed and the aliphatic bond C-C is converted into a C=C bond (Fig. 17.7). Because this reaction requires only one mole of reducing equivalent (H2), as opposed to two moles of H2 in hydrogenolysis, its energy balance is more favorable. However, while dichloroelimination should prevail over hydrogenolysis under hydrogen-limiting conditions, addition of easily available reducing equivalents may decrease this advantage by making competition for reducing equivalents less important (Dolfing 1999).

Examples of (reductive) redox dehalogenation reactions occurring under anaerobic conditions
From a thermodynamic point of view, dehalogenation via routes other than reductive dehalogenation is also feasible. Anaerobic microorganisms should degrade Clorg by oxidative or fermentative pathways, but so far, evidence for their functionality is rather scarce when compared with the knowledge that has been accumulated on reductive dehalogenation. Let us take chlorinated ethylenes as an example. Organisms can grow on their fermentative degradation in a type of reaction in which the chloro substituent serves as an internal electron acceptor (Bradley and Chapelle 2000; Dolfing 1999). Chlorinated ethylenes can, for instance, be fermented to ethane and CO2 or acetate (Dolfing 1988). The exergonicity of such fermentation implies the potential existence of new types of dechlorinating bacteria that would not depend on the presence of external sources of reducing equivalents. Likewise, oxidative degradation of chlorinated ethylenes to H2, CO2 and HCl is an exergonic reaction (−62 to −545 kJ . mol−1 of Cl) which is energetically independent of the activity of H2-consuming organisms.
3.2 Abiotic Degradation of Chlorinated Compounds
Although abiotic degradation does not require the mediation of microorganisms, the biotic processes (which change pH and redox potential, for instance) are frequently required to stimulate abiotic reactions. Abiotic dechlorination is usually a slow process but plays an important role in the degradation of some of the aliphatic Clorg (mainly chloroalkanes and chloroalkenes) and of a few aromatic Clorg. The main mechanisms are hydrogenolysis, dichloroelimination, hydrolysis and dehydrochlorination (Fig. 17.7) (for review see Tobiszewski and Namiesnik 2012). These mechanisms and the degradation rate strongly depend on the redox conditions found in the surrounding environment.
3.2.1 Reductive Pathways of Degradation
Hydrogenolysis and dichloroelimination are both reductive pathways and occur under anaerobic conditions, though dichloroelimination was also demonstrated under partially aerobic conditions (Chen et al. 1996). Hydrogenolysis reactions can be represented by the formula: RCln + 2H+ + 2e− → RHCln−1 + HCl and dichloroelimination by: RCln + 2H+ + 2e− → RHCln−2 + 2HCl. Hydrogenolysis is the major degradation pathway for highly chlorinated alkenes (Nobre and Nobre 2004). In this reaction, the chlorinated compound, considered as an electron acceptor, is reduced by an electron donor. Among all potential electron donors involved in chlorinated compounds hydrogenolysis, the more efficient are zero valent metals (e.g., Fe0, Zn0, Sn0) (Scherer et al. 1998). However, recently, reductive dechlorination of PCBs, pentachloroethane (PCA), PCE, TCE, and carbon tetrachloride by nano-sized zero valent iron (nFe0) has shown promising application due to its higher reactivity than that of Fe0 (Amir and Lee 2011). Likewise, biogenic or chemogenic ferrous iron (FeII) species have been the focus of many studies due to their enhanced reactivity that favors the reductive transformation of a number of Clorg such as carbon tetrachloride, hexachloroethane, 1,1,1-trichloroethane, pentachloronitrobenzene, pentachlorophenol, dichlorodiphenyltrichloroethane and 2,4-dichlorophenoxyacetic acid through coupled biotic-abiotic or abiotic processes (Fig. 17.8). For example, biogenic magnetite (Fe3O4), created by the iron reducing bacterium Geobacter metallireducens, can cause the abiotic reductive dechlorination of carbon tetrachloride, and carbonate green rust formed by the iron reducing bacteria Shewanella putrefaciens CN32 can cause that of cis-DCE. Because both reactive minerals and microorganisms are usually present in aquatic ecosystems, both abiotic and biotic reductive dechlorinations have the potential to occur simultaneously (Liu et al. 2013; Xu et al. 2014). Therefore, abiotic transformation, though less rapid than microbial, may be crucial if the concentration of reactive minerals is high and/or the activity of dechlorinating bacteria is low (Tobiszewski and Namiesnik 2012).

Main mechanisms of anaerobic chlorinated compounds transformation through abiotic or coupled biotic-abiotic processes involving iron species (Modified from Xu et al. 2014). OM organic matter
3.2.2 Hydrolysis and Dehydrochlorination Pathways
Beside reductive pathways, hydrolysis and dehydrochlorination are two abiotic processes that may degrade chlorinated compounds under either aerobic or anaerobic conditions. Hydrolysis in natural waters is an extremely slow process. The reaction is summarized by the formula: RCl + H2O → ROH + HCl. Generally, this reaction happens when organic molecule reacts with water, resulting in the formation of a new covalent bond with OH− and the cleavage of the covalent bond with chlorines. Regarding dehydrochlorination, chlorinated compounds may also undergo this mechanism in certain conditions. The formula is: RHCCl–CRH2 → RHC = CHR + HCl. In this reaction, HCl is eliminated from the solvent molecule, which results in the formation of double or triple carbon bonds and less saturated and less chlorinated compounds.
The axonic zone of Lake Pavin provides suitable conditions for abiotic reductive dechlorination processes. Indeed, this ecosystem is permanently redox-stratified, with anoxic and ferruginous deep waters (from 60 to 92 m depth) topped by oxic shallow waters (from 0 to 60 m). Lake Pavin contains dissolved Fe(II) up to 1.2 mM below the chemocline at 60 m (Viollier et al. 1995). More details on the iron cycle are provided in Chap. 14. The Fe(II) form is released from the sediment and either diffuses towards the mixolimnion to precipitate as ferric iron (FeIII) at the redox interface (Michard et al. 2003) or reacts with sulfides or phosphates to form FeS colloids and secondary minerals such as pyrite, vivianite or siderite (Bura-Nakic et al. 2009). These chemogenic Fe(II) species may therefore favor the abiotic reductive transformation of a number of Clorg in the water column of Lake Pavin. Moreover, it has been demonstrated that the large concentration gradient associated with negative Fe isotope composition observed below the oxic-anoxic interface could be interpreted as the signature of intense bacterial dissimilatory Fe reduction due to, for instance, the presence of the well-known obligatory Fe(III) reducers Geobacter which are present in the anoxic compartment of this ecosystem (personal data). Therefore, reductive dechlorination of the chlorinated compounds in the anoxic zone of Lake Pavin might also occur through a coupled biotic-abiotic process (Fig. 17.8).
3.3 Biotic Degradation of Chlorinated Compounds
3.3.1 Chlorinated Compounds Degradation Under Aerobic Conditions
Aerobic dehalogenation is one form of biodegradation which enable the microorganisms in soil and water to utilize chlorinated substances as carbon and electron source. Over the past decades, many investigations focused on microbial degradation of aromatic and aliphatic Clorg, leading to the identification of an important diversity of dehalogenation mechanisms and dehalogenase enzymes (Janssen et al. 1994; Fetzner 1998). In these enzymatic processes, chlorine substituents are removed to form non-halogenated intermediates that can then enter into general metabolic pathways, such as the tricarboxylic acid cycle (TCA). The main processes of aerobic dehalogenation involve hydrolytic, reductive and oxidative mechanisms (van Pée and Unversucht 2003). Furthermore, a wide range of Clorg can also be microbially degraded under aerobic conditions by means of co-metabolic transformation reactions (mainly oxidations), yielding no carbon or energy benefits to the transforming cells (Horvath 1972).
3.3.1.1 Enzymatic-Mediated Hydrolytic Dehalogenation
Hydrolytic dehalogenation involves the replacement of a chlorine atom by an OH group derived from water and results in the formation of a primary alcohol (Janssen et al. 1994). There are several hydrolytic dehalogenase families which belong to different protein superfamilies of which the members catalyze diverse reactions, mostly with non-chlorinated compounds.
