
Abstract
Flaviviruses are small, positive-strand RNA viruses in the genus Flavivirus within the Flaviviridae family. The Flavivirus genus consists of nearly 80 viruses, approximately half of which are associated with human disease. The majority of flaviviruses are arthropod-borne viruses, or arboviruses, transmitted from infected to susceptible vertebrate hosts primarily by mosquitoes and ticks. Flavivirus infections cause seasonal disease syndromes corresponding to mosquito and tick activity throughout the temperate and tropical areas of the world. Medically important flaviviruses are associated with three clinical syndromes: encephalitis and meningitis; hemorrhagic fever; or fever, arthralgia, and rash. The neurotropic flaviviruses that cause neuroinvasive disease belong primarily to two groups: mosquito-borne viruses in the Japanese encephalitis serocomplex and tick-borne viruses in the tick-borne encephalitis serogroup. The most important human pathogens in these two groups in terms of number of cases include Japanese encephalitis virus, West Nile virus, St. Louis encephalitis virus, and Murray Valley encephalitis virus in the Japanese encephalitis serocomplex; in the tick-borne encephalitis serocomplex, Powassan virus and tick-borne encephalitis virus subtypes Far Eastern, Siberian, and European. In this chapter the general features of the neurotropic flaviviruses will be reviewed. Clinical disease syndromes, epidemiological and ecological aspects, as well as prevention strategies of specific medically important flaviviruses will be described in individual sections at the end of the chapter, with the exception of Japanese encephalitis virus, which will be presented in chapter “Borna Disease Virus.” Dengue viruses, which usually cause febrile illness or hemorrhagic manifestations, occasionally present as meningoencephalitis, and are discussed in chapter “Neurotropic Influenza Virus Infections.”
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Arbovirus
- Flavivirus
- West Nile virus
- Tick-borne encephalitis viruses
- St. Louis encephalitis virus
- Murray Valley encephalitis virus
- Mosquito transmission
Flaviviruses are small, positive-strand RNA viruses which are transmitted from infected to susceptible vertebrate hosts primarily by arthropods (Burke and Monath 2001; Gubler et al. 2007). Flavivirus infections cause seasonal disease syndromes corresponding to mosquito and tick activity throughout the temperate and tropical areas of the world. These seasonal disease outbreaks have been recognized since the 1800s, although flaviviruses were not identified as the etiological agents and arthropods as the transmission vectors until early in the twentieth century, when virus isolation and characterization techniques were developed (Burke and Monath 2001; Solomon 2004; Monath 1989, 1999).
The genus Flavivirus is within the Flaviviridae family, which also includes the Pestivirus and Hepacivirus genera. The Flavivirus genus consists of nearly 80 viruses, approximately half of which are associated with human disease (Burke and Monath 2001; Calisher and Karabatsos 1988; Roehrig 2003; Lindenbach et al. 2007). The majority of flaviviruses are arthropod-borne viruses , or arboviruses, with over half transmitted by mosquitoes, and approximately one third transmitted by ticks (Fig. 1). Five flaviviruses have no known vector (Burke and Monath 2001). Flaviviruses are hypothesized to have derived from a monophyletic lineage, possibly a plant virus, which entered the transmission cycle of a common ancestor to both ticks and mosquitoes, or to ticks and then later mosquitoes (Burke and Monath 2001; Mackenzie et al. 1996; Mackenzie and Field 2004).

General organization of a selection of flaviviruses based on phylogenetic analysis of complete genome sequences (King et al. 2012; Kuno and Chang 2005). The serological complexes and arthropod vectors are shown on the right (Burke and Monath 2001; Kuno et al. 1998). Nucleotide sequences were aligned using CLUSTALW in MegAlign (Lasergene 12, DNASTAR, Madison, WI); phylogeny generated using maximum likelihood (MEGA5) (Tamura et al. 2011)
Flaviviruses were originally characterized serologically and divided into 8 antigenic complexes and 12 subcomplexes based on cross-neutralization assays with hyperimmune antisera (Burke and Monath 2001; Roehrig 2003; McMinn 1997; Calisher et al. 1989; King et al. 2012). Phylogenetic analyses of genomic sequences from archival and recent flavivirus isolates have, in most cases, confirmed the serologically derived antigenic relationships (Fig. 1) (Burke and Monath 2001; Lindenbach et al. 2007; King et al. 2012; Kuno and Chang 2005). Although there are regions of the flavivirus genome that are highly conserved, particularly the genes that code for the antigenic epitopes in the envelope protein, there is also considerable genetic diversity within the genus, with the most distantly related flaviviruses having only about 40 % sequence homology (Burke and Monath 2001; Calisher et al. 1989).
In regions where multiple flaviviruses co-circulate a person may be at risk of serial flavivirus infections, as a prior flavivirus infection does not prevent infection by a different flavivirus. However, antibodies raised in the primary flavivirus infection may modulate the second infection, resulting in milder or more severe illness (Gubler 1998a, b; Porterfield 1986). Experimental evidence suggests that there may be some degree of cross-protection between flaviviruses, as antibodies elicited in the first flavivirus infection produce an anamnestic response and partially neutralize the second flavivirus, leading to reduced clinical symptoms (Tesh et al. 2002). This is hypothesized to be the mechanism for the lack of West Nile encephalitis cases in Central and South America compared to North America. In Central and South America many flaviviruses are known to co-circulate and serosurveys have shown that a high proportion of the population has been previously exposed to a flavivirus. In comparison, in the United States and Canada, West Nile and St. Louis viruses have limited geographic ranges, and flavivirus seroprevalence is low. In contrast, in secondary dengue infections, the anamnestic response may enhance the entry of the second dengue serotype virus into cells, in a process call antibody-dependent enhancement of infection, which may result in more serious disease syndrome. It has been shown that in areas where transmission of multiple dengue serotypes is occurring, most dengue infections are secondary infections, and there is a higher incidence of the more serious dengue hemorrhagic fever/dengue shock syndrome in comparison to areas where a single dengue virus serotype has recently emerged and infected a naïve population (Gubler 1998a, b; Porterfield 1986).
Medically important flaviviruses are associated with three clinical syndromes : encephalitis and meningitis; hemorrhagic fever; or fever, arthralgia, and rash (Burke and Monath 2001; Gould and Solomon 2008). All flaviviruses are neurotropic to some degree, which is probably due to evolutionary conservation of the regions on the envelope protein involved in host cell receptor interactions (Burke and Monath 2001; McMinn 1997; Monath 1986; Gritsun et al. 1995). The neurotropic flaviviruses that cause neuroinvasive disease belong primarily to two groups: mosquito-borne viruses in the Japanese encephalitis serocomplex and tick-borne viruses in the tick-borne encephalitis serogroup (Fig. 1) (Burke and Monath 2001). The most important human pathogens in these two groups in terms of number of cases include Japanese encephalitis virus, West Nile virus, St. Louis encephalitis virus, and Murray Valley encephalitis virus in the Japanese encephalitis serocomplex; in the tick-borne encephalitis serocomplex, Powassan virus and tick-borne encephalitis virus subtypes Far Eastern, Siberian, and European (Burke and Monath 2001; McMinn 1997). In this chapter the general features of the neurotropic flaviviruses will be reviewed. Clinical disease syndromes, epidemiological and ecological aspects, as well as prevention strategies of specific medically important flaviviruses will be described in individual sections at the end of the chapter, with the exception of Japanese encephalitis virus, which will be presented in the chapter entitled “Japanese Encephalitis Virus.” Dengue viruses, which usually cause febrile illness or hemorrhagic manifestations, occasionally present as meningoencephalitis, and are discussed in the chapter entitled “Japanese Encephalitis Virus. “Note added in proof: Zika virus, previously considered to cause a mild febrile illness, has recently been associated with neurological disease in French Polynesia and the Americas. Much research will be needed to determine the mechanisms of pathogenesis and the resultant clinical disease of this emerging virus.”
Ecology and Epidemiology
Japanese encephalitis serocomplex viruses (Fig. 1) are maintained in enzootic cycles between birds and mosquitoes, primarily ornithophilic Culex spp. Humans become infected when they are bitten by an infected mosquito, but viremia is brief and low, and rarely of sufficient titer to infect a mosquito through a blood meal. Similarly, ticks of the genus Ixodes are the vectors of the tick-borne encephalitis viruses, with small mammals such as rodents serving as vertebrate hosts (Calisher et al. 1989; Kuno et al. 1998; Grard et al. 2006). During transmission season, when either the temperature or rainfall provides favorable mosquito or tick breeding conditions, humans become infected when they are bitten by an infected arthropod vector. A more detailed, complete description of arbovirus transmission and ecology is given in the chapter entitled “Influences of Arthropod Vectors on Encephalitic Arboviruses.” prospective multi-center study; no evidence was. Although in general these viruses are not transmitted in nature directly from host to host, cases of human-to-human transmission have been reported to occur through blood transfusions and organ donations during epidemics of intense transmission activity (Montgomery et al. 2006; Iwamoto et al. 2003; Centers for Disease Control and Prevention 2003, 2004; Cushing et al. 2004). Possible cases of sexual and congenital transmission have also been reported (Musso et al. 2015; Foy et al. 2011; O’Leary et al. 2006)
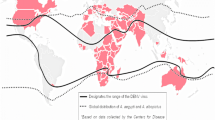
Flaviviruses from the two neurotropic flavivirus groups are distributed widely throughout temperate and tropical regions of the world (Fig. 2). In Asia alone over three billion people are at risk of being infected with Japanese encephalitis virus. Although Japanese encephalitis virus has circulated in Asia for over 100 years, the virus is emerging in new areas where changing agricultural practices have brought arthropod vectors and vertebrate hosts into closer contact with one another, and into contact with naïve susceptible human hosts. As a result, despite the availability of effective, safe vaccines, Japanese encephalitis virus infection is the leading cause of pediatric encephalitis throughout Asia (Campbell et al. 2011; Hills et al. 2014). In addition, flaviviruses such as West Nile virus have emerged for the first time in areas where competent arthropod vectors and susceptible vertebrate hosts have provided the conditions necessary for establishment of virus transmission in new ecological niches. The introduction of West Nile virus into New York in 1999 and the spread of the virus throughout North America since then has resulted in the largest outbreaks of meningitis and encephalitis in the Western Hemisphere (Solomon and Winter 2004). In contrast, although there is evidence of West Nile virus in Central and South America, there have been only sporadic reports of West Nile neuroinvasive disease cases. Powassan virus is being detected with greater frequency in wider geographical regions, probably due to increased laboratory-based surveillance for arboviruses in general since the introduction of West Nile virus .

