
Abstract
It took more than 23 years to propose a defined medium to culture “Pelagibacter ubique” HTCC1062, one of the most dominant clades in the ocean. Although it was first identified in the 1990s by culture-independent approaches based on rRNA gene cloning and sequencing, an artificial seawater enrichment medium has only recently been proposed for this isolate. This success story is a result of the improvement of culture methods, better sensitivity of growth detection, and knowledge of metabolic activities predicted from genome sequences. The new approaches now offer a fraction of 14–40 % that can be cultured. From an optimistic point of view, all uncultured marine microorganisms could now simply be regarded as “not yet cultured”. Culturing is no longer an “old fashioned” technique but an innovative and fast-moving area of research. Technological developments include micro-engineering of ichips, manipulation of single cells, community culture, high-throughput culturing (HTC) processes, and new methods for low biomass detection or targeting specific microorganisms. Culture remains a prerequisite for microbiological studies, as we need to grow microorganisms in the laboratory in order to identify their functions and validate hypotheses deduced from their genomes. The development, improvement, and combination of innovative culture techniques based on information deduced from omics will undoubtedly lead to the isolation and study of presently uncultured marine microorganisms.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Microorganisms populate all marine habitats with an estimated abundance of 104 to 106 cells/ml of seawater. Marine microorganisms play key roles in biogeochemical cycling processes including carbon and nutrient cycling. During the past three decades, the revolution in molecular biology, combined with technological advances and a decrease in sequencing costs, has allowed genomic studies to be performed on ocean microbial communities at a worldwide scale (Kopf et al. 2015; Rusch et al. 2007; Venter et al. 2004). The 16S rRNA gene sequence-based approaches and environmental genomics have revealed the vast diversity of the microbial world. Most of the organisms detected by these approaches had never been described before, even after 120 years of culturing microorganisms. These organisms are known as “not culturable” and represent the dark matter of the microbial domain. More than 99 % of microbial diversity is not yet culturable. The establishment of pure cultures of representatives of all bacterial and archaeal divisions is still a major challenge for microbiologists today. Pure cultures continue to be essential for a true understanding of the physiology of these Bacteria and Archaea and their roles in the environment, and to enable the discovery of new products with biotechnological potentials such as new antibiotics.
During the last two decades, considerable advances have been made in culture approaches making it possible to grow some important “key-player” microorganisms in the laboratory. These advances were not brought about by a revolution or overnight breakthrough in culture methods, but by a gradual improvement in existing methods, and a renewed understanding that the “sea” is an oligotrophic medium with a low abundance of microbial cells, living together.
The aim of this chapter is to provide an overview of the new strategies and technologies used by microbiologists to enhance the isolation and culture of marine microorganisms. Some important aspects, such as the preparation of the growth medium, incubation time, and sensitivity of detection methods for cell growth are first introduced. This is followed by a description of some of the most important technological developments, i.e., high-throughput and automated dilution-to-extinction methods, microencapsulation and immobilization of microorganisms in community cultures, the development of membrane-based cultivation chips for cell enrichment and cultivation, and single-cell techniques for cell isolation.
2 Medium
In 1941, Claude Zobell, a pioneer in the study of marine microorganisms, introduced the marine agar 2216 medium (known as Marine Broth), a nutrient-rich medium, which made possible the isolation of many heterotrophic bacteria, mainly belonging to a small number of genera, including Vibrio, Pseudomonas, Oceanospirillum, Aeromonas, Deleya, Flavobacterium, Alteromonas, and Marinomonas. Using nutrient-rich liquid or solid medium at the enrichment step favors faster-growing bacteria, referred to as “r”-strategists, at the expense of slow-growing, “K”-strategist, bacteria (Watve et al. 2000). K-strategists represent the dominant microorganisms in the pelagic environment. Natural seawater is a complex and living environment and it is difficult to prepare an artificial sea medium that suitably mimics it, providing all the nutrients necessary to sustain microbial growth. With the exception of the precise salt composition, which can be easily achieved, the concentration and sources of organic matter, nitrogen, phosphate, sulfur, trace mineral elements, cofactors, and vitamins and the reconstruction of the carbonate buffer are important factors that often determine the success or failure of enrichment and isolation attempts. Marine Broth 2216 medium has 170 times more dissolved organic carbon than natural seawater, which inhibits the growth of the true oligotrophs that represent the majority of microorganisms present in the ocean. It was only in 1993 when D.K. Button and colleagues started using natural seawater with small amounts of inoculum for isolating marine bacteria. This work resulted in the description of two new oligotrophs, Sphingomonas alaskensis and Cycloclasticus oligotrophus (Button et al. 1998; Schut et al. 1993; Vancanneyt et al. 2001). The concept of using natural seawater as a growth medium and low cell numbers as inoculum was improved upon two decades later in Giovannoni’s laboratory, allowing the isolation of members of the SAR11 clade, one of the most dominant clades of Alphaproteobacteria in the ocean (Cho and Giovannoni 2004; Connon and Giovannoni 2002; Rappe et al. 2002). “Candidatus Pelagibacter ubique” was cultured in an artificial seawater medium with specific nutrients (Carini et al. 2013). The medium was defined based on the metabolic reconstruction of the Ca. P. ubique genome (Giovannoni et al. 2005), which revealed that the genes necessary for assimilatory sulfate reduction were absent (Tripp et al. 2008). The common genes for serine and glycine biosynthesis were also absent and the bacterium was later found to be auxotrophic for the thiamin precursor HMP (Carini et al. 2014). Employing the environment itself as aiding in growing microorganisms has enabled the culture of numerous previously uncultured isolates from different clades.
Marine Crenarchaeota are now recognized as a dominant fraction of the plankton in deep ocean waters. Konneke et al. (2005) used natural seawater from an aquarium as the first step in an attempt to enrich the fraction of Marine Group I Crenarchaeota. After several transfers and the addition of bacterial antibiotics (streptomycin) the fraction of Crenarchaeota increased up to 90 % of the total community. Subsequently, “Nitrosopumilus maritimus” strain SCM1 was isolated in a synthetic medium.
