
Abstract
Pineal parenchymal tumors range from the indolent pineocytoma (PC) to pineal parenchymal tumors of intermediate differentiation (PPTID) and finally to the highly malignant pineoblastoma (PB).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Flexner-Wintersteiner rosettes
- Pineal parenchymal tumor of intermediate differentiation
- Pineoblastoma
- Pineocytoma
1 Overview
-
Pineal parenchy mal tumors range from the indolent pineocytoma (PC) to pineal parenchymal tumors of intermediate differentiation (PPTID) and finally to the highly malignant pineoblastoma (PB).
-
◦ Pineocytoma (ICD-O 9361/1) is a rare, slow-growing pineal neoplasm (WHO grade I) composed of mature-appearing pineocytes and forming pineocytomatous rosettes.
-
◦ Pineal pa renchymal tumor of intermediate differentiation (ICD-O 9362/3) is a rare, intermediate-grade malignant tumor (WHO grade II or III) with diffuse sheets of tumor cells with low or moderate level of mitotic activity.
-
◦ Pineo blastoma (ICD-O 9362/3) is a highly malignant embryonal tumor (WHO grade IV) typically s een in children; it has primitive, blastlike cells and frequent recurrence or dissemination.
-
-
A recently described rare neoplasm, papillary tumor of the pineal, is typically a tumor of adults and will not be discussed in this chapter [1].
2 Clinical Features
-
All primary pineal parenchymal neoplasms, as well as other pineal region masses, may be associated with Parinaud syndrome in the young (dorsal midbrain syndrome), which includes upward gaze paralysis, accommodative paresis, nystagmus, and eyelid retraction.
-
Pineal region masses can also cause significant cerebrospinal fluid (CSF) obstruction leading to headaches, nausea and vomiting, and other signs related to increased intracranial pressure. These symptoms are not specific to pineal parenchymal neoplasms.
-
Pineocyto ma (PC) is less common in the pediatric age group; the rare PC typically presents with signs of increased intracranial pressure and occasionally visual disturbances. The few studies on PC in children report a more frequent local recurrence than in adults, but this finding requires validation with the current WHO grading scheme.
-
Pineal paren chymal tumor of intermediate differentiation (PPTID) is even less frequent in children and its clinical presentation is less well defined; it is considered similar to PC. These tumors have a greater tendency for loc al recurrence, but an objective comparison of recurrence risk between PPTID and PC is not possible on the basis of the available data [2].
-
Pineoblastoma (PB) is the most common pediatric pineal parenchymal tumor, and the majority of tumors occur within the first two decades of life. The interval between initial symptoms and the need for surgical intervention is much shorter than for other pineal parenchymal tumors. Signs and symptoms of intracranial pressure are often present and may worsen rapidly with cerebrospinal dissemination.
3 Neuroimaging
-
Neuroimaging does not provide specific information that can reliably distinguish pineal parenchymal tumo rs from other neoplasms such as germinomas [3].
-
PC is often a well-defined mass that is typically less than 3–4 cm. These tumors are hypointense or isointense on T1-weighted images, and hyperintense on T2-weighted images. The tumors avidly en hance upon gadoli nium administration. Some PCs demonstrate calcifications on CT or MRI (Fig. 34.1).
Fig. 34.1 
Coronal no ncontrast T1-weighted image of a pineal mass in a 17-year-old boy, showing a small cystic pineal gland. The lesion was histologically confirmed as a pineocytoma (PC)
-
PPTID is often indistinguishable from PC and may also demonstrate calcifications or hemorrhage on imaging s tudies. PPTID may be more solid and diffusely enhancing than PC.
-
PB is often a large tumor, rarely seen in association with unilateral or bilateral retinoblastoma. The tumors are often larger, with brighter T2-weighted signals, and are hypointense on T1-weighted images. There is marked gadolinium enhancement, and the tumors are much more variable in their appearance, with calcific ations, necrosis, or hemorrhage. The boundaries of PB may not always be well defined (Fig. 34.2).
Fig. 34.2 
Sagittal, contrast-enhanced T1-weighted MR image of a pineoblastoma (PB) in a 4-year-old child, wit h mass effect and hydrocephalus


4 Histopathology
-
PCs are macroscopically gray, granular, and discrete masses that often fill the pineal recess and the subjacent aqueduct.
-
◦ The tumors are composed of cells with large nuclei, open chromatin density, and ample surrounding cytoplasm (Fig. 34.3).
Fig. 34.3 
Typical histological appearance of pineocytoma (PC). The tumor is sparsely cellular and is compose d of monomorphous, small, mature neuronal cells and neuropil-like background
-
-
The tumor cells are typically monomorphous with only occasional cells with nuclear pleomorphism or hyperchromasia.
-
◦ Necrosis or mitotic figures are exceptionally rare. In some foci, cytological atypia can be interpreted as “degenerative change.”
-
◦ PCs often harbor pineocytomatous rosettes formed by conglomeration of tumor cell processes in a circular or stellate form and are slightly more eosinophilic than the neuropil.
