
Abstract
Over the last decades, the zebrafish has emerged as a powerful vertebrate model for studying development, diseases and behavior, and conducting high-throughput screens for therapeutic development. Large forward genetic screens have led to the generation of multiple mutant lines with developmental or behavioral defects, while transposon-based integration technologies have enabled the creation of transgenic lines essential for the functional analysis of cell and tissue movement, gene regulation, and gene function. The recent development of engineered endonucleases including ZFNs, TALENs and the CRISPR/Cas9 system has revolutionized reverse genetic approaches in zebrafish, allowing for the first time precise genome editing for targeted mutagenesis and transgenesis. In this chapter, we provide an overview of the different approaches used for mutagenesis and transgenesis in zebrafish, with an emphasis on the recent progress in targeted genetic manipulations. Examples of selected mutant and transgenic zebrafish strains are given to illustrate their growing utility for neurobehavioral phenomics and biological psychiatry.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Genome editing
- CRISPR/Cas9
- TALEN
- Zinc-finger nuclease
- Screen
- NHEJ
- Homologous recombination
- Tol2 transposon
- Knock-in
1 Introduction
In the 1960s, George Streisinger and colleagues introduced the zebrafish as a new vertebrate model for the study of developmental genetics. Thanks to its small size, ease of breeding, and external fertilization producing large clutches of transparent embryos, the zebrafish appeared especially suited for morphological observation of developmental processes. Since then, the zebrafish has become a model of choice, not only for studying vertebrate development, but also for modeling human diseases and conducting molecule screening for drug discovery. Comparison of the zebrafish and human genomes indicates that 71.4 % of human protein-coding genes have at least one zebrafish orthologue [1]. Systemic large-scale forward genetic screens combined with the annotation of the zebrafish reference genome have led to the identification of a large variety of mutations affecting embryogenesis, physiology or behavior relevant to human health [2, 3]. More recently, an explosion of new tools and techniques, in particular the development of engineered endonucleases (EENs ), have open a new area for genome editing, allowing the direct manipulation of the zebrafish genome for targeted mutagenesis and transgenesis (Fig. 1). This chapter provides an overview of the different approaches used for mutagenesis and transgenesis in zebrafish, with an emphasis on the recent progress in targeted genetic manipulations, and their ‘translational’ applications to modeling selected brain disorders.

Timeline recapitulating the development of mutagenesis approaches in zebrafish. This historical timeline recapitulates the major advances in the development of mutagenesis approaches in zebrafish over the last 40 years. The first ENU-based mutagenesis screens conducted in the Driever and Nüsslein-Volhard labs have generated several hundreds of mutants that were reported in a dedicated issue of Development in 1996. As such, no author name was reported in the corresponding box
2 Mutagenesis in Zebrafish
2.1 Chemical Genetic Screens
Forward genetic screens allow the identification of genes implicated in a specific biological pathway or process by screening a population of animals in which genome modifications have been randomly induced. Individuals that display a phenotype of interest are identified as carriers of a modified/mutated allele, and subsequent mapping of the modification/mutation responsible for the phenotype identifies the gene involved. Thanks to its external fertilization, its large clutch size, its rapid development and the transparency of its embryos, the zebrafish proved ideal for large-scale forward genetic screens. While initial work used UV light and γ-ray irradiation to trigger chromosomal breaks [4–6], N-ethyl-N-nitrosourea (ENU) has rapidly become the standard choice for chemical mutagenesis [7–9]. ENU is a DNA alkylating agent that mostly triggers point mutations, has a high mutagenic efficiency, and can be directly applied to adult male zebrafish by adding it to the water, making it very easy to use (Fig. 2). The first large-scale ENU screens performed in the Driever lab in Boston and the Nüsslein-Volhard lab in Tubingen have generated several hundreds of mutants with developmental phenotypes that were reported in a dedicated issue of Development in 1996 [10]. Other ENU screens have subsequently identified more genes involved in development [11–16], behavior [17–19] addiction [20], or diseases [21]. While the identification of the mutated alleles by positional cloning is often laborious, whole-genome sequencing at low coverage can now be used to map mutations rapidly [22, 23]. Two approaches using whole genome sequencing have been developed: the bulk-segregant linkage analysis (BSFseq) that involves a mapping cross, and the homozygosity mapping (HMFseq) [24]. Both rely on bioinformatic filtering for mutagenic polymorphisms, and can be analyzed with the open source computational pipeline MegaMapper available at https://wiki.med.harvard.edu/SysBio/Megason/MegaMapper. More affordable approaches using transcriptome sequencing have also been developed, such as Mutation Mapping Analysis Pipeline for Pooled RNA-seq (MMAPPR) [25] and RNA-seq-based bulk segregant analysis [26]. MMAPPR offers the advantage of identifying mutations without sequencing the parental strain or using a SNP database.

Use of ENU mutagenesis in forward genetic screens and TILLING. (a) Overview of a classical ENU-based mutagenesis forward screen. Males are directly treated with ENU added to the water to induce random germline mutations. Mutated males are outcrossed to WT females to generate F1 families. F1 family members are outcrossed to WT to generate F2 families, whose members are randomly incrossed to produce F3 embryos. F3 embryos are examined for phenotypic defects, and their genomes are analyzed for identification of the causative mutation. (b) Overview of ENU-based TILLING. Males are directly treated with ENU to induce random germline mutations and are outcrossed to WT females to generate F1 families. Males from F1 families are sacrificed after cryopreservation of their sperm and sampling of their DNA for analysis. DNA from F1 males is screened for mutations in genes of interest. The cryopreserved sperm from identified carriers is then selected to generate F2 families using in vitro fertilization, and mutant embryos can be identified by genotyping
While originally used in forward screens that identify mutations after phenotypic analysis, ENU mutagenesis has also been applied in reverse genetic approaches, in which mutations are detected first and then associated with a phenotype. Targeting Induced Local Lesions in Genomes (TILLING) was first used to screen for desired mutated alleles in Arabidopsis, and was successfully adapted to the zebrafish 2 years later [27, 28]. In contrast to ENU -based forward genetic screens in which phenotypic analysis is conducted at the F3 generation, DNA analysis and sperm cryopreservation is performed in F1 families in TILLING (Fig. 2b). After mutations have been identified, the cryopreserved sperm is used in in vitro fertilization to generate F2 families, whose carriers are isolated by genotyping. TILLING alleles are currently being generated and distributed to the zebrafish community by the zebrafish TILLING consortium initiated by the Moens lab at the Fred Hutchinson Cancer Research Center and the Solnica-Krezel lab at Washington University School of Medicine in St. Louis. Alleles can be requested online at http://webapps.fhcrc.org/science/tilling/index.php. Complementary to the zebrafish TILLING project, the Zebrafish Mutation Project from the Wellcome Trust Sanger Institute aims to produce a knockout allele in every protein-coding gene of the zebrafish genome, and has so far generated 26,634 alleles. Mutations are identified after whole exome enrichment and Illumina next generation sequencing, and each allele is analyzed for morphological defects [29]. A list of available lines with mutations is available online at http://www.sanger.ac.uk/sanger/Zebrafish_Zmpbrowse.
2.2 Retroviral and Transposon-Mediated Mutagenesis
While ENU mutagenesis is a powerful approach to generate random mutations at a high rate, it requires a significant degree of effort and commitment to identify the mutations. Insertional mutagenesis using retroviruses or transposons offers the advantage of a fast screening of carriers and a rapid identification of the mutated gene by using the sequence of the insertional element as a “tag” for mapping. Retroviruses and transposons have different insertion site preferences and generate null or hypomorphic alleles, or have no effect, depending on where they integrate in the genome.
Retroviral-mediated mutagenesis was the first insertional mutagenesis carried out in zebrafish in the early 1990s [30]. It used a pseudo-type retrovirus derived from the Moloney murine leukemia virus (MoMLV), with the envelop protein replaced by the glycoprotein from the vesicular stomatitis virus (VSV). Like in human cells, this modified retrovirus was shown to preferentially integrate in regions close to transcriptional starts in the zebrafish genome [31, 32]. It was used by the Hopkins lab at the MIT and others to carry out several large insertional forward screens that led to the generation of hundreds of lines with development defects [33–37]. Like ENU mutagenesis and TILLING, retroviral mutagenesis has also been used in reverse genetics by injecting high-titer retroviruses into embryos [32]. Sperm from F1 males is cryopreserved, and mutations are mapped by identifying the genomic sequences flanking the insertion site, or by high-throughput Illumina sequencing to generate a proviral insertion library. Most retroviral insertions have been located in introns, with insertions into the first intron of genes often leading to a decrease in gene expression. Using this approach, the Lin lab at UCLA and the Burgess lab at NHGRI/NIH have generated the Zebrafish Insertion Collection (ZInC) , in which 3054 mutations in genes have been isolated from 6144 F1 fish [38, 39]. Mutant lines can be searched for with the ZInC database (http://research.nhgri.nih.gov/ZInC/?mode=search) and requested through the Zebrafish international Resource Center (ZIRC) (http://zebrafish.org/home/guide.php).