Countries with historic reports of disease cases or other virus activity as of August 2015. Includes human disease cases, virus-specific antibodies in humans or other animals, or virus or viral RNA detected in mosquitoes or vertebrate animals. Geographical distribution by state or province of (a) West Nile virus, (b) St. Louis encephalitis virus , (c) Murray Valley encephalitis virus , and (d) tick-borne encephalitis flaviviruses. The distribution of St. Louis virus in Canada in Ontario, Manitoba, and Quebec was based on a single North American outbreak in 1975–1976
Flavivirus Structure and Replication
Flaviviruses are small, spherical viruses with icosahedral symmetry, approximately 50 nm in diameter (Lindenbach and Rice 2001; Chambers et al. 1990) (Fig. 3). The virion is smooth, with no spikes or surface projections and comprises viral envelope and membrane proteins arranged in head-to-tail heterodimers, embedded in a host cell-derived lipid bilayer, surrounding a nucleocapsid core. The nucleocapsid consists of multiple copies of the capsid protein, arranged in an icosahedral, anchoring the RNA genome (Lindenbach et al. 2007; Kuhn et al. 2002; Mukhopadhyay et al. 2003). The single-stranded, positive-sense RNA genome, approximately 11-kb in length, functions as an mRNA, with a single open reading frame. The genome is flanked at both the 5′ and 3′ ends by untranslated regions, and capped at the 5′ end (Fig. 4). Viral proteins are translated in a polyprotein that is co- and posttranslationally cleaved by cellular and viral proteases and glycosylated by cellular glycosyltransferases into three structural proteins: capsid, premembrane, and envelope; and seven nonstructural (NS) proteins: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 (Lindenbach and Rice 2001; Zhang et al. 2003). The structural proteins make up the virus particle; the nonstructural proteins function primarily in viral replication and virion assembly, together with host cell factors (Lindenbach and Rice 2001; Chambers et al. 1990). The envelope glycoprotein is the major surface protein of the viral particle. It interacts with specific host cell receptors in the initial binding of the virus to the cell surface and is involved with membrane fusion and entry; thus it is an important determinant of tissue tropism and virulence (Roehrig 2003; Heinz and Roehrig 1990). The envelope protein is also the major viral antigen against which host protective antibodies are elicited (Roehrig 2003; Heinz and Roehrig 1990; Guirakhoo et al. 1992). Comparison of flavivirus envelope protein sequences has shown both highly conserved and highly variable subregions (Burke and Monath 2001; Kuno and Chang 2005). However, despite the lack of proofreading in RNA viruses during replication, the envelope gene is one of the most slowly evolving sites, which is probably due to the selective pressure of infecting and replicating in both vertebrate and arthropod cells.

Negative staining electron micrograph of West Nile virions, approximately 45 nm in diameter (×216,000). The virus was isolated in Vero E6 cells from an organ transplant recipient who died in 2002 (Cushing et al. 2004) (From C. Goldsmith, CDC Public Health Image Library)

Flavivirus genome organization. The single-stranded, positive sense RNA genome functions as a messenger RNA consisting of a single open reading frame that codes for a single polyprotein, flanked by noncoding regions at the 5′- and 3′-terminal ends. The polyprotein is co- and posttranslationally cleaved by cellular and viral proteases into three structural and seven nonstructural proteins. The prM is cleaved upon virus particle release through the cell plasma membrane. C capsid, E envelope, NCR noncoding region, NS nonstructural, prM premembrane
Flavivirus replication takes place in the host cell cytoplasm (Fig. 5). The virus binds to the surface of host cells through an interaction between the envelope protein and specific cellular receptors (Lindenbach and Rice 2001; Chambers et al. 1990; Seligman and Bucher 2003; Koschinski et al. 2003). Following attachment, fusion and entry of the virus is carried out via receptor-mediated endocytosis, where the envelope protein undergoes an acid-catalyzed conformational change, resulting in membrane fusion, uncoating, and release of the nucleocapsid into the cytoplasm (Lindenbach and Rice 2001; Heinz and Roehrig 1990). Following translation and processing of the viral proteins, a viral replicase is assembled from viral nonstructural proteins and host factors. Viral replication is initiated with the synthesis of a genome-length minus-strand RNA intermediate, which serves as a template for the synthesis of genomic RNA (Lindenbach and Rice 2001). Virion morphogenesis is hypothesized to occur in association with intracellular membranes, probably the endoplasmic reticulum (Lindenbach and Rice 2001; Chambers et al. 1990). Immature virions assemble in membrane-bound vesicles in a premembrane and envelope protein heterodimer conformation which prevents envelope protein fusion activity. Virions accumulate in the rough endoplasmic reticulum and are transported to the cell surface in acidic vesicles through the host secretory pathway, possibly that used for synthesis of host plasma membrane glycoproteins (Lindenbach and Rice 2001). The virions fuse with the plasma membrane and are released by exocytosis after the membrane protein is cleaved from the precursor premembrane protein in the Golgi by a Golgi protease, signaling virus release and envelope protein activation (Fig. 4) (Guirakhoo et al. 1992).

Flavivirus life cycle [Modified from Lindenbach et al. (Lindenbach et al. 2007)]
Pathogenesis and Immune Response
Flavivirus infection of arthropod cells in culture may show cytopathic effects such as syncytia formation, but infection of mosquito cells is generally persistent, not cytopathic (see “Influences of Arthropod Vectors on Encephalitic Arboviruses”). Virus infection of vertebrate cells in culture shows cytopathic effects such as cellular rounding, and as virions accumulate in the rough endoplasmic reticulum, proliferation, hypertrophy, and fragmentation of the membranes. Mitochondrial damage, rarefaction of cytoplasm, formation of vacuoles and inclusion bodies, and an increase in lysosomal enzymes have also been shown to occur in cultured cells following flavivirus infection (Burke and Monath 2001; Lindenbach and Rice 2001).
In natural infections, an infected arthropod inoculates the flavivirus into vertebrate skin along with the saliva during a bite. Initial virus replication occurs at the site of inoculation in keratinocytes, newly recruited neutrophils, and skin dendritic cells, specifically Langerhans cells (Johnston et al. 2000). Virus is then transported in migrating dendritic Langerhans cells and neutrophils to lymph nodes and from the lymphatic system to the thoracic duct and into the bloodstream (Burke and Monath 2001; Monath 1986; Johnston et al. 2000). This primary viremia is the source of infection of peripheral tissue such as spleen, liver, and kidney. Viremia continues for several days due to release of virus from these tissues back into the bloodstream (Burke and Monath 2001).
The vast majority of infections by neurotropic flaviviruses are self-limiting and the person is either asymptomatic or may have a mild subclinical fever syndrome. In the primary immune response viremia is of low titer and brief, modulated by macrophages and then cleared following the rise of the humoral immune response, usually within a week of infection (Fig. 6) (Burke and Monath 2001). In addition, the T-cell helper and cytotoxic immune response is elicited against infected lymphoblastoid cells (Roehrig 2003; Seligman and Bucher 2003). Virus is usually not detectable in serum collected at the time of onset of symptoms.

Graphical representation of the course of viremia and IgM and IgG antibody immune response in a (a) primary and (b) secondary neurotropic flavivirus infection
Neuroinvasive disease occurs in approximately 1 in 100 to 1 in 1000 neurotropic flavivirus infections and is dependent on viral factors, vector competence, and host factors. The viral factors that are hypothesized to contribute to neuroinvasive disease include the level of viremia and the genetic differences in virus strain neurovirulence (Burke and Monath 2001; McMinn 1997; Monath 1986; Gritsun et al. 1995). Single mutations in the envelope gene have been shown to alter neurovirulence phenotype (McMinn 1997). Arthropod vector competence is another factor that contributes to neurovirulence, and is described in detail in “Influences of Arthropod Vectors on Encephalitic Arboviruses.” In the host, age, gender, genetic susceptibility, pre-existing herpesvirus or heterologous flavivirus infection or immunization, and concomitant parasite infection are factors that contribute to susceptibility to infection and disease severity (Burke and Monath 2001; Libraty et al. 2002). Generally, the highest proportion of neuroinvasive disease is seen in the very young and the elderly. In areas where Japanese encephalitis and Murray Valley viruses are endemic, children make up the largest proportion of cases, and it has been demonstrated experimentally that younger neurons are more susceptible to virus infection. However, in areas with low flavivirus seroprevalence, such as North America, the risk of St. Louis encephalitis and West Nile virus infections resulting in neuroinvasive disease is higher in those over 55 years of age (Burke and Monath 2001; Sejvar and Marfin 2006). The reasons for this are unclear, but may include factors such as the impaired integrity of the blood–brain barrier caused by cardiovascular or other age-related diseases (Solomon 2004).
Most of the data regarding the regions of the central nervous system infected by flaviviruses come from postmortem studies of pediatric Japanese encephalitis cases in Asia, West Nile cases from North America, and experimental infections in animal models. The exact mechanisms by which flaviviruses enter the central nervous system have not been definitively identified, but hypothesized pathways include (1) infection of cerebral endothelial cells and migration across the cell to the brain parenchyma; (2) migration of infected leukocytes through the tight junction formed by endothelial cells; (3) direct choroidal virus shedding; (4) axonal transport up the olfactory nerve; (5) increased permeability due to tumor necrosis factor α induction by attachment of double-stranded RNA to Toll-like receptors; or (6) retrograde transport along peripheral nerve axons (Burke and Monath 2001; Gubler et al. 2007; McMinn 1997; Sejvar and Marfin 2006; Kramer 2007; Hayes et al. 2005; Campbell et al. 2002). Whether the virus enters at a single site or at multiple locations is also unknown.
Once in the central nervous system, the virus replicates and spreads rapidly. Pathogenesis is due to direct, virally mediated damage to neurons and glial cells, cytotoxic immune response to infected cells, the inflammatory immune response in perivascular tissue, and microglial nodule formation (Burke and Monath 2001; Kuno and Chang 2005; Campbell et al. 2002; Solomon 2003). Virus tropism for specific brain areas may vary and could explain the different clinical presentations. In histopathological studies West Nile virus has been shown to directly infect and destroy neurons in the brain stem, deep nuclei of the brain, and anterior horn cells in the spinal cord (Fig. 7) (Cushing et al. 2004). The inflammatory immune response of natural killer cells, macrophages, and T-lymphocytes results in lysis of neuronal tissue and diffuse perivascular inflammation of the brain stem and anterior horn cells of the spinal cord, and immune-mediated damage to bystander nerve cells, glial cells, as well as other surrounding tissue (Gubler et al. 2007; Hayes et al. 2005). Damage to these neuronal cells is characterized by central chromatolysis, cytoplasmic eosinophilia, cell shrinkage, and neuronophagia, and by the formation of cellular nodule formation composed of activated microglia and mononuclear cells (Burke and Monath 2001; Sejvar and Marfin 2006; Kramer 2007; Hayes et al. 2005; Campbell et al. 2002). Apoptosis of motor neurons in the anterior horn of the spinal cord results in flaccid paralysis.

