
Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late adult onset neurodegenerative disorder that mainly affects male carriers of an allele of 55–200 CGG repeats in the FMR1 gene (premutation). FXTAS symptoms include progressive intention tremor, gait ataxia, neuropathy, psychiatric symptoms, cognitive decline, and autonomic dysfunctions. Neuropathological features of FXTAS include global cerebral and cerebellar atrophy, spongiform changes of white matter, marked Purkinje cell dropout and presence of ubiquitin-positive intranuclear inclusions throughout the brain. In contrast to fragile X Syndrome (FXS), FXTAS is associated with elevated expression of repeat containing FMR1 mRNA, which binds to and sequesters specific RNA binding proteins and impedes their normal functions. In addition, the CGG repeat expansion triggers a non-canonical translational initiation event to produce a polyglycine containing protein (FMRpolyG) that accumulates in patients brains. Here we discuss these and other putative molecular mechanisms for FXTAS pathogenesis with a focus on recent findings.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Although the developmental disorder, fragile X syndrome (FXS) , is almost always caused by CGG repeat expansions exceeding 200 CGG repeats (full mutation range), with hypermethylation of the promoter region and consequent transcriptional silencing (Fu et al. 1991; Oberlé et al. 1991; Pieretti et al. 1991; Verkerk et al. 1991; Yu et al. 1991), the adult onset neurodegenerative disorder, fragile X-associated tremor/ataxia syndrome (FXTAS), almost exclusively affects carriers of premutation alleles (55–200 CGG repeats) with nearly all cases having alleles that exceed ~65–70 CGG repeats (Jacquemont et al. 2006). Premutation alleles are generally unmethylated but several studies have reported on the presence of premutation alleles partial methylated even in the lower premutation size range and in some case the methylation status correlated with the severity of the observed clinical involvement (Allingham-Hawkins et al. 1996; Pretto et al. 2014c; Tassone et al. 1999). Furthermore, several studies have reported FXTAS in individuals with a partially methylated full mutation (Loesch et al. 2012; Pretto et al. 2014a; Santa Maria et al. 2013) and also in individuals with smaller alleles in the “gray zone” or intermediate range (45–54 CGG repeats; normal <45 CGG repeats) (Hall et al. 2012; Liu et al. 2013).
Concerning instability of the CGG repeat , alleles in the normal, gray, and premutation size ranges generally have one or more AGG trinucleotides within the CGG repeat tract, typically spaced with intervals of 8–11 intervening CGG repeats (e.g., 9-10-10 pattern). However, these intervening repeats are generally lost with larger CGG repeat sizes within the premutation size range (Kunst and Warren 1994; Zhong et al. 1996), which may result in the loss of repeat length stability (Zhong et al. 1995). Interestingly, expansion from a premutation allele to a full mutation allele occurs only during maternal transmission; that is, transmission of a full mutation allele never occurs from the father, even if the father is a carrier of a full mutation allele (Nolin et al. 2003). Although the presence of AGG interruptions within the CGG repeat does not affect either the expression of the FRM1 mRNA (Yrigollen et al. 2012, 2014) or its translation (Ludwig et al. 2009), it has been demonstrated to play a key role in the stability of the repeat (Nolin et al. 2013; Yrigollen et al. 2012, 2014). Importantly, Yrigollen et al. demonstrated that the risk of expansion from premutation to full mutation alleles during maternal transmission decreases by increasing number of AGG interruptions. In addition, the risk of instability of an intermediate or premutation allele during maternal or paternal transmission is reduced with the presence of AGG interruptions, as is the magnitude of size change that occurs during transmission (Nolin et al. 2013; Yrigollen et al. 2014). Using logistic regression of 710 maternal transmissions and considering the total length of the CGG repeat allele, the number of AGG interruptions, and the age of the mother at childbirth, a model for measuring the risk of expansion to a full mutation during maternal transmission was calculated. This model was determined to be more suitable than that which considered pure CGG stretch instead of total length, confirming the previous data on a smaller sample (Yrigollen et al. 2012).
The prevalence of full mutation is approximately 1 in 5000 males and 1 in 2500–8000 females (reviewed in (Tassone et al. 2012a)). However, based on a recent systematic review and meta-analysis, approximately 1/7000 males and 1/11,000 females carry the full mutation (Hunter et al. 2014).
Reported frequencies of the premutation allele vary among studies of various populations. Canadian studies reported a premutation frequency of approximately 1 in 250 for females and 1 in 800 for males (Dombrowski et al. 2002; Rousseau et al. 1995). However, the frequency of the premutation in females in Israel is substantially higher, at approximately 1 in 113 (Toledano-Alhadef et al. 2001), and lower in an Asian population with a rate of 1 in 1674 premutation males in Taiwan (Tzeng et al. 2005). In a recent report, using the known frequency for premutation females (using the average of 1 in 126 from (Pesso et al. 2000; Toledano-Alhadef et al. 2001)) to calculate the expected number of full mutations, Hagerman (2008) determined that the expected frequency for premutation males would be 1 in 282 and for full mutation (both males and females) would be 1 in 2355. Interestingly, the former value is in agreement with what was observed in recent larger screening studies in which the prevalence of premutation alleles ranged between 150 and 210 in females and 290 and 430 in males (Hunter et al. 2014; Maenner et al. 2013; Tassone et al. 2012a) .
Clinical Involvement in FMR1 Premutation Carriers
Over the last several years, there has been increasing awareness of the spectrum of the phenotypes associated with premutation alleles of the FMR1 gene (Hagerman and Hagerman 2013). While the presence of a full mutation allele (>200 CGG repeats) generally result in FXS, with autism spectrum disorders in over 60 % of subjects (Harris et al. 2008), smaller repeat expansions in the premutation range give rise to several distinct forms of clinical involvement : (a) fragile X-associated tremor/ataxia syndrome (FXTAS) mainly in older males; (b) fragile X-associated primary ovarian insufficiency FXPOI in approximately 20 % of all carrier females; and may give rise to (c) behavioral, physical, emotional, and cognitive problems in some children who are premutation carriers, particularly at repeat sizes in the upper end of the premutation range.
FXTAS is mainly observed in males as demonstrated by the absence of significant differences between carrier females and controls when rated by CRST, ICARS, or UPDRS (Berry-Kravis et al. 2003; Jacquemont et al. 2004), or FXTAS Rating Scale (Leehey et al. 2007)—for details of use and findings with these rating scales in FXTAS the reader can see Chaps. 1 and 7. However, a percentage of female carriers clearly present clinical and neuropathological signs of FXTAS (Berry-Kravis et al. 2005; Jacquemont 2005; Peters et al. 2006; Zuhlke et al. 2004). In contrast to fragile X syndrome, both FXPOI and FXTAS appear to be generally confined to the premutation range with some exceptions (Liu et al. 2013; Loesch et al. 2012; Pretto et al. 2014c; Santa Maria et al. 2013). Moreover, while full mutations are characterized by the absence of FMR1 mRNA, and consequently by the absence of FMRP, premutation carriers possess elevated levels of FMR1 mRNA, with the extent of the increase depending upon the size of the premutation repeat expansion (Sellier et al. 2014; Tassone et al. 2000a). For the largest premutation alleles, FMRP levels are moderately reduced (up to twofold) due to a relative impediment to translation, demonstrated in both human and mouse (Ludwig et al. 2014; Primerano et al. 2002). A diagram of the relative levels of FMR1 mRNA and proteins (FMRP) as a function of the number of CGG repeats and the association of clinical phenotypes is shown in Fig. 6.1.

FXTAS and FXPOI are largely confined to the premutation range and are thought to occur through an RNA toxic gain of function due to excess FMR1 mRNA; however, patients with partially methylated full mutation alleles continue to express mRNA and are potentially at risk of developing FXTAS. By contrast, fragile X syndrome is caused by reduced/absent FMRP, due to silencing of the FMR1 gene in the full mutation range. Features of the fragile X syndrome spectrum may also occur in the upper premutation range due to reduced protein production. Dashed lines for FMR1 mRNA levels in the full mutation range reflect variations in degree of methylation; FMRP levels are reduced due to both lower mRNA levels and reduction in translation efficiency from the upper premutation range
The expanded CGG repeat, located in the 5′ UTR region of the FMR1 gene, is now believed to cause the cellular toxicity in FXTAS, whereas FMRP is not thought to play a role in the neurodegenerative syndrome , since full mutation alleles (where FMRP is absent) are not associated with FXTAS. It is, however, possible that the decreased levels of FMRP could somehow act in concert with the effects of the toxic mRNA, particularly in the upper end of the premutation range, where lower FMRP levels are detected; however, many cases of FXTAS are observed for CGG repeats in the 80–100 range, where FMRP levels are not substantially reduced. Thus, it appears clear that FXS and FXTAS are caused by a CGG expansion in the FMR1 gene but with completely different molecular mechanisms (RNA Gain of Function for FXTAS; Loss of FMRP function for FXS).
As noted above, FXTAS is more frequent in males, as would be expected for an X-linked disorder (Adams et al. 2007; Berry-Kravis et al. 2007; Coffey et al. 2008; Greco et al. 2008; Hagerman and Hagerman 2004b; Hagerman et al. 2004, 2007). In particular, female carriers are less likely to develop the disorder due to the protective effect of the expression of the normal allele in a portion of cells. Moreover, for those females who do experience features of FXTAS, expression of the normal FMR1 allele is also likely to be responsible for the less severe clinical outcome (Hagerman and Hagerman 2004b; Zuhlke et al. 2004). X-inactivation effects are therefore predicted to have a protective effect toward the neurological features of FXTAS. Several reports, in which a correlation between the severity of clinical signs and the X-inactivation ratio has been observed, seem to be supportive of such theory (Berry-Kravis et al. 2005; Coffey et al. 2008; Jacquemont 2005). However, females can present with significant higher rates of muscle pain, thyroid problems, hypertension, and fibromyalgia (Coffey et al. 2008; Hagerman and Hagerman 2013; Hundscheid et al. 2003; Rodriguez-Revenga et al. 2009).