Insertional mutagenesis using transposons has also been used for gene inactivation. Transposons, or “jumping genes”, are mobile DNA sequences that can change their position within the genome, thereby altering it and creating mutations. Insertions and excisions require the activity of the transposase enzyme. Several transposable elements including Sleeping beauty, Ac/Ds and Tol2 have been used in zebrafish for both mutagenesis and transgenesis, Tol2 being the most common [40–42]. Transposons have been favored over retroviruses due to their ease of use and their ability to integrate large transgenes. Transposon-based gene-breaking constructs have been improved over the years to simultaneously inactivate a gene (“gene trap”) and insert transgenes of interest such as fluorescent proteins (“protein trap”) (Fig. 3a and b), or to inactivate a gene in a conditional manner. The FlipTrap cassette, for instance, allows conditional mutagenesis thanks to the insertion of loxP and FRT sites for Cre-mediated and Flp-mediated recombination, respectively [43]. When integrated into an intron of a gene, the FlipTrap cassette forms a fusion protein of citrine and the endogenous protein, thereby revealing the expression profile of the targeted gene when it is expressed. Exposure to Cre recombinase removes the citrine sequence and a splice donor sequence associated to it, thereby inducing a truncation of the gene. Flp-mediated recombination allows the exchange of the cassette with any DNA sequence after the integration has occurred. Several FlipTrap lines have been made available through the FlipTrap database (http://www.fliptrap.org/static/anatomies.html). Other efficient gene-breaking constructs such as the RP2 cassette [44], the FlEx cassette [45] or a recently developed bipartite Gal4-containing vector [46] also function as conditional alleles thanks to the presence of loxP and/or FRT sites flanking the mutagenic cassette. Transposon-based cassettes have been used to perform several mutagenesis screens [32, 47–49]. A list of gene trap fish lines obtained from the Kawakami lab is provided through the zTrap database (http://kawakami.lab.nig.ac.jp/ztrap/). The zTrap database allows the search for gene trap insertions located within or near genes of interest [50].

Overview of transposon-based gene-trap, protein trap and enhancer trap approaches . All transposon-based gene trap, protein trap and enhancer trap vectors contain transposable elements (TE) that mediate random integration of the vector in the genome. (a) In a gene trap approach, the vector contains a splice acceptor site (SA) upstream of a reporter sequence with a stop codon and a polyA (pA) signal at its 3′ end. Because the reporter does not have any start codon, its transcription depends on the regulation of the endogenous gene by the upstream regulatory element (enhancer). Proper splicing of the SA to the 5′ exon of the gene integrates the reporter into the transcript and generates a truncated protein. (b) In a protein trap approach, the vector contains both a SA and a splice donor site (SD) flanking the reporter sequence. The reporter is devoid of start and stop codons, allowing the fusion between the reporter and the endogenous transcript when integration in an intron is in the correct orientation and proper reading frame. (c) In an enhancer trap, the vector contains a basal promoter with minimal activity upstream of a reporter sequence with a start codon, a stop codon and a pA signal. When the vector integrates near an endogenous transcriptional enhancer, its basal promoter becomes regulated by it and drives the expression of the reporter without any mutagenic effect
2.3 Targeted Mutagenesis
While ENU, retroviruses and transposon-based constructs are powerful mutagenesis tools for forward genetic screens, the genome modifications they generate are random, making it effortful and time-consuming to isolate a mutant for a gene of interest. For many years, methods for engineering specific loci in the genome were restricted to organisms like the mouse, in which embryonic stem cells can be manipulated in a precise way through homologous recombination (HR) . In zebrafish, gene knockdown was transiently achieved by injecting antisense morpholino oligonucleotides (MOs) designed to block the splicing or translation of a targeted mRNA [51]. MOs have been widely used to test gene function, but have recently raised some concerns regarding their specificity [52, 53]. A comparative study looking at more than 80 genes notably reported that around 80 % of the phenotypes observed in MO-injected embryos (“morphants”) could not be detected in the corresponding mutants [54]. These differences have led to the assumption that MO-induced phenotypes often result from off-target effects, and that mutants should become the standard model to describe gene function. On the other hand, deleterious mutations have recently been shown to activate genetic compensatory mechanisms [55]. Further investigation will likely be required to explain the discrepancies between morphant and mutant phenotypes for a specific gene.
The ability to precisely manipulate the zebrafish genome has remained a long-standing quest that was only recently resolved by the discovery of sequence-specific endonucleases and their engineering as genome editing tools. All engineered endonucleases (EENs) consist of a sequence-specific DNA targeting component (protein domain or RNA) and a double-stranded DNA cleaving endonuclease (catalytic domain) that introduces double-strand breaks (DSBs) in the genome. DSBs can be repaired by two different pathways: non-homologous end joining (NHEJ) and homology-directed repair (HDR ). NHEJ can ligate the cleaved DNA double strands without any template but introduces insertions or deletions (indels) at the cut site. HDR, on the other hand, uses a homologous template of DNA to repair DSBs. NHEJ is ten times more active than HDR or HR during zebrafish development [56–58]. This error-prone repair mechanism is exploited to introduce a frameshift mutation leading to a non-functional protein. Indel mutations generated by NHEJ are easily detected by analyzing the formation of heteroduplexes between mutant and wild-type (WT) alleles, either by a mobility assay, in which heteroduplexes and homoduplexes have different electrophoretic migration profiles [59], by using enzymes like the endonucleases Surveyor or Cel-I or the bacteriophage resolvase T7E1 that recognize and cut mismatches [60–62], or by high resolution melt curve analysis (HRMA) [63–65]. The nature of indels can be further characterized by directly analyzing Sanger sequencing data with the poly peak parser software available at http://yost.genetics.utah.edu/software.php [66]. Several EENs including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and RNA-guided nucleases (CRISPR/Cas9) have been used successfully in zebrafish for targeted mutagenesis (Fig. 4), each of them presenting its own advantages (Table 1). A searchable database, EENdb, collects reported TALENs, ZFNs and CRISPR/Cas systems for different organisms including zebrafish and can be accessed at http://eendb.zfgenetics.org/ [67]. Another software, ZiFit, can be used to design ZFNs, TALENs, or CRIPSRs and is available at http://zifit.partners.org/ZiFiT/Introduction.aspx [68].

Engineered endonucleases for targeted mutagenesis . (a) ZFNs are composed of zinc finger arrays (ZFAs) fused to the catalytic domain of the FokI endonuclease. Each ZFA generally consists in three fingers that each recognizes and binds to a specific 3 nt DNA sequence. Since FokI becomes active upon dimerization, ZFNs work in pairs, cleaving DNA only after each of them has bound to its target sequence. (b) TALENs are constructed by fusing the catalytic domain of FokI to the DNA-binding transcription activator-like effector (TALE) proteins. Each TALE contains an N-terminal translocation domain that recognizes a 5′-T (in red in the DNA sequence), a DNA-binding central repeat domain, and a C-terminal sequence. The central domain contains repeat units composed of 33–35 conserved amino acids, with differences at amino acids 12 and 13 that form the repeat variable di-residue (RVD). Each RVD recognizes and binds to a single specific nucleotide and is therefore responsible for the DNA binding specificity of each repeat unit. Like ZFNs, TALENs function by pairs to cleave DNA. (c) In the CRISPR/Cas9 system, a single guide RNA (sgRNA) recruits the endonuclease Cas9 to the genomic sequence it complements. The sgRNA is composed of 20 nt sequence that directly matches the DNA target sequence, followed by 72–80 nt of the bacterial crRNA/tracrRNA sequence that are required for the formation of hairpin loops stabilizing the sgRNA. Cas9 has two catalytic domains, RuvC and HNH, that each cleaves a DNA strand. The presence of NGG as a protospacer adjacent motif (PAM) is required in 3′ of the target sequence for DNA recognition by Cas9
2.3.1 ZFNs
First described in 1996 [69], ZFNs are chimeric proteins composed of a DNA-binding zinc finger array (ZFA ) fused to the catalytic domain of the non-specific bacterial endonuclease FokI that becomes active upon dimerization (Fig. 4a). Each ZFA generally contains three small Cys2His2 zinc fingers derived from natural transcription factors (“Cys2His2” corresponds to the four residues that coordinate the zinc atom), with each finger recognizing and binding to a specific 3 bp DNA sequence. Many fingers recognizing 5′-GNN, 5′-ANN and 5′-CNN triplets (with N being any base) have been isolated using phage display, and a catalogue of fingers and their binding preferences has been generated [70–77]. While in theory, fingers can be assembled into any combination to construct a ZFA against any sequence of interest, designing ZFAs with specific and efficient DNA binding activities has been a challenge, as the interaction of each finger with DNA is context dependent. Several methods involving direct assembly or screening strategies have been developed to generate efficient ZFAs. Modular assembly (MA) directly ligates fingers that recognize different triplets, but does not take into account the context-dependent effects of the DNA sequence, leading to a rather high failure rate [78]. Best success has been achieved using targets composed of 5′-GNN [79]. In contrast to MA, oligomerized pooled engineering (OPEN) uses a bacterial two-hybrid selection method to identify ZFAs with high efficiencies and high affinities from a combinatorial library of multi-finger arrays recognizing 9 bp sequences [80, 81]. A similar approach with a one-hybrid selection system has also been used [82]. While more efficient, these approaches require expertise in constructing libraries and are quite labor-intensive. A more recent and easier method named Context-dependent assembly (CoDA) assembles three-finger arrays by selecting N- and C-terminal fingers from known ZFAs containing a common middle finger, thereby accounting for context-dependent effects between adjacent fingers [83].