Only a small proportion of viable cells present in microbial communities can be grown using growth media based on agar (or other gelling agents), an enigma known as the “great plate count anomaly” (Staley and Konopka 1985). The diversity of marine microorganisms obtained after streaking on solid medium may depend on the gelling agent used for their isolation (Joint et al. 2010). A hidden pitfall was revealed in the common preparation of solid medium. The simultaneous addition of phosphate and agar prior to autoclaving causes the formation of hydrogen peroxide in the medium, which inhibits the growth of microorganisms. The separation of phosphate and agar before the autoclave step allows the development of numerous colonies, among which over 30 % previously uncultured organisms (Tanaka et al. 2014).
Finally, it appears that medium based on natural seawater taken on the sampling site, in the first step of enrichment/isolation improves the growth of presently untamed microorganisms. The number of “uncultured” species domesticated in artificial media is also increased by several prior-growing periods in natural seawater (Nichols et al. 2010).
3 Incubation Time
An important aspect of the culturing of abundant and dominant marine microorganisms is incubation time. Long incubation times are needed to allow the growth of key players such as members of the SAR11 clade. Song et al. (2009) demonstrated that after 20–24 weeks of incubation, 64–82 % of the total number of isolates belongs to SAR11. Members of the Roseobacter clade are abundant in seawater and have been detected in many marine habitats. A new genus, Planktomarina gen. nov., of the Roseobacter clade was described, after it was isolated after an incubation time of 7 weeks (Giebel et al. 2013). Hahnke et al. (2015) incubated samples for three months after an algal bloom and isolated novel abundant bacterioplankton species. The autotrophic ammonia-oxidizing archaeon strain SCM1 required an enrichment period of 6 months before being isolated. Under optimal conditions, strain SCM1 reaches the stationary phase after 20 days of incubation, which indicates that long incubation times are necessary to culture members of this clade.
A long period of incubation is an important factor in the success of isolating environmentally relevant microorganisms, meaning that microbiologists have to be patient, although such time requirements are at odds with academic programs and increasing pressure to publish rapidly.
4 Measurement of Microbial Growth and Detection Sensitivity
Methods to measure cell density vary in terms of sensitivity and time requirements. For a long time, microbiologists assessed microbial growth based on the turbidity they observed in the inoculated liquid medium. Measurement of turbidity is fast and easy to perform. It is therefore still frequently used, but the disadvantage of this technique is the rather low sensitivity. Many cultures have been discarded because they apparently displayed no visible growth, due to poor cell density. Microscopic measurements are more sensitive but, particularly in the case of epifluorescence microscopy, they are time-consuming. Flow cytometry has a high sensitivity, the counting procedure is fast and simple, but requires strong expertise and a careful setting up of the equipment. During the last few decades, cheaper and easy-to-use flow cytometers have been introduced and are now common equipment in research laboratories.
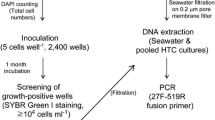
New protocols have been developed to access high-throughput isolation and culture microorganisms on a microplate format. In Giovannoni’s laboratory, two approaches have been used: first, a 48-microarray filter device was developed in order to decrease the growth detection limit and to permit cell numeration with densities as low as 103 cells/ml using 200-µl aliquots of culture (Connon and Giovannoni 2002). Hahnke et al. (2015) then improved this method using a 96-well blotting manifold (Bio-Dot, Bio-Rad, Munich, Germany) and a vacuum pump (Millipore, Billerica, MA, USA) under low, non-disruptive pressure (<5 mm Hg). Formaldehyde-fixed samples were filtered directly onto 4-mm polycarbonate filters with a pore size of 0.2 μm (GTTP, Millipore, Billerica, MA, USA), and stained with either 1 × SYBR Green or 1 μg/ml DAPI and mounted on glass slides with Citifluor and VectaShield (4:1).
In 2007, Stingl used a flow cytometer to enumerate cell densities after transferring samples of 200 µl into a 96-well plate and staining with SYBR Green1, the detection limit was of the same order (Stingl et al. 2007). This method is fast, as it takes around 10 min to analyze a 96-well microplate. On the Cocagne platform (described below), the SYBR Green labeling method developed in 2006 by Martens-Habbena is used to quantify cell numbers. An aliquot of 100 µl from a 96-well microplate is transferred to a black microplate containing 50 µl of a SYBR Green solution (Martens-Habbena and Sass 2006). After 3 h of incubation in the dark, a microplate reader can detect the relative fluorescence units (RFU) at 485 nm in each well of the microplate. RFU is correlated with cell density. This method is also fast that it takes less than one minute to read a microplate with a detection limit at 2 × 105 cells/ml in seawater medium.
5 Targeting Microorganisms of Interest
High-throughput culturing (HTC) using dilution-to-extinction, culture chip, and single-cell isolation allows large number of isolates to be recovered. In order to screen the numerous cultures, different methods can be used to dereplicate the isolates or to target specific groups of microorganisms. Dereplication using restriction fragment length polymorphism (RFLP) after amplification of the 16S rRNA gene is convenient for clustering the isolates and identifying mixed cultures (Cho and Giovannoni 2004; Connon and Giovannoni 2002). The 48-array developed in Giovannoni’s laboratory allowed Rappe et al. (2002) to use labeled oligonucleotide probes targeting the SAR11 cells (FISH) directly on the array filter. Hahnke et al. (2015) developed CARD-FISH on a 96-polycarbonate membrane to quantify and to identify the presence of different taxa with specific labeled probes.