Although details of the medical, cognitive, psychiatric, and neurological phenotypes of FXTAS and in premutation carriers in general, are discussed more extensively in other chapters of this book, a few additional comments regarding phenotype are pertinent here. Borderline to mild cognitive deficits generally are observed when FMRP levels are moderately reduced, as is often the case for individuals with alleles in the high premutation range (>150 CGG repeats) or for individuals with alleles in the low full mutation range that remain unmethylated and therefore retain transcriptional and somewhat translational activity (Tassone et al. 2000b). Numerous phenotypic problems have been reported in premutation carriers that seem to begin in many cases in childhood. The most consistent deficits seen in such carriers of a large premutation include shyness, anxiety, social deficits, ADHD, and executive function deficits (Aziz et al. 2003; Chonchaiya et al. 2012; Cornish et al. 2005; Farzin et al. 2006). Thus, for the larger premutation alleles , cognitive involvement may reflect the combined effects of both RNA toxicity and lowered FMRP levels (Farzin et al. 2006; Goodlin-Jones et al. 2004; Tassone et al. 2000a). The occurrence of seizures in about 8–13 % of those with the premutation (Bailey et al. 2008; Chonchaiya et al. 2012; Hagerman and Stafstrom 2009), appears to be associated with the development of autism spectrum disorder (ASD) in males with the premutation (Chonchaiya et al. 2012). Further, immune mediate disorders, including hypothyroidism and fibromyalgia, sleep apnea, hypertension, and migraine are more common in premutation carriers than controls (Hagerman and Hagerman 2013), Finally, very recent studies of early development in infant premutation carriers has evidentiated the presence of processing deficits similar to infants with FXS. Specifically, a visual discrimination deficit in babies with the premutation that is similar to what seen in babies with the full mutation and that is significantly different from visual discrimination in age- and sex-matched control babies has been recently reported. These observation have suggested that spatiotemporal processing impairment may constitute an endophenotype in infant premutation carriers (Gallego et al. 2014). In addition, examination of cognitive, communication, and social–behavioral profiles has demonstrated subtle developmental differences in premutation infants as young as 12 months of age (Wheeler et al. 2015).
A number of studies have reported on the correlation between the length of the CGG repeat within the premutation range and the severity of clinical involvement in premutation carriers. These correlations are particularly strong with the neurological (FXTAS) phenotypes including the age of onset of tremor and ataxia (Tassone et al. 2007a), overall motor impairment (Leehey et al. 2007), severity of white matter disease and degree of brain atrophy (Cohen et al. 2006; Loesch et al. 2005), severity of neuropathic signs (Berry-Kravis et al. 2007), degree of neuropathy as measured by nerve conduction studies (Soontarapornchai et al. 2008), reduced cerebellar volume (Adams et al. 2007), and the percent of inclusions and age at death (Greco et al. 2006). These associations are discussed further in Chap. 5.
FMR1 Gene Structure
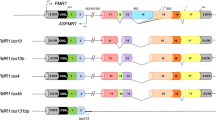
The FMR1 gene consists of 17 exons spanning 38 kb of Xq27.3 (Eichler et al. 1993). The gene is expressed in many tissues; however, the highest expression of the 4.4 kb transcript is observed in brain, placenta, testis, lung, and kidney (Hinds et al. 1993). In both human, including fetal brain neurons, and mouse, extensive alternative splicing of the FMR1 gene has been demonstrated by RT-PCR analysis , and several FMRP isoforms have been observed on Westerns (Ashley et al. 1993; Huang et al. 1996; Sittler et al. 1996; Verheij et al. 1993; Verkerk et al. 1993) due to alternative splicing in the carboxy-terminal half of the FMR1 gene. Published studies have suggested that alternative splicing in the FMR1 gene does not seem to be tissue specific, as similar ratios of transcripts were found in several fetal tissues, including brain and testis (Verkerk et al. 1993); however, quantitative approaches were not taken. The main splice variants observed in the FMR1 gene involve the use of alternative splice acceptors in exons 12, 14, 15, and 17 (Ashley Ct et al. 1993; Verkerk et al. 1993). The existence of 16 different mRNA isoforms has been recently described in different tissues (Pretto et al. 2015). Although the relative abundance of these isoforms was reported to be significantly increased in premutations, interestingly the abundance of isoforms spliced at both exons 12 and 14, specifically Iso10 and Iso10b, containing the complete exon 15 and differing only in splicing in exon 17 was four to sixfold increased compared to controls. Based on their findings, the authors suggested that RNA toxicity is likely to arise from an increased level of all FMR1 mRNA isoforms and such increase may have a functional relevance in the pathology of FMR1-associated disorders. Although the specific function of the splice variants is not known, four or five different FMRP isoforms have been described using FMRP-specific antibodies (Verheij et al. 1995) some of which act as shuttling proteins that transports their mRNA targets from the nucleus to the cytoplasm (Dury et al. 2013).
In addition to the presence of alternative splicing of the FMR1 gene, a detailed analysis of the CG-rich, TATA-less, promoter region of the FMR1 gene has revealed an influence of the CGG repeat with respect to initiation or the start of initiation. The existence of three distinct groups of transcriptional initiation sites , and the distribution of these start sites, which is modulated by the number of CGG repeats in the downstream (5′ UTR) region, has been demonstrated in different tissues including lymphocytes and neuronal cells (Beilina et al. 2004; Tassone et al. 2011). While premutation alleles appear to preferentially express the longer FMR1 mRNA (50 bp longer compared to the most frequent transcription initiation site in the normal alleles) (site II or site III), normal alleles appear to preferentially use the shorter transcripts initiating at site I (Beilina et al. 2004; Tassone et al. 2011). Interestingly, the nucleotide sequence of all three transcriptional initiation sites was found to be highly similar to the consensus sequence of pyrimidine-rich initiator (Inr) elements (consensus sequence YYAN(T/A)YY) (Javahery et al. 1994), which are usually located near the start site and have been implicated in transcription initiation in TATA-less genes (Chow et al. 1995). In addition, a fourth transcription initiation site, located between the previously identified sites I and II, was identified in brain tissues, suggesting the presence of a brain-specific transcription start site (Tassone et al. 2011). The dependency of alternative transcription initiation site usage on the CGG repeats length has also been observed in mice (Tassone et al. 2011).
Alternative polyadenylation site usage in the 3′UTR, which is implicated in the regulation of gene expression of many genes, has also been observed in the FMR1 mRNA. Several different mRNA transcripts through the usage of different polyadenylation signals were identified in both human and mouse brain tissue (Tassone et al. 2011). Thus, the combination of both alternative 5′ and 3′ UTRs in addition the complexity of expression of the FMR1 isoforms suggest their potential role in neuronal physiology, as well as in FMR1-associated disorders; however, it needs to be investigated.
Importantly, the differential usage of initiators in normal and premutation alleles may imply a possible variation in translational efficiency (post-transcriptional regulation) due to variation in 5′ structure and sequence. Indeed, the FMRP deficit observed, especially in the upper premutation range, is CGG dependent and is due to decreased translational efficiency. Reduced translational efficiency was observed both in cell lines and in transient transfection experiments using expanded alleles spanning the entire premutation range (Chen et al. 2003; Primerano et al. 2002). Similar findings are observed in two independently generated knock-in mouse models of FXTAS (Brouwer et al. 2008; Entezam et al. 2007; Iliff et al. 2013; Ludwig et al. 2014; Willemsen et al. 2003). To date, the precise mechanism by which the expanded CGG repeat impedes translation is not clear. Translation from the larger premutation alleles is expected to be severely inhibited by the predicted free energies of stabilization of the CGG repeat element. One interesting feature of the transition from normal to the premutation range is the gradual loss of AGG interruptions within the CGG element located in the 5′ UTR region of the FMR1 mRNA.
As mentioned earlier, a few studies have shown that both transcription and translation expression levels (Ludwig et al. 2009; Tassone et al. 2007b; Yrigollen et al. 2012) of the FMR1 mRNA are not influenced by AGG interruptions . Therefore, even if the presence of AGG repeats may modify the secondary/tertiary structure of the UTR region of the FMR1 gene, such alterations do not seem to have an effect on transcription or represent a rate-limiting step in translational initiation.
Higher FMR1 Transcription Rate in Premutation Alleles
While hypermethylation of the FMR1 gene in full mutation alleles leads to transcriptional silencing, premutation alleles are generally unmethylated and, therefore, transcriptionally active. Moreover, FMR1 message levels are two to tenfold higher than normal, particularly in the upper end of the premutation range (~100–200 CGG repeats), despite their reduced protein (FMRP) levels (Sellier et al. 2014). Both elevated mRNA levels (up to fivefold) and FMRP deficit were also observed in some fragile X males with a partially methylated full mutation, even for alleles greater than 300 CGG repeats (Tassone et al. 2000b).