Since the FokI endonuclease domain must dimerize to be active [84], ZFNs function by pairs, cleaving DNA only after each of them has bound to its target sequence (Fig. 4a). Obligate heterodimer modifications have been introduced in the FokI catalytic domain to increase ZFN efficacy and reduce off target cleavages [85, 86]. The spacer that separates the ZFA target sequences is relatively short, of variable size and has no sequence requirement. Two to five amino acids can be introduced between the ZFA and the FokI as an inter-domain linker to accommodate the variable size of the spacer in the DNA sequence [87, 88]. While the requirement of two ZFNs to target a sequence provides good specificity and limits off target effects, finding a target sequence in the 5′ region of a gene to generate a null mutation can be limited by the context-dependent affinity of each zinc finger within a ZFA. Nonetheless, ZFNs have been successfully employed for gene targeting in zebrafish since the first reports of their use [82, 89, 90]. ZFN target sites can be identified in several organisms including zebrafish with ZFNgenome, a comprehensive open source accessible at http://bindr.gdcb.iastate.edu/ZFNGenome/ [91]. mRNAs encoding ZFNs are then injected at one-cell stage after ZFNs have been assembled.
2.3.2 TALENs
While useful for targeted mutagenesis, ZFNs have rapidly been challenged by the development of TALENs, which appear to be more mutagenic in zebrafish [92, 93]. TALENs are chimera proteins obtained by fusing the DNA-binding transcription activator-like effectors (TALEs) to the catalytic domain of FokI (Fig. 4b). TALEs were originally identified in the bacterial plant pathogen Xanthomonas and were named for their ability to trigger the expression of genes promoting infection in the host cell [94].
Each TALE is composed of an N-terminal translocation domain that recognizes a 5′-T, a DNA-binding central repeat domain, and a C-terminal sequence. The central domain contains 15.5–19.5 repeat units composed of 33–35 conserved amino acids, with differences at amino acids 12 and 13 forming the repeat variable di-residue (RVD) (the last repeat unit contains only 20 amino acids and is referred to as a half repeat). Each RVD recognizes and binds to a single specific nucleotide and is therefore responsible for the DNA binding specificity of each repeat unit. The RVDs NI, HD and NG are commonly used to target the nucleotides A, C and T, respectively, while NN, NK and NH can be employed for targeting a guanine, with NK and NH binding more specifically but with a weaker affinity [95–97]. In contrast to ZFNs, whose efficiency is context-dependent, TALENs do not have much requirement in terms of the targeted sequence besides a 5′-T and a minimum length of 11 RDVs for the binding domain [98]. Several online tools such as TALE-NT [99] (https://tale-nt.cac.cornell.edu/node/add/talen), Mojo Hand [100] (http://www.talendesign.org/), or idTALE [101] (http://omictools.com/idtale-s5415.html) can be used to identify the optimal TALEN target sequence within a gene of interest.
Like ZFNs, TALENs are engineered with the FokI endonuclease catalytic domain as an obligate heterodimer and must therefore work by pairs to cleave DNA. The optimal spacer length seems to depend on the scaffold of the TALEN, with TALENs containing short C-terminal lengths (17–28 amino acids) being more efficient with shorter spacers (12–14 bp). Several methods have been developed for constructing TALENs and dictate the scaffold used. The Golden Gate cloning strategy uses restriction digest of a TALE plasmid library by type II endonucleases followed by ligation [102]. This approach is theoretically a one-step assembly that can construct TALE repeats in one digest and ligation reaction. While several Golden Gate derived methods have been generated and some commercially available, they do not always use the same TALE scaffold and are not always compatible [103–107]. Other approaches such as the Unit assembly method [108, 109] and the restriction enzyme and ligation (REAL) method [110] rely on standard molecular cloning using hierarchical restriction digests and ligations. While effective, these methods are labor-intensive and do not allow the construction of TALENs in a large scale. High-throughput can be achieved with the fast ligation-based automatable solid-phase high-throughput (FLASH) or the iterative capped assembly (ICA) methods that use solid-phase ligation on magnetic beads instead of the time-consuming transformation and growing of bacteria [111, 112].
Due to their higher mutation frequencies, the rarity of off-target effects, and the presence of target sequences in almost every gene, TALENs have quickly become the method of choice for mutagenesis in zebrafish since their first application in 2011–2012 [64, 109, 113–115]. As for ZFNs, mRNAs encoding TALENs are injected at one-cell stage. TALEN efficiency can be assessed the day after by analyzing heteroduplex formation in injected embryos.
2.3.3 CRISPR/Cas9 System
The CRISPR (clustered regularly interspaced short palindromic repeats) / Cas (CRISPR associated proteins) system was originally identified as a defense mechanism used by bacteria and archae against the introduction of foreign nucleotides form bacteriophage and exogenous plasmids [116–118]. Invading nucleic acids are first recognized as foreign and integrated as spacers between short DNA repeats (the CRISPR locus) in the host genome, thereby forming CRISPR arrays. Transcription of these CRISPR arrays generates primary transcripts, or pre CRISPR RNAs (pre-crRNA) that are subsequently cleaved into small CRISPR RNAs (crRNAs ). Upon infection, the crRNAs whose spacers have a sequence close to the invading nucleic acids bind to them, and recruit a second non-coding RNA with partial complementarity to the crRNA named auxiliary trans-activating crRNA (tracrRNA ). The complex tracrRNA/crRNA in turn recruits nucleases associated with the CRISPR locus named Cas to degrade the intruder nucleic acids and prevent pathogen invasion. Of particular interest is Cas9, an endonuclease that introduces DSB s in the target DNA thanks to its two nuclease active sites, RuvC and HNH, that each cleaves a DNA strand (Fig. 4c). Several groups saw the genome-editing possibilities offered by the CRISPR/Cas9 system and adapted it for its use in eukaryotic cells. The crRNA and tracrRNA of Streptococcus pyogenes were fused into a single guide RNA (sgRNA) named for its ability to recruit and activate Cas9 [119]. On the other hand, the sequence of the Streptococcus pyogenes Cas9 has been modified by codon optimization and the introduction of nuclear localization signals to promote its use in eukaryotic cells [119–121].
Since the targeting properties of the CRISPR/Cas9 system only rely on the sequence of the sgRNA , it has become very easy to target any sequence of interest in the genome. Each sgRNA is composed of a 20 nt sequence that directly matches the target sequence, followed by 72–80 nt of the 3′ crRNA/tracrRNA sequence that are required for the formation of hairpin structures stabilizing the sgRNA [122, 123]. The only constraint for the design of sgRNAs is the presence of NGG at the 3′ end of the target site that acts as a protospacer adjacent motif (PAM) required for DNA recognition by Cas9 and Cas9 subsequent activation (Fig. 4c) [124–127]. The requirement of NGG as a PAM currently limits the number of sequences recognized by Cas9, but a recent study has successfully engineered efficient Cas9 derivatives with altered PAM specificities, thereby expanding the repertoire of PAMs needed [128]. Several servers and online softwares have been specifically developed for the design of sgRNAs, including CRISPRdirect (http://crispr.dbcls.jp/) [129], the Optimized CRISPR Design (http://crispr.mit.edu/) from the Zhang lab, the Cas9 Online Designer (http://cas9.wicp.net/) developed by Dayong Guo, Cas-Designer (http://rgenome.net/cas-designer/) [130], sgRNACas9 (http://www.biootools.com/) [131], and CHOPCHOP (https://chopchop.rc.fas.harvard.edu/) [132].