6 Dilution-to-Extinction Method and High-Throughput Isolation
The dilution-to-extinction method introduced by Button et al. in 1993 was improved a decade later in Giovannoni’s laboratory (Connon and Giovannoni 2002). The aim was to drastically decrease the number of cells inoculated per well to around 1–5 cells in a small volume of medium, with the idea that only one cell would grow in a well of a microplate and a pure culture would thus be obtained. The theoretical number of pure cultures was estimated by the equation µ = −n(1 − p) ln(1 − p) and the estimation of culturability was given by V = −ln(1 − p)/X, where µ is an estimation of the expected number of pure cultures, n is the number of inoculated wells, V is the estimated culturability, p is the proportion of wells positive for growth (wells positive for growth/total inoculated wells), and X is the initial inoculum of cells added per well. Utilization of microplates for culturing has introduced high throughput into the culture process. In a single round, Connon and Giovannoni (2002) incubated approximately 2500 extinction cultures. A culturability of up to 14.3 % was observed after three weeks of incubation, which is 120 times higher than obtained with classical methods. Cell densities were in the range of 1.3 × 103 to 1.6 × 106 cells/ml; densities that would be impossible to detect by measurement of turbidity. Many of the microorganisms that were cultured using this approach were unique cell lineages and included cultures related to the clones SAR11 (Alphaproteobacteria), OM43 (Betaproteobacteria, related to the methylotroph Methylophilus methylotrophus), SAR92 (Gammaproteobacteria), and OM60/OM241 (Gammaproteobacteria).
The Laboratory of Microbiology of Extreme Environments (UMR 6197 LM2E, Plouzané, France) has automated the high-throughput dilution-to-extinction method (Cocagne platform). A pipetting robot (Starlet, Hamilton) dispenses the medium (500 µl/well) and the cells (1–3 cells/well) into the wells of 20 microplates loaded onto the deck of the pipetting robot. Filter-sterilized natural seawater is used as growth medium, amended with micromole traces of nitrogen, phosphate, and a carbon source. A column with deep wells filled with sterile medium serves as control. The microplates are sealed with silicone caps and incubated at in situ temperature for at least two months. After incubation, the detection of growth in deep-well plates is done by labeling growing cells with SYBR Green followed by the detection of relative fluorescence compared to blanks using a microplate reader (Infinite® 200 PRO, Tecan). For this purpose, deep-well plates and dark microplates (Corning) are loaded on the robot’s deck and the detection method programmed in the Hamilton software is started. All steps are automatically performed according to the program instructions. The pipetting robot first distributes 50 µl/well of the SYBR Green solution into the dark microplates (Corning), and then it transfers an aliquot (100 µl/well) of cell suspension from the deep-well plates to the dark microplates. After 3 h of incubation in the dark, the dark microplates are read on the microplate reader at 485 nm. Wells are considered to be positive when their relative fluorescence is higher than the average of the blanks (sterile medium) plus three times the value of the standard deviation of the blanks. The level of the relative fluorescence correlates with cell concentration. The positive wells of the twenty inoculated microplates, i.e., the wells with microbial growth, are then redistributed into new microplates with fresh medium. These newly inoculated deep-well plates are incubated under the same conditions as described above. A DNA extraction protocol, using the NucleoSpin 96 Tissue Kit (Macherey-Nalgel) is programmed in the robot’s software. The 16S rRNA gene is then amplified and sequenced. The Cocagne platform has been used on samples of coastal seawater of Brittany. Twenty microplates, in other words 1760 extinction cultures, were incubated in natural local seawater at 15 °C for three months. Two cells were inoculated per well and the culturability obtained was 14 %. DNA was extracted and partial sequences of the 16S rRNA gene from one hundred and fifty isolates were analyzed. The results indicate the presence of numerous novel genera and species affiliated to Alpha- and Gammaproteobacteria (L’Haridon, unpublished).
7 Cell Immobilization for Isolation and Cultivation of Marine Bacteria and Archaea
Microbial cell immobilization is a method that aims to fix microorganisms on the surface of or within specific carriers. This technique is commonly used in many fields including food, pharmaceutical, agricultural, therapeutic, environmental, and research applications (Cassidy et al. 1996). Many different strategies are employed. These include immobilization by binding (physical adsorption, ionic binding, or covalent binding) on an inert carrier, self-aggregation (flocculation or chemical cross-linking), encapsulation or entrapment (membrane entrapment or entrapment within the network of a polymer matrix). Here we present applications of whole cell immobilization by entrapment in different polymer matrices for the isolation and culture of marine heterotrophic Bacteria and Archaea.
Cell entrapment in natural polymer matrices is the most frequently used technique because it is one of the gentlest protocols, inducing the highest cell viability (Kanasawud et al. 1989). The polymer matrix allows the diffusion of small molecules that sustain the viability, activity, and growth of the entrapped cells. In addition, cells are protected against attacks by bacteriophages, and from shear forces, abiotic stresses, and potential inhibitors that may be present in the culture medium. Moreover, entrapped cells are easily recovered from their growth environment (D’Souza 2002; Nussinovitch 2010). Polymer gels can be shaped in the form of beads, spherical geometry of which increases mass transfer compared to biofilms. Various methods, such as extrusion and emulsion, can be used to produce gel beads. In the extrusion method, an aqueous mixture containing polymers and microorganisms is extruded through a syringe to form beads that fall into a hardening solution. Bead size depends on the syringe orifice diameter, the concentration, temperature, viscosity and flow rate of the polymer solution, and the distance between the orifice and the hardening solution. The emulsion technique is based on a dispersion process in a two-phase system in which an aqueous solution containing polymers and microorganisms is dispersed in an oil/organic phase under agitation to form a water-in-oil emulsion. Temperature decrease induces the polymerization of beads that are then placed in a hardening solution (Kourkoutas et al. 2004; Rathore et al. 2013). Depending on the polymer, the hardening solution contains different types of cross-linking agents. The emulsion technique allows the production of gel beads of different diameters (5–5000 µm) depending on the emulsion conditions and agitation speed. Beads can be roughly separated into microbeads and macrobeads according to their size; macrobeads being greater than 100 µm in diameter (John et al. 2011). Because of the cell immobilization, free and immobilized populations do not have the same environmental conditions. As a result, immobilized cells have a different physiology and growth capacity compared to free-living cells (Doleyres et al. 2004; Rathore et al. 2013).