Elevated FMR1 mRNA levels observed in premutation carriers appear to result not from increased mRNA stability (Tassone et al. 2000a) but rather from increased transcriptional activity of the FMR1 gene. This was best demonstrated by increased levels of run-on transcription in premutation alleles compared to normal (Tassone et al. 2007a). Nuclear retention of expanded repeat FMR1 mRNA was not observed by RNA in situ hybridization experiments in patient derived lymphocytes (Tassone et al. 2007a) although this has not yet been evaluated in neurons. Elevated levels of FMR1 messenger RNA and a deficit in translation efficiency in expanded alleles has been evaluated using in vitro translation experiments with a luciferase reporter mRNA containing the 5′UTR of the FMR1 gene containing varying numbers of CGG repeats. Specifically, the translation efficiency gradually decreases with increasing CGG repeat number (Chen et al. 2003).
The augmented transcription of FMR1 mRNA is associated with epigenetic alterations induced by the CGG repeat expansion itself (Todd et al. 2010). Specifically, ectopically expressed CGG repeat expansions are sufficient to elicit chromatin changes in a Drosophila model of the disease and similar changes are observed in patient derived cell lines. Interestingly, these alterations in local chromatin structure are dynamic and modifiable by genetic and pharmacologic means, suggesting that agents aimed at chromatin remodeling might have therapeutic potential in FXTAS (Todd et al. 2010). How exactly the repeat leads to these epigenetic alterations is unclear. Long tracts of CGG repeats are known to exclude nucleosomes in vitro (Wang et al. 1996). Should this also occur in vivo it could lead to enhanced transcription by enhancing access of transcription factors to the promoter. Alternatively, recent reports demonstrate that transcribed CGG repeats form R-loop structures that could also potentially trigger increased FMR1 expression (Groh et al. 2014; Loomis et al. 2014; Usdin et al. 2014). Future work will be required to define the sequential pathway and proximal events that lead to transcriptional up-regulation.
ASFMR1: The Antisense Transcript at the FMR1 Locus
Using RACE analysis and strand-specific RT-PCR, Ladd and collaborators (Ladd et al. 2007) identified an antisense transcript of the FMR1 gene (ASFMR1) overlapping with the CGG repeat region. The ASFMR1 is widely expressed in human tissues, with a higher expression in brain and similarly to the FMR1 gene, the expression appears to be influenced by the CGG repeat number as it is upregulated in peripheral blood leukocytes of premutation carriers and silenced in full mutation alleles. Moreover, ASFMR1 transcript is alternatively spliced, polyadenylated, exported to the cytoplasm, and contains an open reading frame encompassing the CCG repeat that potentially encodes a polyproline peptide (Ladd et al. 2007). Shortly after this report, Khalil and collaborators described a second antisense transcript to FMR1, FMR4, which originates upstream of the FMR1 start site and covers 2.4 kb of sequence (Khalil et al. 2008). Expression of FMR4, like that of ASFMR1 and FMR1, is increased in brain from premutation individuals and silenced in individuals with the full mutation (Khalil et al. 2008). Importantly, FMR4 overexpression increases cell proliferation while FMR4 down-regulation induces apoptosis in vitro (Khalil et al. 2008). More recently, two novel transcripts arising from the FMR1 locus, FMR5 and FMR6, were identified (Pastori et al. 2013). FMR5 is a sense-oriented long non-coding RNA (lncRNA) transcribed from approximately 1 kb upstream of the FMR1 transcription start site. In contrast to FMR1, the FMR5 transcript is not differentially expressed in human brain from unaffected individuals compared to full and premutation patients, suggesting that its transcription is independent of CGG repeat expansion (Pastori et al. 2013). FMR6 is a spliced 600 nt long antisense transcript whose sequence is complementary to the 3′ region of FMR1. FMR6 begins in the 3′UTR, ends in exon 15 of FMR1, and uses the same splice junctions as FMR1. Unexpectedly, the expression of FMR6 is reduced in premutation carriers suggesting that abnormal transcription and/or chromatin remodeling occurs toward the distal end of the locus. However, the chromatin marks associated with the 3′ end of FMR1 in premutation carriers are yet to be described and further studies are needed to determine the potential contribution of these long non-coding RNA to the variable clinical phenotypes associated with FXTAS and to the FMR1-associated disorders.
RNA Toxic Gain-of-Function Model
As stated above, FXTAS has been observed, with rare exceptions, only in premutation carriers (Loesch et al. 2012; Pretto et al. 2014c; Santa Maria et al. 2013). The absence of FXTAS in carriers of fully silenced FMR1 alleles has led to the hypothesis that FXTAS is the result of a toxic gain-of-function of the FMR1 mRNA itself (Hagerman and Hagerman 2004a; Hagerman et al. 2001). This hypothesis is based on a precedent set by the myotonic dystrophies , DM1 and DM2, in which expression of mutant RNAs containing hundreds to thousands of CUG (DM1) or CCUG (DM2) repeats accumulate in nuclear RNA aggregates that sequester specific RNA-binding proteins (the Muscleblind-like proteins). Depletion of the free pool of MBNL proteins leads to specific RNA metabolisms changes, which ultimately results in the symptoms of myotonic dystrophy (Nelson et al. 2013; Ranum and Cooper 2006).
Extending this RNA gain-of-function model to FXTAS predicts that the mutant FMR1 RNA containing expanded CGG repeats may sequester specific proteins resulting in loss of their normal functions, which would ultimately cause the symptoms of FXTAS (Hagerman and Hagerman 2004a). Consistent with this hypothesis, Iwahashi and collaborators identified more than 20 proteins from inclusions purified from brains of FXTAS patients (Iwahashi et al. 2006). Among these, two RNA-binding proteins were of special interest, hnRNP A2/B1, that is mutated in families with inherited degeneration affecting muscle, brain, bone and motor neurons (Kim et al. 2013), and MBNL1, the RNA-binding protein that is sequestered in myotonic dystrophy (Kanadia et al. 2003; Miller et al. 2000). However, a role for MBNL1 in FXTAS appears unlikely, since no genetic interaction between MBNL1 and CGG-mediated neurodegeneration was observed in fly model of FXTAS (Sofola et al. 2007), and no misregulation of splicing events regulated by MBNL1 were observed in brain samples from FXTAS patients (Sellier et al. 2010).
Association of hnRNP A2/B1 to RNA containing expanded CGG repeats was confirmed by several independent analyses (Jin et al. 2007; Sellier et al. 2010; Sofola et al. 2007). Specifically, the interaction of hnRNP A2/B1 with RNA containing expanded CGG repeats was observed in cytoplasmic cerebellar lysates (Sofola et al. 2007), and the importance of the titration of the cytoplasmic pool of hnRNP A2/B1 by expanded CGG repeats was demonstrated by the impaired dendritic delivery of the BC1 RNA, a known target of hnRNP A2/B1 (Muslimov et al. 2011). In contrast, nuclear hnRNP A2/B1 exhibited little binding to CGG RNA (Sofola et al. 2007), and alternative splicing regulated by nuclear hnRNP A2/B1 were not altered in brain samples of FXTAS patients (Sellier et al. 2010). These data suggest that some modifications of hnRNP A2/B1, either in the nucleus or in the cytoplasm, may modify the ability of hnRNP A2/B1 to bind to CGG RNA, so that expanded CGG repeats may recruit and deplete the cytoplasmic pool of hnRNP A2/B1, but not the nuclear pool of hnRNP A2/B1. In addition, the ability of hnRNP A proteins to unfold RNA structures formed by expanded CGG repeats (Khateb et al. 2004; Ofer et al. 2009) raises the interesting hypothesis that hnRNP A2/B1 may also act as an RNA chaperone that destabilizes the RNA structures formed by expanded CGG repeats.
hnRNP A2/B1 recruits other proteins, such as the CUGBP1 RNA-binding protein , to RNA containing expanded CGG repeats (Sofola et al. 2007). Overexpression of either hnRNP A2/B1 or CUGBP1 rescues the neurodegeneration in a Drosophila expressing 90 CGG repeats, highlighting the potential importance of hnRNP A2/B1 and CUGBP1 to FXTAS pathology (Jin et al. 2007; Sofola et al. 2007). Similarly, hnRNP A2/B1 interacts with the heterochromatin protein 1 (HP1) to silence the expression of specific Drosophila retrotransposons, such as gypsy or copia, and the titration of hnRNP A2/B1 by expanded CGG repeats results in increased expression of these retrotransposons in fly model of FXTAS (Tan et al. 2012). Knockdown of gypsy RNA expression, but not of copia, suppress the toxicity induced by expanded CGG repeats in Drosophila (Tan et al. 2012). Whether the expression of retrotransposons is altered in FXTAS patients remains to be determined, but it is interesting to note that increased levels of 5-hydroxymethylcytosine (5hmc) have been identified in repetitive elements DNA in a mouse model of FXTAS (Yao et al. 2014).
In addition to hnRNP A2/B1, proteomic analyses revealed that SAM68 , a splicing regulator encoded by the KHDRBS1 gene, was also found in CGG RNA aggregates (Sellier et al. 2010). However, overexpression of SAM68 was not sufficient to rescue neuronal cell death induced by expression of expanded CGG repeats. Similarly to hnRNP A2/B1 that recruits CUGBP1 through protein–protein interactions, SAM68 did not bind directly to CGG repeats and recruitment of SAM68 within CGG RNA aggregates occurred in trans.
Sellier and collaborators (2013) found that DROSHA-DGCR8 , the enzymatic complex that processes pri-microRNAs into pre-miRNAs, associates directly with expanded CGG repeats. Sequestration of DROSHA-DGCR8 within CGG RNA aggregates resulted in reduced processing of pri-miRNAs in neuronal cells expressing expanded CGG repeats, and overexpression of DGCR8 rescued neuronal cell death induced by expression of CGG repeats (Sellier et al. 2013). DROSHA-DGCR8 also recruits SAM68 to CGG repeats. Other studies also demonstrated alteration of the expression of specific miRNAs in blood samples of FXTAS patients and in Drosophila expressing CGG repeat (Alvarez-Mora et al. 2013; Tan et al. 2012). These data suggest that misregulation of the processing and/or of the expression of miRNA may be of importance for the pathogenicity of FXTAS.