Because of its ease of use and affordability, the CRISPR/cas9 system has rapidly been applied in zebrafish, with mutagenesis rates comparable or superior to those obtained with TALENs [133–136]. sgRNAs are obtained by in vitro transcription from plasmids or oligos and co-injected with Cas9 mRNA at one-cell stage to induce DSB s in the target sequence. Some studies have also directly injected the Cas9 protein with sgRNAs to increase mutagenic activity [137–139]. A major advantage of the CRISPR/Cas9 system is that its high efficiency is sometimes sufficient to introduce extensive biallelic mutations causing phenotypes in injected embryos, a feature not often seen using TALENs [140]. In addition, it offers the possibility to simultaneously target multiple sequences at once by co-injecting several sgRNAs , or by using a plasmid with multiple sgRNA cassettes under the control of U6 or H1 promoters, a process named CRISPR multiplexing [140–143]. Multiplexing has recently been employed in a high-throughput mutagenesis set-up to successfully generate mutations in 83 different genes in the zebrafish genome [144]. At lower scale, multiplexing can be very useful to generate double or triple mutants in related genes for which single mutants would lack a phenotype due to compensatory mechanisms. It can also be employed to study the role of non-coding RNA genes that are not affected by changing the frame of translation. The identification of optimal targets in multiple locus has been facilitated by the recent development of specialized softwares such as CRISPRseek (http://www.bioconductor.org/packages/release/bioc/html/CRISPRseek.html) [145] or CRISPR MultiTargeter (http://www.multicrispr.net/) [146]. Finally, a last advantage of the CRISPR/Cas9 system is the possibility of disrupting gene function in a spatially controlled manner by injecting a modular vector that contains an sgRNA cassette under the control of a U6 promoter and a Cas9 cassette under the control of a tissue- or cell-specific promoter [147].
Altogether, the CRISPR/Cas9 system offers so many advantages that it has quickly become the method of choice for targeted mutagenesis in zebrafish. A major drawback, though, is the rather high frequency of off-target effects observed in various models [122, 123, 148–150]. The short length of the target sequence, and the tolerance of Cas9 for mismatches between the target sequence and the sgRNA, can lead to the mutation of secondary targets in the genome that would need repeated outcrossing to be eliminated. Choosing unique target sequences using the specific softwares mentioned above is thus important. Specificity can be further improved by using truncated sgRNAs (17 nt) that have a decreased mutagenesis rate at off-target sites [151]. Finally, Cas9 variants possessing only one nuclease catalytic site instead of two can be used [122, 152, 153]. These Cas9 “nickases” introduce nicks in one DNA strand only, and must be used in pairs with two sgRNAs to introduce DSBs. This system is thus analogous to TALENs or ZFNs by requiring a dual recognition of the targeted DNA sequence.
2.4 Chromosomal Deletions and Inversions
In addition to generating small indel mutations in a gene of interest, large genomic deletions or inversions can be introduced by injecting several TALEN pairs or multiple sgRNAs. DSB s are introduced simultaneously at two separate sites, leading to the loss, or more rarely the inversion, of the DNA fragment in between. In zebrafish, genomic deletions with sizes ranging from several hundred bases to 1 Mb have been reported [141, 154, 155]. Introducing large deletions has proved useful in different systems to study the role of cis-regulatory sequences [156, 157], or to recreate translocations similar to those found in human diseases [158–160].
3 Transgenesis in Zebrafish
Transgenesis is defined as the introduction of exogenous genes, or “transgenes” , into the genome of a living organism. The first zebrafish transgenic lines were obtained by the random integration of transgenes and regulatory promoters in the genome using retroviruses or transposon-based systems. By expressing transgenes such as fluorescent proteins or genes with dominant negative mutations, and by providing a spatial and/or temporal control of gene activation, these transgenic lines proved to be powerful tools for observing the fate and behavior of cells and tissues, studying gene regulation, and testing gene function in development, behavior and diseases. More recently, TALENs and the CRISPR/Cas9 system have revolutionized zebrafish research by allowing the insertion of sequences into specific loci in the genome, making the generation of knock-ins (KIs) finally possible.
3.1 Transposon-Based Transgenesis with Random Insertion
3.1.1 Enhancer Traps and Protein Traps
As mentioned previously for transposon-based mutagenesis, transposon-based constructs have been used in gene trap, enhancer trap or protein trap configurations (Fig. 3) in the context of high throughput screens. All constructs possess transposable elements (TE) derived from Sleeping beauty or Tol2 transposons that allow random integration in the genome. While gene traps are used for insertional mutagenesis (discussed earlier in section 2.2.), enhancer traps have a limited mutagenic effect and are designed to report the transcriptional activity of enhancers located nearby their site of integration (Fig. 3c). Proteins traps are constructed to create a fusion between the full-length trapped gene and the reporter, allowing the visualization of protein expression in the embryo (Fig. 3b).
Numerous enhancer trap screens have been conducted in zebrafish and have led to the creation of a large library of transgenic lines expressing fluorescent reporters or drivers in specific cells and tissues. Several basal promoters have been employed, including keratin 4 (krt4) and keratin 8 (krt8), gata2, hsp70, c-fos, Eb1, ef1a, thymidine kinase, the carp β-actin promoter (TKBA), and the medaka edar locus [161–170]. These basal promoters have various trapping efficiencies and can drive different expression profiles based on their sensitivity to the genomic enhancer regulating them [163]. Although all basal promoters have been useful to reveal specific patterns of expression during development, some have a bias for traps with expression in specific structures (for instance, the E1b promoter has a strong bias for cranial ganglia), while others can drive non-specific background expression is tissues such as the muscles or the dermis. Several reporters have also been used, the most common being fluorescent reporters like EGFP to monitor transcriptional activity and follow the movement or differentiation of the cells labeled, and Gal4, to drive effector gene expression where Gal4 is expressed using the Gal4/UAS system. Several Gal4 enhancer trap lines have been generated in combination with a UAS:EGFP or UAS:Kaede reporter, where the photo-convertible Kaede fluorescent protein can be used for mapping neural circuits or cell lineages [163, 164]. A collection of enhancer trap lines is described in the ZETRAP 2.0 database available at http://plover.imcb.a-star.edu.sg/webpages/home.html [171].
In parallel to enhancer traps, protein traps have been developed to generate an in-frame fusion between the full-length trapped gene and the reporter (Fig. 3b). By retaining all the regulatory sequences of the endogenous genes, this approach allows detailed studies on the expression of the protein trapped and its regulation as well as its localization within cells. As mentioned previously, protein traps have mostly been combined with gene-traps to allow simultaneous gene inactivation and protein inactivation [43].
3.1.2 The Gateway System for Easy Transgenesis
While enhancer and protein trap constructs have been instrumental for visualizing developing tissues or protein localization, they are not particularly suited for overexpressing a gene of interest in a temporally or spatially controlled manner. Transgenesis is an essential tool for testing gene and cell function, but has been historically laborious in zebrafish due to technical limitations such as laborious conventional cloning and low rates of germline transmission when using supercoiled or linear DNA [172, 173]. To overcome these limitations, the Tol2Kit system (that uses the recombination-based cloning of multiple DNA fragments) was designed to easily generate expression constructs for transgenesis [174]. This multisite Gateway technology relies on the att site specific recombination system from the λ phage [175], and uses different engineered att sites that recombine specifically to assemble up to five DNA fragments in a directional manner. Three different “entry” clones containing a promoter, a coding sequence of interest, and a polyA or a tag, respectively, are recombined into a “destination” vector that also possesses Tol2 recombination elements for integration in the genome with high efficiency. The plasmid hence generated is co-injected with transposase mRNA at one-cell stage for transient or stable transgenesis. Carriers of the transgene are usually easily identified by the expression of a reporter gene. A main advantage of this approach is its modularity that allows the generation of libraries of entry clones with promoters or genes of interest. For instance, entry clones with the promoter element from the hsp70 gene [176] or a UAS promoter have been generated for conditional expression. A list of essential Tol2Kit clones can be requested online at http://tol2kit.genetics.utah.edu/index.php/Main_Page. By providing a simple, affordable and flexible system to generate transgenesis constructs, the Tol2Kit has largely facilitated zebrafish research, promoting the sharing of clones within the zebrafish community and making transgenesis available for any lab. Several labs have expanded the number of clones using the gateway technology and made their resources available (http://lawsonlab.umassmed.edu/gateway.html). To date, a list of 14,524 transgenic lines generated with either “trap” or Gateway constructs can be viewed on Zfin at http://zfin.org/action/fish/search.
3.2 Targeted Transgenesis and the Generation of Knock-Ins
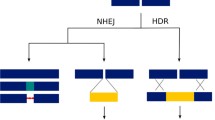
By providing an efficient approach to integrate DNA constructs into the genome, transposon-derived elements have been instrumental for the study of gene function and tissue morphogenesis in zebrafish. However, transposon-mediated integrations occur randomly, precluding precise genome editing. TALENs and CRISPR/Cas9, on the other hand, allow targeted engineering and have been recently employed to insert small or large sequences at precise loci into the genome. Several methods involving the NHEJ or the HDR pathways have successfully led to the generation of the first KIs in zebrafish.
3.2.1 Integration via the HDR Pathway
DNA integration mediated by HDR has been achieved with both TALENs and the CRISPR/Cas9 system. Several templates including linearized plasmid and single-stranded DNA (ssDNA ) have been used with various integration efficiencies.