Photomicrograph of immunohistochemical staining of brain tissue from a fatal West Nile encephalitis case, showing West Nile antigen-positive neurons and neuronal processes in the brain stem and anterior horn cells (in red) (From W.-J. Shieh and S. Zaki, CDC Public Health Image Library)
Persistent and long-term pathological changes are often seen following neuroinvasive flavivirus infection, such as residual neurological deficits, electroencephalographic changes, and psychiatric disturbances (Burke and Monath 2001). Long-term follow-up studies in Japanese encephalitis cases in children have shown neuronal loss and dense microglial scarring resulting in recurrent neurological disease (Solomon 2003, 2004; Monath 1986). Chronic progressive encephalitis has been observed years after infection with tick-borne encephalitis virus (Burke and Monath 2001; Monath 1986). In West Nile virus encephalitis patients in North America, the majority experience long-term neurological deficits, with only 13 % reporting full recovery in physical cognitive and functional abilities 1 year after illness (Sejvar 2014; Sejvar et al. 2003a, 2010; Staples et al. 2014).
Clinical Presentation
The majority of flavivirus infections are asymptomatic or subclinical (Solomon 2004; Kramer 2007; Sejvar 2014). Clinical disease ranges from a mild febrile illness to a severe neurological syndrome following an incubation period of 2–14 days (Mackenzie et al. 1996). Febrile illness is characterized by fever, chills, headache, back pain, myalgia, and anorexia, as well as eye pain, pharyngitis, nausea, vomiting, and diarrhea (Mackenzie et al. 1996; Campbell et al. 2002). A transient maculopapular rash over the trunk and limbs is also common. Acute illness usually lasts from 3 days to several weeks. Most patients with uncomplicated fever completely recover within days to months, but prolonged fatigue is often seen (Solomon 2004; Sejvar and Marfin 2006; Campbell et al. 2002).
Fever symptoms may be followed in 1–4 days by acute or subacute appearance of meningeal and neurological signs (Table 1). The neurological syndrome depends on the part of the nervous system that is infected: the parenchyma of the brain, which causes encephalitis; the meninges, which causes meningitis; or the anterior horn cells of the spinal cord, which causes myelitis (Burke and Monath 2001; Solomon 2004; Sejvar and Marfin 2006; Hayes et al. 2005; Sejvar 2014; Hayes and Gubler 2005; Petersen and Marfin 2002; Petersen et al. 2002; Sejvar et al. 2003b) The primary clinical presentations may overlap, and include a reduced level of consciousness, often associated with seizures, flaccid paralysis, and parkinsonian movement disorders (Solomon 2004; Halstead and Jacobson 2003). Encephalitis is more common than meningitis, with 50–85 % of patients presenting with encephalitis, compared to 5–50 % with meningitis (Solomon 2004; Sejvar 2014). Seizures are more common in children, with approximately 85 % Japanese encephalitis or Murray Valley encephalitis pediatric patients and 10 % of adult West Nile encephalitis patients experiencing seizures (Solomon 2004; Mackenzie et al. 1996; Solomon et al. 2000). Motor weakness occurs in 10–50 % of flavivirus neuroinvasive cases, with acute asymmetric flaccid paralysis similar to that seen in poliomyelitis (Solomon 2004; Hayes et al. 2005). Coma occurs in approximately 15 % of patients.
The case fatality rate among hospitalized patients with neuroinvasive disease ranges from approximately 9 % of those infected with North American West Nile virus to 30 % of pediatric Japanese encephalitis cases. The cause of death is primarily due to neuronal dysfunction, respiratory failure, and cerebral edema (Campbell et al. 2002; Sejvar et al. 2010). Multiple or prolonged seizures in Japanese encephalitis patients are associated with a poor outcome, as are changes in respiratory pattern, flexor and extensor posturing, and pupillary and oculocephalic reflex abnormalities (Solomon 2004; Solomon et al. 2000). About one half of survivors have long-term neurological sequelae, including motor deficits, cognitive and language impairment, and convulsions, with children making up the largest proportion of this group (Hayes et al. 2005; Solomon et al. 2000; Douglas et al. 2006). In addition, even those who were considered to have good recovery may have subtle long-term effects such as learning disorders, behavior problems, and other neurological deficits. Follow-up studies of pediatric Japanese encephalitis cases show that a high proportion of patients experience persistent movement disorders 3–5 years later (Murgod et al. 2001). Many patients with poliomyelitis do not recover, although limb strength may improve over time (Hayes et al. 2005; Staples et al. 2014). Since the introduction of West Nile virus in 1999, in follow-up studies on short- and long-term outcomes of West Nile neuroinvasive infections, <50 % of patients had full cognitive and functional recovery after 1 year. Frequent complaints included fatigue, muscle aches, and difficulties with memory and concentration, suggesting a subcortical type of cognitive dysfunction based on prominent thalamic and basal ganglia involvement (Sejvar 2014). Case fatality rate of hospitalized patients with West Nile disease is 4–16 %, with a further two- to threefold increase in mortality observed up to 3 years after acute illness, and possibly longer (Staples et al. 2014).
The clinical course of tick-borne encephalitis virus infections may be distinct from the mosquito-borne flaviviruses, with many infections taking a biphasic course. Onset of illness may be more gradual, with patients experiencing an influenza-like illness for approximately 1 week, with fever, headache, malaise, and myalgia, followed by an asymptomatic period of up to a week (Gresikova and Calisher 1989; Leonova et al. 2014; Lindquist and Vapalahti 2008). A second phase involving the central nervous system includes clinical symptoms ranging from mild meningitis to severe encephalitis, with or without myelitis and spinal paralysis (Burke and Monath 2001; Gresikova and Calisher 1989). Generally, symptoms are more severe in adults than in children (Lindquist 2008; Logar et al. 2000). Long-term neurological problems are similar to those resulting from other neuroinvasive flavivirus infections (Lindquist and Vapalahti 2008).
Laboratory Diagnosis
Flavivirus infections may present with clinical symptoms similar to those of other virological or treatable bacterial infections, such as a flu-like illness, encephalitis, or polio-like myelitis (Burke and Monath 2001). In addition, vaccine preventable diseases such as Japanese encephalitis and tick-borne encephalitis may be clinically indistinguishable and cause similar disease syndromes to neurotropic flavivirus infections for which there is no effective vaccine. Therefore, laboratory diagnosis is necessary to identify etiology and differentiate between other bacterial or viral pathogens, guide effective clinical management and/or treatment, as well consider public health responses such as vaccination. Virus isolation or viral RNA detection in serum, cerebral spinal fluid, or tissue is the gold standard for diagnosis of a viral infection, but it is not sensitive in neurotropic flavivirus infections, as low levels (≤100 infectious particles per mL) of viremia are usually cleared by the onset of illness (Fig. 6).
Diagnosis is usually made serologically by detection of virus-specific antibody, ideally from paired acute and convalescent specimens, with the rise in antibody titer indicative of a recent infection (Martin et al. 2000; Nasci et al. 2002). In practice, however, only a single acute specimen is usually obtained. In these cases, specific immunoglobulin M (IgM) antibody-capture enzyme-linked immunosorbent assay (ELISA) can be used for rapid detection of acute flaviviral infections , as IgM antibody is produced early in infection, rises rapidly to detectable levels, and is less cross-reactive than IgG antibodies (Fig. 6) (Iwamoto et al. 2003; Martin et al. 2000; Johnson et al. 2000, 2005a; Wong et al. 2003, 2004).
CSF is the preferable specimen for diagnosis of flavivirus neuroinvasive infections, as antiflavivirus IgM antibody may be present in serum, but may not be the cause of encephalitis, such as in inapparent or mild infections or following a recent flavivirus vaccination (Burke et al. 1982, 1985). This is especially prevalent in areas where there is high background immunity in the population, such as in Asia where a large proportion of the population have been exposed to or vaccinated against Japanese encephalitis virus. Anti-Japanese encephalitis IgM antibody has been shown to be detectable in serum as much as 6 months following vaccination with the live attenuated vaccine virus (Roehrig et al. 2003). However, IgM antibodies elicited in non-neuroinvasive flavivirus infections or following flavivirus vaccination do not enter the CSF ; therefore by testing the CSF, the effect of background IgM antibodies in the serum is eliminated (Mackenzie et al. 1996; Martin et al. 2000, 2002; Johnson et al. 2000; Burke et al. 1982; Chanama et al. 2005). IgM antibody is usually detectable in the CSF by onset of illness or within a few days thereafter, except in very acute, sudden-onset encephalitis, when the IgM antibodies may not have reached detectable levels at hospital admission, in which case the IgM ELISA may result in a false negative (Fig. 6) (Johnson et al. 2000; Burke et al. 1982). Therefore it is critical for diagnosis that a second specimen be collected and tested if possible 7 days after onset of illness or at hospital discharge (Martin et al. 2000; Lindsey et al. 2012).
Cross-reactivity in serological assays , including the IgM ELISA, is a problem in flavivirus diagnostics. Antibodies within a flavivirus serocomplex are highly cross-reactive; those between serocomplexes are less cross-reactive but still may confound accurate diagnosis (Martin et al. 2002). This is due to the heterologous population of antibodies produced in the infection against different epitopes of the flaviviral envelope protein, some of which are virus species specific, and others of which are conserved across the serocomplex or flavivirus genus. Antibodies elicited against these conserved regions cross-react in serological assays with other flaviviral antigens and cause false-positive results in the IgM ELISA . For example, dengue and West Nile viruses, which are not in the same serocomplex, co-circulate in Africa and Asia. Dengue virus does not cause encephalitis but may have a clinical presentation of encephalopathy, and a specimen from a dengue patient submitted for West Nile testing may have a positive result in a West Nile virus IgM ELISA (Hogrefe et al. 2004; Niedrig et al. 2007). The plaque-reduction neutralization test is a more specific assay and is used to confirm or differentiate conflicting IgM ELISA results in primary flavivirus infections.
In secondary flavivirus infections, an anamnestic reaction may occur, in which antibodies from the first flavivirus infection are elicited and cross-react in the IgM ELISA with antigens to the most recent infection, producing false-positive results (Porterfield 1986; Johnson et al. 2005b). In addition, the plaque neutralization test has less specificity in secondary flavivirus infections, as neutralizing antibody from the primary flavivirus infection rises quickly and the titer may be equal to or higher than that of the neutralizing antibody titer elicited in the acute flavivirus infection (Johnson et al. 2005b). In addition, IgM antibody to the second infecting virus may not rise to levels detectable by the IgM ELISA , producing a false-negative result (Fig. 6b).
Diagnostic assays such as the microsphere immunoassays have been shown to have improved specificity and sensitivity, and can be used for differential diagnosis (Johnson et al. 2005a; Wong et al. 2003, 2004). These assays, based on Luminex™ technology, can be run in a one-well multiplex format, which reduces the sometimes very limited specimen volume needed, and have a statistical-based results interpretation.
Detection of flavivirus antigen in brain tissue by immunohistochemistry is useful for diagnosis in fatal cases, as these patients may not have detectable IgM or IgG antibodies in serum or CSF (Cushing et al. 2004). In addition, viral nucleic acid detection in CSF has also proved useful in up to 50 % of very acute infections, which when used together with IgM ELISA enhances sensitivity (Tilley et al. 2006; Lanciotti et al. 2000; Lanciotti and Kerst 2001).