Proteomic analysis performed by Jin and collaborators (2007) showed that Purα (purine-rich binding protein α) binds to RNA containing expanded CGG repeats. Purα is a single-stranded cytoplasmic DNA- and RNA-binding protein that has been implicated in many biological processes, including RNA transport and translation. Importantly, overexpression of Purα rescued neurodegeneration in a Drosophila model of FXTAS (Jin et al. 2007). However, the presence of Purα within nuclear aggregates in FXTAS brain samples was not consistently observed. Jin et al. (2007) found Purα in cytoplasmic inclusions in Drosophila expressing 90 CGG repeats. In contrast, Purα was not identified within the intranuclear inclusions in brain sections of mouse premutation models, or in hippocampal or cortical brain sections derived from patients with FXTAS (Galloway et al. 2014; Iwahashi et al. 2006; Sellier et al. 2013). These results indicate that composition of the inclusions may vary from one model organism to the other.
As with hnRNP A2/B1 that recruits CUGBP1, Purα recruits Rm62, the Drosophila ortholog of the RNA helicase P68/DDX5 (Qurashi et al. 2011). Overexpression of Rm62 rescued neurodegeneration in flies expressing 90 CGG repeats, highlighting the potential importance of P68/DDX5 to FXTAS pathology (Qurashi et al. 2011). Of interest, the RNA helicases P68/DDX5 and DDX6 have been recently reported to be involved in the unfolding of expanded CUG repeats in myotonic dystrophy (Laurent et al. 2012; Pettersson et al. 2014). These data raise the hypothesis that regulating the RNA structures of CUG expanded repeats in DM, or of CGG repeats in FXTAS may be of therapeutic interest (Disney et al. 2012; Tran et al. 2014; Yang et al. 2015).
Finally, simultaneous studies by Peter Todd and David Nelson’s groups demonstrated that overexpression of TDP-43 , encoded by the TARDBP gene, suppresses the neurodegeneration induced by expanded CGG repeats in Drosophila (Galloway et al. 2014; He et al. 2014). Interestingly, the expression of Tardbp is down-regulated in Purkinje neurons of a mouse model of FXTAS expressing the 5′UTR of FMR1 with 90 CGG repeats under the Pcp2 promoter (Galloway and Nelson 2009). TDP-43 is of special interest for neurodegenerative diseases since protein inclusions of TDP-43 in neurons is a histopathological hallmark of amyotrophic lateral sclerosis (ALS) (Neumann et al. 2006), and mutation of TARDBP leads to amyotrophic lateral sclerosis and frontotemporal dementia (ALS-FTD) (Kunst and Warren 1994). Interestingly, TDP-43 is not found within the RNA aggregates of CGG repeats, but requires the hnRNP A2/B1 homologues Hrb87F and Hrb98DE to modulate the CGG repeat triggered toxicity (He et al. 2014). Overall, these observations suggest that mutant FMR1 RNA containing expanded CGG repeats could be pathogenic by sequestering specific RNA-binding proteins, resulting in loss of their normal functions, ultimately leading to neuronal cell dysfunction and death. In that aspect, it remains to be determined whether expression of Purα, hnRNP A2/B1, P68/DDX5, DROSHA-DGCR8, CUGBP1, or TDP-43 rescues any phenotype in mouse models expressing expanded CGG repeats. Such functional rescue experiments will be instrumental to determine the importance of these candidate RNA-binding proteins to FXTAS pathology.
Inclusion Formation
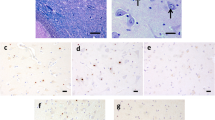
The presence of eosinophilic intranuclear inclusions, broadly distributed throughout the brain and brainstem of affected individuals (Greco et al. 2002, 2006; Tassone et al. 2004) represent the neuropathological hallmark of FXTAS. These inclusions are present as single, spherical (~2–5 μm of diameter) particles that are found exclusively within the nuclei. Inclusions have been identified in both neurons and astrocytes throughout the cerebrum and the brainstem, including cells of the ependymal and sub-ependymal layers (Tassone et al. 2004). They are most numerous in the hippocampus and are rarely seen in Purkinje cells of the cerebellum (Greco et al. 2002, 2006). Outside of the CNS, rare inclusions have been observed in both the pineal and the posterior and anterior pituitary glands but also in the pancreas, in the heart, in the thyroid and adrenal glands, in the Leydig cells of the testis, in ganglia and in periadrenal fat tissue (Greco et al. 2007; Hunsaker et al. 2011). Inclusions have also been observed in postmortem brain tissue of females with FXTAS (Hagerman et al. 2004; Tassone et al. 2012b).
Pioneering work from Hagerman and Tassone’s laboratories demonstrated that these inclusions are ubiquitin-positive and contain various chaperone proteins such as Hsp27, Hsp70, and αB-crystallin, but are negative for tau proteins, α-synuclein, or polyglutamine (Greco et al. 2002; Iwahashi et al. 2006). Importantly, these inclusions also stain positive for the FMR1 RNA messenger as well as for its polyglycine RAN-translated product (Buijsen et al. 2014; Tassone et al. 2004; Todd et al. 2010) suggesting that expression of mutant FMR1 mRNA may trigger inclusion formation. In support of this hypothesis, the formation of inclusions can be recapitulated in neuronal cell cultures using a minimal construct expressing an expansion of 99 CGG repeats embedded within the 5′UTR of FMR1 (Arocena et al. 2005; Sellier et al. 2010; Todd et al. 2010). Of interest, plasmids harboring either no expanded CGG repeats or expanded CGG repeat but under an inactive promoter (no RNA) did not lead to inclusion formation. Also, intranuclear inclusions formed even when the portion encoding FMRP was deleted (Arocena et al. 2005; Todd et al. 2010). Thus, expression of the 5′UTR of FMR1 containing a CGG repeat expansion is necessary and sufficient for the formation of these inclusions.
These observations in neuronal cell cultures are consistent with the work of Jin et al. (2003), who showed that expression in Drosophila of a construct containing the 5′UTR of FMR1 with 90 CGG repeat induces the formation of cytoplasmic ubiquitin-positive inclusions associated with neurodegeneration and eye pathology (Jin et al. 2003). Similarly, a knock-in (KI) mouse model, in which the endogenous eight CGG repeats of the murine Fmr1 were replaced with an expansion containing ~100 CGG repeats of human origin, demonstrated ubiquitin-positive nuclear inclusions and mild neuromotor and behavioral disturbances (Brouwer et al. 2008; Van Dam et al. 2005; Willemsen et al. 2003). A result confirmed by recent mouse models where heterologous expression of the sole 5′UTR of FMR1 containing expanded CGG repeats under strong chimeric promoters is sufficient to cause cellular toxicity, despite the presence of normal Fmr1 alleles and unaltered expression of Fmrp (Hashem et al. 2009; Hukema et al. 2014). These animal models demonstrate that the expression of FMR1 mRNA containing expanded CGG repeats is both necessary and sufficient to cause the pathological features of human FXTAS (Berman et al. 2014).
RAN Translation Produces a Polyglycine-Containing Protein in FXTAS
What drives inclusion formation in FXTAS was unclear until recently. The working hypothesis in the field was that the CGG RNA repeats were serving as a nidus for inclusion formation by nucleating together a set of RNA-binding proteins. However, while some inclusions in brain samples of FXTAS patients contain mutant FMR1 RNA with expanded CGG repeats (Tassone et al. 2004), mouse models in which express expanded CGG repeats show numerous ubiquitin-positive inclusions but only rarely RNA aggregates, suggesting that other factors might trigger inclusion formation (Sellier et al. 2013). Moreover, the inclusions observed in FXTAS are ubiquitin positive, which is a characteristic not commonly observed in other RNA-dominant disorders such as Myotonic Dystrophy.
A hint for how nucleotide repeats in the 5′UTR of a messenger RNA might trigger inclusion formation came with the discovery of repeat associated non-AUG (RAN) translation by Laura Ranum and colleagues (Zu et al. 2011). They found that expression of either CAG in cells outside the context of an AUG-initiated open reading frame were capable of supporting translational initiation and production of homopolymeric proteins. Surprisingly, this RAN translation occurred in all three possible reading frames to produce polyglutamine, polyalanine, and polyserine containing proteins. Initiation was largely independent of the surrounding sequence context but was influenced by the cell type in which the repeats were expressed and was somewhat different for different repeat frames. They then established that these alternative “RAN” products accumulated in tissues in animal models of Spinocerebellar Ataxia type 8 and in lymphoblasts from patients with DM1 (where a CAG repeat is expressed as an antisense transcript). Since these initial observations, RAN translation has been reported in association with CUG repeats (as seen in DM1) and with both GGGGCC and CCCCGG repeat expansions in C9 or f72 that cause ALS and Frontotemporal Dementia (Ash et al. 2013; Gendron et al. 2013; Mori et al. 2013a, b; Zu et al. 2011, 2013).