-
◦ The structure of a pineocytomatous rosette is similar to a pineoblastic or Homer-Wright rosette, but it is conspicuously less cellular and larger; the majority of cells have ample cytoplasm.
-
◦ PCs can show isolated or clustered large ganglion-like cells and focal islands of neuropil, but these findings are rar e (Fig. 34.4).
Fig. 34.4 
Histolog ical features of pineocytoma. Less common features include a rich, neuropil-like matrix and rare ganglion-like cells
-
-
PPTID shows focally diffuse or lobular architecture. Some PPTIDs resemble pineocytoma with more hypercellularity and occasional mitoses and abundant neuropil, but others look like small blue r ound cells wit h no mitoses and intervening neuropil (Fig. 34.5).
Fig. 34.5 
Pineal parenchymal tumor of intermediate differentiation (PPTID) composed of a mixture of small and ma ture cells presenting a mixed appearance between pineocytoma and pineoblastoma
-
◦ The tumors have high or moderate cellularity with focally overlapping nuclei, cells with scant cytoplasm, and scattered mitotic figures.
-
◦ PPTID may harbor focal PC-like areas admixed with sheets of tumor cells or lobules with increased cellularity (Fig. 34.5).
-
◦ Rarely, these tumors contain giant cells or moderate nuclear atypia (Fig. 34.6), and occasionally Homer Wright–type rosettes or large ganglion-like cells.
Fig. 34.6 
Cytol ogic atypia in PPTID showing rare multin ucleated cells and large, bizarre yet degenerative-appearing nuclei
-
◦ Grading of PPTID is controversial, and a clear distinction between grade II and grade III neoplasms has not emerged. The use of proliferation rate and a grading scheme to segregate WHO grade II from grade III tumors showed no survival difference [4].
-
◦ A grading scheme based on mitotic rate and neurofilament staining was proposed earlier [5]:
-
WHO grade II PPTID has fewer than six mitoses per ten high power fields and strong immunostaining with neurofilament antibody.
-
WHO grade III PPTID has more than six mitoses per ten high power fields and little or no staining with neurofilament antibody.
-
-
-
PB is macroscopically soft, gelatinous, and focally hemorrhagic and necrotic. These irregular neoplas ms can be quite large.
-
◦ PB has been described as similar to medulloblastoma or supratentorial primitive neuroectodermal tumor (PNET). The most recent WHO classification has abandoned the term PNET, and refer to such tumors as Embryonal Tumor, NOS as of 2016. The tumors are composed of small, blue round cells with scant cytoplasm and hyperchromatic nuclei (Fig. 34.7).
Fig. 34.7 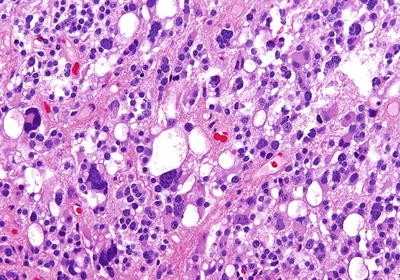
Pine oblastoma composed of highly cellular, small blue round cells with numerous mitotic figures
-
◦ The cells are often arranged in sheets or discohesive clusters. The tumor cells typically form Flexner-Wintersteiner rosettes in which the cells are radially arranged around a small luminal s tructure (Fig. 34.8).
Fig. 34.8 
Retinobla stic (Flexner-Wintersteiner) rosette in pineoblastoma (arrow)
-
◦ In addition, neuroblastic-type Homer-Wright rosettes identical to those seen in medulloblastomas can be observed (Fig. 34.9).
Fig. 34.9 
Neurob lastic (Homer-Wright) rosettes in pineoblastoma
-
◦ PB has conspicuous mitotic figures, which can be numerous in some areas. Occasional giant cells, hemorrhage, and calcification are also commonly present.
-
◦ Rare tumor cel ls may harbor melanin pigment, but the pigment more often is hemosiderin.
-







5 Immunohistochemistry
-
As a general rule, with the increasing grade of the tumor, neurofilament immunostaining decreases and Ki-67 proliferation rate increases [6]. A WHO grade III PPTID has only focal neurofilament staining (Fig. 34.10).
Fig. 34.10 
Immunohistochemical staining for neurofilament protein in PPTID, with only patchy stainin g. With a high rate of mitotic figures, this tumor was graded as WHO grade III, but this patient did not have a recurrence following near-total gross resection
-
PC typically demonstrates strong positivity for S-100 protein, neuron-specific enolase (NSE), synaptophysin, and chromogranin, with variable immunoreactivity to neurofilament proteins [7].
-
◦ The tumors are also focally positive for glial fibrillary acidic protein (GFAP), retinal-S antigen, and rhodopsin.
-
-
PPT ID is similar to PC, with strong positivity for synaptophysin, NSE, and S-100 protein. Antibodies for neurofilament proteins stain the tumors focally, and in some cases isol ated cells within the tumor show intense staining.