Use of the hemagglutination inhibition and the complement fixation assays has decreased in recent years as they require paired specimens and lack specificity. The CT scan has not been shown to be an effective diagnostic method for identifying flavivirus encephalitis cases, and the MRI yields characteristic abnormal results in only 25–35 % of cases, and these may be nonspecific (Sejvar and Marfin 2006; Hayes et al. 2005; Campbell et al. 2002; Sejvar et al. 2003a; Solomon et al. 2000).
Treatment
Treatment of encephalitis is supportive and includes pain control for headaches, rehydration, antiemetic treatment for nausea and vomiting, reduction of intracranial pressure, and control of seizures (Solomon 2006). In patients with paralysis, the airway is managed to reduce aspiration and obstruction. Ventilation support may be required in patients with neuromuscular respiratory failure. Antivirals and other treatments such as ribavirin interferon-α, and immunoglobulin have not been found to be effective. High-dose corticosteroid treatment may be contraindicated because of the risk of depressing the immune system before the virus is cleared (Hayes et al. 2005).
Prevention
Vector control programs up to the early 1970s successfully eradicated mosquitoes such as Aedes aegypti, the primary mosquito vector of dengue and yellow fever viruses, from most of Central and South America (see “Influences of Arthropod Vectors on Encephalitic Arboviruses”). However, these programs were not sustainable, and as a result Aedes aegypti has re-infested these areas and dengue and yellow fever epidemics have reemerged. Most vector control programs are organized on a local rather than national level and it becomes difficult to maintain the funding for these programs in the absence of cases, once the epidemics have passed. Insecticide treatment is expensive, must be periodically applied, and may have deleterious effects on other species, including humans, and resistance to classes of insecticides develops quickly in the arthropod vectors, which complicates this effort. In addition, barrier systems such as bed nets, which have been used successfully to disrupt transmission of malaria, are not effective methods with Culex spp. mosquitoes, as these mosquitoes typically feed at dusk when human activity is high.
Vaccination remains the most effective method to prevent flavivirus infection. Vaccination has been used successfully to prevent Japanese encephalitis and tick-borne encephalitis and will be described in more detail in the chapter entitled “Japanese Encephalitis Virus” and below, respectively (Solomon 2006; Marfin et al. 2005; Monath 2001, 2002).
Brief Descriptions of Specific Neurotropic Flaviviruses
West Nile Virus
West Nile virus was first isolated from the blood of a febrile patient in Uganda in 1937, and is considered a common childhood infection in Africa. The virus is separated into multiple lineages based on phylogenetic analysis of the complete West Nile virus genomes; lineage 1 and 2 strains have been most often associated with outbreaks in humans and equines (Lanciotti et al. 1999, 2002; Donadieu et al. 2013). Lineage 1 consists of strains from Western Africa, Eastern Europe, the Middle East, and recently, North America, and includes Kunjin virus from Australia (Fig. 2a) (Lanciotti et al. 2002). West Nile lineage 1 virus strains have caused encephalitis epidemics throughout western Africa, Eastern Europe, the Middle East, and North America and epizootics with high equine mortality in Europe and North America (Solomon 2004; Sejvar and Marfin 2006). In West Nile virus endemic areas of Africa, children are most likely to become infected, with a small percentage of infections developing symptoms of West Nile fever and very few cases of West Nile encephalitis, a similar pattern to that of Japanese encephalitis virus in Asia (Burke and Monath 2001). However, a higher proportion of adults may become infected when West Nile virus moves to new areas such as North America, or when susceptible adults, particularly travelers, enter an area of transmission activity. Lineage 2 strains historically have been considered less pathogenic than lineage 1 strains, causing febrile illness, but until recently have not been associated with neuroinvasive disease. The geographical range of West Nile lineage 2 strains was thought to be restricted to sub-Saharan Africa and Madagascar. Recently however, lineage 2 strains have been shown to be responsible for outbreaks in humans and equines in South Africa, as well as in eastern and southern Europe (Donadieu et al. 2013).
West Nile virus is transmitted in an enzootic cycle between mosquitoes, primarily Culex spp., and birds, particularly water birds and birds in the corvid family (see “Influences of Arthropod Vectors on Encephalitic Arboviruses”) Volume 2. Humans and horses become infected through the bite of an infected mosquito, and viremia is generally low and brief. Most cases occur in the mid to late summer in temperate regions, which corresponds to the transmission activity of the mosquito vector (Campbell et al. 2002; Nasci et al. 2002).
West Nile virus was identified as the cause of a meningoencephalitis epidemic of 59 cases and 7 deaths in New York that began in August in 1999 (Campbell et al. 2002). The North American West Nile virus is most closely related to and probably originated from a lineage 1 strain from Israel (Lanciotti et al. 1999). Since its introduction, the range of West Nile virus has extended across the United States, north into Canada, and into Central and South America (Gonzalez-Reiche et al. 2010; Morales et al. 2006; Morales-Betoulle et al. 2013). Through 2013, nearly 40,000 West Nile infections have been reported throughout the continental United States . Approximately 17,000 of those are neuroinvasive disease cases, and there have been over 1500 deaths (http://www.cdc.gov/westnile/statsMaps/index.html) (Hayes et al. 2005). In Canada there have been more than 5000 West Nile infections reported since the first cases were detected in Ontario in 2002, approximately 240 of which caused neuroinvasive disease (http://www.phac-aspc.gc.ca/wnv-vwn/mon-hmnsurv-archive-eng.php#a2002_07). Since 2006, probable human West Nile cases based on serology have been identified in Central and South America, and West Nile virus has been isolated from mosquitoes in Guatemala and equines in Argentina, but large West Nile encephalitis epidemics have not been reported from these areas (Gonzalez-Reiche et al. 2010; Morales et al. 2006; Komar and Clark 2006; Komar et al. 2005; Mattar et al. 2005). In Europe, epizootics resulting from autochthonous transmission of West Nile virus have occurred for over 20 years, with human cases reported sporadically (Sambri et al. 2013; Rossini et al. 2008).
Since its introduction in 1999, the ecology, epidemiology, and pathology of North American West Nile virus have been intensely studied. The many competent mosquito vectors and susceptible vertebrate hosts, as well as the virulent North American West Nile virus strain, may have contributed to the rapid spread and establishment of the virus in North America (see “Influences of Arthropod Vectors on Encephalitic Arboviruses”) (Brault et al. 2004). In addition, the combination of flood irrigation practices in the Great Plains with the preponderance of the highly competent vector Culex tarsalis in this region has resulted in the highest annual and cumulative incidence of neuroinvasive disease in the northern Great Plains states, along with the Louisiana–Mississippi Gulf region. The multiple determinants of vector-borne disease make it difficult to predict over the long term whether the epidemic cycle of West Nile virus will be similar to that of St. Louis virus, which is characterized by discrete epidemics followed by long periods of senescence, or to Japanese encephalitis virus, with annual epidemics. To date there has been a seasonal epidemic pattern of continuous cases similar to that of Japanese encephalitis virus, but the total case count varies considerably from year to year. Seroprevalence in North America and Europe remains low, between 2 and 3 % a year in areas where outbreaks have occurred, which is similar to that of St. Louis virus in North America (Campbell et al. 2002).
Similar to other flaviviruses in the Japanese encephalitis serocomplex, the majority of West Nile virus infections are asymptomatic, with 1 in 5 experiencing a mild illness characterized by acute onset of fever, headache, stiff neck, fatigue, malaise, muscle pain and weakness, gastrointestinal symptoms, and a transient macular rash on the trunk and extremities. Symptoms usually resolve within 60 days; however, long-term effects have been reported (Hayes et al. 2005; Campbell et al. 2002; Petersen and Marfin 2002). The higher reported rates of West Nile fever, compared to those of Japanese encephalitis and St. Louis encephalitis virus infections, may be due to the increased awareness of the disease in the United States (Sejvar 2014; Staples et al. 2014).
Neuroinvasive disease , including encephalitis, meningitis, paralysis, and seizures, develops in approximately 1 in 140 infections, with encephalitis making up the largest proportion, which is similar to that of other Japanese encephalitis antigenic complex virus infections (Campbell et al. 2002; Sejvar 2014). Acute asymmetric flaccid paralysis has been reported in approximately 13 % of patients with West Nile neuroinvasive disease (Hayes et al. 2005). Although the range of illness is found across all age groups, younger persons tend to have milder West Nile fever, whereas the elderly are more likely to proceed to the more severe West Nile encephalitis (Sejvar and Marfin 2006; Sejvar et al. 2003b). The case fatality rate is approximately 4–18 % of hospitalized patients, with mortality higher in the elderly and in immunocompromised persons (Sejvar and Marfin 2006; Petersen et al. 2003). Long-term neurological problems in survivors and muscle weakness patients with paralysis have been reported (Sejvar and Marfin 2006; Hayes et al. 2005; Sejvar et al. 2003a, 2010).
Human-to-human transmission of West Nile virus through transfusion of blood products and transplantation of solid organs was identified in 2002 among asymptomatic donors in areas where there was intense West Nile virus transmission (Montgomery et al. 2006; Centers for Disease Control and Prevention 2009). A probable case of transplacental infection has also been reported (Sejvar and Marfin 2006). As a result, routine blood and organ screening by highly sensitive and specific nucleic acid amplification tests has been implemented (Centers for Disease Control and Prevention 2003, 2004).
Vaccines to protect against West Nile virus infection have been developed for the veterinary market and are commercially available for horses and birds (Davis et al. 2001). Consideration of the risk–benefit ratio of human West Nile virus vaccination is ongoing but the development of a commercial vaccine is unlikely, because the percentage of the population at risk is very low, and the elderly, which are the most likely to have severe clinical manifestations and thus the group which would most benefit from a vaccine, are also the most likely to have higher risk of adverse effects from the vaccine. The pattern of future outbreaks of West Nile virus in North America will be an important component of West Nile virus vaccine development.
St. Louis Encephalitis Virus
St. Louis encephalitis virus was first identified as the causative agent in encephalitis epidemics in Illinois and Missouri in 1932 and 1933, and was first recognized in South America in the 1960s (Gubler et al. 2007; Tsai and Mitchell 1989; Brinker et al. 1979). St. Louis encephalitis virus is widely dispersed throughout the Americas (Fig. 2b). The largest epidemic in the United States, with nearly 2000 cases, occurred throughout the Midwest in 1975. In the most recent outbreak in 1990 there were nearly 250 cases, primarily in Florida and Texas. An outbreak of 46 human cases of St. Louis encephalitis was reported in Argentina in 2005 and in the same year, St. Louis encephalitis virus was isolated from the serum of a suspected dengue case in Brazil (Spinsanti et al. 2008; Rocco et al. 2005). St. Louis encephalitis virus activity has been characterized by periodic outbreaks interspersed with long periods of sporadic cases (Brinker et al. 1979). Since the introduction of West Nile virus to the United States in 1999, surveillance of arboviruses has increased, and subsequently, St. Louis encephalitis cases not associated with epidemics have been identified. There have been approximately 90 cases reported since 2004, the majority of which were neuroinvasive disease (http://www.cdc.gov/sle). This may be a result of increased surveillance rather than a true increase in St. Louis encephalitis cases.