Todd and colleagues (2013) independently discovered that placement of CGG repeats upstream of a fluorescent reporter protein (GFP) in certain reading frames led to aggregation of GFP in transfected cells and in Drosophila. This aggregation was associated with production of a higher molecular weight species that resulted from translational initiation within the 5′UTR just above the CGG repeat. They were able to demonstrate by mass spectroscopy and biochemical analysis that this unconventional translational initiation (CGG RAN translation) occurred at one of several “near” AUG codons (one nucleotide different from AUG) to produce products in two reading frames, producing a polyglycine containing protein (FMRpolyG) and a polyalanine containing protein (FMRpolyA) (Todd et al. 2013). Production of FMRpolyG was confirmed in patients by use of antibodies generated against the predicted C terminus of this novel protein. The FMRpolyG protein was found in a fraction of ubiquitinated inclusions in brain autopsy samples from patients (Todd et al. 2013). Recent studies with a second set of antibodies covering different N-terminal and C-terminal epitopes revealed that most of the ubiquitin-positive inclusions observed in FXTAS brain sections are also FMRpolyG positive (Buijsen et al. 2014).
A role for FMRpolyG in disease pathogenesis is supported by work in Drosophila. In both transfected cells and in flies, it was demonstrated that moving the repeat to the 3′UTR or placing a stop codon just above the repeat prevented FMRpolyG production and suppressed CGG repeat associated toxicity. In contrast, placing an AUG start codon above the repeat-enhanced FMRpolyG production and led to greater lethality in flies and cells. However, the mechanism by which FMRpolyG induces toxicity is still unclear. Impairment of the ubiquitin proteasome system (UPS) enhances CGG repeat associated toxicity (Handa et al. 2005; Oh et al. 2015; Todd et al. 2013) and expression of chaperone proteins such as HSP-70 suppresses CGG repeat associated toxicity in flies (Jin et al. 2003). This genetic interaction between UPS impairment and CGG toxicity is largely driven by RAN translation (Oh et al. 2015). Moreover, RAN translation is capable of triggering UPS impairment in transfected cells (Oh et al. 2015). Together these studies suggest that failures in protein quality control are likely one method by which RAN translation cause toxicity, but future work will be needed to define how this unusual process contributes to FXTAS pathogenesis.
Disruption of Lamin A/C Architecture
Among the various proteins that Iwahashi and collaborators identified within the inclusions in brain samples of FXTAS patients (Iwahashi et al. 2006), the lamin A/C proteins are of special interest. Lamin proteins constitute a matrix of protein that is located next to the inner nuclear membrane and that is involved in nuclear stability, chromatin structure, and gene expression. Mutations in the LMNA gene, which encodes both lamin A and C splicing forms, are associated with several diseases called laminopathies , including limb girdle and Emery–Dreifuss muscular dystrophies, dilated cardiomyopathy, lipodystrophy, Charcot–Marie–Tooth disease and Hutchinson–Gilford progeria syndrome. Interestingly, nuclear organization of lamin A/C is disrupted in MEF of knock-in mice expressing ~200 CGG repeats, as well as in skin fibroblasts of FXTAS patients with loss of the typical architecture of lamin A/C within the nuclear ring (Garcia-Arocena et al. 2010). These alterations of the lamin A/C structure can be recapitulated in cell cultures upon transfection of a minimal construct expressing expanded CGG repeats embedded within the 5′UTR of FMR1 (Arocena et al. 2005; Hoem et al. 2011; Sellier et al. 2010). Also, the expression of Lmna mRNA is decreased in Purkinje neurons of a mouse model of FXTAS expressing 90 CGG repeats under the Pcp2 promoter (Galloway et al. 2014). In contrast, the expression of LMNA mRNA is increased while the amounts of soluble protein of Lamin A and C are slightly reduced in brain samples of FXTAS patients (Garcia-Arocena et al. 2010). Overall, these results suggest that lamin A/C architecture and expression are altered in FXTAS; however, it remains to determine the molecular mechanism leading to these alterations. It is therefore intriguing that recent data on GGGGCC repeat expansions in C9orf72 which cause ALS and FTD are associated with changes in nuclear envelope architecture and nuclear pore complex function (Freibaum et al. 2015; Jovicic et al. 2015; Zhang et al. 2015). Thus, the pathological consequences of lamin alterations in FXTAS are an exciting area of research in need of further exploration.
Mitochondrial Dysfunction
Mitochondrial biogenesis plays a central role in neurogenesis and neuroplasticity and several studies have demonstrated significant mitochondrial dysfunction in premutation carriers (Kaplan et al. 2012; Napoli et al. 2011; Ross-Inta et al. 2010), both with and without fragile X-associated tremor ataxia syndrome (FXTAS). Specifically, altered biochemical characteristics pointing to a lower ATP production, including decreased NAD- and FAD-linked oxygen uptake rates, decreased mitochondrial protein expression, increased oxidative/nitrative stress, and altered zinc bioavailability were demonstrated in both cultured dermal fibroblasts and brain samples from premutation carriers with and without FXTAS, within a wide range of CGG repeats (Napoli et al. 2011; Ross-Inta et al. 2010). Kaplan et al. (2012) observed fewer mitochondria and greatly reduced mitochondria mobility in hippocampal neuronal culture from preCGG KI mice, expressing low FMRP levels and higher Fmr1 mRNA than that measured in wild type during the early stages of development. In addition, preCGG neurons presented with abnormal metabolic function including higher rates of basal oxygen consumption, ATP production, and proton leak. The authors suggested that deficits in mitochondrial trafficking and metabolic function could play a role in the early pathophysiology observed in premutation carriers and also potentially contribute to the risk of developing FXTAS.
In addition, an altered Ca2+ regulation was reported in both neuronal cultures from the premutation mouse model (Fmr1 preCGG mouse) (Cao et al. 2012) but also in iPSC derived from premutation carriers (Liu et al. 2012). The impaired Ca2+ regulation was associated with alteration of the most abundant excitatory neurotransmitter in the vertebrate CNS, the Glutammate, with a decrease in the expression of GLT-1 and GLAST Glutamate uptake and of their expression levels in astrocytes cultures from preCGG mice and in postmortem cerebellum of PM carriers with FXTAS compared with age-matched controls. Higher CGG repeat number had the greatest reductions in both proteins (Cao et al. 2013; Pretto et al. 2014b). Abnormal clustered burst (CB) firing electrical activity and abnormal patterns of synchronized calcium oscillations in the cytosol were also observed with the premutation mouse neurons, which, interestingly, were reversed with mGluR5 antagonists or the GABAA receptor positive modulator allopregnanolone (Cao et al. 2012) making it a potential candidate for a beneficial therapeutic approach in FXTAS.
Finally, shorter telomeres have been observed in a number of neurodegenerative conditions including Alzheimer disease, cell senescence, and also in Down syndrome (Jenkins et al. 2006; Panossian et al. 2003). The role of telomeres includes protection against degeneration; they also appear to be essential for chromosomal stability and could lead to cell senescence. Telomere shortening has also been observed in a small sample of male carriers of the premutation (Jenkins et al. 2008) with FXTAS and with or without dementia. However, further studies are warranted to establish if telomere shortening could be considered a biomarker for the cellular dysfunction observed in FXTAS.
The above observations suggest that many triggering events can be associated with CGG expansion leading to cellular dysregulation and dysfunction and ultimately, potentially, to neurodegeneration and FMR1-associated disorders.
Summary
Individuals with expanded repeat of 55–200 CGG repeats are at risk to develop the late onset disease FXTAS, which might be the most common progressing neurological disorder associated with a single gene defect in males. FXTAS is likely to be the consequence of the increased expression of mutant FMR1 mRNA containing expanded (55–200) CGG repeats. Despite recent and important advances, the molecular mechanisms that lead to both inclusion formation and cell toxicity in FXTAS remain to be clarified. Several nonexclusive models for RNA-triggered pathogenesis in FXTAS can be hypothesized (summarized in Fig. 6.2

Diagram showing the molecular alterations of premutation expanded alleles including increased FMR1 mRNA expression levels, R-Loop formation, sequestration of CGG binding proteins, miRNA dysregulation and RAN translation
). First, it is possible, as in the case of myotonic dystrophy, that expanded CGG repeats toxicity, derives from sequestration of CGG RNA-binding proteins, which would limit the availability of those proteins to carry out their normal cellular functions. Alternatively, interactions between yet to be defined proteins and expanded CGG repeats could lead to activation of downstream signaling cascades potentially harmful for the cell. A second mechanism involves noncanonical translation of the expanded CGG repeats in a small protein composed of polyglycine, which is prone to aggregation. However, whether and how FMRpolyG alters neuronal function in mammalian systems requires further elucidation. Finally, mild decreased expression of the FMRP protein may contribute to FXTAS pathogenesis.
In conclusion, contributions to pathology from multiple pathogenic mechanisms may converge to explain the great variability in clinical presentations of FXTAS. Thus, more studies are warranted to improve our understanding of this multifaceted disorder and to design and develop appropriate targeted therapeutic approaches.