-
◦ The staining for GFAP and retinal-S antigen is variable; some tumors may be negative for these two markers.
-
-
PB exhibits a le ss pronounced retinoblastomatous differentiation but is often equally positive for synaptophysin as the other pineal parenchymal tumors.
-
◦ Chromogranin is positive in only a small percentage of tumors and in a limited number of tumor cells (Fig. 34.11).
Fig. 34.11 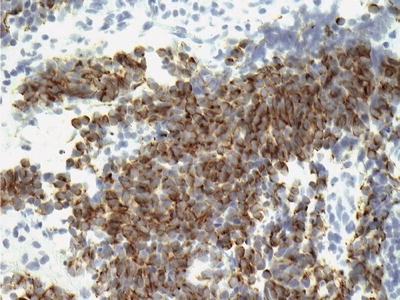
Immunohist ochemical staining for chromogranin in pineoblastoma. Note the patchy staining of the cytoplasm
-
-
S-100 protein and retinal-S antigen are typically negative, with rare weak positivity only in an occasional tumor.


6 Molecular Pathology
-
The RB1 gene encodes a protein that acts as a cell cycle checkpoint at the G1 phase. Some patie nts suffering from retinoblastoma and PB harbor germline losses of the RB1 gene.
-
Trilateral retinoblastoma, a heritable form of bilateral retinoblastoma in association with PB, has a rather dismal prognosis. The disease is associated with germline deletion of the RB1 gene from chromosome 13.
-
A subgroup of PB demonstrates DICER1 mutations, including some patients with germline mutations of this gene [8].
7 Prognosis
-
The following characteristics were found to influence outcome in pineal neoplasms [9]:
-
◦ Tumor histological grade
-
◦ Tumor volume
-
-
Tumors with a pineoblastomatous component have a much worse overall survival than the other histological types.
-
Gross total resection seems to portend a better prognosis, even for patients with PB [10].
-
Aggressi ve chemotherapy and radiotherapy has been suggested to increase survival for patients with PB [11].
References
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds). WHO classification of tumours of the central nervous system (Revised 4th Edition) International Agency for Research on Cancer (IARC); Lyon 2016.
Ito T, Kanno H, Sato K, Oikawa M, Ozaki Y, Nakamura H, et al. Clinicopathologic study of pineal parenchymal tumors of intermediate differentiation. World Neurosurg. 2014;81:783–9.
Kakigi T, Okada T, Kanagaki M, Yamamoto A, Fushimi Y, Sakamoto R, et al. Quantitative imaging values of CT, MR, and FDG-PET to differentiate pineal parenchymal tumors and germinomas: are they useful? Neuroradiology. 2014;56:297–303.
Fevre-Montange M, Vasiljevic A, Frappaz D, Champier J, Szathmari A, Aubriot Lorton MH, et al. Utility of Ki67 immunostaining in the grading of pineal parenchymal tumours: a multicentre study. Neuropathol Appl Neurobiol. 2012;38:87–94.
Jouvet A, Saint-Pierre G, Fauchon F, Privat K, Bouffet E, Ruchoux MM, et al. Pineal parenchymal tumors: a correlation of histological features with prognosis in 66 cases. Brain Pathol. 2000;10:49–60.
Arivazhagan A, Anandh B, Santosh V, Chandramouli BA. Pineal parenchymal tumors—utility of immunohistochemical markers in prognostication. Clin Neuropathol. 2008;27:325–33.
Coca S, Vaquero J, Escandon J, Moreno M, Peralba J, Rodriguez J. Immunohistochemical characterization of pineocytomas. Clin Neuropathol. 1992;11:298–303.
de Kock L, Sabbaghian N, Druker H, Weber E, Hamel N, Miller S, et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol. 2014;128:583–95.
Fauchon F, Jouvet A, Paquis P, Saint-Pierre G, Mottolese C, Ben Hassel M, et al. Parenchymal pineal tumors: a clinicopathological study of 76 cases. Int J Radiat Oncol Biol Phys. 2000;46:959–68.
Mandera M, Marcol W, Kotulska K, Olakowska E, Golka D, Malinowska I, et al. Childhood pineal parenchymal tumors: clinical and therapeutic aspects. Neurosurg Rev. 2010;34:191–6.
Farnia B, Allen PK, Brown PD, Khatua S, Levine NB, Li J, et al. Clinical outcomes and patterns of failure in pineoblastoma: a 30-year, single-institution retrospective review. World Neurosurg. 2014;82:1232–41.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Tihan, T. (2016). Pineal Parenchymal Tumors. In: Adesina, A., Tihan, T., Fuller, C., Poussaint, T. (eds) Atlas of Pediatric Brain Tumors. Springer, Cham. https://doi.org/10.1007/978-3-319-33432-5_34
Download citation
DOI: https://doi.org/10.1007/978-3-319-33432-5_34
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33430-1
Online ISBN: 978-3-319-33432-5
eBook Packages: MedicineMedicine (R0)