Maintained in enzootic cycle between birds and Culex spp. mosquitoes, St. Louis encephalitis virus also infects horses and humans; however neither horses nor humans play a primary role in the transmission cycle, and there is no morbidity or mortality associated with St. Louis encephalitis virus infection in horses (Tsai and Mitchell 1989). Despite an expansive geographical range throughout the temperate and tropical regions in the Americas, rates of St. Louis encephalitis virus transmission to humans are low, although intense mosquito–bird transmission can presage epidemics. Historically St. Louis encephalitis has been considered a disease of rural agricultural areas, although there have been urban outbreaks; in the 1975 outbreak the largest number of cases occurred in the Chicago metropolitan area. However, even in these cases vegetated parkland areas were shown to be the sites of transmission (Burke and Monath 2001). Transmission is seasonal, corresponding to mosquito activity during the late summer months (see 26”Influences of Arthropod Vectors on Encephalitic Arboviruses”).
St. Louis encephalitis is in the Japanese encephalitis serocomplex, and genetically closely related to West Nile virus (Fig. 1) (Tsai and Mitchell 1989). In Central and South America, where St. Louis encephalitis and West Nile viruses co-circulate, it may not be possible to differentiate between the two viruses in serological assays due to the cross-reactivity, although this is the primary method of diagnosis, as virus isolation from either West Nile or St. Louis virus infected human patients is rare.
The majority of human St. Louis encephalitis virus infections are subclinical, with the ratio of encephalitis cases to asymptomatic infections approximately 1:85 to 1:800 in adults and children, respectively. Adults are more likely to become infected in North America due to low seroprevalence rates. There are three clinical syndromes associated with St. Louis encephalitis neuroinvasive infections: encephalitis, aseptic meningitis, and febrile headache. Case fatality rates increase with age, from 2 % in young adults to 22 % in elderly patients. From 30 to 50 % of survivors experience slow, complete recovery, whereas 20 % experience long-term neurological symptoms, including gait and speech disturbances, sensorimotor impairment, psychoneurotic complaints, and tremors (Tsai and Mitchell 1989). Vaccines have been developed to protect against St. Louis encephalitis virus infection. However, given the long time between epidemics, the low seroprevalence rate, and the cost of bringing a vaccine to market, commercial vaccine development is unlikely.
Murray Valley Encephalitis Virus
Outbreaks of encephalitis were reported in the Murray Valley of Australia in the early 1900s, although the virus was not identified or characterized as being distinct from Japanese encephalitis virus until 1951 (Burke and Monath 2001; Solomon 2004; Marshall 1988). Like Japanese encephalitis and West Nile viruses, Murray Valley encephalitis virus infections can cause polio-like acute flaccid paralysis, and early Murray Valley encephalitis virus outbreaks were thought to be poliomyelitis.
Murray Valley encephalitis virus is transmitted in an enzootic cycle between water birds and the principal mosquito vector Culex annulirostris, which breeds in transient pools (see “Influences of Arthropod Vectors on Encephalitic Arboviruses”) (Marshall 1988). Similar to Japanese encephalitis serogroup viruses, large water birds such as herons, egrets, and pelicans are the primary vertebrate hosts (Mackenzie et al. 1996; Marshall 1988). Mammals such as kangaroos and rabbits also may be significant viremic hosts in the transmission cycle (Burke and Monath 2001). The range of Murray Valley encephalitis virus extends throughout the tropical northern parts of Australia and New Guinea, and in these areas, Murray Valley encephalitis virus infection is the most common cause of viral encephalitis (Fig. 2c). Similar to the other neurotropic flaviviruses, humans are infected incidentally and probably do not contribute to the transmission cycle (Douglas et al. 2006). Outbreaks of Murray Valley encephalitis in 1951 (45 cases), 1974 (58 cases), and 2011 (17 cases) have been interspersed with sporadic cases; approximately 40 cases have been reported in the last 25 years (Selvey et al. 2014).
Febrile illness due to Murray Valley encephalitis infection is not reported, but the ratio of subclinical infections to encephalitis cases is estimated to be 1:1000 (Douglas et al. 2006). The clinical patterns of the disease include rapid onset of fatal encephalitis, flaccid paralysis, tremor, or encephalitis with complete recovery, similar to those of the other Japanese encephalitis complex viruses. Clinical illness is generally seen in young children and nonimmune adults (Solomon 2004; Douglas et al. 2006).
(Solomon 2004; Selvey et al. 2014). The case fatality rate is 31 %; a third of survivors experience long-term neurological deficits (Douglas et al. 2006). Because of the large proportion of inapparent infections and high seroprevalence in adults, large-scale vaccination programs against Murray Valley encephalitis virus have not been considered necessary or economically feasible.
Tick-Borne Encephalitis Viruses
Scandinavian church records from the eighteenth century describe a tick-borne encephalitis-like illness occurring annually in spring time. Russian spring-summer encephalitis was first described as a clinical illness in the far-eastern region of the Soviet Union in 1935. Tick-borne encephalitis virus was isolated from a human brain and ticks were shown to be the arthropod vectors in 1937 (Burke and Monath 2001; Gresikova and Calisher 1989; Lindquist and Vapalahti 2008; Gritsun et al. 2003). Since then three subtypes of tick-borne encephalitis virus have been identified: Far Eastern (Russian spring-summer), Siberian, and European (Lindquist and Vapalahti 2008). The geographical ranges of the tick-borne encephalitis viruses extend from western Europe to the east coast of Japan, corresponding to those of the tick hosts (Fig. 2d) (Burke and Monath 2001; Calisher et al. 1989; King et al. 2012; Grard et al. 2006; Lindenbach and Rice 2001; Gresikova and Calisher 1989; Tsai and Mitchell 1989; Reid 1988).
Tick-borne encephalitis virus is transmitted to humans through the bite of an infected tick of the Ixodes spp., primarily I. ricinus for the European subtype and I. persulcatus for the Siberian and Far Eastern subtypes. In addition to being vectors of the virus, and because of their longer life span compared to mosquitoes, ticks are also the main virus reservoir. Small rodents are the primary vertebrate amplifying, reservoir, and overwintering hosts (see “Influences of Arthropod Vectors on Encephalitic Arboviruses”) (Lindquist and Vapalahti 2008). Although not amplifying hosts, cattle, sheep, and goats infected with tick-borne encephalitis viruses may excrete virus in their milk, so that humans can become infected by ingesting raw milk or cheese (Gresikova and Calisher 1989). Transmission has been infrequently reported during slaughtering of viremic animals, and directly person-to-person, through blood transfusion and breastfeeding.
The risk of acquiring tick-borne encephalitis in forested areas through activities such as camping, hiking, or military training peaks in early and late summer when ticks are active; however, cases have been reported during the hot summer months despite lower tick activity, as people increasingly come into contact with ticks during outdoor activities. The risk is negligible for people who remain in urban or unforested areas (Lindquist and Vapalahti 2008).
Tick-borne encephalitis viruses are the most medically important arbovirus in Europe, with more than 10,000 cases of tick-borne encephalitis occurring annually, 3000 of which require hospital treatment (Lindquist and Vapalahti 2008). Western Siberia has the largest annual number of reported cases, but the endemic area extends throughout Eurasia (Fig. 2d) (Palo 2014; Yun et al. 2011). During the past two decades new endemic foci and increased cases have been reported in Europe, and because of its emergence, tick-borne encephalitis needs to be considered outside the traditional endemic areas (Lindquist and Vapalahti 2008).
Clinical disease is biphasic and disease severity varies between the three tick-borne encephalitis virus subtypes (Burke and Monath 2001; Calisher et al. 1989; King et al. 2012; Grard et al. 2006; Lindenbach and Rice 2001; Gresikova and Calisher 1989; Tsai and Mitchell 1989; Reid 1988; Belikov et al. 2014). The European subtype is associated with milder disease. After an incubation period of 3–7 days, in the first phase, patients may experience an influenza-like illness for approximately 1 week, with fever, headache, malaise, and myalgia (Gresikova and Calisher 1989; Leonova et al. 2014; Lindquist and Vapalahti 2008). Following an asymptomatic period of up to a week, 20–30 % experience a second phase involving the central nervous system, with the clinical symptoms ranging from mild meningitis to severe encephalitis, with or without myelitis and spinal paralysis (Burke and Monath 2001; Gresikova and Calisher 1989). The reported case fatality rate ranges from 1–5 %, and 10–20 % of survivors, generally those with the more severe clinical symptoms, have long-term neurological problems (Lindquist and Vapalahti 2008).
The more severe course of disease results from infection by the Far-Eastern and Siberian subtypes (Gresikova and Calisher 1989; Belikov et al. 2014). The prodromal phase may consist of symptoms similar to those of the Japanese encephalitis complex virus infections: fever, headache, anorexia, nausea, vomiting, and photophobia. In the second phase, infection of the brain stem and upper cervical cord produces stiff neck, ataxia, sensorial changes, convulsions, and in about 20 % of cases, flaccid paralysis (Lindquist and Vapalahti 2008). The case fatality rate is approximately 20 %; 30–60 % of survivors experience residual neuronal damage (Gresikova and Calisher 1989; Kaiser 2002).
Tick-borne encephalitis vaccines are commercially available in Europe, and routine vaccination is recommended for children in many European countries (Gresikova and Calisher 1989; Smit and Postma 2014). Vaccinations may be required for travelers to endemic areas in eastern Russia, where the seroprevalence rate may be as high as 51 % (Wittermann et al. 2015). No tick-borne encephalitis vaccines are licensed or available in the United States .
Powassan Virus
Powassan virus was first isolated in Ontario, Canada from a pediatric case of encephalitis in 1958, retrospectively from ticks collected in Colorado in 1952, and in Russia from ticks in 1996. Powassan virus, the only member of the tick-borne encephalitis antigenic complex found in North America, is widely distributed in temperate regions in the northern hemisphere (Fig. 2d) (Burke and Monath 2001; Gubler et al. 2007; Artsob 1989). North American Powassan cases are concentrated in New York, Ontario, and Quebec (Centers for Disease Control and Prevention 2001). However, Powassan virus infection has been diagnosed with increasing frequency both within the known range and in areas where Powassan cases had not been previously reported (Ebel 2010). Whether this is due to heightened awareness and increased surveillance for arboviruses following the introduction of West Nile virus into North America, or because Powassan is an emerging virus is unknown.