Many natural polymers (e.g., κ-carrageenan, gellan gum, agarose, agar, gelatin, alginate, chitosan) can be used for cell entrapment (Cachon et al. 1997). These polymers are usually cheap and can be used alone or in combination. However, they vary in toxicity, gelling properties, rheological behavior, and mechanical stability according to the conditions during the bead formation and incubation. The selection of the right polymer and encapsulation method is therefore critical depending on the type of application and microorganisms to be immobilized, in order to ensure cell viability while preserving the beads’ mechanical stability, depending on culture conditions (especially salt concentration and temperature).
7.1 Immobilization for the Isolation of Marine Microorganisms
An original application of whole cell immobilization is the microencapsulation method developed by Zengler et al. (2002) for high-throughput culture and isolation of marine aerobic mesophilic microorganisms. The principle behind this method is the physical separation and massive parallel culture of cells that are individually entrapped in gel microdroplets (GMDs) of agarose (60 µm diameter) and incubated as a continuous community culture (flow rate 13 ml/h) in an HPLC column under low nutrient flux conditions for long incubation periods. This GMD community culture allows the exchange of metabolites and/or signaling molecules between the micro-colonies that grow within the GMDs, which are physically separated from each other. At the end of the continuous culture, GMDs containing micro-colonies are sorted by flow cytometry and further incubated in deep-well microplates (one GMD per well) for clonal enrichment in a rich organic medium. This technique was applied to different habitats and has provided more than 10,000 bacterial and fungal isolates per natural sample, including novel marine bacteria from previously uncultured groups (Zengler et al. 2002, 2005). It is a promising technique, although the critical step is the positive GMD sorting by flow cytometry, which may decrease cell viability. The use of a suitable cell sorter that can directly disperse the positive GMDs into microplate-wells is therefore important at this stage. In Zengler’s publications (Zengler et al. 2002, 2005) GMDs were directly sorted and distributed into microplates by a FACSAria® flow cytometer.
The Laboratory of Microbiology of Extreme Environments (UMR 6197 LM2E, Plouzané, France) has automated the Zengler method using a Hamilton pipetting robot (Cocagne platform) for GMD distribution into deep-well microplates and growth detection by labeling of growing cells with SYBR Green, followed by the detection of the RFU compared with blanks using a microplate reader. Moreover, a protocol for the microencapsulation (60 µm beads) and culture of thermophilic marine cells in heat-stable microbeads made of gellan and agarose was also developed (Fig. 15.1).

Development of a bacterial colony inside a GMD at 65 °C
The same microencapsulation method was also used for the isolation of slow-growing marine bacteria from mixed samples. Akselband et al. (2006) developed a new strategy based on the growth detection of individual cells encapsulated in agarose GMDs (30–50 µm in diameter). After 6 h growth in mixed culture, the slow-growing micro-colonies were sorted from fast growing ones by measuring their size within the GMD with a flow cytometer (EPICS EliteTM, Coulter) after LIVE/DEAD staining (Bac Light Bacterial Viability Kit), and transferred to fresh medium. Akselband et al. (2006) suggest the use of the same strategy for targeting specific activities using fluorogenic substrates for GMD sorting. In their study, they also showed that 75 % of the marine isolates grew faster in GMDs than in liquid medium.
Ben-Dov and collaborators developed another strategy for the isolation of marine aerobic mesophilic microorganisms using a macro-encapsulation technique (Ben-Dov et al. 2009). After dilution of environmental samples, microbial cells were individually entrapped in agar beads (1–3 mm), which were then coated with a porous polysulfonic polymeric membrane. This type of membrane prevents agar dehydration but enables the exchange of molecules (nutrients and waste). After sterilization of the surfaces with alcohol, the beads were placed in a suitable simulated or natural environment for long periods of incubation (at least 3 weeks). Then, the beads were recovered and sterilized by flaming, and the embedded agar spheres were removed, flattened, and observed under the microscope for the presence of microbial colonies. In order to enrich and isolate microorganisms, the agar beads were diluted and remixed with warm agar to form new beads. The coated beads were then repeatedly incubated in the appropriate environment. Following every two to three transfers, colonies were streaked onto appropriate agar plates. This labor-intensive technique was successful in growing several novel microorganisms from different environments, including the mucus layer of the Red Sea coral Fungia granulosa.
7.2 Immobilization for Increasing Cell Growth Efficiency and Stability
Entrapment of mesophilic aerobic marine microorganisms within polymer matrices (usually alginate) in pure culture or as consortia is used in environmental applications (such as biosensor and in bioremediation) (Cassidy et al. 1996; Futra et al. 2014) and in biotechnological applications in bioreactors (biomass and metabolite production and waste water treatment) (Roy 2015). Cell entrapment has been used for the culture of thermophilic strains (Kanasawud et al. 1989; Klingeberg et al. 1990; Norton and Lacroix 2000) but never for marine (hyper)thermophilic anaerobic microorganisms because of the difficulties of gelling properties in a saline environment. However, cell entrapment offers numerous advantages over free-cell cultures in bioreactors (Bustard et al. 2000). Cell entrapment induces a much more stable and robust continuous culture system because it prevents cell washout, improves genetic stability, protects organisms against shear forces and possible presence of toxic compounds in the culture medium whilst increasing cell numbers and product yields (Rathore et al. 2013). Moreover, immobilization facilitates separation between biomass and products and makes possible the reusability of the beads with immobilized cells.