3.3.1.1.1 Haloalkane Dehalogenases
These enzymes are the best studied hydrolytic dehalogenases. They belong to the α/β hydrolase superfamily and contain a catalytic triad consisting of one histine and two aspartate residues which are involved in the nucleophilic substitution of Cl− by water (Ollis et al. 1992). Although these haloalkane dehalogenases can convert a broad range of chlorinated alkanes and alkenes to their corresponding alcohols, they can also dehalogenate aromatic compounds, such as the LinB enzyme of Sphingomonas paucimobilis UT26 which catalyzes chlorinated cyclic dienes degradation (Marek et al. 2000).
3.3.1.1.2 Haloacid Dehalogenases
These enzymes are the second most common group of aliphatic dehalogenases. These enzymes are dimers and define the so-called HAD (haloacid dehalogenase) superfamily of hydrolases. They are responsible of the hydrolysis of chlorinated carboxylic acids. Like haloalkane dehalogenases, they use an aspartate-based catalytic mechanism but there is no histidine residue in the catalytic site to activate a nucleophilic water molecule.
Representatives of these two enzyme families have been isolated from various bacteria including Moraxella, Mycobacterium, Pseudomonas, Xanthobacter, Sphingomonas, Sphingobium and Rhodococcus species (van Pée and Unversucht 2003).
3.3.1.1.3 Hydrolytic Dehalogenases Specific to Chlorinated Aromatics
At present, only two hydrolytic dehalogenases targeting specifically chlorinated aromatics have been reported. The first is the 4-chlorobenzoyl-CoA dehalogenase (CbzA), belonging to the enoyl hydratase superfamily. It is a component of the 4-chlorobenzoate degradation system. This system comprises three separate enzymes and requires CoA and ATP as cofactors (Benning et al. 1998; Pieper et al. 2010). In the reaction, the compound is first conjugated to CoA and further dechlorinated by CbzA which catalyzes the displacement of chlorine by a nucleophilic addition-elimination mechanism. This process was demonstrated in few species belonging to Pseudomonas, Rhodococcus and Acinetobacter genera (Janssen et al. 2005). The second enzyme specific to chlorinated aromatics is the chlorothalonil hydrolytic dehalogenase (Chd) of Pseudomonas sp. CTN-3 strain. This enzyme contains a conserved domain of metallo-β-lactamase superfamily and catalyzes the dechlorination of chlorothalonil through a not yet described mechanism independent of CoA and ATP (Wang et al. 2010).
3.3.1.2 Reductive Dehalogenation
Reductive dehalogenation of chlorinated compounds can occur under aerobic conditions. This process can be mediated by two major enzyme families, i.e. glutathion-S-transferase and cytochrome P450 type enzymes.
Glutathion-S-transferase (GST) Some aliphatic and aromatic chlorinated compounds can be reductively dehalogenated by thiolytic substitution in the presence of glutathione. During this reaction, the chlorine atoms are displaced by the nucleophilic attack of the thiolate anion of glutathione through the GST activity (Wilce and Parker 1994). The most studied enzyme of this group involved in chlorinated aliphatics degradation is the dichloromethane dehalogenase (DcmA). It catalyses the conversion of DCA to HCl and formaldehyde, a central intermediate of methylotrophic growth used for biomass and energy production (Kayser et al. 2002). This enzyme can be found in a large variety of methylotrophic bacteria belonging to the Proteobacteria phylum such as Methylophilus, Methylorhabdus, Methylobacterium, Hyphomicrobium, Methylopila, Albibacter, Paracoccus, Ancylobacter and Pseudomonas species (Fetzner and Lingens 1994; Torgonskaya et al. 2011). A glutathione-dependent biotransformation was also reported for various chlorinated aromatics including the chlorothalonil and the tri-/tetrachloro-p-hydroquinones, through the action of the bacterial GST of species belonging to the Ochrobactrum, Flavobacterium and Spingbium genera (Kim et al. 2004).
C-type cytochromes Respiratory c-type cytochromes such as cytochrome P-450CAM may also mediate reductive dechlorination of chlorinated aliphatics but only under very low O2 concentrations. These cytochromes have been proposed to mediate hexachloroethane, pentachloroethane and tetrachloromethane reduction (Walsh et al. 2000). This mechanism can be found in Pseudomonas sp. and in the purple nonsulfur bacteria Rhodopseudomonas and Rhodospirillum.
3.3.1.3 Oxidative Dehalogenation
Microbial oxygenases are key enzymes for aerobic biodegradation of chlorinated compounds, which are thus used as carbon and energy source (Pérez-Pantoja et al. 2012). Oxidative dehalogenation involves the replacement of a chlorine atom by an OH group whose oxygen atom is derived from O2. Two classes of oxygenases type dehalogenases have been identified: (i) the monooxygenases which incorporate one oxygen atom to chlorinated compounds to remove chlorine atom whereas the second oxygen atom is reduced to water and (ii) the dioxygenases which add two atoms of O2 into the substrate to remove the chlorine atom (for review see Arora et al. 2010). Though the majority of dehalogenating oxygenases have been demonstrated for aromatic substrates (Fetzner and Lingens 1994), some have also been shown for aliphatic relatives such as vinyl chloride (VC) and dichloroethene (DCE) (Bradley and Chapelle 2000).
3.3.1.3.1 The Monooxygenases
These enzymes are classified into two subclasses depending on the cofactor they used. Flavin-dependent monooxygenases contain flavin as prostethic group and require NADP or NADPH as coenzyme (Arora et al. 2010). Examples of well-studied enzymes of this type are the pentachlorophenol-4-monooxygenase (PcpB), the chlorophenol 4-monooxygenase and the 2,4-dichlorophenol monooxygenase (Fetzner 1998; Arora et al. 2010). These enzymes, mainly involved in polychlorinated compound dehalogenation, have been characterized in various microorganisms including Pseudomonas, Sphingobium, Sphingomonas, Burkholderia, Ralstonia and Azotobacter genera. The second subclass is represented by the P450 monooxygenases whose best example is the P-450CAM monooxygenase system, already mentioned in the reductive dehalogenation but which can also catalyze oxidative dehalogenation reactions under O2-rich conditions. From Nishino et al. (2013), this monooxygenase might also be involved in the DCE dechlorination in Polaromonas chloroethenica JS666. Finally, a third mechanism results in the VC and ethene assimilation through epoxidation. It is catalyzed by an alkene monooxygenase (AkMO), followed by conversion of the (chloro)epoxide to a 2-hydroxyalkyl coenzyme M by an epoxyalkane coenzyme M transferase (EaCoMT) with the release of the chlorine atom (Coleman and Spain 2003). The by-product is then metabolized through the TCA cycle. Bacteria involved in this mechanism belong to Mycobacterium, Nocardioides, Pseudomonas, Ochrobactrum and Ralstonia genera.
3.3.1.3.2 The Dioxygenases
The aerobic degradation of aromatic compounds is frequently initiated by Rieske non-heme iron oxygenases. These enzymes are multicomponent complexes composed of a terminal oxygenase component (iron-sulfur protein) and of different electron transport proteins. They catalyze the incorporation of two oxygen atoms into the aromatic ring to form arene cis-diols which spontaneously rearomatize along with chloride elimination, yielding a (chloro)catechol product. This compound then undergoes the ortho-cleavage pathway to form β-ketoadipate (β-KAP), a central metabolite in the degradation of aromatic compounds. This molecule, presumed to have lost Cl− during one of the above listed-reactions, is then subjected to further transformations before entering the TCA cycle (Ogawa et al. 2003). Nowadays, two-component dioxygenase enzymes that dehalogenate 4-chlorophenylacetate and 2-halobenzoate but also a three-component dioxygenase system that dehalogenates both Z-chlorobenzoate and 2,4-dichlorobenzoate have been described in different strains of Pseudomonas (Copley 1997). Likewise, biphenyl 2,3-dioxygenases (BphAs) are of crucial importance for the successful metabolism of PCBs in environment. These enzymes are able to oxidize a wide range of PCB congeners, from mono-chlorobiphenyls to 2,3,4,5,2′,5′-hexachlorobiphenyl, with an increased preference for dioxygenation linked to a decrease of chlorine atom number (Pieper and Seeger 2008; Pieper et al. 2010). This reaction has been observed in a variety of microorganisms belonging to Alcaligenes, Acinetobacter, Rhodococcus and Burkholderia genera (Fetzner 1998; Pérez-Pantoja et al. 2012).