Powassan virus comprises two closely related lineages: the Powassan virus prototype lineage and the deer tick virus lineage (Ebel 2010). The prototype Powassan virus is principally maintained between Ixodes cookei ticks and the groundhog (Marmota monax) or striped skunk (Mephitis mephitis) ; the deer tick virus is believed to be maintained between Ixodes scapularis ticks and the white-footed mouse (Peromyscus leucopus) (Ei Khoury et al. 2013). Both lineages have been linked to human disease, although bites to humans by I. cookei ticks are rare, whereas I. scapularis tick bites are common (Ebel 2010).
In the United States, disease caused by Powassan virus has occurred sporadically, primarily in the late spring, early summer, and mid-fall when ticks are most active. Approximately 80 Powassan virus infections have been reported since the 1950s, with over 80 % causing neuroinvasive disease. The case fatality rate is approximately 10 % (Ebel 2010). As with other flaviviruses, the majority of Powassan infections are asymptomatic (Gubler et al. 2007). Clinical symptoms can include fever, headache, vomiting, weakness, confusion, loss of coordination, speech difficulties, seizures, and memory loss. Powassan virus can infect the central nervous system and cause encephalitis and meningitis. At least half of survivors have long-lasting sequelae, such as recurrent headaches, muscle wasting, and memory problems .
Other Flaviviruses Causing Encephalitis
Other flaviviruses generally associated with enzootic transmission may cause sporadic encephalitis cases, or may be emerging. The etiological agent may be difficult to identify by serological assays in regions where there are multiple flaviviruses circulating, due to the cross-reactivity in serological assays, and the infrequency of obtaining a virus isolate. These viruses may emerge or reemerge as significant human pathogens as deforestation and changing agricultural practices bring humans into areas of enzootic transmission cycles.
Rocio virus : Rocio virus was first isolated from a fatal human case during an epidemic of encephalitis in southeastern Brazil in 1975 (Iversson 1989). Between 1975 and 1976, there were over 1000 cases reported, with a 10 % fatality rate, and neurological sequelae were observed in 20 % of survivors. Male adults working in or near the forests were shown to have the highest risk of infection (Iversson 1989).
The transmission cycle of Rocio virus has not been clearly established, although wild birds are believed to be the primary vertebrate host, similar to the other mosquito-borne neurotropic flaviviruses. The virus replicates in Culex mosquitoes in the laboratory; however in nature the Rocio virus has been most often isolated from Psorophora mosquitoes (Iversson 1989).
Since 1976, despite the continuation of sporadic cases of encephalitis in southeastern Brazil and serological evidence of Rocio virus infections in horses and humans, only one case of human Rocio virus infection has been identified (Gubler et al. 2007; Silva et al. 2014). This could be due to the difficulty of differentiating by clinical symptoms or serology alone those encephalitis cases caused by Rocio virus from those caused by the closely related St. Louis encephalitis virus , which co-circulates in the same geographical area (Fig. 1) (Burke and Monath 2001; Figueiredo 2000; Medeiros et al. 2007). Likely Rocio virus infections are an under-recognized cause of neuroinvasive illness in the Americas.
Louping ill virus : In Scotland, louping ill neurological disease has been recognized in sheep since the 1700s, and the virus was isolated there in 1929 (Jeffries et al. 2014). The first probable human case was reported in 1934 (Gubler et al. 2007).
Louping ill virus is genetically most closely related to the European subtype of tick-borne encephalitis virus, and similarly is transmitted by Ixodes spp. (Fig. 1) (King et al. 2012; Moureau et al. 2015). The geographical range of louping ill virus is in upland grazing areas throughout the United Kingdom, Ireland, Norway, Spain, Greece, and Turkey (Fig. 2d). However, recently the virus has been detected in Scandinavia, and Negishi virus, classified as a genotype of Louping ill virus, was isolated from a human in Japan (Gubler et al. 2007). The natural vertebrate hosts of louping ill virus are speculated to be rodents, deer, and hares, but sheep and cattle are the most important enzootic hosts from an agricultural perspective (Jeffries et al. 2014). Louping ill virus causes neurological disease in sheep, and to a lesser degree, cattle, yet interestingly, the other tick-borne encephalitis viruses do not (Jeffries et al. 2014). Most human infections have occurred through occupational exposure to infected ticks on livestock, such as stockmen, abattoir workers, butchers, and veterinarians (Lindquist and Vapalahti 2008).
There have been 44 reports of human disease caused by louping ill virus, with one fatal case. No human encephalitis cases of been identified since 1991, possibly due to lack of awareness among clinicians for this “forgotten” disease and subsequent lack of specific testing (Jeffries et al. 2014). Serosurveys suggest that at-risk groups are exposed to louping ill virus, but that most infections are asymptomatic or result in an influenza-like illness. Clinical disease is characterized by fever, headache, and some muscle stiffness, which may be followed by more severe neurological signs. Four cases have presented as poliomyelitis-like disease (Jeffries et al. 2014). Vaccination successfully protects livestock but does not eliminate persistence in ticks or virus transmission in wildlife hosts.
Modoc virus : Modoc virus was first isolated from a deer mouse in Modoc County, California in 1958, and subsequently in other regions in western United States and Canada (Burke and Monath 2001). Modoc virus was reported as the etiological agent in one human aseptic meningitis case (Gubler et al. 2007). Rodents are the primary host and the virus is hypothesized to be maintained in nature by virus shedding in persistently infected rodents and through horizontal transmission (Adams et al. 2013). No arthropod vector has been identified (Fig. 1).
References
Adams AP, Travassos da Rosa AP, Nunes MR, Xiao SY, Tesh RB (2013) Pathogenesis of Modoc virus (Flaviviridae; Flavivirus) in persistently infected hamsters. Am J Trop Med Hyg 88:455–460
Artsob H (1989) Powassan virus. In: Monath TP (ed) The arboviruses: epidemiology and ecology, vol IV. CRC Press, Boca Raton, pp 29–49
Belikov SI, Kondratov IG, Potapova UV, Leonova GN (2014) The relationship between the structure of the tick-borne encephalitis virus strains and their pathogenic properties. PLoS One 9, e94946
Brault AC, Langevin SA, Bowen RA, Panella NA, Biggerstaff BJ, Miller BR, Komar N (2004) Differential virulence of West Nile strains for American crows. Emerg Infect Dis 10:2161–2168
Brinker KR, Paulson G, Monath TP, Wise G, Fass RJ (1979) St Louis encephalitis in Ohio, September 1975: clinical and EEG studies in 16 cases. Arch Intern Med 139:561–566
Burke D, Monath TP (2001) Flaviviruses. In: Knipe D, Howley P (eds) Fields virology, vol 1, 4th edn. Lippincott Williams and Wilkins, Philadelphia, pp 1043–1125
Burke DS, Nisalak A, Ussery MA (1982) Antibody capture immunoassay detection of japanese encephalitis virus immunoglobulin m and g antibodies in cerebrospinal fluid. J Clin Microbiol 16:1034–1042
Burke DS, Nisalak A, Ussery MA, Laorakpongse T, Chantavibul S (1985) Kinetics of IgM and IgG responses to Japanese encephalitis virus in human serum and cerebrospinal fluid. J Infect Dis 151:1093–1099
Calisher C, Karabatsos N (1988) Arbovirus serogroups: definition and geographic distribution. In: Monath TP (ed) The arboviruses: epidemiology and ecology, vol I. CRC Press, Boca Raton, pp 19–57
Calisher CH, Karabatsos N, Dalrymple JM, Shope RE, Porterfield JS, Westaway EG, Brandt WE (1989) Antigenic relationships between flaviviruses as determined by cross-neutralization tests with polyclonal antisera. J Gen Virol 70(Pt 1):37–43
Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ (2002) West Nile virus. Lancet Infect Dis 2:519–529
Campbell GL, Hills SL, Fischer M, Jacobson JA, Hoke CH, Hombach JM, Marfin AA, Solomon T, Tsai TF, Tsu VD, Ginsburg AS (2011) Estimated global incidence of Japanese encephalitis: a systematic review. Bull World Health Organ 89(766–774):774A–774E
Centers for Disease Control and Prevention (2009) West Nile virus transmission via organ transplantation and blood transfusion—Louisiana, 2008. MMWR Morb Mortal Wkly Rep 58:1263–1267
Centers for Disease Control and Prevention (2003) Update: detection of West Nile virus in blood donations—United States, 2003. [erratum appears in MMWR Morb Mortal Wkly Rep. 2003 Oct 3;52(39):942]. MMWR Morb Mortal Wkly Rep 52:916–919
Centers for Disease Control and Prevention (2004) Update: West Nile virus screening of blood donations and transfusion-associated transmission—United States, 2003. MMWR Morb Mortal Wkly Rep 53:281–284
Centers for Disease Control and Prevention (2001) Outbreak of Powassan encephalitis—Maine and Vermont, 1999–2001. MMWR Morb Mortal Wkly Rep 50:761–764
Chambers TJ, Hahn CS, Galler R, Rice CM (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688
Chanama S, Sukprasert W, Sa-ngasang A, An A, Sangkitporn S, Kurane I, Anantapreecha S (2005) Detection of Japanese encephalitis (JE) virus-specific IgM in cerebrospinal fluid and serum samples from JE patients. Jpn J Infect Dis 58:294–296
Cushing MM, Brat DJ, Mosunjac MI, Hennigar RA, Jernigan DB, Lanciotti R, Petersen LR, Goldsmith C, Rollin PE, Shieh WJ, Guarner J, Zaki SR (2004) Fatal West Nile virus encephalitis in a renal transplant recipient. Am J Clin Pathol 121:26–31
Davis BS, Chang GJ, Cropp B, Roehrig JT, Martin DA, Mitchell CJ, Bowen R, Bunning ML (2001) West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J Virol 75:4040–4047