The Laboratory of Microbiology of the Extreme Environments (UMR 6197 LM2E, Plouzané, France) has developed a cell entrapment protocol for (hyper)thermophilic marine microorganisms in order to improve the cultivation of deep-sea hydrothermal microbial communities. An experimental design, set to evaluate different incubation conditions (pH, temperature, salt, and sulfur), showed that beads (1–2 mm diameter) made of a mixture of heat-stable polymers (gellan and xanthan gums) were resistant to most incubation conditions. After 5 weeks of incubation, beads showed good resistance to all tested conditions (Fig. 15.2), except those simultaneously including high temperature (100 °C), low NaCl concentration (<30 g/l), and extreme pH (≤4 or ≥8). Batch experiments under nitrogen gas using Ravot medium (Gorlas et al. 2013) showed that thermophilic (Thermosipho sp. DSM 101094) and hyperthermophilic (Thermococcus sp. KOD1) microorganisms were effectively immobilized in beads and grew to high cell numbers. Cell counting by regular methods is impossible in polymer beads. Therefore, a protocol based on cellular ATP measurement was developed and applied to beads and culture medium. The correlation between cell counting using a Thoma cell counting chamber and ATP values was determined for each tested sample in order to determine the number of cells/ml for each strain. The ATP content of bacterial suspensions in the liquid culture media was determined using a Kikkoman Lumitester C-110 (Isogen Life Science) in combination with the BacTiter-Glo Microbial Cell Viability assay (Promega) according to the manufacturer’s instructions. In the case of beads, ca 100 mg of beads was placed in a pre-weighted sterile hemolysis tube (Gosselin). Subsequently, the beads were washed thrice with 100 µl of sterile degassed saline solution. For ATP measurement, 100 µl of sterile distilled water was added to the beads, which were vortexed for 10 s before adding 100 µl of BacTiter-Glo buffer. As for liquid medium, internal calibration was performed with 10 µl of a 100 nm ATP solution and maximal fluorescence emission values were considered. The number of active cells was 2.4 × 105 cells/g of beads for Thermosipho sp. (DSM 101094) and 2.3 × 106 cells/g of beads for T. kodakarensis (KOD1) just after immobilization, which corresponds to 2.7 and 54 % of cells surviving the immobilization step, respectively. After a few hours, cells grew within the beads as well as in the liquid medium. The cultures reached cell densities of 6.1 × 108 cells/ml in liquid medium and 2.9 × 107 cells/g in beads after 24 h incubation at 65 °C for Thermosipho sp. (DSM 101094), compared to 2.5 × 108 cells/ml for free-cell culture under the same conditions. In the case of T. kodakarensis (KOD1) the highest cell densities were obtained at 70 °C with 3.3 × 108 cells/ml and 4.8 × 107 cells/g in liquid medium and in beads, respectively, after 24 h incubation compared to 1.4 × 108 cells/ml in free-cell suspension medium. The high percentage of survival together with the high cell densities of both strains in beads and liquid medium showed that entrapment and culture of immobilized anaerobic thermophilic and hyperthermophilic marine strains is possible at high temperature (Landreau et al. submitted).

Beads observation using a binocular magnifying glass (magnification × 2.4) after immobilization 5-week incubation
Using the same entrapment protocol, the proof of concept was established in a continuous culture that was performed for 42 days in a gas-lift bioreactor flushed with nitrogen and containing 40 % (v/v) of freshly inoculated beads with different thermophilic and hyperthermophilic deep-sea microorganisms. Gel beads proved to be highly resistant to mechanical and chemical degradation during the 42 days of continuous culture. Moreover, cell quantification, organic acid concentrations, and ATP monitoring showed that polymer beads and effluents were highly colonized and that the microorganisms were reactive to culture conditions.
8 Enrichment Chambers and Culture Chips
The environment is often an excellent growth medium (Ferrari et al. 2008; Kaeberlein et al. 2002) and this forms the basis of many laboratory cultivation techniques. We can build better and smaller culturing devices. These devices are laboratory-on-a-chip (LOC) or micro-engineering (MEMS) approaches for which the technologies are in part derived from the electronics industry that is entering life sciences (Ingham and van Hylckama 2008; Weibel et al. 2007). Therefore, microbiologists have access to improved manufacturing and design capabilities leading to the creation of sampling and culture devices for the laboratory or for implantation in the natural environment. This is both intuitively obvious and experimentally supported. Logically, we want to either move the laboratory into the environment or the environment into the laboratory in order to recreate the missing factors that act as roadblocks to cultivation. Here we deal with culture chambers (laboratory into environment) and culture chips (environment into laboratory) as examples of technological advances that use this idea.
8.1 Porous In Situ Cultivation Chambers
Porous cages and chambers allow the culture system to be placed in the appropriate environment. Whilst even this measure is not a perfect recreation, such systems have shown great value in culturing otherwise refractory microorganisms. One simple but effective version is a diffusion chamber bounded by two membranes (Fig. 15.3a). This chamber is filled with agar lacking nutrients and inoculated with an environmental sample and then sealed (Kaeberlein et al. 2002). The diffusion chamber can be placed in the sea or in soil, or a close simulation of the environment (e.g., sediment in an aquarium), and the external conditions rapidly equilibrate with the small volume of the interior. Micro-colonies trapped within the agar can often then be cultured conventionally, whilst closely positioned micro-colonies can be investigated for the possibility that culturing required interactions between different species. This technique has resulted in the enrichment of previously uncultured organisms, up to 30 % success in culturing microorganisms from marine and littoral habitats. A logical extension of this idea is the iChip, a highly multiplexed version of the diffusion chamber (Fig. 15.3b). The iChip allows multiple polyoxymethylene (a hydrophobic plastic) chambers to be enclosed with two polycarbonate membranes (Nichols et al. 2010). The chambers are loaded with agarose before the second membrane is put in place, dipped in the appropriate environmental sample to inoculate, sealed and implanted into soil.