3.3.1.4 Biodegradation by Co-metabolism
Besides specific dehalogenation pathways, aerobic co-metabolism represents a significant process responsible for aliphatic and aromatic chlorinated compound biodegradation in the environment (Field and Sierra-Alvarez 2008; Jechorek et al. 2003; Little et al. 1988). It involves microbial oxygenase enzymes, e.g. methane monooxygenases (MMOs), toluene mono- and dioxygenases, ammonia monooxygenases and biphenyl monooxygenases among others and does not provide any benefit to the microorganisms (Furukawa 2000; Hazen 2010). In general, these enzymes have low specificity to chlorinated compounds but this limitation is overcome by the increase in enzyme expression levels in presence of the growth substrate for the microorganism. MMOs have been the most widely studied and are present under two forms, the soluble form (sMMO) found in a few selected methanotrophs and the particulate form (pMMO) found in most methanotrophs. These enzymes can oxidize more than 300 different compounds, most of them chemically distinct from methane, the natural substrate of the enzyme (Hazen 2010).
3.3.1.4.1 Co-metabolism of Chlorinated Aliphatics
Various chlorinated aliphatic compounds including VC, DCE and trichloroethene (TCE) can be metabolized through a co-metabolism process in the presence of growth-substrates such as methane, butane, propane, ammonium, ethylene, ethane, phenol, benzene, isopropylene or toluene (Hazen 2010; Nzila 2013). For instance, TCE can be converted by the bias of the MMOs in a non-stable epoxide which is spontaneously degraded into stable water-soluble products (acetic, formic, oxalic, glyoxylic, and mono-, di-, and trichloroacetic acids) and CO2 (Little et al. 1988). In this reaction, chlorine atoms are liberated under the Cl− form. Some bacteria can also use VC as primary substrate to co-metabolize DCE and to a lesser extent, TCE (Broholm et al. 2005). Likewise, other aliphatic molecules such as chloroform and DCA have been reported to be co-metabolized by bacteria using alkane (methane or butane) or ammonia as growth substrates (Hamamura et al. 1997). Biodegradation of chlorinated aliphatic compounds through co-metabolic reactions have been shown for many bacteria belonging to the Xanthobacter, Rhodococcus, Nitrosomonas, Pseudomonas, Mycobacterium, Methylobacterium, Methylocystis, Ralstonia genera.
3.3.1.4.2 Co-metabolism of Chlorinated Aromatics
Like aliphatic compounds, various chlorinated aromatic compounds can be biodegraded in the context of co-metabolism by a wide variety of microorganisms (Hazen 2010). Thus, co-metabolism of chlorophenols has been reported in bacteria using phenol (Aktas and Cecen 2009) but also glucose or dextrose (Ziagova et al. 2009) as growth substrates. This mechanism has also been described for the oxidation of chlorobenzenes, chlorobenzoates and PCBs in presence of acetate, glucose, fluorobenzenes, but also of low chlorinated chlorobenzoates and PCBs as growth substrates. Bacteria involved in chlorinated aromatics degradation include members of the Burkholderia, Alcaligenes, Arthrobacter, Nocardia, Acinetobacter, Pseudomonas and Staphylococcus genera among others. In general, aerobic biodegradation pathways of aromatic compounds are catalyzed, by dioxygenases, though involvement of MMOs has been reported for some chlorobenzenes (Jechorek et al. 2003; Hazen 2010).
At present, although there is no evidence of chlorinated compounds biodegradation in the oxic zone of Lake Pavin, most of microorganisms involved in the different mechanisms responsible for dechlorination have been detected in the water column of this ecosystem (Biderre-Petit et al. 2011a; Lehours et al. 2007). It is particularly the case of methylotrophic bacteria (Borrel et al. 2011), a common group characterized in most freshwaters. We can thus hypothesize that some of these methylotrophs might also carry enzymes related, for instance, to the DcmA dehalogenase and thus be involved in chlorinated aliphatics biodegradation through the aerobic reductive dehalogenation pathway. Moreover, a recent study focusing on methane cycling in this ecosystem has revealed the presence of a wide diversity of methanotrophs harboring genes which encode various pMMOs (Biderre-Petit et al. 2011b). Consequently, these bacteria could represent potential key-players in the halogen cycle in this environment by performing co-metabolism oxidation.
3.3.2 Chlorinated Compounds Degradation Under Anaerobic Conditions
Although abiotic processes might be involved in reductive transformations of chlorinated compounds under anoxic conditions, the majority of these reactions are biologically catalyzed. Reductive dehalogenation has been reported for many aliphatic (e.g. chloromethanes, chloroethanes, chloroethenes, chlorinated acetic acids) and aromatic (e.g. chlorobenzoates, chlorophenols, chlorobenzenes, dioxins, PCBs) chlorinated compounds. These molecules can serve in three metabolic functions in different anaerobic bacteria: (i) as carbon or energy source or both, (iii) as terminal electron acceptor in anaerobic respiration process and (ii) as substrate for co-metabolic activity. The respiration process is the most widespread in environment but also the best documented because its wide use in bioremediation strategies (Holliger et al. 1998b; Hiraishi 2008).
3.3.2.1 Chlorinated Compounds as Carbon or/and Energy Source
Some microorganisms have ability to grow under anaerobic conditions by using the chlorinated compounds as an electron donor and/or carbon source (Kuntze et al. 2011). These reactions may result in the complete mineralization of these molecules to CO2 or in their fermentation in products such as acetate and formate. Only very little is known about the degradation pathways and enzymes involved. Anaerobic growth on chlorinated compounds as electron donors or carbon source was clearly the least common form of biodegradability. Indeed, evidence for this type of metabolism was limited to five aliphatic compounds, i.e. chloromethane, DCA, VC, cis-DCE and chloroacetic acids and to only few aromatic compounds in chlorobenzoate and chlorophenol categories. For both chloromethane and DCA degradation, the reaction involves corrinoid proteins (vitamin B12 containing proteins) and the transfer of the methyl or methylene group, depending on the substrate, onto tetrahydrofolate which is an important coenzyme of chlorine-metabolizing organisms (Magli et al. 1998; Harper 2000). This mechanism has been shown in few isolated microorganisms (e.g. Acetobacterium dehalogenans and Dehalobacterium formicoaceticum), but also in mixed cultures (methanogenic/acetogenic). DCA degradation can also occur under denitrifying conditions by Acinetobacter and Hyphomicrobium species. Finally, chloroacetic acids can be used as sole substrate for growth by the phototrophic anaerobe, Rhodospirillum photometricum. For chloroethenes and chlorinated aromatics, their mineralization into CO2 and Cl− was essentially observed in mixed cultures under methanogenic, iron-reducing, humus-reducing and manganese-reducing conditions (for review see Field and Sierra-Alvarez 2004). Moreover, in previous studies Thauera chlorobenzoica strain 3CB-1 was reported to utilize 3-chlorobenzoate as sole sources of cell carbon and energy growth substrate under denitrifying conditions (Song et al. 2001; Kuntze et al. 2011).
3.3.2.2 Reductive Dehalogenation Through Organohalide Respiration
Organohalide respiration (OHR) is the more efficient and the well-studied biodegradation pathway of chlorinated compounds under anaerobic conditions. In this respiratory process, bacteria use chlorinated species as the terminal electron acceptors during electron-based energy conservation (Holliger et al. 1998b). Highly reduced anaerobic environments (indicated by a low redox potential), typical for methanogenesis and sulfate-reduction, have been found to be a requisite for the reductive dechlorination of halogenated compounds (i.e., the substitution of halogen atoms by hydrogen atoms) (Stuart et al. 1999; Olivas et al. 2002). Almost all chlorinated compounds, whatever the molecular structure and the number of substitutions, can be degraded through this microbial process.