Donadieu E, Bahuon C, Lowenski S, Zientara S, Coulpier M, Lecollinet S (2013) Differential virulence and pathogenesis of West Nile viruses. Viruses 5:2856–2880
Douglas MW, Stephens DP, Burrow JN, Anstey NM, Talbot K, Currie BJ (2006) Murray Valley encephalitis in an adult traveller complicated by long-term flaccid paralysis: case report and review of the literature. Trans R Soc Trop Med Hyg 101(3):284–288
Ebel GD (2010) Update on Powassan virus: emergence of a North American tick-borne flavivirus. Annu Rev Entomol 55:95–110
Ei Khoury MY, Camargo JF, Wormser GP (2013) Changing epidemiology of Powassan encephalitis in North America suggests the emergence of the deer tick virus subtype. Expert Rev Anti Infect Ther 11:983–985
Figueiredo LT (2000) The Brazilian flaviviruses. Microbes Infect 2:1643–1649
Foy BD, Kobylinski KC, Chilson Foy JL, Blitvich BJ, Travassos da Rosa A, Haddow AD, Lanciotti RS, Tesh RB (2011) Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis 17:880–882
Gonzalez-Reiche AS, Monzon-Pineda Mde L, Johnson BW, Morales-Betoulle ME (2010) Detection of West Nile viral RNA from field-collected mosquitoes in tropical regions by conventional and real-time RT-PCR. Methods Mol Biol 630:109–124
Gould EA, Solomon T (2008) Pathogenic flaviviruses. Lancet 371:500–509
Grard G, Moureau G, Charrel RN, Lemasson JJ, Gonzalez JP, Gallian P, Gritsun TS, Holmes EC, Gould EA, de Lamballerie X (2006) Genetic characterization of tick-borne flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. Virology 361(1):80–92
Gresikova M, Calisher C (1989) Tick-borne encephalitis. In: Monath T (ed) The arboviruses: epidemiology and ecology, vol IV. CRC Press, Boca Raton, pp 177–202
Gritsun TS, Holmes EC, Gould EA (1995) Analysis of flavivirus envelope proteins reveals variable domains that reflect their antigenicity and may determine their pathogenesis. Virus Res 35:307–321
Gritsun TS, Lashkevich VA, Gould EA (2003) Tick-borne encephalitis. Antiviral Res 57:129–146
Gubler DJ (1998a) Dengue and dengue hemorrhagic fever. Clin Microbiol Rev 11:480–496
Gubler DJ (1998b) The global pandemic of dengue/dengue haemorrhagic fever: current status and prospects for the future. Ann Acad Med Singapore 27:227–234
Gubler D, Kuno G, Markoff L (2007) Flaviviruses, 5th edn. Lippincott Williams and Wilkins, Philadelphia
Guirakhoo F, Bolin RA, Roehrig JT (1992) The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology 191:921–931
Halstead SB, Jacobson J (2003) Japanese encephalitis. Adv Virus Res 61:103–138
Hayes E, Gubler D (2005) West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med 57:181–194
Hayes EB, Sejvar JJ, Zaki SR, Lanciotti RS, Bode AV, Campbell GL (2005) Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis 11:1174–1179
Heinz FX, Roehrig JT (1990) Flaviviruses. Elsevier Science Publishing BV, Amsterdam
Hills S, Martin R, Marfin A, Fischer M (2014) Control of Japanese encephalitis in Asia: the time is now. Expert Rev Anti Infect Ther 12:901–904
Hogrefe WR, Moore R, Lape-Nixon M, Wagner M, Prince HE (2004) Performance of immunoglobulin G (IgG) and IgM enzyme-linked immunosorbent assays using a West Nile virus recombinant antigen (preM/E) for detection of West Nile virus- and other flavivirus-specific antibodies. J Clin Microbiol 42:4641–4648
Iversson LB (1989) Rocio encephalitis. In: Monath TP (ed) The arboviruses: epidemiology and ecology, vol IV. CRC Press, Boca Raton, pp 77–92
Iwamoto M, Jernigan DB, Guasch A, Trepka MJ, Blackmore CG, Hellinger WC, Pham SM, Zaki S, Lanciotti RS, Lance-Parker SE, DiazGranados CA, Winquist AG, Perlino CA, Wiersma S, Hillyer KL, Goodman JL, Marfin AA, Chamberland ME, Petersen LR (2003) Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med 348:2196–2203
Jeffries CL, Mansfield KL, Phipps LP, Wakeley PR, Mearns R, Schock A, Bell S, Breed AC, Fooks AR, Johnson N (2014) Louping ill virus: an endemic tick-borne disease of Great Britain. J Gen Virol 95:1005–1014
Johnson AJ, Martin DA, Karabatsos N, Roehrig JT (2000) Detection of anti-arboviral immunoglobulin G by using a monoclonal antibody-based capture enzyme-linked immunosorbent assay. J Clin Microbiol 38:1827–1831
Johnson AJ, Noga AJ, Kosoy O, Lanciotti RS, Johnson AA, Biggerstaff BJ (2005a) Duplex microsphere-based immunoassay for detection of anti-West Nile virus and anti-St. Louis encephalitis virus immunoglobulin m antibodies. Clin DiagnLab Immunol 12:566–574
Johnson BW, Kosoy O, Martin DA, Noga AJ, Russell BJ, Johnson AA, Petersen LR (2005b) West Nile virus infection and serologic response among persons previously vaccinated against yellow fever and Japanese encephalitis viruses. Vector Borne Zoonotic Dis 5:137–145
Johnston LLJ, Halliday GGM, King NNJ (2000) Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J Invest Dermatol 114:560–568
Kaiser R (2002) Tick-borne encephalitis (TBE) in Germany and clinical course of the disease. Int J Med Microbiol 291(Suppl 33):58–61
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (2012). Virus taxonomy: classification and nomenclature of viruses. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego.
Komar N, Clark GG (2006) West Nile virus activity in Latin America and the Caribbean. Rev Panam Salud Publica 19:112–117
Komar O, Robbins MB, Contreras GG, Benz BW, Klenk K, Blitvich BJ, Marlenee NL, Burkhalter KL, Beckett S, Gonzalvez G, Pena CJ, Peterson AT, Komar N (2005) West Nile virus survey of birds and mosquitoes in the Dominican Republic. Vector Borne Zoonotic Dis 5:120–126
Koschinski A, Wengler G, Wengler G, Repp H (2003) The membrane proteins of flaviviruses form ion-permeable pores in the target membrane after fusion: identification of the pores and analysis of their possible role in virus infection. J Gen Virol 84:1711–1721
Kramer LD (2007) West Nile virus. Lancet Neurol 6:171–181
Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH (2002) Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725
Kuno G, Chang GJ (2005) Biological transmission of arboviruses: reexamination of and new insights into components, mechanisms, and unique traits as well as their evolutionary trends. Clin Microbiol Rev 18:608–637
Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB (1998) Phylogeny of the genus Flavivirus. J Virol 72:73–83
Lanciotti RS, Kerst AJ (2001) Nucleic acid sequence-based amplification assays for rapid detection of West Nile and St. Louis encephalitis viruses. J Clin Microbiol 39:4506–4513
Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Crise B, Volpe KE, Crabtree MB, Scherret JH, Hall RA, MacKenzie JS, Cropp CB, Panigrahy B, Ostlund E, Schmitt B, Malkinson M, Banet C, Weissman J, Komar N, Savage HM, Stone W, McNamara T, Gubler DJ (1999) Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286:2333–2337
Lanciotti RS, Kerst AJ, Nasci RS, Godsey MS, Mitchell CJ, Savage HM, Komar N, Panella NA, Allen BC, Volpe KE, Davis BS, Roehrig JT (2000) Rapid detection of west nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J Clin Microbiol 38:4066–4071
Lanciotti RS, Ebel GD, Deubel V, Kerst AJ, Murri S, Meyer R, Bowen M, McKinney N, Morrill WE, Crabtree MB, Kramer LD, Roehrig JT (2002) Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 298:96–105
Leonova GN, Maystrovskaya OS, Kondratov IG, Takashima I, Belikov SI (2014) The nature of replication of tick-borne encephalitis virus strains isolated from residents of the Russian Far East with inapparent and clinical forms of infection. Virus Res 189:34–42
Libraty DH, Nisalak A, Endy TP, Suntayakorn S, Vaughn DW, Innis BL (2002) Clinical and immunological risk factors for severe disease in Japanese encephalitis. Trans R Soc Trop Med Hyg 96:173–178
Lindenbach B, Rice C (2001) Flaviviridae: the viruses and their replication. In: Knipe D, Howley P (eds) Fields virology, vol 1, 4th edn. Lippincott William and Wilkins, Philadelphia, pp 991–1043
Lindenbach B, Heinz-Jurgen T, Rice C (2007) Flaviviridae: the viruses and their replication. In: Knipe D, Howley P (eds) Fields virology, vol 1, 5th edn. Lippincott William and Wilkins, Philadelphia, pp 1101–1152
Lindquist L (2008) Tick-borne encephalitis (TBE) in childhood. Acta Paediatr 97:532–534
Lindquist L, Vapalahti O (2008) Tick-borne encephalitis. Lancet 371:1861–1871
Lindsey NP, Sejvar JJ, Bode AV, Pape WJ, Campbell GL (2012) Delayed mortality in a cohort of persons hospitalized with West Nile virus disease in Colorado in 2003. Vector Borne Zoonotic Dis 12:230–235
Logar M, Arnez M, Kolbl J, Avsic-Zupanc T, Strle F (2000) Comparison of the epidemiological and clinical features of tick-borne encephalitis in children and adults. Infection 28:74–77
Mackenzie JS, Field HE (2004) Emerging encephalitogenic viruses: lyssaviruses and henipaviruses transmitted by frugivorous bats. Arch Virol Suppl 18:97–111
Mackenzie J, Poidninger M, Lindsay M, Hall R, Sammels L (1996) Molecular epidemiology and evolution of mosquito-borne flaviviruses and alphaviruses enzootic in Australia. Virus Genes 11:225–237
Marfin AA, Eidex RS, Kozarsky PE, Cetron MS (2005) Yellow fever and Japanese encephalitis vaccines: indications and complications. Infect Dis Clin North Am 19(151–168):ix
Marshall I (1988) Murray Valley and Kunjin encephalitis. In: Monath TP (ed) The arboviruses: epidemiology and ecology, vol III. CRC Press, Boca Raton, pp 151–189
Martin DA, Muth DA, Brown T, Johnson AJ, Karabatsos N, Roehrig JT (2000) Standardization of immunoglobulin M capture enzyme-linked immunosorbent assays for routine diagnosis of arboviral infections. J Clin Microbiol 38:1823–1826
Martin DA, Biggerstaff BJ, Allen B, Johnson AJ, Lanciotti RS, Roehrig JT (2002) Use of immunoglobulin M cross-reactions in differential diagnosis of human flaviviral encephalitis infections in the United States. Clin Diagn Lab Immunol 9:544–549
Mattar S, Edwards E, Laguado J, Gonzalez M, Alvarez J, Komar N (2005) West Nile virus antibodies in Colombian horses. Emerg Infect Dis 11:1497–1498
McMinn PC (1997) The molecular basis of virulence of the encephalitogenic flaviviruses. J Gen Virol 78(Pt 11):2711–2722