Cultivation chambers for enrichment and near natural culture conditions. a In situ cultivation chamber filled with agar, then placed back in natural or simulated environment for cultivation of microorganisms in the interior. Scale bar (top panel a) indicates 1 cm for all panels. b iChip, an array of cultivation chambers similar to those in panel. Top view of whole chip. Middle cross section showing microbial growth in compartments. Bottom Plating an indicator strain on the upper surface reveals antimicrobial activity from the bacteria contained within compartment X. c Selective, asymmetric capture chamber for mycelial organisms. BEV, Bird’s Eye View. XS Cross section. The upper membrane (m) is a barrier for all microorganisms, the larger pore membrane can be penetrated by actinomycetes (represented by chains of green ovoids, growing from sediment) better than other bacteria, so tends to trap and enrich selectively. d Variant culture chamber with slowly degrading material (P, polymer) or microbial population in central chamber. Whilst bounded by two membranes, only the upper membrane (em, experimental membrane) communicates with inner chamber, the lower (cm, control membrane) does not. Therefore, the two microbial populations growing on the outer surfaces of em and cm can be expected to differ. e Porous tube containing bacteria (light blue) for exceptionally high surface area to volume ratio, allows recirculation of contents and extremely rapid exchange with the environment (darker blue). f Closing cage system of entrapment. Left part of this panel shows unfolded cage (uc) with a mesh hundreds of micrometers. Center cage now folded into a cube (fc) trapping a multicellular microbe or marine invertebrate. Right Assembly of multiple cages to create synthetic tissues or communities. The height in the XS of panels A, B and E is exaggerated circa two-fold to show detail. A blue background indicates marine use, a beige background that the main validation to date has been in the soil, but that the technology is applicable to the marine environment
A notable success of the iChip was the use of these devices in a screening of up to 10,000 micro-colonies in order to isolate an example of a new class of antibiotics, teixobactin, from Eleftheria terrae, a group of Bacteria not previously known to be a good source of antimicrobials (Ling et al. 2015). The iChip uses an additional strategy when looking for antimicrobials. The first line of activity screening is performed by recovering the device, then spread plating the upper membrane with indicator bacteria. In the first instance, the microorganisms grow inside the micro-wells, sandwiched between two membranes allowing entry of small molecular weight compounds such as nutrients and quorum sensing molecules that support growth. In the second stage, antimicrobials diffuse from the trapped micro-colonies to an indicator strain on the outer surface to be assayed. The high frequency of “hits” from a highly mined environment is promising and there is every reason for thinking this approach will adapt well to the less well-explored marine environment. So far, the approach is not selective—what grows and subsequently assayed. Enrichment culture, for example with poorly soluble polymers, would be one way to bias the method towards isolating microorganisms with a desired phenotype, such as polymer degradation. A variant of this diffusion chamber system that possesses selectivity uses asymmetric membranes (Fig. 15.3c). The upper membrane cannot be penetrated by microorganisms and the lower membrane has a pore size that enriches a particular group by selectively permitting growth into the chamber (Gavrish et al. 2008). When the pore size of the lower chamber is set to 0.45 µm, filamentous bacteria (particularly actinomycetes) showed an advantage in penetrating the chamber and could therefore be enriched and isolated. Precise modulation of pore size usefully excluded fungi and other microorganisms, which frequently overgrow actinomycetes in more conventional environmental screenings. Given that actinomycetes are major antibiotic producers but the number of new useful antibiotics is falling, selective entrapment of filamentous bacteria is a promising technique. If membranes can be fabricated that are selectively porous based on other properties (e.g., surface charge or hydrophobicity or surface molecules) this selective entrapment technique could become even more powerful.
Another variant of the culture chamber places the enrichment material in the central chamber and allows microorganisms to grow on the exterior of the membrane (Fig. 15.3d; MicroDish BV, unpublished). The advantage of this strategy is allowing a controlled experiment to take place. Most culturing chambers just trap what grows; they are a form of sampling rather than experimentation. In this scheme, microorganisms grow on the outer surface of a membrane exposed to the marine environment as well as the contents of an inner chamber (separated by a porous membrane, the inner chamber can contain nutrients or other microorganisms)—this acts as the experiment. A second membrane within the same chamber has equal exposure to the environment but no communication with the central chamber; this is the control. Therefore, a metagenomics experiment or targeted culturing approach should allow analysis of the differences in microorganism abundance (or nucleic acid sequence) between the experimental and control conditions. More complex geometries in culture chambers are possible, permitting multivariable experimentation in situ. An alternative format for porous enclosure is a hollow fiber (Fig. 15.3e), a tubular membrane which has an exceptionally high area in contact with the environment (Aoi et al. 2009). A porous polyvinylidene fluoride (PVDF) membrane (0.1 µm pores) contains the microorganisms of interest. This system shows a significant enhancement of culturability over agar-based methods, tested with three microbial populations derived from marine and industrial environments. Additionally, the total volume of the system can be increased (simply by extending the tubing), allowing rather larger populations of microorganisms to be enriched than is the case with micro-colony-based methods.
The diffusion chambers described above are sealed by hand under aseptic conditions and then placed in the environment. A particularly interesting development for the marine environment is the self-closing cultivation chamber (Fig. 15.3f), particularly if these could be closed in hard to reach environments, such as the deep-sea. One such approach is the creation of flat boxes that self-assemble (Leong et al. 2008). The sides to the box are all porous; when folded they form a cage. The assembled boxes can be sorted and are amenable to manipulation by magnetic fields. These properties offer the potential to stack boxes and therefore the creation of artificially structured communities of cells, again moving from sampling to experiments. Later developments have created self-folding structures, created purely from polymers that are optically transparent and capable of trapping microorganisms including eukaryotic cells (Azam et al. 2011).
8.2 Cultivation Chips
Starting with the Petri dish, there are a number of possible improvements that might be envisaged that would aid marine culturing. These include miniaturization, a greater potential for automation, the absence of gel polymers as a matrix (such as agar that may contain inhibitory compounds), and suitability for imaging or other detection methods. An early attempt at “the better Petri dish” is the hydrophobic grid membrane (HGM), a porous filter subdivided into hundreds of growth areas by wax barriers. HGM offers flexibility (can be placed on agar or non-agar surfaces and can be moved). The HGM has a greater dynamic range for counting colony-forming units compared to an equivalent area of agar (Sharpe and Michaud 1974). This is explained by a better segregation of colonies and effective statistics derived from the distribution of colonies segregated between a large number of compartments.