OHR is performed by organohalide-respiring bacteria (OHRBs) that have been identified in various taxa including the δ-Proteobacteria, ε-Proteobacteria, Firmicutes and Chloroflexi. These bacteria are divided into two categories, i.e. facultative and obligate OHRBs. Besides Clorg, facultative OHRBs are able to use a wide diversity of electron acceptors (e.g. fumarate, nitrate, sulfate, thiosulfate, Fe(III), Mn(IV), U(VI), S0, selenate, arsenate) but also several electron donors (H2, pyruvate, acetate, lactate, butyrate, succinate, formate, ethanol, glycerol, crotonate). They include members of the Desulfomonile, Geobacter, Sulfurospirillum and Desulfitobacterium genera (Bradley and Chapelle 2000; Smidt and de Vos 2004; Hiraishi 2008). The second category of ORHBs, and possibly the most intriguing, is composed of extremely specialized bacteria requiring an organohalide molecule as terminal electron acceptor, H2 or acetate as electron donor and vitamin B12 as cofactor to perform OHR. Currently, obligate OHRBs are present in the Chloroflexi phylum with representatives of the “Dehalococcoidia” class such as Dehalococcoides and Dehalogenimonas species (Löffler et al. 2013) and in the Firmicutes, within the Dehalobacter genus (Holliger et al. 1998a).
OHR reactions are catalyzed by the reductive dehalogenases (RDase), an iron-sulfur and corrinoid containing family of enzymes, which is very diverse and whose number is continually growing, suggesting that the diversity of this protein family is much deeper than is currently accounted for (Hug and Edwards 2013). Reductive dehalogenase genes typically comprise an operon containing rdhA (gene for catalytically active enzyme), rdhB (gene for a putative membrane-anchoring protein) and sometimes one or more associated genes (rdhTKZECD) (Smidt et al. 2000). The rdhA genes have been identified in a wide variety of strictly anaerobic microorganisms, in which a unique archaeal Ferroglobus species (Hug and Edwards 2013; Hug et al. 2013). Genomic analysis and genome comparisons between obligate and facultative OHRBs recently revealed specific features such as the presence of multiple putative RDase genes (up to 39) in the genome of obligate OHRBs. These genes are generally localized in high plasticity regions suggesting an important role of gene transfer and/or genomic rearrangement in the adaptation of these microorganisms (Kube et al. 2005; McMurdie et al. 2009; Kruse et al. 2013; Richardson 2013).
3.3.2.3 Dehalogenation by Anaerobic Co-metabolism
Anaerobic co-metabolism results in the partial or complete reductive dehalogenation of chlorinated compounds. This kind of metabolism has been shown for a wide variety of lightly and heavily chlorinated aliphatics while it is more rarely shown for chlorinated aromatics (for review see Field and Sierra-Alvarez 2004). Regarding aliphatics, the majority of studies reveal that they can be slowly co-metabolized by pure or mixed cultures involving mainly methanogens but also acetogenic, fermentative, sulfate-reducing and iron-reducing bacteria. Rapid anaerobic co-metabolism was observed in a few cases (e.g. pentachloroethane, chloroethenes) due to an enzymatic reduction by a reductive dehalogenase expressed for another chlorinated compound. However, the more common, slow anaerobic co-metabolism results from the direct reaction of the chlorinated compound with commonly occurring reduced enzyme cofactors (e.g. vitamin B12, a common cofactor of strict anaerobes, especially those involved in chlorine metabolism, or yet the nickel containing coenzyme F430 of methanogens). Regarding aromatic compounds, anaerobic co-metabolism degradation has essentially been observed for chlorobenzenes and PCBs. For the formers, best examples are tetrachlorobenzene and hexachlorobenzene dehalogenation by Staphylococcus epidermidis (Tsuchiya and Yamaha 1984) but also in anaerobic sewage sludge respectively (Yuan et al. 1999). About PCBs, only a few congeners were demonstrated to be subject to co-metabolic reductive dechlorination as long as more highly chlorinated parent compounds were present in the non-methanogenic mixed culture (May et al. 2006).
Methanogenesis is a very active process in Lake Pavin. Although CH4 production has been demonstrated in the anoxic water layers, CH4 concentration is mainly due to CH4 flux from the sediment. More details on methane cycle and the related prokaryotic actors are provided in Chap. 19. As described in the above paragraph, OHR generally occurs under methanogenic conditions in most environments (Vogel and McCarty 1985); hence Lake Pavin provides ideal conditions for this process. Likewise, OM degradation in the water column can provide a wide variety of electron donors such as short-chain fatty acids (e.g. acetate, butyrate, propionate) and H2 which are essential to anaerobic microbial respiratory processes such as OHR. Nowadays, various microorganisms able to synthesize these molecules have been characterized in the anoxic zone of Lake Pavin (Biderre-Petit et al. 2011a) e.g. Syntrophus species which are hydrogen-producing partners. These species are known to live in syntrophic association with hydrogen-using microorganisms, such as OHRBs of the class Dehalococcoidia (Bunge et al. 2007). Moreover, in Lake Pavin, Syntrophus species co-occur with several phylotypes closely affiliated with representing obligate OHRBs of this class (Biderre-Petit et al. 2011a) (Fig. 17.9). All together, these data strongly support the possible occurrence of both abiotic and biotic reductive dehalogenation of chlorinated compounds in this lake although this remains to be confirmed by further studies.

Phylogenetic trees showing the position of obligate (a) and facultative OHRBs (b) 16S rRNA genes sequences recovered from water column of Lake Pavin (unpublished data). The trees were based on the neighbour-joining algorithm within the MEGA v6 package (Tamura et al. 2013). Nodes with bootstraps values ≥50 % are indicated. Scale bar represents 5 % sequence divergence
4 Conclusions and Perspectives
Research works conducted over the last decade on most ecosystems revealed that natural chlorination and dechlorination of OM were much more extensive and ubiquitous than previously suggested. As reviewed in this Chapter, a vast array of biotic and abiotic processes can lead to the transformation of Cl− into Clorg, resulting in the production of thousands of natural chlorinated compounds, and vice versa. Moreover, the high abundance of a natural organic Cl pool in the environment implies that the Cl transformation and cycling are of fundamental and general importance to most living-organisms and to maintain ecosystem properties. However, though the existence of Cl cycle is now well accepted, its regulation remains poorly understood. Likewise, while knowledge is now available for a few terrestrial ecosystems, data from freshwater systems are still scarce.
Lake Pavin, with (i) its volcanic rocks that may contain high Cl contents, (ii) its dense surrounding vegetation suggesting a high chlorinated OM load and degradation within the water column, (iii) its division into three distinct vertical zones (oxic, suboxic and permanently anoxic zones) favourable both to oxic and anoxic processes of chlorination and dechlorination, (iv) its redox conditions, Fe(II) species concentrations and CH4 content in the bottom layers, all favorable to reductive dechlorination processes, and finally (v) its important microbial diversity (fungi and prokaryotes), constitutes an ideal natural field laboratory to study this biogeochemical cycle.
Thus far, the Cl cycle in Lake Pavin is largely unknown due to lack of data. But this is also true for most, if not all, aquatic environments for which the most important aspects regarding this cycling – including Cl− and Clorg pools in sediment and water, are largely missing. In Lake Pavin, the only data available is about the measurement of Cl− concentrations along the water column (Viollier et al. 1995). Another study, aiming at describing microbial community composition along the water column, provides evidence that putative dehalogenating phylotypes are present in the monimolimnion (Fig. 17.9). Among other, members of the Dehalococcoidia class, methanogenic Archaea and methylotrophic bacteria are the most promising candidates to take part directly or indirectly at the transformation of Clorg.
Future works aiming at identifying Clorg present in Lake Pavin as well as characterizing microbial populations and enzymatic mechanisms responsible of Cl transformation will help to fill these gaps. In addition to provide a better understanding of the Cl cycle in freshwater ecosystems, it will certainly results in the discovery of new reaction mechanisms and enzymes with many potential applications, for instance, in bioremediation.