One of the first reports of gene targeting via HR in zebrafish used TALENs and a linearized DNA vector containing the cassette to be inserted flanked by homologous sequences to the genomic target of around 800 and 900 bp on each side [177]. Several cassettes with loxP, eGFP, or eGFP-stop sequences were used to modify three different loci in the genome. Authors co-injected the linearized donor plasmids with TALEN mRNAs at one cell stage and were able to detect HR between the donor plasmids and the endogenous loci, with transmission to the germline in one case, albeit with low efficiency (about 1.5 %). Subsequent studies demonstrated that the length of homology arms as well as the configuration of the targeting construct have a significant impact on the efficiency of HDR [178]. In particular, increasing the length of the left and right arms to 1 and 2 kb, and introducing a DSB in the shorter homology arm, were shown to greatly improve efficient HR and germline transmission (over 10 %).
HDR has also been achieved using ssDNA with short homology arms as a template together with TALENs [113] or the CRISPR/Cas9 system [133, 134, 136]. Short fragments encoding restriction sites (6 bp) or loxP sites (34 bp) have been successfully integrated after co-injecting TALEN mRNAs and ssDNA oligonucleotides with short homology arms of 20 and 18 bp [113]. Interestingly in that case, increasing the length of homology arms seemed to reduce the frequency of HDR integrations. While germline transmission of the integrated DNA could be observed in 10 % of the cases, a major drawback of this approach was the frequent imprecise integration of the donor DNA with additional indel mutations. Similar results were obtained after co-injecting ssDNA oligonucleotides with sgRNAs and Cas9 mRNA [133, 134, 136]. All studies reported so far achieved precise integration of the template in the targeted genome location with various efficiencies. However, in all cases, imprecise repair events were frequently detected as a probable result of NHEJ . Inhibit the NHEJ pathway by blocking the activity of endogenous DNA ligase IV with the Scr7 inhibitor has recently been shown to increase the efficiency of HDR-mediated genome editing in mammalian cells and mice [179, 180], and might lead to similar improvement in zebrafish in the future.
3.2.2 Integration via the NHEJ Pathway
Considering the prevalence of NHEJ in zebrafish during development [56–58], recent studies have exploited the NHEJ pathway to elicit targeted integration of large donor DNAs [181, 182]. In this approach, the donor vector contains a short sequence bearing the TALEN or the CRISPR target site upstream the cassette to be integrated. Co-injection of this donor plasmid with sgRNA and Cas9 mRNA (or alternatively TALEN mRNAs) lead to the concurrent cleavage of the plasmid and the genomic target, and the subsequent integration of the linearized plasmid at the genomic target by an NHEJ repair mechanism. Alternatively, two different sgRNAs can be used, one for cleaving the genome target, and the other for cleaving the donor plasmid. This method has proved to be very efficient for generating KIs allele, with rates of germline transmission over 30 %. However, since the integration of the donor plasmid can occur in both forward or reverse orientation and three different frames, screening efforts are necessary to isolate the appropriate lines. Introducing short homologous sequences flanking the sgRNA target site into the donor seems to improve the precision of integration by involving both NHEJ and HDR mechanisms [183]. Alternatively, selecting an sgRNA target in the intron of the gene sequence, and using a donor vector with a homologous arm spanning from that sgRNA site to the 3′ region of the targeted gene, can be employed to circumvent the requirement of in-frame insertions and increase KI efficiency [184].
4 Conclusions and Future Directions
Overall, the last 40 years have witnessed major advances in zebrafish research, from the first mutagenesis screens to the use of EENs for targeted genome editing . With precise genomic manipulations now available, the zebrafish has caught-up with other vertebrate organisms, combining genomic approaches previously restricted to the mouse with screening and high-resolution imaging techniques only possible in fish. This progress has promoted the development and use of mutant and transgenic lines in a wide number of research areas, and in neurobehavioral phenotyping research in particular. The following examples illustrate the wide spectrum of neural phenotypes studied, and their relevance to selected human brain disorders (see also Tables 2 and 3 for additional examples).
The Allan–Herndon–Dudley syndrome (AHDS ) syndrome is a rare developmental nervous system disorder characterized by severe intellectual disability, muscle hypotonia and spastic paraplegia. It is caused by mutations in the mct8 (slc16a2) gene located on the X chromosome that encodes a thyroid hormone receptor. Impaired Mct8 function is thought to prevent the entry of the active T3 hormone into neurons, leading to abnormal neurological development. While the MCT8 knockout mouse recapitulates the metabolic and endocrine defects seen in patients, they do not have any neurological or behavioral phenotype. In order to determine the functions of Mct8 in AHDS, Zada and colleagues used ZFNs to generate an mct8 zebrafish mutant line [185]. Video-tracking behavioral imaging as well as time-lapse imaging of neuronal circuits showed that Mct8 zebrafish mutants had a reduced locomotor activity that correlated with defects in synaptic density of motoneuron arbors and abnormal axonal branching in sensory neurons. Additional behavioral defects were observed, including increased and more fragmented sleep, and altered responses to light variations. Thus, the use of ZFNs to induce targeted mutation in Mct8 in zebrafish lead to the development of the first vertebrate model of AHDS that recapitulates the full spectrum of defects seen in patients. As zebrafish is particularly suited for large-throughput approaches, this mct8 mutant line could further be used in pharmacological screens for therapeutic development.
In addition to modeling neurological developmental diseases , zebrafish mutant lines have been used to study complex behaviors. Sleep, for instance, is an evolutionary conserved state that is essential to all organisms, but whose regulation remains poorly understood. In particular, the factors that transmit circadian information to regulate sleep are largely unknown. Using TALENs as a mutagenesis approach, Gandhi and colleagues generated a new zebrafish line harboring a null mutation in the aanat2 gene that encodes an enzyme essential for melatonin synthesis [186]. Videotracking assays revealed that aanat2 mutants had a normal sleep pattern during daytime but a reduced sleep and a longer sleep latency during night. Importantly, circadian rhythms were not disrupted in mutants, revealing for the first time that melatonin is not required to initiate or maintain the circadian clock. Altogether, the use of TALENs is this example allowed the generation of the first genetic loss-of-function model for melatonin in a diurnal vertebrate, and lead to the discovery of the endogenous functions of melatonin in sleep regulation.
Complementary to mutants, transgenic lines have proved very helpful to decipher the mechanisms underlying complex behaviors and neurological disorders (see Table 3). Narcolepsy, for example, is a rare chronic sleep disorder involving excessive daytime sleepiness, sleep fragmentation and paralysis at night, hypnagogic hallucinations and cataplexy. It is caused by the selective degeneration of hypothalamic hypocretin/orexin (HCRT) neurons, whose activity is known to regulate several other behaviors including food intake, reward or drug addiction. To generate a zebrafish model of narcolepsy, Elbaz and colleagues used Tol2-mediated transgenesis to establish a stable transgenic line expressing the nitroreductase nfsB gene under the control of the hcrt promoter [187]. Exposing transgenic embryos to the drug metrodinazole induces the apoptosis of cells expressing nfsB, providing an inducible method to selectively ablate HCRT neurons at specific times. As expected, transgenic embryos lacking HCRT neurons recapitulated the defects seen in narcoleptic patients, including increased sleep time and transitions between wake and sleep states. They further had altered locomotor responses to light and sound, suggesting a broader function of HCRT neurons in mediating behavioral responses to external stimuli.
Transgenesis not only allows the ablation or silencing of a specific class of neurons regulating complex behaviors, but can also be used to express specific mutations in genes causing human disorders. Amyotrophic lateral sclerosis (ALS ) is an adult-onset lethal neurodegenerative disease characterized by the progressive loss of motor neurons. While the majority of ALS is sporadic, around 10 % of cases are familial and caused by mutations in certain genes. Among them, mutations in the superoxide dismutase Sod1 gene have been associated with 20 % of familial ALS (fALS) . Several zebrafish transgenic lines expressing the SOD1-G93A mutation have been generated using Tol2-mediated transgenesis [188, 189]. They all showed defects associated with fALS including abnormal motor neuron outgrowth and branching, loss of neuromuscular junctions, muscle atrophy and motor neuron cell loss leading to premature death. These new transgenic lines thus provide an additional system for observing the progression of ALS directly in vivo in an intact organism, and isolating new effective compounds in therapeutics screens.
Overall, targeted mutagenesis and transgenesis have broaden the field of zebrafish research in many areas. The development of conditional mutant and targeted transgenic lines is now under way and will expand the repertoire of lines and resources currently available. For instance, one recent study has introduced attP site at specific loci into the genome for future recombination-mediated site-specific transgenesis [190]. Other new approaches that could be adapted to zebrafish research include Cas9 engineering to regulate transcription [191–193]. The zebrafish model is looking at a bright future, one that George Streisinger would be proud of.