References
Adams JS, Adams PE, Nguyen D, Brunberg JA, Tassone F, Zhang W, Koldewyn K, Rivera SM, Grigsby J, Zhang L, Decarli C, Hagerman PJ, Hagerman RJ (2007) Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS). Neurology 69(9):851–859
Allingham-Hawkins DJ, Brown CA, Babul R, Chitayat D, Krekewich K, Humphries T, Ray PN, Teshima IE (1996) Tissue-specific methylation differences and cognitive function in fragile X premutation females. Am J Med Genet 64(2):329–333
Alvarez-Mora MI, Rodriguez-Revenga L, Madrigal I, Torres-Silva F, Mateu-Huertas E, Lizano E, Friedlander MR, Marti E, Estivill X, Mila M (2013) MicroRNA expression profiling in blood from fragile X-associated tremor/ataxia syndrome patients. Genes Brain Behav 12(6):595–603
Arocena DG, Iwahashi CK, Won N, Beilina A, Ludwig AL, Tassone F, Schwartz PH, Hagerman PJ (2005) Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet 14(23):3661–3671
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, Van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77(4):639–646
Ashley Ct SJ, Cb K, Ha L, Ee E, Dl N, Warren S (1993) Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nat Genet 4(3):244–251
Ashley CT Jr, Wilkinson KD, Reines D, Warren ST (1993) FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262(5133):563–566
Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, Willemsen R, Patton M (2003) Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet 121B(1):119–127
Bailey DB Jr, Raspa M, Olmsted M, Holiday DB (2008) Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet 146A(16):2060–2069
Beilina A, Tassone F, Schwartz PH, Sahota P, Hagerman PJ (2004) Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum Mol Genet 13(5):543–549
Berman RF, Buijsen RA, Usdin K, Pintado E, Kooy F, Pretto D, Pessah IN, Nelson DL, Zalewski Z, Charlet-Bergeurand N, Willemsen R, Hukema RK (2014) Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome. J Neurodev Disord 6(1):25
Berry-Kravis E, Lewin F, Wuu J, Leehey M, Hagerman R, Hagerman P, Goetz CG (2003) Tremor and ataxia in fragile X premutation carriers: blinded videotape study. Ann Neurol 53(5):616–623
Berry-Kravis E, Potanos K, Weinberg D, Zhou L, Goetz CG (2005) Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann Neurol 57(1):144–147
Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, Grigsby J, Bourgeois JA, Finucane B, Jacquemont S, Brunberg JA, Zhang L, Lin J, Tassone F, Hagerman PJ, Hagerman RJ, Leehey MA (2007) Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord 22(14):2018–2030
Brouwer JR, Severijnen E, De Jong FH, Hessl D, Hagerman RJ, Oostra BA, Willemsen R (2008) Altered hypothalamus-pituitary-adrenal gland axis regulation in the expanded CGG-repeat mouse model for fragile X-associated tremor/ataxia syndrome. Psychoneuroendocrinology 33(6):863–873
Buijsen RA, Sellier C, Severijnen LA, Oulad-Abdelghani M, Verhagen RF, Berman RF, Charlet-Berguerand N, Willemsen R, Hukema RK (2014) FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun 2:162
Cao Z, Hulsizer S, Tassone F, Tang HT, Hagerman RJ, Rogawski MA, Hagerman PJ, Pessah IN (2012) Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet 21(13):2923–2935
Cao Z, Hulsizer S, Cui Y, Pretto DL, Kim KH, Hagerman PJ, Tassone F, Pessah IN (2013) Enhanced asynchronous Ca(2+) oscillations associated with impaired glutamate transport in cortical astrocytes expressing Fmr1 gene premutation expansion. J Biol Chem 288(19):13831–13841
Chen LS, Tassone F, Sahota P, Hagerman PJ (2003) The (CGG)n repeat element within the 5′ untranslated region of the FMR1 message provides both positive and negative cis effects on in vivo translation of a downstream reporter. Hum Mol Genet 12(23):3067–3074
Chonchaiya W, Au J, Schneider A, Hessl D, Harris SW, Laird M, Mu Y, Tassone F, Nguyen DV, Hagerman RJ (2012) Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet 131(4):581–589
Chow CW, Clark MP, Rinaldo JE, Chalkley R (1995) Multiple initiators and C/EBP binding sites are involved in transcription from the TATA-less rat XDH/XO basal promoter. Nucleic Acids Res 23(16):3132–3140
Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, Yuhas J, Borodyanskaya M, Grigsby J, Doerflinger M, Hagerman PJ, Hagerman RJ (2008) Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A 146A(8):1009–1016
Cohen S, Masyn K, Adams J, Hessl D, Rivera S, Tassone F, Brunberg J, Decarli C, Zhang L, Cogswell J, Loesch D, Leehey M, Grigsby J, Hagerman PJ, Hagerman RJ (2006) Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology 67(8):1426–1431
Cornish K, Kogan C, Turk J, Manly T, James N, Mills A, Dalton A (2005) The emerging fragile X premutation phenotype: evidence from the domain of social cognition. Brain Cogn 57(1):53–60
Disney MD, Liu B, Yang WY, Sellier C, Tran T, Charlet-Berguerand N, Childs-Disney JL (2012) A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem Biol 7(10):1711–1718
Dombrowski C, Levesque ML, Morel ML, Rouillard P, Morgan K, Rousseau F (2002) Premutation and intermediate-size FMR1 alleles in 10 572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet 11(4):371–378
Dury AY, El Fatimy R, Tremblay S, Rose TM, Cote J, De Koninck P, Khandjian EW (2013) Nuclear fragile X mental retardation protein is localized to Cajal bodies. PLoS Genet 9(10):e1003890
Eichler EE, Richards S, Gibbs RA, Nelson DL (1993) Fine structure of the human FMR1 gene [published erratum appears in Hum Mol Genet 1994 Apr;3(4):684–5]. Hum Mol Genet 2(8):1147–1153
Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K (2007) Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 395(1–2):125–134
Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R (2006) Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr 27(2 Suppl):S137–S144
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders N, Valentine M, Miller BL, Wong PC (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525(7567):129–133
Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG Jr, Warren ST, Oostra BA, Nelson DL, Caskey CT (1991) Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67(6):1047–1058
Gallego P, Burris J, Rivera S (2014) Visual motion processing deficits in infants with the fragile X premutation. J Neurodev Disord 6:29
Galloway JN, Nelson DL (2009) Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol 4(6):785
Galloway JN, Shaw C, Yu P, Parghi D, Poidevin M, Jin P, Nelson DL (2014) CGG repeats in RNA modulate expression of TDP-43 in mouse and fly models of fragile X tremor ataxia syndrome. Hum Mol Genet 23(22):5906–5915. doi:10.1093/hmg/ddu314
Garcia-Arocena D, Yang JE, Brouwer JR, Tassone F, Iwahashi C, Berry-Kravis EM, Goetz CG, Sumis AM, Zhou L, Nguyen DV, Campos L, Howell E, Ludwig A, Greco C, Willemsen R, Hagerman RJ, Hagerman PJ (2010) Fibroblast phenotype in male carriers of FMR1 premutation alleles. Hum Mol Genet 19(2):299–312
Gendron TF, Cosio DM, Petrucelli L (2013) c9RAN translation: a potential therapeutic target for the treatment of amyotrophic lateral sclerosis and frontotemporal dementia. Expert Opin Ther Targets 17(9):991–995
Goodlin-Jones BL, Tassone F, Gane LW, Hagerman RJ (2004) Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr 25(6):392–398
Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ (2002) Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 125(Pt 8):1760–1771
Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, Hessl D, Becker EJ, Papazian J, Leehey MA, Hagerman RJ, Hagerman PJ (2006) Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 129(Pt 1):243–255
Greco CM, Soontarapornchai K, Wirojanan J, Gould JE, Hagerman PJ, Hagerman RJ (2007) Testicular and pituitary inclusion formation in fragile X associated tremor/ataxia syndrome. J Urol 177(4):1434–1437
Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, Vartanian TK, Brunberg JA, Hagerman PJ, Hagerman RJ (2008) Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol 65(8):1114–1116
Groh M, Lufino MM, Wade-Martins R, Gromak N (2014) R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10(5):e1004318
Hagerman PJ (2008) The fragile X prevalence paradox. J Med Genet 45(8):498–499
Hagerman PJ, Hagerman RJ (2004a) The fragile-X premutation: a maturing perspective. Am J Hum Genet 74(5):805–816
Hagerman PJ, Hagerman RJ (2004b) Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment Retard Dev Disabil Res Rev 10(1):25–30
Hagerman R, Hagerman P (2013) Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol 12(8):786–798
Hagerman PJ, Stafstrom CE (2009) Origins of epilepsy in fragile X syndrome. Epilepsy Curr 9(4):108–112
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ (2001) Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57:127–130
Hagerman RJ, Leavitt BR, Farzin F, Jacquemont S, Greco CM, Brunberg JA, Tassone F, Hessl D, Harris SW, Zhang L, Jardini T, Gane LW, Ferranti J, Ruiz L, Leehey MA, Grigsby J, Hagerman PJ (2004) Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet 74(5):1051–1056
Hagerman RJ, Coffey SM, Maselli R, Soontarapornchai K, Brunberg JA, Leehey MA, Zhang L, Gane LW, Fenton-Farrell G, Tassone F, Hagerman PJ (2007) Neuropathy as a presenting feature in fragile X-associated tremor/ataxia syndrome. Am J Med Genet A 143(19):2256–2260
Hall D, Tassone F, Klepitskaya O, Leehey M (2012) Fragile X–associated tremor ataxia syndrome in FMR1 gray zone allele carriers. Mov Disord 27(2):297–301
Handa V, Goldwater D, Stiles D, Cam M, Poy G, Kumari D, Usdin K (2005) Long CGG-repeat tracts are toxic to human cells: implications for carriers of Fragile X premutation alleles. FEBS Lett 579(12):2702–2708
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ (2008) Autism profiles of males with fragile X syndrome. Am J Ment Retard 113(6):427–438