References
Aeschbach-Hertig W, Hofer M, Schmid M, Kipfer R, Imboden D (2002) The physical structure and dynamics of a deep, meromictic crater lake (Lac Pavin, France). Hydrobiologia 487:111–136
Aktas O, Cecen F (2009) Cometabolic bioregeneration of activated carbons loaded with 2-chlorophenol. Bioresour Technol 100:4604–4610
Alberty RA (2003) Thermodynamics of biochemical reactions. Wiley, Hoboken
Allain EJ, Hager LP, Deng L, Jacobsen EN (1993) Highly enantioselective epoxidation of disubstituted alkenes with hydrogen peroxide catalyzed by chloroperoxidase. J Am Chem Soc 115:4415–4416
Amir A, Lee W (2011) Enhanced reductive dechlorination of tetrachloroethene by nano-sized zero valent iron with vitamin B12. Chem Eng J 170:492–497
Arora PK, Srivastava A, Singh VP (2010) Application of monooxygenases in dehalogenation, desulphurization, denitrification and hydroxylation of aromatic compounds. J Biorem Biodegrad 1:1–8
Asplund G, Grimvall A (1991) Organohalogens in nature. Environ Sci Technol 25:1346–1350
Assayag N, Jézéquel D, Ader M, Viollier E, Michard G, Prévot F, Agrinier P (2008) Hydrological budget, carbon sources and biogeochemical processes in Lac Pavin (France): constraints from δ18O of water and δ13C of dissolved inorganic carbon. Appl Geochem 23:2800–2816
Bantleon R, Altenbuchner J, van Pee KH (1994) Chloroperoxidase from Streptomyces lividans: isolation and characterization of the enzyme and the corresponding gene. J Bacteriol 176:2339–2347
Bastviken D, Thomsen F, Svensson T, Karlsson S, Sandén P, Shaw G, Matucha M, Öberg G (2007) Chloride retention in forest soil by microbial uptake and by natural chlorination of organic matter. Geochim Cosmochim Acta 71:3182–3192
Bastviken D, Svensson T, Sandén P, Kylin H (2013) Chlorine cycling and fates of 36Cl in terrestrial environments. In Svensk Kärnbränslehantering AB Swedish nuclear fuel and waste management co technical report TR-13-26
Bayer K, Scheuermayer M, Fieseler L, Hentschel U (2013) Genomic mining for novel FADH2-dependent halogenases in marine sponge-associated microbial consortia. Mar Biotechnol 15:63–72
Bengtson P, Bastviken D, De Boer W, Öberg G (2009) Possible role of reactive chlorine in microbial antagonism and organic matter chlorination in terrestrial environments. Environ Microbiol 11:1330–1339
Bengtson P, Bastviken D, Oberg G (2013) Possible roles of reactive chlorine II: assessing biotic chlorination as a way for organisms to handle oxygen stress. Environ Microbiol 15:991–1000
Benning MM, Wesenberg G, Liu RQ, Taylor KL, Dunaway-Mariano D, Holden HM (1998) The three-dimensional structure of 4-hydroxybenzoyl-CoA thioesterase from Pseudomonas sp. strain CBS-3. J Biol Chem 273:33572–33579
Bernhardt P, Okino T, Winter JM, Miyanaga A, Moore BS (2011) A stereoselective vanadium-dependent chloroperoxidase in bacterial antibiotic biosynthesis. J Am Chem Soc 133:4268–4270
Biderre-Petit C, Boucher D, Kuever J, Alberic P, Jézéquel D, Chebance B, Borrel G, Fonty G, Peyret P (2011a) Identification of sulfur-cycle prokaryotes in a low-sulfate lake (Lake Pavin) using aprA and 16S rRNA gene markers. Microb Ecol 61:313–327
Biderre-Petit C, Jézéquel D, Dugat-Bony E, Lopes F, Kuever J, Borrel G, Viollier E, Fonty G, Peyret P (2011b) Identification of microbial communities involved in the methane cycle of a freshwater meromictic lake. FEMS Microbiol Ecol 77:533–545
Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL (2006) Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature 440:368–371
Borrel G, Jezequel D, Biderre-Petit C, Morel-Desrosiers N, Morel JP, Peyret P, Fonty G, Lehours AC (2011) Production and consumption of methane in freshwater lake ecosystems. Res Microbiol 162:832–847
Bradley PM, Chapelle FH (2000) Aerobic microbial mineralization of dichloroethene as sole carbon substrate. Environ Sci Technol 34:221–223
Broholm K, Ludvigsen L, Jensen TF, Ostergaard H (2005) Aerobic biodegradation of vinyl chloride and cis-1,2-dichloroethylene in aquifer sediments. Chemosphere 60:1555–1564
Bunge M, Kleikemper J, Miniaci C, Duc L, Muusse MG, Hause G, Zeyer J (2007) Benzoate-driven dehalogenation of chlorinated ethenes in microbial cultures from a contaminated aquifer. Appl Microbiol Biotechnol 76:1447–1456
Bura-Nakic E, Viollier E, Jézéquel D, Thiam A, Ciglenecki I (2009) Reduced sulfur and iron species in anoxic water column of meromictic crater Lake Pavin (Massif Central, France). Chem Geol 266:311–317
Burd W, Yourkevich O, Voskoboev AJ, van Pee KH (1995) Purification and properties of a non-haem chloroperoxidase from Serratia marcescens. FEMS Microbiol Lett 129:255–260
Butler A, Sandy M (2009) Mechanistic considerations of halogenating enzymes. Nature 460:848–854
Chen C, Puhakka JA, Ferguson JF (1996) Transformations of 1,1,2,2-tetrachloroethane under methanogenic conditions. Environ Sci Technol 30:542–547
Coleman NV, Spain JC (2003) Epoxyalkane: coenzyme M transferase in the ethene and vinyl chloride biodegradation pathways of mycobacterium strain JS60. J Bacteriol 185:5536–5545
Copley SD (1997) Diverse mechanistic approaches to difficult chemical transformations: microbial dehalogenation of chlorinated aromatic compounds. Chem Biol 4:169–174
de Jong RM, Dijkstra BW (2003) Structure and mechanism of bacterial dehalogenases: different ways to cleave a carbon-halogen bond. Curr Opin Struct Biol 13:722–730
Dismukes GC (1986) The metal centers of the photosynthetic oxygen-evolving complex. Photochem Photobiol 43:99–115
Dolfing J (1988) Acetogenesis. In: Zehnder AJB (ed) Biology of anaerobic microorganisms. Wiley Interscience, New York
Dolfing J (1990) Reductive dechlorination of 3-chlorobenzoate is coupled to ATP production and growth in an anaerobic bacterium, strain DCB-1. Arch Microbiol 153:264–266
Dolfing J (1999) Comment on “Methane as a product of chloroethene biodegradation under methanogenic conditions”. Environ Sci Technol 33:2302–2303
Dolfing J (2000) Energetics of anaerobic degradation pathways of chlorinate aliphatic compounds. Microb Ecol 40:2–7
Dolfing J (2003) Thermodynamic consideration for dehalogenation. In: Haggblom MM (ed) Dehalogenation: microbial processes and environmental applications. Kluwer academic publishers, Secaucus, pp 89–114
Dolfing J, Harrisson BK (2000) Gibbs free energy of formation of halogenated aromatic compounds and their potential role as electron acceptors in anaerobic environments. Environ Sci Technol 26:2213–2218
Dolfing J, Janssen DB (1994) Estimates of Gibbs free energies of formation of chlorinated aliphatic compounds. Biodegradation 5:21–28
Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH (2004) Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature 427:561–565
Dong C, Flecks S, Unversucht S, Haupt C, van Pèe KH, Naismith JH (2005) Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 309:2216–2219
Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH (2004) Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol 11:817–833
Enell M, Wennberg L (1991) Distribution of halogenated organic-compounds (AOX) – Swedish transport to surrounding sea areas and mass balance studies in 5 drainage systems. Water Sci Technol 24:385–395
Eriksson E (1960) The yearly circulation of chloride and sulfur in nature; metoerological, geochemical and pedological implications. Part II. Tellus 12:63–109