Medeiros DB, Nunes MR, Vasconcelos PF, Chang GJ, Kuno G (2007) Complete genome characterization of Rocio virus (Flavivirus: Flaviviridae), a Brazilian flavivirus isolated from a fatal case of encephalitis during an epidemic in Sao Paulo state. J Gen Virol 88:2237–2246
Monath TP (1986) Pathobiology of the flaviviruses. Plenum, New York
Monath TP (1989) Yellow fever. In: Monath TP (ed) The arboviruses:epidemiology and ecology, vol 5. CRC Press, Boca Raton, pp 139–231
Monath T (1999) Yellow fever. In: Plotkin S, Orenstein W (eds) Vaccines, 3rd edn. WB Saunders, Philadelphia
Monath TP (2001) Prospects for development of a vaccine against the West Nile virus. Ann N Y Acad Sci 951:1–12
Monath TP (2002) Japanese encephalitis vaccines: current vaccines and future prospects. Curr Top Microbiol Immunol 267:105–138
Montgomery SP, Brown JA, Kuehnert M, Smith TL, Crall N, Lanciotti RS, Macedo de Oliveira A, Boo T, Marfin AA (2006) Transfusion-associated transmission of West Nile virus, United States 2003 through 2005. Transfusion 46:2038–2046
Morales MA, Barrandeguy M, Fabbri C, Garcia JB, Vissani A, Trono K, Gutierrez G, Pigretti S, Menchaca H, Garrido N, Taylor N, Fernandez F, Levis S, Enria D (2006) West Nile virus isolation from equines in Argentina, 2006. Emerg Infect Dis 12:1559–1561
Morales-Betoulle ME, Komar N, Panella NA, Alvarez D, Lopez MR, Betoulle JL, Sosa SM, Muller ML, Kilpatrick AM, Lanciotti RS, Johnson BW, Powers AM, Cordon-Rosales C, Arbovirus Ecology Work G (2013) West Nile virus ecology in a tropical ecosystem in Guatemala. Am J Trop Med Hyg 88:116–126
Moureau G, Cook S, Lemey P, Nougairede A, Forrester NL, Khasnatinov M, Charrel RN, Firth AE, Gould EA, de Lamballerie X (2015) New insights into flavivirus evolution, taxonomy and biogeographic history, extended by analysis of canonical and alternative coding sequences. PLoS One 10, e0117849
Mukhopadhyay S, Kim BS, Chipman PR, Rossmann MG, Kuhn RJ (2003) Structure of West Nile virus. Science 302:248
Murgod UA, Muthane UB, Ravi V, Radhesh S, Desai A (2001) Persistent movement disorders following Japanese encephalitis. Neurology 57:2313–2315
Musso D, Roche C, Robin E, Nhan T, Teissier A, Cao-Lormeau VM (2015) Potential sexual transmission of Zika virus. Emerg Infect Dis 21:359–361
Nasci RS, Komar N, Marfin AA, Ludwig GV, Kramer LD, Daniels TJ, Falco RC, Campbell SR, Brookes K, Gottfried KL, Burkhalter KL, Aspen SE, Kerst AJ, Lanciotti RS, Moore CG (2002) Detection of West Nile virus-infected mosquitoes and seropositive juvenile birds in the vicinity of virus-positive dead birds. Am J Trop Med Hyg 67:492–496
Niedrig M, Sonnenberg K, Steinhagen K, Paweska JT (2007) Comparison of ELISA and immunoassays for measurement of IgG and IgM antibody to West Nile virus in human sera against virus neutralisation. J Virol Methods 139:103–105
O’Leary DR, Kuhn S, Kniss KL, Hinckley AF, Rasmussen SA, Pape WJ, Kightlinger LK, Beecham BD, Miller TK, Neitzel DF, Michaels SR, Campbell GL, Lanciotti RS, Hayes EB (2006) Birth outcomes following West Nile Virus infection of pregnant women in the United States: 2003–2004. Pediatrics 117:e537–e545
Palo RT (2014) Tick-borne encephalitis transmission risk: its dependence on host population dynamics and climate effects. Vector Borne Zoonotic Dis 14:346–352
Petersen LR, Marfin AA (2002) West Nile virus: a primer for the clinician. Ann Intern Med 137:173–179
Petersen LRLR, Roehrig JTJT, Hughes JMJM (2002) West Nile virus encephalitis. N Engl J Med 347:1225–1226
Petersen LR, Marfin AA, Gubler DJ (2003) West Nile virus. JAMA 290:524–528
Porterfield JS (1986) Antibody-dependent enhancement of viral infectivity. Adv Virus Res 31:335–355
Reid H (1988) Louping-ill. In: Monath TP (ed) The arboviruses: epidemiology and ecology, vol III. CRC Press, Boca Raton, pp 117–135
Rocco IM, Santos CL, Bisordi I, Petrella SM, Pereira LE, Souza RP, Coimbra TL, Bessa TA, Oshiro FM, Lima LB, Cerroni MP, Marti AT, Barbosa VM, Katz G, Suzuki A (2005) St. Louis encephalitis virus: first isolation from a human in Sao Paulo State, Brazil. Rev Inst Med Trop Sao Paulo 47:281–285
Roehrig JT (2003) Antigenic structure of flavivirus proteins. Adv Virus Res 59:141–175
Roehrig JT, Nash D, Maldin B, Labowitz A, Martin DA, Lanciotti RS, Campbell GL (2003) Persistence of virus-reactive serum immunoglobulin m antibody in confirmed west nile virus encephalitis cases. Emerg Infect Dis 9:376–379
Rossini G, Cavrini F, Pierro A, Macini P, Finarelli A, Po C, Peroni G, Di Caro A, Capobianchi M, Nicoletti L, Landini M, Sambri V (2008) First human case of West Nile virus neuroinvasive infection in Italy, September 2008—case report. Euro Surveill. 13(41)
Sambri V, Capobianchi M, Charrel R, Fyodorova M, Gaibani P, Gould E, Niedrig M, Papa A, Pierro A, Rossini G, Varani S, Vocale C, Landini MP (2013) West Nile virus in Europe: emergence, epidemiology, diagnosis, treatment, and prevention. Clin Microbiol Infect 19:699–704
Sejvar JJ (2014) Clinical manifestations and outcomes of West Nile virus infection. Viruses 6:606–623
Sejvar JJ, Marfin AA (2006) Manifestations of West Nile neuroinvasive disease. Rev Med Virol 16:209–224
Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA, Fleischauer A, Leis AA, Stokic DS, Petersen LR (2003a) Neurologic manifestations and outcome of West Nile virus infection. Jama 290:511–515
Sejvar JJ, Leis AA, Stokic DS, Van Gerpen JA, Marfin AA, Webb R, Haddad MB, Tierney BC, Slavinski SA, Polk JL, Dostrow V, Winkelmann M, Petersen LR (2003b) Acute flaccid paralysis and West Nile virus infection. Emerg Infect Dis 9:788–793
Sejvar JJ, Davis LE, Szabados E, Jackson AC (2010) Delayed-onset and recurrent limb weakness associated with West Nile virus infection. J Neurovirol 16:93–100
Seligman SJ, Bucher DJ (2003) The importance of being outer: consequences of the distinction between the outer and inner surfaces of flavivirus glycoprotein E. Trends Microbiol 11:108–110
Selvey LA, Dailey L, Lindsay M, Armstrong P, Tobin S, Koehler AP, Markey PG, Smith DW (2014) The changing epidemiology of Murray Valley encephalitis in Australia: the 2011 outbreak and a review of the literature. PLoS Negl Trop Dis 8:e2656
Silva JR, Romeiro MF, Souza WM, Munhoz TD, Borges GP, Soares OA, Campos CH, Machado RZ, Silva ML, Faria JL, Chavez JH, Figueiredo LT (2014) A Saint Louis encephalitis and Rocio virus serosurvey in Brazilian horses. Rev Soc Bras Med Trop 47:414–417
Smit R, Postma MJ (2014) Review of tick-borne encephalitis and vaccines: clinical and economical aspects. Expert Rev Vaccines 14(5):737–747
Solomon T (2003) Recent advances in Japanese encephalitis. J Neurovirol 9:274–283
Solomon T (2004) Flavivirus encephalitis. N Engl J Med 351:370–378
Solomon T (2006) Control of Japanese encephalitis—within our grasp? N Engl J Med 355:869–871
Solomon T, Winter PM (2004) Neurovirulence and host factors in flavivirus encephalitis—evidence from clinical epidemiology. Arch Virol Suppl 18:161–170
Solomon T, Dung NM, Kneen R, Gainsborough M, Vaughn DW, Khanh VT (2000) Japanese encephalitis. J Neurol Neurosurg Psychiatry 68:405–415
Spinsanti LI, Diaz LA, Glatstein N, Arselan S, Morales MA, Farias AA, Fabbri C, Aguilar JJ, Re V, Frias M, Almiron WR, Hunsperger E, Siirin M, Da Rosa AT, Tesh RB, Enria D, Contigiani M (2008) Human outbreak of St. Louis encephalitis detected in Argentina, 2005. J Clin Virol 42:27–33
Staples JE, Shankar MB, Sejvar JJ, Meltzer MI, Fischer M (2014) Initial and long-term costs of patients hospitalized with West Nile virus disease. Am J Trop Med Hyg 90:402–409
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tesh RB, Travassos da Rosa AP, Guzman H, Araujo TP, Xiao SY (2002) Immunization with heterologous flaviviruses protective against fatal West Nile encephalitis. Emerg Infect Dis 8:245–251
Tilley PA, Fox JD, Jayaraman GC, Preiksaitis JK (2006) Nucleic acid testing for west nile virus RNA in plasma enhances rapid diagnosis of acute infection in symptomatic patients. J Infect Dis 193:1361–1364
Tsai T, Mitchell C (1989) St. Louis encephalitis. In: Monath TP (ed) The arboviruses: epidemiology and ecology, vol IV. CRC Press, Boca Raton, pp 113–143
Wittermann C, Izu A, Petri E, Gniel D, Fragapane E (2015) Five year follow-up after primary vaccination against tick-borne encephalitis in children. Vaccine 33(15):1824–1829
Wong SJ, Boyle RH, Demarest VL, Woodmansee AN, Kramer LD, Li H, Drebot M, Koski RA, Fikrig E, Martin DA, Shi PY (2003) Immunoassay targeting nonstructural protein 5 to differentiate West Nile virus infection from dengue and St. Louis encephalitis virus infections and from flavivirus vaccination. J Clin Microbiol 41:4217–4223
Wong SJ, Demarest VL, Boyle RH, Wang T, Ledizet M, Kar K, Kramer LD, Fikrig E, Koski RA (2004) Detection of human anti-flavivirus antibodies with a west nile virus recombinant antigen microsphere immunoassay. J Clin Microbiol 42:65–72
Yun SM, Kim SY, Ju YR, Han MG, Jeong YE, Ryou J (2011) First complete genomic characterization of two tick-borne encephalitis virus isolates obtained from wild rodents in South Korea. Virus Genes 42:307–316
Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Strauss JH, Kuhn RJ, Rossmann MG (2003) Structures of immature flavivirus particles. Embo J 22:2604–2613
Acknowledgments
I would like to thank Jennifer Leyman of the CDC Division of Vector-borne Diseases for providing the flavivirus distribution maps; Cynthia Goldsmith, CDC, for providing the West Nile electron micrograph; Sherif Zaki, CDC, for providing the immunohistochemical staining photomicrograph; and Mary Crabtree for construction of the flavivirus phylogenetic tree.
Note: The findings and conclusions in this chapter are those of the author and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Johnson, B.W. (2016). Neurotropic Flaviviruses. In: Reiss, C. (eds) Neurotropic Viral Infections. Springer, Cham. https://doi.org/10.1007/978-3-319-33133-1_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-33133-1_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33131-7
Online ISBN: 978-3-319-33133-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)