Abbreviations
- Cas:
-
CRISPR-associated
- CRISPR:
-
Clustered regulatory interspaced short palindromic repeats
- DSB:
-
Double strand break
- EENs:
-
Engineered endonucleases
- ENU:
-
N-ethyl-N-nitrosourea
- HDR:
-
Homology-directed repair
- HR:
-
Homologous recombination
- KI:
-
Knock-in
- NHEJ:
-
Non homologous end joining
- ssDNA:
-
Single-stranded DNA
- TALEN:
-
Transcription-activator like effector nuclease
- WT:
-
Wild-type
- ZFN:
-
Zinc-finger nuclease
References
Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503.
Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46.
Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36.
Chakrabarti S, Streisinger G, Singer F, Walker C. Frequency of gamma-ray induced specific locus and recessive lethal mutations in mature germ cells of the zebrafish, Brachydanio rerio. Genetics. 1983;103:109–23.
Walker C, Streisinger G. Induction of mutations by gamma-rays in pregonial germ cells of zebrafish embryos. Genetics. 1983;103:125–36.
Grunwald DJ, Streisinger G. Induction of mutations in the zebrafish with ultraviolet light. Genet Res. 1992;59:93–101.
Grunwald DJ, Streisinger G. Induction of recessive lethal and specific locus mutations in the zebrafish with ethyl nitrosourea. Genet Res. 1992;59:103–16.
Mullins MC, Hammerschmidt M, Haffter P, Nusslein-Volhard C. Large-scale mutagenesis in the zebrafish: in search of genes controlling development in a vertebrate. Curr Biol. 1994;4:189–202.
Solnica-Krezel L, Schier AF, Driever W. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics. 1994;136:1401–20.
Nusslein-Volhard C. The zebrafish issue of development. Development. 2012;139:4099–103.
Xiao T, Roeser T, Staub W, Baier H. A GFP-based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development. 2005;132:2955–67.
Pogoda HM, Sternheim N, Lyons DA, Diamond B, Hawkins TA, Woods IG, et al. A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev Biol. 2006;298:118–31.
Kuhlman J, Eisen JS. Genetic screen for mutations affecting development and function of the enteric nervous system. Dev Dyn. 2007;236:118–27.
Covassin LD, Siekmann AF, Kacergis MC, Laver E, Moore JC, Villefranc JA, et al. A genetic screen for vascular mutants in zebrafish reveals dynamic roles for Vegf/Plcg1 signaling during artery development. Dev Biol. 2009;329:212–26.
Lee J, Cox BD, Daly CM, Lee C, Nuckels RJ, Tittle RK, et al. An ENU mutagenesis screen in zebrafish for visual system mutants identifies a novel splice-acceptor site mutation in patched2 that results in Colobomas. Invest Ophthalmol Vis Sci. 2012;53:8214–21.
Dai ZX, Yan G, Chen YH, Liu W, Huo ZJ, Wen ZH, et al. Forward genetic screening for zebrafish mutants defective in myelopoiesis. Nan Fang Yi Ke Da Xue Xue Bao. 2010;30:1230–3.
Muto A, Orger MB, Wehman AM, Smear MC, Kay JN, Page-McCaw PS, et al. Forward genetic analysis of visual behavior in zebrafish. PLoS Genet. 2005;1:e66.
Baraban SC, Dinday MT, Castro PA, Chege S, Guyenet S, Taylor MR. A large-scale mutagenesis screen to identify seizure-resistant zebrafish. Epilepsia. 2007;48:1151–7.
Wolman MA, Jain RA, Marsden KC, Bell H, Skinner J, Hayer KE, et al. A genome-wide screen identifies PAPP-AA-mediated IGFR signaling as a novel regulator of habituation learning. Neuron. 2015;85:1200–11.
Darland T, Dowling JE. Behavioral screening for cocaine sensitivity in mutagenized zebrafish. Proc Natl Acad Sci U S A. 2001;98:11691–6.
Kim SH, Wu SY, Baek JI, Choi SY, Su Y, Flynn CR, et al. A post-developmental genetic screen for zebrafish models of inherited liver disease. PLoS One. 2015;10:e0125980.
Voz ML, Coppieters W, Manfroid I, Baudhuin A, Von Berg V, Charlier C, et al. Fast homozygosity mapping and identification of a zebrafish ENU-induced mutation by whole-genome sequencing. PLoS One. 2012;7:e34671.
Bowen ME, Henke K, Siegfried KR, Warman ML, Harris MP. Efficient mapping and cloning of mutations in zebrafish by low-coverage whole-genome sequencing. Genetics. 2012;190:1017–24.
Obholzer N, Swinburne IA, Schwab E, Nechiporuk AV, Nicolson T, Megason SG. Rapid positional cloning of zebrafish mutations by linkage and homozygosity mapping using whole-genome sequencing. Development. 2012;139:4280–90.
Hill JT, Demarest BL, Bisgrove BW, Gorsi B, Su YC, Yost HJ. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013;23:687–97.
Miller AC, Obholzer ND, Shah AN, Megason SG, Moens CB. RNA-seq-based mapping and candidate identification of mutations from forward genetic screens. Genome Res. 2013;23:679–86.
Wienholds E, Schulte-Merker S, Walderich B, Plasterk RH. Target-selected inactivation of the zebrafish rag1 gene. Science. 2002;297:99–102.
Wienholds E, van Eeden F, Kosters M, Mudde J, Plasterk RH, Cuppen E. Efficient target-selected mutagenesis in zebrafish. Genome Res. 2003;13:2700–7.
Kettleborough RN, Busch-Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496:494–7.
Lin S, Gaiano N, Culp P, Burns JC, Friedmann T, Yee JK, et al. Integration and germ-line transmission of a pseudotyped retroviral vector in zebrafish. Science. 1994;265:666–9.
Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–51.
Wang D, Jao LE, Zheng N, Dolan K, Ivey J, Zonies S, et al. Efficient genome-wide mutagenesis of zebrafish genes by retroviral insertions. Proc Natl Acad Sci U S A. 2007;104:12428–33.
Gaiano N, Amsterdam A, Kawakami K, Allende M, Becker T, Hopkins N. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature. 1996;383:829–32.
Amsterdam A, Burgess S, Golling G, Chen W, Sun Z, Townsend K, et al. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–24.
Golling G, Amsterdam A, Sun Z, Antonelli M, Maldonado E, Chen W, et al. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet. 2002;31:135–40.
Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–7.
Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–93.
Varshney GK, Huang H, Zhang S, Lu J, Gildea DE, Yang Z, et al. The Zebrafish Insertion Collection (ZInC): a web based, searchable collection of zebrafish mutations generated by DNA insertion. Nucleic Acids Res. 2013;41:D861–4.
Varshney GK, Lu J, Gildea DE, Huang H, Pei W, Yang Z, et al. A large-scale zebrafish gene knockout resource for the genome-wide study of gene function. Genome Res. 2013;23:727–35.
Kawakami K, Shima A, Kawakami N. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proc Natl Acad Sci U S A. 2000;97:11403–8.
Davidson AE, Balciunas D, Mohn D, Shaffer J, Hermanson S, Sivasubbu S, et al. Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Dev Biol. 2003;263:191–202.
Emelyanov A, Gao Y, Naqvi NI, Parinov S. Trans-kingdom transposition of the maize dissociation element. Genetics. 2006;174:1095–104.
le Trinh A, Hochgreb T, Graham M, Wu D, Ruf-Zamojski F, Jayasena CS, et al. A versatile gene trap to visualize and interrogate the function of the vertebrate proteome. Genes Dev. 2011;25:2306–20.
Clark KJ, Balciunas D, Pogoda HM, Ding Y, Westcot SE, Bedell VM, et al. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat Methods. 2011;8:506–15.
Ni TT, Lu J, Zhu M, Maddison LA, Boyd KL, Huskey L, et al. Conditional control of gene function by an invertible gene trap in zebrafish. Proc Natl Acad Sci U S A. 2012;109:15389–94.
Balciuniene J, Nagelberg D, Walsh KT, Camerota D, Georlette D, Biemar F, et al. Efficient disruption of Zebrafish genes using a Gal4-containing gene trap. BMC Genomics. 2013;14:619.
Kawakami K, Takeda H, Kawakami N, Kobayashi M, Matsuda N, Mishina M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell. 2004;7:133–44.
Petzold AM, Balciunas D, Sivasubbu S, Clark KJ, Bedell VM, Westcot SE, et al. Nicotine response genetics in the zebrafish. Proc Natl Acad Sci U S A. 2009;106:18662–7.
Seiler C, Gebhart N, Zhang Y, Shinton SA, Li YS, Ross NL, et al. Mutagenesis screen identifies agtpbp1 and eps15L1 as essential for T lymphocyte development in zebrafish. PLoS One. 2015;10:e0131908.