Eustaquio AS, Pojer F, Noel JP, Moore BS (2008) Discovery and characterization of a marine bacterial SAM-dependent chlorinase. Nat Chem Biol 4:69–74
Fahimi IJ, Keppler F, Schöler HF (2003) Formation of chloroacetic acids from soil, humic acid and phenolic moieties. Chemosphere 52:513–520
Fetzner S (1998) Bacterial dehalogenation. Appl Microbiol Biotechnol 50:633–657
Fetzner S, Lingens F (1994) Bacterial dehalogenases: biochemistry, genetics, and biotechnological applications. Microbiol Rev 58:641–685
Field JA, Sierra-Alvarez R (2004) Biodegradability of chlorinated solvents and related chlorinated aliphatic compounds. Rev Environ Sci Biotechnol 3:185–254
Field JA, Sierra-Alvarez R (2008) Microbial transformation and degradation of polychlorinated biphenyls. Environ Pollut 155:1–12
Finley SD, Broadbelt LJ, Hatzimanikatis V (2009) Thermodynamic analysis of biodegradation pathways. Biotechnol Bioeng 103:532–541
Fujimori DG, Walsh CT (2007) What’s new in enzymatic halogenations. Curr Opin Chem Biol 11:553–560
Furukawa K (2000) Engineering dioxygenases for efficient degradation of environmental pollutants. Curr Opin Biotechnol 11:244–249
Godduhn A, Duffy LK (2003) Multi-generational health risks of persistent organic pollution in the Far North. Environ Sci Policy 6:341–353
Goldberg RN, Tewari YB, Bhat TN (2004) Thermodynamics of enzyme-catalyzed reactions – a database for quantitative biochemistry. Bioinformatics 20:2874–2877
Graedel TE, Keene WC (1996) The budget and cycle of earth’s natural chlorine. Pure Appl Chem 68:1689–1697
Gribble GW (2003) The diversity of naturally produced organohalogens. Chemosphere 52:289–297
Gribble GW (2004) Amazing organohalogens. Am Sci 92:342–349
Gribble GW (2010) Naturally occurring organohalogen compounds-a comprehensive update. In: Progress in the chemistry of organic natural products. Springer, Wien, pp 256–327
Grisham MB (1991) Chloroperoxidase: a review. In: Everse J, Everse KE, Grisham MB (eds) Peroxidase in chemistry and biology. CRC Press, Boca Raton, pp 85–137
Guerrero R, Berlanga M (2006) Life’s unity and flexibility: the ecological link. Int Microbiol 9:225–235
Gustavsson M, Karlsson S, Öberg G, Sandén P, Svensson T, Valinia S, Thiry Y, Bastviken D (2012) Organic matter chlorination rates in different boreal soils: the role of soil organic matter content. Environ Sci Technol 46:1504–1510
Hager LP, Morris DR, Brown FS, Eberwein H (1966) Chloroperoxidase. II. Utilization of halogen anions. J Biol Chem 241:1769–1777
Hamamura N, Page C, Long T, Semprini L, Arp DJ (1997) Chloroform cometabolism by butane-grown CF8, Pseudomonas butanovora, and Mycobacterium vaccae JOB5 and methane-grown Methylosinus trichosporium OB3b. Appl Environ Microbiol 63:3607–3613
Hamilton JTG, McRoberts WC, Keppler F, Kalin RM, Harper DB (2003) Chloride methylation by plant pectin: an efficient environmentally significant process. Science 301:206–209
Harper DB (2000) The global chloromethane cycle: biosynthesis, biodegradation and metabolic role. Nat Prod Rep 17:337–348
Hazen TC (2010) Cometabolic bioremediation. In: Handbook of hydrocarbon and lipid microbiology. Springer, Berlin Heidelberg, pp 2505–2514
Herczeg AL, Leaney FW (2011) Review: environmental tracers in arid-zone hydrology. Hydrogeol J 19:17–29
Hiraishi A (2008) Biodiversity of dehalorespiring bacteria with special emphasis on polychlorinated biphenyl/dioxin dechlorinators. Microbes Environ 23:1–12
Hjelm O, Johansson MB, Öberg-Asplund G (1995) Organically bound halogens in coniferous forest soil – distribution pattern and evidence of in situ production. Chemosphere 30:2353–2364
Holliger C, Hahn D, Harmsen H, Ludwig W, Schumacher W, Tindall B, Vazquez F, Weiss N, Zehnder AJ (1998a) Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra- and trichloroethene in an anaerobic respiration. Arch Microbiol 169:313–321
Holliger C, Wohlfarth G, Diekert G (1998b) Reductive dechlorination in the energy metabolism of anaerobic bacteria. FEMS Microbiol Rev 22:383–398
Horvath RS (1972) Microbial co-metabolism and the degradation of organic compounds in nature. Bacteriol Rev 36:146–155
Hruška J, Oulehle F, Šamonil P, Šebesta J, Tahovská K, Hleb R, Houška J, Šikl J (2012) Longterm forest soil acidification, nutrient leaching and vegetation development: linking modelling and surveys of a primeval spruce forest in the Ukrainian Transcarpathian Mts. Ecol Model 244:2–37
Hu S, Hager LP (1999) Highly enantioselective propargylic hydroxylations catalyzed by chloroperoxidase. J Am Chem Soc 121:872–873
Hug LA, Edwards EA (2013) Diversity of reductive dehalogenase genes from environmental samples and enrichment cultures identified with degenerate primer PCR screens. Front Microbiol 4:1–16
Hug LA, Maphosa F, Leys D, Löffler FE, Smidt H, Edwards EA, Adrian L (2013) Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos Trans R Soc Lond B Biol Sci 368:20120322
Isidorov VA (1990) Chapter 3: Organic chemisty of the earth’s atmosphere. In: Natural source of organic components of the atmosphere. Springer, Berlin
Janssen DB, Pries F, van der Ploeg JR (1994) Genetics and biochemistry of dehalogenating enzymes. Annu Rev Microbiol 48:163–191
Janssen DB, Oppentocht JE, Poelarends GJ (2001) Microbial dehalogenation. Curr Opin Biotechnol 12:254–258
Janssen DB, Dinkla IJ, Poelarends GJ, Terpstra P (2005) Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities. Environ Microbiol 7:1868–1882
Jechorek M, Wendlandt KD, Beck M (2003) Cometabolic degradation of chlorinated aromatic compounds. J Biotechnol 102:93–98
Jézéquel D, Sarazin G, Prévot F, Viollier E, Groleau A, Michard G (2012) Bilan hydrique du lac Pavin. In Le lac Pavin. Revue des Sciences Naturelles d’Auvergne 74–75: 73–96
Jobard M, Rasconi S, Solinhac L, Cauchie HM, Sime-Ngando T (2012) Molecular and morphological diversity of fungi and the associated functions in three European nearby lakes. Environ Microbiol 14:2480–2494
Johansson E, Sandén P, Öberg G (2003) Organic chlorine in deciduous and coniferous forest soils in southern Sweden. Soil Sci 168:347–355
Jordan A, Harnisch J, Borchers R, Le Guern F, Shinohara H (2000) Volcanogenic halocarbons. Environ Sci Technol 34:1122–1124
Kayser MF, Ucurum Z, Vuilleumier S (2002) Dichloromethane metabolism and C1 utilization genes in Methylobacterium strains. Microbiology 148:1915–1922
Keller S, Wage T, Hohaus K, Holzer M, Eichhorn E, van Pèe KH (2000) Purification and partial characterization of tryptophan 7-halogenase (PrnA) from Pseudomonas fluorescens. Angew Chem Int Ed 39:2300–2302
Keppler F, Eiden R, Niedan V, Pracht J, Scholer HF (2000) Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature 403:298–301
Kim EJ, Oh JE, Chang YS (2003) Effects of forest fire on the level and distribution of PCDD/Fs and PAHs in soil. Sci Total Environ 311:177–189
Kim Y-M, Park K, Joo G-J, Jeong E-M, Kim J-E, Rhee I-K (2004) Glutathione-dependent biotransformation of the fungicide chlorothalonil. J Agric Food Chem 52:4192–4196
Kirchner JW, Feng X, Neal C (2000) Fractal stream chemistry and its implications for contaminant transport in catchments. Nature 403:524–527
Kirk O, Conrad LS (1999) Metal-free haloperoxidases: fact or artifact? Angew Chem Int Ed Engl 38:977–979
Kirner S, Krauss S, Sury G, Lam ST, Ligon JM, van Pee KH (1996) The non-haem chloroperoxidase from Pseudomonas fluorescens and its relationship to pyrrolnitrin biosynthesis. Microbiology 142:2129–2135