Hashem V, Galloway JN, Mori M, Willemsen R, Oostra BA, Paylor R, Nelson DL (2009) Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet 18(13):2443–2451
He F, Krans A, Freibaum BD, Taylor JP, Todd PK (2014) TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum Mol Genet 23(19):5036–5051
Hinds HL, Ashley CT, Sutcliffe JS, Nelson DL, Warren ST, Housman DE, Schalling M (1993) Tissue specific expression of FMR-1 provides evidence for a functional role in fragile X syndrome [see comments] [published erratum appears in Nat Genet 1993 Nov;5(3):312]. Nat Genet 3(1):36–43
Hoem G, Raske CR, Garcia-Arocena D, Tassone F, Sanchez E, Ludwig AL, Iwahashi CK, Kumar M, Yang JE, Hagerman PJ (2011) CGG-repeat length threshold for FMR1 RNA pathogenesis in a cellular model for FXTAS. Hum Mol Genet 20(11):2161–2170
Huang T, Li LY, Shen Y, Qin XB, Pang ZL, Wu GY (1996) Alternative splicing of the FMR1 gene in human fetal brain neurons. Am J Med Genet 64(2):252–255
Hukema RK, Riemslagh FW, Melhem S, Van Der Linde HC, Severijnen LA, Edbauer D, Maas A, Charlet-Berguerand N, Willemsen R, Van Swieten JC (2014) A new inducible transgenic mouse model for C9orf72-associated GGGGCC repeat expansion supports a gain-of-function mechanism in C9orf72-associated ALS and FTD. Acta Neuropathol Commun 2:166
Hundscheid RD, Smits AP, Thomas CM, Kiemeney LA, Braat DD (2003) Female carriers of fragile X premutations have no increased risk for additional diseases other than premature ovarian failure. Am J Med Genet A 117(1):6–9
Hunsaker MR, Greco CM, Spath MA, Smits AP, Navarro CS, Tassone F, Kros JM, Severijnen LA, Berry-Kravis EM, Berman RF, Hagerman PJ, Willemsen R, Hagerman RJ, Hukema RK (2011) Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol 122(4):467–479
Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J (2014) Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A 164A(7):1648–1658
Iliff AJ, Renoux AJ, Krans A, Usdin K, Sutton MA, Todd PK (2013) Impaired activity-dependent FMRP translation and enhanced mGluR-dependent LTD in Fragile X premutation mice. Hum Mol Genet 22(6):1180–1192
Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain 129(Pt 1):256–271
Jacquemont S (2005) Screening for FXTAS. Eur J Hum Genet 13(1):2–3
Jacquemont S, Farzin F, Hall D, Leehey M, Tassone F, Gane L, Zhang L, Grigsby J, Jardini T, Lewin F, Berry-Kravis E, Hagerman PJ, Hagerman RJ (2004) Aging in individuals with the FMR1 mutation. Am J Ment Retard 109(2):154–164
Jacquemont S, Leehey MA, Hagerman RJ, Beckett LA, Hagerman PJ (2006) Size bias of fragile X premutation alleles in late-onset movement disorders. J Med Genet 43(10):804–809
Javahery R, Khachi A, Lo K, Zenzie-Gregory B, Smale ST (1994) DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol Cell Biol 14(1):116–127
Jenkins EC, Velinov MT, Ye L, Gu H, Li S, Jenkins EC Jr, Brooks SS, Pang D, Devenny DA, Zigman WB, Schupf N, Silverman WP (2006) Telomere shortening in T lymphocytes of older individuals with Down syndrome and dementia. Neurobiol Aging 27(7):941–945
Jenkins EC, Ye L, Gu H, Ni SA, Duncan CJ, Velinov M, Pang D, Krinsky-Mchale SJ, Zigman WB, Schupf N, Silverman WP (2008) Increased “absence” of telomeres may indicate Alzheimer’s disease/dementia status in older individuals with Down syndrome. Neurosci Lett 440(3):340–343
Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST (2003) RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 39(5):739–747
Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST (2007) Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55(4):556–564
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18(9):1226–1229
Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS (2003) A muscleblind knockout model for myotonic dystrophy. Science 302(5652):1978–1980
Kaplan ES, Cao Z, Hulsizer S, Tassone F, Berman RF, Hagerman PJ, Pessah IN (2012) Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J Neurochem 123(4):613–621
Khalil AM, Faghihi MA, Modarresi F, Brothers SP, Wahlestedt C (2008) A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One 3(1):e1486
Khateb S, Weisman-Shomer P, Hershco I, Loeb LA, Fry M (2004) Destabilization of tetraplex structures of the fragile X repeat sequence (CGG)n is mediated by homolog-conserved domains in three members of the hnRNP family. Nucleic Acids Res 32(14):4145–4154
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, Maclea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495(7442):467–473
Kunst CB, Warren ST (1994) Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell 77(6):853–861
Ladd PD, Smith LE, Rabaia NA, Moore JM, Georges SA, Hansen RS, Hagerman RJ, Tassone F, Tapscott SJ, Filippova GN (2007) An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet 16(24):3174–3187
Laurent FX, Sureau A, Klein AF, Trouslard F, Gasnier E, Furling D, Marie J (2012) New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res 40(7):3159–3171
Leehey MA, Berry-Kravis E, Min SJ, Hall DA, Rice CD, Zhang L, Grigsby J, Greco CM, Reynolds A, Lara R, Cogswell J, Jacquemont S, Hessl DR, Tassone F, Hagerman R, Hagerman PJ (2007) Progression of tremor and ataxia in male carriers of the FMR1 premutation. Mov Disord 22(2):203–206
Liu J, Koscielska KA, Cao Z, Hulsizer S, Grace N, Mitchell G, Nacey C, Githinji J, Mcgee J, Garcia-Arocena D, Hagerman RJ, Nolta J, Pessah IN, Hagerman PJ (2012) Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet 21(17):3795–3805
Liu Y, Winarni T, Zhang L, Tassone F, Hagerman R (2013) Fragile X-associated tremor/ataxia syndrome (FXTAS) in grey zone carriers. Clin Genet 84(1):74–77
Loesch DZ, Litewka L, Brotchie P, Huggins RM, Tassone F, Cook M (2005) Magnetic resonance imaging study in older fragile X premutation male carriers. Ann Neurol 58(2):326–330
Loesch DZ, Sherwell S, Kinsella G, Tassone F, Taylor A, Amor D, Sung S, Evans A (2012) Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin Genet 82(1):88–92
Loomis EW, Sanz LA, Chedin F, Hagerman PJ (2014) Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet 10(4):e1004294
Ludwig AL, Raske C, Tassone F, Garcia-Arocena D, Hershey JW, Hagerman PJ (2009) Translation of the FMR1 mRNA is not influenced by AGG interruptions. Nucleic Acids Res 37(20):6896–6904
Ludwig AL, Espinal GM, Pretto DI, Jamal AL, Arque G, Tassone F, Berman RF, Hagerman PJ (2014) CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum Mol Genet 23(12):3228–3238
Maenner MJ, Baker MW, Broman KW, Tian J, Barnes JK, Atkins A, Mcpherson E, Hong J, Brilliant MH, Mailick MR (2013) FMR1 CGG expansions: prevalence and sex ratios. Am J Med Genet B Neuropsychiatr Genet 162B(5):466–473
Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19(17):4439–4448
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, Van Der Zee J, Cruts M, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D (2013a) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126(6):881–893
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013b) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125):1335–1338
Muslimov IA, Patel MV, Rose A, Tiedge H (2011) Spatial code recognition in neuronal RNA targeting: role of RNA-hnRNP A2 interactions. J Cell Biol 194(3):441–457
Napoli E, Ross-Inta C, Wong S, Omanska-Klusek A, Barrow C, Iwahashi C, Garcia-Arocena D, Sakaguchi D, Berry-Kravis E, Hagerman R, Hagerman PJ, Giulivi C (2011) Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum Mol Genet 20(15):3079–3092
Nelson DL, Orr HT, Warren ST (2013) The unstable repeats—three evolving faces of neurological disease. Neuron 77(5):825–843
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, Mccluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133
Nolin SL, Brown WT, Glicksman A, Houck GE, Gargano AD, Sullivan A, Biancalana V, Brondum-Nielsen K, Hjalgrim H, Holinski-Feder E, Kooy F, Longshore J, Macpherson J, Mandel JL, Matthijs G, Rousseau F, Steinbach P, Vaisanen ML, Von Koskull H, Sherman S (2003) Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet 72:454–464
Nolin SL, Sah S, Glicksman A, Sherman SL, Allen E, Berry-Kravis E, Tassone F, Yrigollen C, Cronister A, Jodah M, Ersalesi N, Dobkin C, Brown WT, Shroff R, Latham GJ, Hadd AG (2013) Fragile X AGG analysis provides new risk predictions for 45–69 repeat alleles. Am J Med Genet A 161(4):771–778
Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252(5010):1097–1102
Ofer N, Weisman-Shomer P, Shklover J, Fry M (2009) The quadruplex r(CGG)n destabilizing cationic porphyrin TMPyP4 cooperates with hnRNPs to increase the translation efficiency of fragile X premutation mRNA. Nucleic Acids Res 37(8):2712–2722
Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, Todd PK (2015) RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum Mol Genet 24(15):4317–4326
Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, Masterman D, Cummings JL, Effros RB (2003) Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiol Aging 24(1):77–84
Pastori C, Peschansky VJ, Barbouth D, Mehta A, Silva JP, Wahlestedt C (2013) Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Hum Genet 133(1):59–67
Pesso R, Berkenstadt M, Cuckle H, Gak E, Peleg L, Frydman M, Barkai G (2000) Screening for fragile X syndrome in women of reproductive age. Prenat Diagn 20(8):611–614
Peters N, Kamm C, Asmus F, Holinski-Feder E, Kraft E, Dichgans M, Bruning R, Gasser T, Botzel K (2006) Intrafamilial variability in fragile X-associated tremor/ataxia syndrome. Mov Disord 21:98–102
Pettersson OJ, Aagaard L, Andrejeva D, Thomsen R, Jensen TG, Damgaard CK (2014) DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res 42(11):7186–7200
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66(4):817–822
Pretto D, Yrigollen CM, Tang HT, Williamson J, Espinal G, Iwahashi CK, Durbin-Johnson B, Hagerman RJ, Hagerman PJ, Tassone F (2014a) Clinical and molecular implications of mosaicism in FMR1 full mutations. Front Genet 5:318