Kawakami K, Abe G, Asada T, Asakawa K, Fukuda R, Ito A, et al. zTrap: zebrafish gene trap and enhancer trap database. BMC Dev Biol. 2010;10:105.
Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–20.
Schulte-Merker S, Stainier DY. Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology. Development. 2014;141:3103–4.
Stainier DY, Kontarakis Z, Rossi A. Making sense of anti-sense data. Dev Cell. 2015;32:7–8.
Kok FO, Shin M, Ni CW, Gupta A, Grosse AS, van Impel A, et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev Cell. 2015;32:97–108.
Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–3.
Hagmann M, Bruggmann R, Xue L, Georgiev O, Schaffner W, Rungger D, et al. Homologous recombination and DNA-end joining reactions in zygotes and early embryos of zebrafish (Danio rerio) and Drosophila melanogaster. Biol Chem. 1998;379:673–81.
Dai J, Cui X, Zhu Z, Hu W. Non-homologous end joining plays a key role in transgene concatemer formation in transgenic zebrafish embryos. Int J Biol Sci. 2010;6:756–68.
Liu J, Gong L, Chang C, Liu C, Peng J, Chen J. Development of novel visual-plus quantitative analysis systems for studying DNA double-strand break repairs in zebrafish. J Genet Genomics. 2012;39:489–502.
Ota S, Hisano Y, Muraki M, Hoshijima K, Dahlem TJ, Grunwald DJ, et al. Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells. 2013;18:450–8.
Oleykowski CA, Bronson Mullins CR, Godwin AK, Yeung AT. Mutation detection using a novel plant endonuclease. Nucleic Acids Res. 1998;26:4597–602.
Qiu P, Shandilya H, D’Alessio JM, O’Connor K, Durocher J, Gerard GF. Mutation detection using surveyor nuclease. Biotechniques. 2004;36:702–7.
Vouillot L, Thelie A, Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda). 2015;5:407–15.
Parant JM, George SA, Pryor R, Wittwer CT, Yost HJ. A rapid and efficient method of genotyping zebrafish mutants. Dev Dyn. 2009;238:3168–74.
Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, et al. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8:e1002861.
Xing L, Quist TS, Stevenson TJ, Dahlem TJ, Bonkowsky JL. Rapid and efficient zebrafish genotyping using PCR with high-resolution melt analysis. J Vis Exp. 2014:e51138.
Hill JT, Demarest BL, Bisgrove BW, Su YC, Smith M, Yost HJ. Poly peak parser: method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev Dyn. 2014;243:1632–6.
Xiao A, Wu Y, Yang Z, Hu Y, Wang W, Zhang Y, et al. EENdb: a database and knowledge base of ZFNs and TALENs for endonuclease engineering. Nucleic Acids Res. 2013;41:D415–22.
Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38:W462–8.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–60.
Choo Y, Klug A. Selection of DNA binding sites for zinc fingers using rationally randomized DNA reveals coded interactions. Proc Natl Acad Sci U S A. 1994;91:11168–72.
Choo Y, Klug A. Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc Natl Acad Sci U S A. 1994;91:11163–7.
Greisman HA, Pabo CO. A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science. 1997;275:657–61.
Segal DJ, Dreier B, Beerli RR, Barbas 3rd CF. Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci U S A. 1999;96:2758–63.
Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas 3rd CF. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2001;276:29466–78.
Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A, et al. Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2005;280:35588–97.
Liu Q, Xia Z, Zhong X, Case CC. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J Biol Chem. 2002;277:3850–6.
Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–88.
Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–5.
Carroll D, Morton JJ, Beumer KJ, Segal DJ. Design, construction and in vitro testing of zinc finger nucleases. Nat Protoc. 2006;1:1329–41.
Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301.
Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4:1471–501.
Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26:695–701.
Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods. 2011;8:67–9.
Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000;28:3361–9.
Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–85.
Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–93.
Handel EM, Alwin S, Cathomen T. Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol Ther. 2009;17:104–11.
Wilson KA, McEwen AE, Pruett-Miller SM, Zhang J, Kildebeck EJ, Porteus MH. Expanding the repertoire of target sites for zinc finger nuclease-mediated genome modification. Mol Ther Nucleic Acids. 2013;2:e88.
Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–8.
Foley JE, Yeh JR, Maeder ML, Reyon D, Sander JD, Peterson RT, et al. Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN). PLoS One. 2009;4:e4348.
Reyon D, Kirkpatrick JR, Sander JD, Zhang F, Voytas DF, Joung JK, et al. ZFNGenome: a comprehensive resource for locating zinc finger nuclease target sites in model organisms. BMC Genomics. 2011;12:83.
Chen S, Oikonomou G, Chiu CN, Niles BJ, Liu J, Lee DA, et al. A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res. 2013;41:2769–78.
Moore FE, Reyon D, Sander JD, Martinez SA, Blackburn JS, Khayter C, et al. Improved somatic mutagenesis in zebrafish using transcription activator-like effector nucleases (TALENs). PLoS One. 2012;7:e37877.
Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol. 2010;13:394–401.
Christian ML, Demorest ZL, Starker CG, Osborn MJ, Nyquist MD, Zhang Y, et al. Targeting G with TAL effectors: a comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS One. 2012;7:e45383.
Cong L, Zhou R, Kuo YC, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun. 2012;3:968.
Streubel J, Blucher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat Biotechnol. 2012;30:593–5.
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12.
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:W117–22.
Neff KL, Argue DP, Ma AC, Lee HB, Clark KJ, Ekker SC. Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinf. 2013;14:1.
Li L, Piatek MJ, Atef A, Piatek A, Wibowo A, Fang X, et al. Rapid and highly efficient construction of TALE-based transcriptional regulators and nucleases for genome modification. Plant Mol Biol. 2012;78:407–16.
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82.
Morbitzer R, Elsaesser J, Hausner J, Lahaye T. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011;39:5790–9.
Sakuma T, Hosoi S, Woltjen K, Suzuki K, Kashiwagi K, Wada H, et al. Efficient TALEN construction and evaluation methods for human cell and animal applications. Genes Cells. 2013;18:315–26.
Ding Q, Lee YK, Schaefer EA, Peters DT, Veres A, Kim K, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–51.
Ma AC, Lee HB, Clark KJ, Ekker SC. High efficiency In Vivo genome engineering with a simplified 15-RVD GoldyTALEN design. PLoS One. 2013;8:e65259.
Uhde-Stone C, Gor N, Chin T, Huang J, Lu B. A do-it-yourself protocol for simple transcription activator-like effector assembly. Biol Proced Online. 2013;15:3.
Huang P, Xiao A, Tong X, Zu Y, Wang Z, Zhang B. TALEN construction via “Unit Assembly” method and targeted genome modifications in zebrafish. Methods. 2014;69:67–75.
Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011;29:699–700.
Reyon D, Khayter C, Regan MR, Joung JK, Sander JD. Engineering designer transcription activator-like effector nucleases (TALENs) by REAL or REAL-Fast assembly. Curr Protoc Mol Biol. 2012;Chapter 12:Unit 12 5.
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30:460–5.
Briggs AW, Rios X, Chari R, Yang L, Zhang F, Mali P, et al. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012;40:e117.
Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug 2nd RG, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–8.
Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, et al. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol. 2011;29:697–8.
Cade L, Reyon D, Hwang WY, Tsai SQ, Patel S, Khayter C, et al. Highly efficient generation of heritable zebrafish gene mutations using homo- and heterodimeric TALENs. Nucleic Acids Res. 2012;40:8001–10.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12.
Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–5.
Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–97.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–8.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32.
Shah SA, Erdmann S, Mojica FJ, Garrett RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013;10:891–9.
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–7.
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997.
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–49.
Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–5.
Naito Y, Hino K, Bono H, Ui-Tei K. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics. 2015;31:1120–3.
Park J, Bae S, Kim JS. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–6.
Xie S, Shen B, Zhang C, Huang X, Zhang Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS One. 2014;9:e100448.
Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–7.
Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, et al. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development. 2013;140:4982–7.
Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, et al. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS One. 2013;8:e68708.
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–9.
Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465–72.
Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One. 2014;9:e98186.
Sung YH, Kim JM, Kim HT, Lee J, Jeon J, Jin Y, et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res. 2014;24:125–31.
Kotani H, Taimatsu K, Ohga R, Ota S, Kawahara A. Efficient multiple genome modifications induced by the crRNAs, tracrRNA and Cas9 protein complex in zebrafish. PLoS One. 2015;10:e0128319.
Jao LE, Wente SR, Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci U S A. 2013;110:13904–9.
Ota S, Hisano Y, Ikawa Y, Kawahara A. Multiple genome modifications by the CRISPR/Cas9 system in zebrafish. Genes Cells. 2014;19:555–64.
Ranganathan V, Wahlin K, Maruotti J, Zack DJ. Expansion of the CRISPR-Cas9 genome targeting space through the use of H1 promoter-expressed guide RNAs. Nat Commun. 2014;5:4516.