Microfabrication allows further subdivision of growth areas to the point where custom-built disposables with thousands of compartments are available (Incom, USA). Micro-engineering allows a series of configurations of what is effectively highly multiplexed multiwall plates. However, miniaturizing multiwall plates beyond a certain threshold creates problems as well as advantages. There may be issues of aeration (liquid trapped in capillaries cannot be shaken to introduce oxygen), imaging (hard to focus on cells in capillaries), assay sensitivity in low volumes and dehydration (nanoliter wells dry out unless the humidity is carefully controlled), cross contamination, and dependence on robotics that microbiology laboratories often lack. There are solutions and work-around possibilities for many of these issues. Simply optimizing geometry, shaking speed and angle, and media components within conventional multiwall plates can considerably help oxygenation (Duetz et al. 2000). Oxygen can be delivered to 96-well plates through an oxygen permeable membrane, leading to 96-well microtiter plates with oxygen transfer rates comparable to Erlenmeyer flasks (Microflask System, Applikon Biotechnology, NL). Additionally, the capillarity of micro- or nanoliter wells has been exploited to capture droplets in stackable microcapillary arrays.
Porous ceramics make a good basis for culture chips (Fig. 15.4), with the advantages of flatness, porosity, low autofluorescence, biocompatibility, and despite inertness, good ability to conjugate biomolecules (Ingham et al. 2007; Microdish BV, NL). The downside is brittleness of the ceramic, requiring reinforcement for good handling. Micro-engineering techniques can create micro-wells (7–300 μm across, 10–40 μm deep, fixed or variable geometries). Similar to culturing chambers, these can be used, in combination with sediments or other samples, to culture microorganisms that were refractory to culturing with conventional techniques. Additionally, the small volume of culture medium allows the use of high cost reagents or additives. Control of compartment size, combined with spacing allows fine-tuning of application. Furthermore, a common task in microbiology is the replication of microorganisms using a velvet pad (Lederberg and Lederberg 1952) or a simple printing device, such as a 96-pin array with a pin spacing of ~0.5 mm. It is possible to deploy bacteria using non-contact printing devices; even ones modified from conventional inkjet printers (Flickinger et al. 2007). In terms of contact printing, miniaturized arrays of pins fabricated from elastopolymers allow a similar process but on a finer scale. A pin spacing of 80 μm is usable to replicate between micro-wells of a culture chip (Ingham et al. 2010).

MDCC. a Cultivation chip (MDCC180.10, 8 × 36 mm), floating on water, with PAO base and more than 4000 180-μm-diameter compartments in a hexagonal array coated with platinum. b Image of an array of 200-μm-diameter micro-colonies, visualized by green fluorescent protein and captured by a hand held digital camera. c Chip with variably spaced wells (MDCC180.10VAR) inoculated with fungi to look at the effect of colony density and size on growth. d Single 180-μm-diameter, 10-μm deep well. e Section of an MDCC20.10 chip with 20-μm2 wells in an orthogonal array (125,000 wells per chip) used to culture bacteria, subsequently stained with a fluorescent dye and imaged by fluorescence microscopy. The four sections of this panel show how image processing moves the raw image from a gray scale photograph to a binary image—the final image used to score whether a compartment supports growth and therefore is scored as a CFU. f Image of a single colony from a screening of culture from arctic sediment. g Image of fungal mycelia growing out of 180-μm-diameter compartments as mycelial bundles. White scale bar (panel b) indicates 6 mm when applied to panel a, 1 mm for b, 2.5 mm for c, 40 µm for d, 150 µm for e, 420 µm for f and 800 µm when applied to G
Suspension cultivation is also possible in a highly multiplexed and miniaturized device. For example, arrays of microfluidic capillaries can sustain microbial growth using peristaltic pumps to move fluid and introduce oxygen (Gan et al. 2011). Moreover, micro-fabricated chips can be used to address questions such as to the organization of microbial communities (Keymer et al. 2006) or to approximate to the function of chemostats (Balagadde et al. 2005). We can expect further inventiveness in terms of microbial cultivation chips in the future (Lok 2015), and also hope that low cost manufacturing will make these devices affordable for the marine microbiologists. Therefore, these micro-fabricated chips will play a significant part in increasing the culturing members of the marine microbiome.
9 Single-Cell Techniques
In order to establish a pure culture, microbiologists have to isolate a viable cell and maintain its physical isolation whilst the cell divides to form a colony. Often, a pure culture is achieved by statistical means: either a sample is diluted until on average there is only a single viable cell left in the cultivation chamber, or cells are spread over a surface or in small volumes until on average there is a single isolated cell in a given location. Either way, when employing these methods there is little or no control over which individual cell in the original sample goes where. A fundamentally different approach involves the direct selective isolation of single cells from a sample, combined with a method for directing the cell to a known location, for example on an agar plate, a well in a multi-well plate, a micro-well, or micro-chamber in a microchip. Having isolated the cell and defined its position we can then alter its microenvironment to improve its culturability.
The isolation, handling, and analysis of single cells is nowadays a topic of growing interest because of the potential to target rare cells and obtain information on the heterogeneity of cultures (including gene expression) (Blainey 2013; Ishii et al. 2010; Stepanauskas 2012; Yun et al. 2013). A variety of techniques is either already available or under development to achieve this which will be discussed here. Many of the techniques were originally developed for use with mammalian cells, but could be or in some cases already have been adapted for use with marine microorganisms.
9.1 Cell Sorting
Sorting techniques can either be active or passive. Active systems generally use external fields (e.g. mechanical, acoustic, electric, magnetic, optical, hydrodynamic) to impose forces to displace cells, whereas passive systems use inertial forces, filters, and adhesion mechanisms to purify cell populations (Wyatt Shields IV et al. 2015). For the selective isolation of cells active sorting is generally preferred.