Kruse T, Maillard J, Goodwin L, Woyke T, Teshima H, Bruce D, Detter C, Tapia R, Han C, Huntemann M, Wei CL, Han J, Chen A, Kyrpides N, Szeto E, Markowitz V, Ivanova N, Pagani I, Pati A, Pitluck S, Nolan M, Holliger C, Smidt H (2013) Complete genome sequence of Dehalobacter restrictus PER-K23(T.). Stand Genomic Sci 8:375–388
Kube M, Beck A, Zinder S, Kuhl H, Reinhardt R, Adrian L (2005) Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1. Nat Biotechnol 23:1269–1273
Kuntze K, Kiefer P, Baumann S, Seifert J, von Bergen M, Vorholt JA, Boll M (2011) Enzymes involved in the anaerobic degradation of meta-substituted halobenzoates. Mol Microbiol 82:b758–b769
Lang A, Polnick S, Nicke T, William P, Patallo EP, Naismith JH, van Pèe KH (2011) Changing the regioselectivity of the tryptophan 7-halogenase PrnA by site-directed mutagenesis. Angew Chem Int Ed Engl 50:2951–2953
Laturnus F, Taselmann KFH, Borch T, Groen C (2002) Terrestrial natural sources of trichloromethane (chloroform, CHCl3) – An overview. Biogeochemistry 60:121–139
Lehours AC, Evans P, Bardot C, Joblin K, Fonty G (2007) Phylogenetic diversity of archaea and bacteria in the anoxic zone of a meromictic lake (Lake Pavin, France). Appl Environ Microbiol 73:2016–2019
Little CD, Palumbo AV, Herbes SE, Lidstrom ME, Tyndall RL, Gilmer PJ (1988) Trichloroethylene biodegradation by a methane-oxidizing bacterium. Appl Environ Microbiol 54:951–956
Liu Y, Li F-B, Xia W, Xu J-M, Yu X-S (2013) Association between ferrous iron accumulation and pentachlorophenol degradation at the paddy soil-water interface in the presence of exogenous low-molecular-weight dissolved organic carbon. Chemosphere 91:1547–1555
Löffler FE, Tiedje JM, Sanford RA (1999) Fraction of electrons consumed in electron acceptor reduction and hydrogen thresholds as indicators of halorespiratory physiology. Appl Environ Microbiol 65:4049–4056
Löffler FE, Yan J, Ritalahti KM, Adrian L, Edwards EA, Konstantinidis KT, Muller JA, Fullerton H, Zinder SH, Spormann AM (2013) Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int J Syst Evol Microbiol 63:625–635
Lovett GM, Likens GE, Buso DC, Driscoll CT, Bailey SW (2005) The biogeochemistry of chlorine at Hubbard Brook, New Hampshire, USA. Biogeochemistry 72:191–232
Magli A, Messmer M, Leisinger T (1998) Metabolism of dichloromethane by the strict anaerobe Dehalobacterium formicoaceticum. Appl Environ Microbiol 64:646–650
Marek J, Vévodova J, Smatanova IK, Nagata Y, Svensson LA, Newman J, Takagi M, Damborsky J (2000) Crystal structure of the haloalkane dehalogenase from Sphingomonas paucimobilis UT26. Biochemistry 39:14082–14086
May HD, Cutter LA, Miller GS, Milliken CE, Watts JE, Sowers KR (2006) Stimulatory and inhibitory effects of organohalides on the dehalogenating activities of PCB-dechlorinating bacterium o-17. Environ Sci Technol 40:5704–5709
Mayer T, Snodgrass WJ, Morin D (1999) Spatial characterization of the occurrence of road salts and their environmental concentrations as chlorides in Canadian surface waters and benthic sediments. Water Qual Res J Can 34:545–574
McCulloch A (2003) Chloroform in the environment: occurrence, sources, sinks and effects. Chemosphere 50:1291–1308
McMurdie PJ, Behrens SF, Muller JA, Goke J, Ritalahti KM, Wagner R, Goltsman E, Lapidus A, Holmes S, Löffler FE, Spormann AM (2009) Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides. PLoS Genet 5, e1000714
Men Y, Seth EC, Yi S, Allen RH, Taga ME, Alvarez-Cohen L (2014) Sustainable growth of Dehalococcoides mccartyi 195 by corrinoid salvaging and remodeling in defined lactate-fermenting consortia. Appl Environ Microbiol 80:2133–2141
Michard G, Viollier E, Jézéquel D, Sarazin G (1994) Geochemical study of a crater lake: Pavin Lake, France — identification, location and quantification of the chemical reactions in the lake. Chem Geol 115:103–115
Michard G, Jézéquel D, Viollier E (2003) Vitesses de réaction de dissolution et précipitation au voisinage de l’interface oxydo-réducteur dans un lac méromictique : le lac Pavin (Puy de Dôme, France). Revue des sciences de l’eau 16:199–218
Monchy S, Sanciu G, Jobard M, Rasconi S, Gerphagnon M, Chabé M, Cian A, Meloni D, Niquil N, Christaki U, Viscogliosi E, Sime-Ngando T (2011) Exploring and quantifying fungal diversity in freshwater lake ecosystems using rDNA cloning/sequencing and SSU tag pyrosequencing. Environ Microbiol 13:1433–1453
Murphy CD (2006) Recent developments in enzymatic chlorination. Nat Prod Rep 23:147–152
Neumann CS, Fujimori DG, Walsh CT (2008) Halogenation strategies in natural product biosynthesis. Chem Biol 15:99–109
Ni X, Hager LP (1998) cDNA cloning of Batis maritima methyl chloride transferase and purification of the enzyme. Proc Natl Acad Sci U S A 95:12866–12871
Nishino SF, Shin KA, Gossett JM, Spain JC (2013) Cytochrome P450 initiates degradation of cis-dichloroethene by Polaromonas sp. dtrain JS666. Appl Environ Microbiol 79:2263–2272
Nobre RC, Nobre MM (2004) Natural attenuation of chlorinated organics in a shallow sand aquifer. J Hazard Mater 110:129–137
Nzila A (2013) Update on the cometabolism of organic pollutants by bacteria. Environ Pollut 178:474–482
Öberg G (2002) The natural chlorine cycle-fitting the scattered pieces. Appl Microbiol Biotechnol 58:565–581
Öberg G (2003) The biogeochemistry of chlorine in soil. In: Gribble G (ed) The handbook of environmental chemistry. The natural production of organohalogen compounds, vol 3, part P. Springer, Berlin, pp 43–62
Öberg G, Holm M, Sandén P, Svensson T, Parikka M (2005) The role of organic-matter-bound chlorine in the chlorine cycle: a case study of the Stubbetorp catchment, Sweden. Biogeochemistry 75:241–269
Ogawa N, Miyashita K, Chakrabarty AM (2003) Microbial genes and enzymes in the degradation of chlorinated compounds. Chem Rec 3:158–171
Olivas Y, Dolfing J, Smith GB (2002) The influence of redox potential on the degradation of halogenated methanes. Environ. Toxicol Chem 21:493–499
Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A (1992) The α/β hydrolase fold. Protein Eng 5:197–211
Ortiz-Bermúdez P, Hirth KC, Srebotnik E, Hammel KE (2007) Chlorination of lignin by ubiquitous fungi has a likely role in global organochlorine production. Proc Natl Acad Sci U S A 104:3895–3900
Pelletier I, Pfeifer O, Altenbuchner J, van Pee KH (1994) Cloning of a second non-haem bromoperoxidase gene from Streptomyces aureofaciens ATCC 10762: sequence analysis, expression in Streptomyces lividans and enzyme purification. Microbiology 140:509–516
Pérez-Pantoja D, Donoso R, Agulló L, Córdova M, Seeger M, Pieper DH, González B (2012) Genomic analysis of the potential for aromatic compounds biodegradation in Burkholderiales. Environ Microbiol 14:1091–1117
Pieper DH, Seeger M (2008) Bacterial metabolism of polychlorinated biphenyls. J Mol Microbiol Biotechnol 15:121–138
Pieper DH, González B, Cámara1 B, Pérez-Pantoja D, Reineke W (2010) Aerobic degradation of chloroaromatics. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin/Heidelberg, pp 839–864
Rai GP, Sakai S, Florez AM, Mogollon L, Hager LP (2001) Directed evolution of chloroperoxidase for improved epoxidation and chlorination catalysis. Adv Synth Catal 343:638–645
Redon P-O, Abdesselam A, Bastviken D, Cecchini S, Nicolas M, Thiry Y (2011) Chloride and organic chlorine in forest soils: storage, residence times, and influence of ecological conditions. Environ Sci Technol 45:7202–7208