Yin L, Maddison LA, Li M, Kara N, LaFave MC, Varshney GK, et al. Multiplex conditional mutagenesis using transgenic expression of Cas9 and sgRNAs. Genetics. 2015;200:431–41.
Varshney GK, Pei W, LaFave MC, Idol J, Xu L, Gallardo V, et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015;25:1030–42.
Zhu LJ, Holmes BR, Aronin N, Brodsky MH. CRISPRseek: a bioconductor package to identify target-specific guide RNAs for CRISPR-Cas9 genome-editing systems. PLoS One. 2014;9:e108424.
Prykhozhij SV, Rajan V, Gaston D, Berman JN. CRISPR multitargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PLoS One. 2015;10:e0119372.
Ablain J, Durand EM, Yang S, Zhou Y, Zon LI. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell. 2015;32:756–64.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6.
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–43.
Liu D, Wang Z, Xiao A, Zhang Y, Li W, Zu Y, et al. Efficient gene targeting in zebrafish mediated by a zebrafish-codon-optimized cas9 and evaluation of off-targeting effect. J Genet Genomics. 2014;41:43–6.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84.
Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–41.
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9.
Gupta A, Hall VL, Kok FO, Shin M, McNulty JC, Lawson ND, et al. Targeted chromosomal deletions and inversions in zebrafish. Genome Res. 2013;23:1008–17.
Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141.
Li Y, Rivera CM, Ishii H, Jin F, Selvaraj S, Lee AY, et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 2014;9:e114485.
Wang S, Sengel C, Emerson MM, Cepko CL. A gene regulatory network controls the binary fate decision of rod and bipolar cells in the vertebrate retina. Dev Cell. 2014;30:513–27.
Piganeau M, Ghezraoui H, De Cian A, Guittat L, Tomishima M, Perrouault L, et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013;23:1182–93.
Blasco RB, Karaca E, Ambrogio C, Cheong TC, Karayol E, Minero VG, et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014;9:1219–27.
Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–7.
Balciunas D, Davidson AE, Sivasubbu S, Hermanson SB, Welle Z, Ekker SC. Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics. 2004;5:62.
Parinov S, Kondrichin I, Korzh V, Emelyanov A. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev Dyn. 2004;231:449–59.
Scott EK, Baier H. The cellular architecture of the larval zebrafish tectum, as revealed by gal4 enhancer trap lines. Front Neural Circuits. 2009;3:13.
Scott EK, Mason L, Arrenberg AB, Ziv L, Gosse NJ, Xiao T, et al. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat Methods. 2007;4:323–6.
Asakawa K, Kawakami K. The Tol2-mediated Gal4-UAS method for gene and enhancer trapping in zebrafish. Methods. 2009;49:275–81.
Asakawa K, Suster ML, Mizusawa K, Nagayoshi S, Kotani T, Urasaki A, et al. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci U S A. 2008;105:1255–60.
Distel M, Wullimann MF, Koster RW. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Proc Natl Acad Sci U S A. 2009;106:13365–70.
Poon KL, Liebling M, Kondrychyn I, Garcia-Lecea M, Korzh V. Zebrafish cardiac enhancer trap lines: new tools for in vivo studies of cardiovascular development and disease. Dev Dyn. 2010;239:914–26.
Levesque MP, Krauss J, Koehler C, Boden C, Harris MP. New tools for the identification of developmentally regulated enhancer regions in embryonic and adult zebrafish. Zebrafish. 2013;10:21–9.
Otsuna H, Hutcheson DA, Duncan RN, McPherson AD, Scoresby AN, Gaynes BF, et al. High-resolution analysis of central nervous system expression patterns in zebrafish Gal4 enhancer-trap lines. Dev Dyn. 2015;244:785–96.
Kondrychyn I, Teh C, Garcia-Lecea M, Guan Y, Kang A, Korzh V. Zebrafish enhancer TRAP transgenic line database ZETRAP 2.0. Zebrafish. 2011;8:181–2.
Stuart GW, McMurray JV, Westerfield M. Replication, integration and stable germ-line transmission of foreign sequences injected into early zebrafish embryos. Development. 1988;103:403–12.
Stuart GW, Vielkind JR, McMurray JV, Westerfield M. Stable lines of transgenic zebrafish exhibit reproducible patterns of transgene expression. Development. 1990;109:577–84.
Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–99.
Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–95.
Halloran MC, Sato-Maeda M, Warren JT, Su F, Lele Z, Krone PH, et al. Laser-induced gene expression in specific cells of transgenic zebrafish. Development. 2000;127:1953–60.
Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods. 2013;10:329–31.
Shin J, Chen J, Solnica-Krezel L. Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development. 2014;141:3807–18.
Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–8.
Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–42.
Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014;24:142–53.
Kimura Y, Hisano Y, Kawahara A, Higashijima S. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci Rep. 2014;4:6545.
Hisano Y, Sakuma T, Nakade S, Ohga R, Ota S, Okamoto H, et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci Rep. 2015;5:8841.
Li J, Zhang BB, Ren YG, Gu SY, Xiang YH, Du JL. Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res. 2015;25:634–7.
Zada D, Tovin A, Lerer-Goldshtein T, Vatine GD, Appelbaum L. Altered behavioral performance and live imaging of circuit-specific neural deficiencies in a zebrafish model for psychomotor retardation. PLoS Genet. 2014;10:e1004615.
Gandhi AV, Mosser EA, Oikonomou G, Prober DA. Melatonin is required for the circadian regulation of sleep. Neuron. 2015;85:1193–9.
Elbaz I, Yelin-Bekerman L, Nicenboim J, Vatine G, Appelbaum L. Genetic ablation of hypocretin neurons alters behavioral state transitions in zebrafish. J Neurosci. 2012;32:12961–72.
Sakowski SA, Lunn JS, Busta AS, Oh SS, Zamora-Berridi G, Palmer M, et al. Neuromuscular effects of G93A-SOD1 expression in zebrafish. Mol Neurodegener. 2012;7:44.
Ramesh T, Lyon AN, Pineda RH, Wang C, Janssen PM, Canan BD, et al. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis Model Mech. 2010;3:652–62.
Mosimann C, Puller AC, Lawson KL, Tschopp P, Amsterdam A, Zon LI. Site-directed zebrafish transgenesis into single landing sites with the phiC31 integrase system. Dev Dyn. 2013;242:949–63.
Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–6.
Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–71.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83.
Mahmood F, Fu S, Cooke J, Wilson SW, Cooper JD, Russell C. A zebrafish model of CLN2 disease is deficient in tripeptidyl peptidase 1 and displays progressive neurodegeneration accompanied by a reduction in proliferation. Brain. 2013;136:1488–507.
Da Costa MM, Allen CE, Higginbottom A, Ramesh T, Shaw PJ, McDermott CJ. A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis Model Mech. 2014;7:73–81.
Singh C, Oikonomou G, Prober DA. Norepinephrine is required to promote wakefulness and for hypocretin-induced arousal in zebrafish. Elife. 2015;4:e07000.
Elbaz I, Lerer-Goldshtein T, Okamoto H, Appelbaum L. Reduced synaptic density and deficient locomotor response in neuronal activity-regulated pentraxin 2a mutant zebrafish. FASEB J. 2015;29:1220–34.
Yelin-Bekerman L, Elbaz I, Diber A, Dahary D, Gibbs-Bar L, Alon S, et al. Hypocretin neuron-specific transcriptome profiling identifies the sleep modulator Kcnh4a. Elife. 2015;4:e08638.
Godoy R, Noble S, Yoon K, Anisman H, Ekker M. Chemogenetic ablation of dopaminergic neurons leads to transient locomotor impairments in zebrafish larvae. J Neurochem. 2015;135:249–60.
Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119:1382–95.
Lee A, Mathuru AS, Teh C, Kibat C, Korzh V, Penney TB, et al. The habenula prevents helpless behavior in larval zebrafish. Curr Biol. 2010;20:2211–6.
Yokogawa T, Hannan MC, Burgess HA. The dorsal raphe modulates sensory responsiveness during arousal in zebrafish. J Neurosci. 2012;32:15205–15.
Bergeron SA, Carrier N, Li GH, Ahn S, Burgess HA. Gsx1 expression defines neurons required for prepulse inhibition. Mol Psychiatry. 2015;20:974–85.
Acknowledgements
We apologize to authors whose work was omitted from this article due to space limitations. Work in the F.E.P. laboratory is supported in part by R00NS083714 from the National Institute of Neurological Disorders and Stroke at the National Institute of Health.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Poulain, F.E. (2017). Mutagenesis and Transgenesis in Zebrafish. In: Kalueff, A. (eds) The rights and wrongs of zebrafish: Behavioral phenotyping of zebrafish. Springer, Cham. https://doi.org/10.1007/978-3-319-33774-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-33774-6_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33773-9
Online ISBN: 978-3-319-33774-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)