Pretto DI, Kumar M, Cao Z, Cunningham CL, Durbin-Johnson B, Qi L, Berman R, Noctor SC, Hagerman RJ, Pessah IN, Tassone F (2014b) Reduced excitatory amino acid transporter 1 and metabotropic glutamate receptor 5 expression in the cerebellum of fragile X mental retardation gene 1 premutation carriers with fragile X-associated tremor/ataxia syndrome. Neurobiol Aging 35(5):1189–1197
Pretto DI, Mendoza-Morales G, Lo J, Cao R, Hadd A, Latham GJ, Durbin-Johnson B, Hagerman R, Tassone F (2014c) CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet 51(5):309–318
Pretto DI, Eid JS, Yrigollen CM, Tang HT, Loomis EW, Raske C, Durbin-Johnson B, Hagerman PJ, Tassone F (2015) Differential increases of specific FMR1 mRNA isoforms in premutation carriers. J Med Genet 52(1):42–52
Primerano B, Tassone F, Hagerman RJ, Hagerman PJ, Amaldi F, Bagni C (2002) Reduced FMR1 mRNA translation efficiency in Fragile X patients with premutations. RNA 8(12):1482–1488
Qurashi A, Li W, Zhou JY, Peng J, Jin P (2011) Nuclear accumulation of stress response mRNAs contributes to the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet 7(6):e1002102
Ranum LP, Cooper TA (2006) RNA-mediated neuromuscular disorders. Annu Rev Neurosci 29:259–277
Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xuncla M, Badenas C, Kulisevsky J, Gomez B, Mila M (2009) Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet 17(10):1359–1362
Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman RJ, Hagerman PJ, Giulivi C (2010) Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J 429(3):545–552
Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K (1995) Prevalence of carriers of premutation-size alleles of the FMRI gene—and implications for the population genetics of the fragile X syndrome. Am J Hum Genet 57(5):1006–1018
Santa Maria L, Pugin A, Alliende M, Aliaga S, Curotto B, Aravena T, Tang HT, Mendoza-Morales G, Hagerman R, Tassone F (2013) FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin Genet 86(4):378–382
Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Adams JS (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29(7):1248–1261
Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, Tassone F, Willemsen R, Disney MD, Hagerman PJ, Todd PK, Charlet-Berguerand N (2013) Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 3(3):869–880
Sellier C, Usdin K, Pastori C, Peschansky VJ, Tassone F, Charlet-Berguerand N (2014) The multiple molecular facets of fragile X-associated tremor/ataxia syndrome. J Neurodev Disord 6(1):23
Sittler A, Devys D, Weber C, Mandel JL (1996) Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of fmr1 protein isoforms. Hum Mol Genet 5(1):95–102
Sofola OA, Jin P, Qin Y, Duan R, Liu H, De Haro M, Nelson DL, Botas J (2007) RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55(4):565–571
Soontarapornchai K, Maselli R, Fenton-Farrell G, Tassone F, Hagerman PJ, Hessl D, Hagerman RJ (2008) Abnormal nerve conduction features in fragile X premutation carriers. Arch Neurol 65(4):495–498
Tan H, Poidevin M, Li H, Chen D, Jin P (2012) MicroRNA-277 modulates the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet 8(5):e1002681
Tassone F, Longshore J, Zunich J, Steinbach P, Salat U, Taylor AK (1999) Tissue-specific methylation differences in a fragile X premutation carrier. Clin Genet 55(5):346–351
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ (2000a) Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 66(1):6–15
Tassone F, Hagerman RJ, Loesch DZ, Lachiewicz A, Taylor AK, Hagerman PJ (2000b) Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet 94(3):232–236
Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ (2004) Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 41(4):e43
Tassone F, Adams J, Berry-Kravis EM, Cohen SS, Brusco A, Leehey MA, Li L, Hagerman RJ, Hagerman PJ (2007a) CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet B Neuropsychiatr Genet 144(4):566–569
Tassone F, Beilina A, Carosi C, Albertosi S, Bagni C, Li L, Glover K, Bentley D, Hagerman PJ (2007b) Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA 13(4):555–562
Tassone F, De Rubeis S, Carosi C, La Fata G, Serpa G, Raske C, Willemsen R, Hagerman PJ, Bagni C (2011) Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res 39(14):6172–6185
Tassone F, Iong KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, Nguyen D, Mu LY, Laffin J, Bailey DB Jr, Hagerman RJ (2012a) FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med 4(12):100
Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, Gane LW, Jacquemont S, Basuta K, Jin LW, Hagerman PJ, Hagerman RJ (2012b) Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav 11(5):577–585
Todd PK, Oh SY, Krans A, Pandey UB, Di Prospero NA, Min KT, Taylor JP, Paulson HL (2010) Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet 6(12):e1001240
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL (2013) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78(3):440–455
Toledano-Alhadef H, Basel-Vanagaite L, Magal N, Davidov B, Ehrlich S, Drasinover V, Taub E, Halpern GJ, Ginott N, Shohat M (2001) Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet 69:351–360
Tran T, Childs-Disney JL, Liu B, Guan L, Rzuczek S, Disney MD (2014) Targeting the r(CGG) repeats that cause FXTAS with modularly assembled small molecules and oligonucleotides. ACS Chem Biol 9(4):904–912
Tzeng CC, Tsai LP, Hwu WL, Lin SJ, Chao MC, Jong YJ, Chu SY, Chao WC, Lu CL (2005) Prevalence of the FMR1 mutation in Taiwan assessed by large-scale screening of newborn boys and analysis of DXS548-FRAXAC1 haplotype. Am J Med Genet A 133(1):37–43
Usdin K, Hayward BE, Kumari D, Lokanga RA, Sciascia N, Zhao XN (2014) Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front Genet 5:226
Van Dam D, Errijgers V, Kooy RF, Willemsen R, Mientjes E, Oostra BA, De Deyn PP (2005) Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS). Behav Brain Res 162(2):233–239
Verheij C, Bakker CE, De Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA (1993) Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 363(6431):722–724
Verheij C, De Graaff E, Bakker CE, Willemsen R, Willems PJ, Meijer N, Galjaard H, Reuser AJ, Oostra BA, Hoogeveen AT (1995) Characterization of FMR1 proteins isolated from different tissues. Hum Mol Genet 4(5):895–901
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, Vanommen GJB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey TC, Nelson DL, Oostra BA, Warren ST (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65(5):905–914
Verkerk AJ, De Graaff E, De Boulle K, Eichler EE, Konecki DS, Reyniers E, Manca A, Poustka A, Willems PJ, Nelson DL, Oostra BA (1993) Alternative splicing in the fragile X gene FMR1. Hum Mol Genet 2(8):1348
Wang Z, Taylor AK, Bridge JA (1996) FMR1 fully expanded mutation with minimal methylation in a high functioning fragile X male. J Med Genet 33(5):376–378
Wheeler AC, Mussey J, Villagomez A, Bishop E, Raspa M, Edwards A, Bodfish J, Bann C, Bailey DB Jr (2015) DSM-5 changes and the prevalence of parent-reported autism spectrum symptoms in Fragile X syndrome. J Autism Dev Disord 45(3):816–829
Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, Schrier M, Van Unen L, Tassone F, Hoogeveen AT, Hagerman PJ, Mientjes EJ, Oostra BA (2003) The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet 12(9):949–959
Yang WY, Wilson HD, Velagapudi SP, Disney MD (2015) Inhibition of Non-ATG translational events in cells via covalent small molecules targeting RNA. J Am Chem Soc 137(16):5336–5345
Yao B, Lin L, Street RC, Zalewski ZA, Galloway JN, Wu H, Nelson DL, Jin P (2014) Genome-wide alteration of 5-hydroxymethylcytosine in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum Mol Genet 23(4):1095–1107
Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, Tassone F (2012) AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med 14(8):729–736
Yrigollen CM, Martorell L, Durbin-Johnson B, Naudo M, Genoves J, Murgia A, Polli R, Zhou L, Barbouth D, Rupchock A, Finucane B, Latham GJ, Hadd A, Berry-Kravis E, Tassone F (2014) AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J Neurodev Disord 6(1):24
Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, Holman K, Mulley JC, Warren ST, Schlessinger D et al (1991) Fragile X genotype characterized by an unstable region of DNA. Science 252(5010):1179–1181
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525(7567):56–61
Zhong N, Yang W, Dobkin C, Brown WT (1995) Fragile X gene instability: anchoring AGGs and linked microsatellites. Am J Hum Genet 57(2):351–361
Zhong N, Ju W, Pietrofesa J, Wang D, Dobkin C, Brown WT (1996) Fragile X “gray zone” alleles: AGG patterns, expansion risks, and associated haplotypes. Am J Med Genet 64(2):261–265
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108(1):260–265
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110(51):E4968–E4977
Zuhlke C, Budnik A, Gehlken U, Dalski A, Purmann S, Naumann M, Schmidt M, Burk K, Schwinger E (2004) FMR1 premutation as a rare cause of late onset ataxia—evidence for FXTAS in female carriers. J Neurol 251(11):1418–1419
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tassone, F., Sellier, C., Charlet-Berguerand, N., Todd, P.K. (2016). The Molecular Biology of Premutation Expanded Alleles. In: Tassone, F., Hall, D. (eds) FXTAS, FXPOI, and Other Premutation Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-33898-9_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-33898-9_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33896-5
Online ISBN: 978-3-319-33898-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)