Redon P-O, Jolivet C, Saby NPA, Abdelouas A, Thiry Y (2013) Occurrence of natural organic chlorine in soils for different land uses. Biogeochemistry 114:413–419
Richardson RE (2013) Genomic insights into organohalide respiration. Curr Opin Biotechnol 24:498–505
Scherer MM, Blako BA, Gallagher DA, Tratnyek PG (1998) Correlation analysis of rate constants for dechlorination by zero valent ion. Environ Sci Technol 32:3026–3033
Shaw PD, Hager LP (1959) Biological chlorination IV. Peroxidative nature of enzymatic chlorination. J Am Chem Soc 81:6527–6528
Smidt H, de Vos WM (2004) Anaerobic microbial dehalogenation. Annu Rev Microbiol 58:43–73
Smidt H, Akkermans AD, van der Oost J, de Vos WM (2000) Halorespiring bacteria-molecular characterization and detection. Enzyme Microb Technol 27:812–820
Song B, Palleroni NJ, Kerkhof LJ, Häggblom MM (2001) Characterization of halobenzoate-degrading, denitrifying Azoarcus and Thauera isolates and description of Thauera chlorobenzoica sp. nov. Int J Syst Evol Microbiol 51:589–602
Song J, Ahn H, Kim H, Song B (2006) Molecular cloning and expression of perhydrolase genes from Pseudomonas aeruginosa and Burkholderia cepacia in Escherichia coli. Biotechnol Lett 28:849–856
Stuart SL, Woods SL, Lemmon TL, Ingle JD (1999) The effect of redox potential changes on reductive dechlorination of pentachlorophenol and the degradation of acetate by a mixed, methanogenic culture. Biotechnol Bioeng 63:69–78
Svensson T, Sandén P, Bastviken D, Öberg G (2007) Chlorine transport in a small catchment in southeast Sweden during two years. Biogeochemistry 82:181–199
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thauer RK, Jungermann K, Decker K (1977) Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180
Tobiszewski M, Namiesnik J (2012) Abiotic degradation of chlorinated ethanes and ethenes in water. Environ Sci Pollut Res Int 19:1994–2006
Torgonskaya ML, Doronina NV, Hourcade E, Trotsenko YA, Vuilleumier S (2011) Chloride-associated adaptive response in aerobic methylotrophic dichloromethane-utilising bacteria. J Basic Microbiol 51:296–303
Trevisan V, Signoretto M, Colonna S, Pironti V, Strukul G (2004) Microencapsulated chloroperoxidase as a recyclable catalyst for the enantioselective oxidation of sulfides with hydrogen peroxide. Angew Chem Int Ed 43:4097–4099
Tröjbom M, Söderbäck B, Kalinowski B (2008) Hydrochemistry of surface water and shallow groundwater. Site descriptive modeling, SDM-Site Laxemar. SKB R-08-46, Svensk Kärnbränslehantering AB
Tsuchiya T, Yamaha T (1984) Reductive dechlorination of 1, 2, 4-trichlorobenzene by Staphylococcus epidermidis isolated from intestinal contents of rats. Agric Biol Chem 48:1545–1550
Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT (2005a) Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature 436:1191–1194
Vaillancourt FH, Yin J, Walsh CT (2005b) SyrB2 in syringomycin E biosynthesis is a nonheme FeII α-ketoglutarate- and O2-dependent halogenase. Proc Natl Acad Sci U S A 102:10111–10116
van Pée KH, Unversucht S (2003) Biological dehalogenation and halogenation reactions. Chemosphere 52:299–312
van Schijndel JW, Vollenbroek EG, Wever R (1993) The chloroperoxidase from the fungus Curvularia inaequalis; a novel vanadium enzyme. Biochim Biophys Acta 1161:249–256
Verhagen FJM, Swarts HJ, Kuyper TW, Wijnberg JBPA, Field JA (1996) The ubiquity of natural adsorbable organic halogen production among basidiomycetes. Appl Microbiol Biotechnol 45:710–718
Viollier E, Jézéquel D, Michard G, Pèpe M, Sarazin G, Alberic P (1995) Geochernical study of a crater lake (Pavin Lake, France): Trace-element behaviour in the monimolimnion. Chem Geol 125:61–72
Vogel TM, McCarty PL (1985) Biotransformation of tetrachloroethylene to trichloroethylene, dichloroethylene, vinyl chloride, and carbon dioxide under methanogenic conditions. Appl Environ Microbiol 49:1080–1083
Wagenknecht HA, Woggon WD (1997) Identification of intermediates in the catalytic cycle of chlororperoxidase. Chem Biol 4:367–372
Wagner C, El Omari M, Konig GM (2009) Biohalogenation: nature’s way to synthesize halogenated metabolites. J Nat Prod 72:540–553
Walsh ME, Kyritsis P, Eady NAJ, Hill HAO, Wong L-L (2000) Catalytic reductive dehalogenation of hexachloroethane by molecular variants of cytochrome P450cam (CYP101). Eur J Biochem 267:5815–5820
Wang G, Li R, Li S, Jiang J (2010) A novel hydrolytic dehalogenase for the chlorinated aromatic compound chlorothalonil. J Bacteriol 192:2737–2745
White PJ, Broadley MR (2001) Chloride in soils and its uptake and movement within the plant: a review. Ann Bot 88:967–988
Wiesner W, van Pee KH, Lingens F (1988) Purification and characterization of a novel bacterial non-heme chloroperoxidase from Pseudomonas pyrrocinia. J Biol Chem 263:13725–13732
Wilce MCJ, Parker MW (1994) Structure and function of glutathione S-transferases. Biochim Biophys Acta 1205:1–18
Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC, Moore BS (2007) Molecular basis for chloronium-mediated meroterpene cyclization: cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J Biol Chem 282:16362–16368
Winterton N (2000) Chlorine: the only green element – towards a wider acceptance of its role in natural cycles. Green Chem 2:173–225
Xu Y, He Y, Feng X, Liang L, Xu J, Brookes PC, Wu J (2014) Enhanced abiotic and biotic contributions to dechlorination of pentachlorophenol during Fe(III) reduction by an iron-reducing bacterium Clostridium beijerinckii Z. Sci Total Environ 473–474:215–223
Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT (2007) Chlorination by a long-lived intermediate in the mechanism of flavindependent halogenases. Biochemistry 46:1284–1292
Yuan SY, Su CJ, Chang BV (1999) Microbial dechlorination of hexachlorobenzene in anaerobic sewage sludge. Chemosphere 38:1015–1023
Yunos NM, Bellomo R, Story D, Kellum J (2010) Bench-to-bedside review: chloride in criticalillness. Crit Care 14:226
Zaks A, Dodds DR (1995) Chloroperoxidase-catalyzed asymmetric oxidations: substrate specificity and mechanistic study. J Am Chem Soc 117:10419–10424
Ziagova M, Kyriakou G, Liakopoulou-Kyriakides M (2009) Co-metabolism of 2,4- dichlorophenol and 4-Cl-m-cresol in the presence of glucose as an easily assimilated carbon source by Staphylococcus xylosus. J Hazard Mater 163:383–390
Acknowledgements
This study includes results from PhD researches supported by various instances: Auvergne Region, CNRS, DGA, Blaise Pascal University and French Ministry of Research and Technology. The study was also granted from different sources: French National Programme INSU-EC2CO (Project EChlore) and ANR Programme PRECODD (Project EVASOL). Finally, we are indebted to Dr Lorenz Adrian, Helmholtz Centre for Environmental Research at Leipzig, for a critical review of the ms.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dugat-Bony, E., Peyret, P., Biderre-Petit, C. (2016). New Insights into the Microbial Contribution to the Chlorine Cycle in Aquatic Ecosystems. In: Sime-Ngando, T., Boivin, P., Chapron, E., Jezequel, D., Meybeck, M. (eds) Lake Pavin. Springer, Cham. https://doi.org/10.1007/978-3-319-39961-4_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-39961-4_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39960-7
Online ISBN: 978-3-319-39961-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)