During the sorting process the cells can be dispersed in a static or moving fluid, or as single cells in micro-droplets or gel microbeads. To obtain a high throughput, the cells need to be suspended in a flow, sufficiently separated from each other to allow easy separation. The cells are then analyzed one at a time as they flow past a sensing system and then actively knocked out of the flow when the cell has the desirable properties. Throughput in static systems is generally lower, and the techniques used often put high demands on the dexterity of the operator. Automation is sometimes possible, for example, through the combined use of advanced control systems and robotics (Lu et al. 2010; Zhang et al. 2012), and can speed up the isolation process. However, flow-through systems can suffer from blockages.
9.2 Facs
The most well known technique used for flow-based cell sorting is the Fluorescence Activated Cell Sorter or FACS. Although traditionally mainly regarded as a tool for the analysis and sorting of large mammalian cells, the technique is increasingly used for microorganisms (Czechowska et al. 2008; Davey and Kell 1996; Mazard et al. 2014; Morono et al. 2013; Winson and Davey 2000). A typical FACS is shown in Fig. 15.5. It includes a stream containing the cells to be sorted, a device for hydrodynamic focusing this stream into a narrow laminar flow for cell analysis, a laser system that enables one to measure the fluorescent and light scattering properties of each single cell, a droplet generator system which produces droplets containing single cells, a method to place an electric charge on a droplet according to whether the cell is to be collected or not, and a method for selectively diverting droplets in an electric field. The droplets are sorted into tubes or wells of microtiter plates, although sorting microorganisms directly onto agar plates is also possible (Fig. 15.6). A standard FACS can achieve high cell throughputs (>104 cells/s), but large samples are needed, not all cells are fluorescent, it is difficult to isolate all individual cells in a sample, and it is often not possible to guarantee that the droplets contain single cells. A number of alternative cell sorters, many of them based upon miniaturization and making use of Microelectromechanical systems (MEMS) and Laboratory-on-a-Chip technologies, has been or is being developed in order to overcome these problems. Often their throughput of these miniaturized cell sorters is lower than FACS, enabling slower and more detailed cell analysis techniques such as image analysis or Raman spectroscopy to be used rather than fluorescence alone.

Sketches of methods of isolation of cells with micropipettes. Cells can be isolated either by aspiration onto the tip of a narrow capillary (a), or into the body of the capillary itself (b). Aspiration onto the tip is more suitable for larger cells and filamentous organisms, but the risk of mechanical damage is greater

Schematic of a fluorescence activated cell sorter (FACS). A FACS is made up of three main systems: fluidics, optics, and electronics. Fluidics: A sample of cells is hydrodynamically focused using a sheath flow. As cells flow along the stream of liquid, a laser scans them. Optics: Laser light scattered by the cell is collected by a detector such as a photo multiplier tube and is used to count cells and to measure the size and granularity of the cell. Laser light is also used to excite fluorescence from specifically labeled sub-populations of cells. Electronics: The light signals are converted into electronic signals and a computer processes the information to determine which cells are to be sorted. The electronics system controls the charging and deflection of particles. Droplets, each containing a single cell, are created at the output nozzle by vibrating the nozzle at an optimal frequency and an electrical charge (−) is applied to the droplets to be collected as they exit the nozzle. The charged droplet is then deflected left towards the positive electrode (+) into a collection tube. Droplets that contain no cells or cells that are not to be collected pass straight through into the waste tube. The collected population is a pure population for the criteria determined when setting up the experiment (for example cells with above a certain threshold of fluorescence emission)
These cell sorters include inkjet printer-based systems with integrated image analysis (Stumpf et al. 2015; Yusof et al. 2011), dielectrophoretic cell sorters (Hu et al. 2005; Zhang et al. 2015), optical force-based cell sorters (Enger et al. 2004; Keloth et al. 2015; Landenberger et al. 2012), and various valve-based microfluidic cell sorters fabricated with soft lithography (Fu et al. 2002; Robert et al. 2011).
9.3 Mechanical Micromanipulation Techniques
The use of mechanical micromanipulation techniques for the isolation of microbial cells has a long history, and reviews on early techniques have been given by Johnstone (1969, 1973), and more recently by Fröhlich and König (2000). Early techniques used micro-needles or microcapillaries for the isolation of microorganisms, but success was limited by the technology then available. The development of improved micromanipulators with accurate pneumatic or hydraulic pressure control systems and improved microscopy techniques have made the isolation of cells with these techniques much more straightforward, and many examples of the use of mechanical micromanipulators for the isolation of microbial cells can now be found in the literature (Fröhlich and König 1999; Ishøy et al. 2006). Either the entire cell is drawn into a micropipette that is much larger than the cell, or held at a pipette tip that has an opening that is smaller than the cell diameter (Lu et al. 2010) (Fig. 15.7). Microfabrication nowadays makes it possible to manipulate single cells with pressure force in a massively parallel way (Nagai et al. 2015), although pressure forces on mechanically isolated cells can be large, and can lead to damage to shear-sensitive cells.

Schematic of optical tweezing for cell isolation (i, ii and iii). Micrographs of optical tweezing (iv and v) and isolation (vi) of single cells. A tightly focused near infrared laser beam is used to trap and move a single cell in three dimensions away from surrounding cells, either in an environmental sample (schematic shown in i) or from a laboratory culture of cyanobacteria (iv). The selected cell is physically removed by optically tweezing the single cell away from the other cells via a narrow meandering channel (ii and v) to a region where there are no other contaminating cells, such as a culture chamber in a customized chip or a microcapillary (iii and vi). Cell selection and isolation can now be automated to decrease the burden on the operator. The isolated cell can be used to start a pure culture or can be joined in the chamber with a second, or any number of selected cells for co-culture
An alternative approach to mechanical cell isolation using needles or pipettes involves the use of an externally applied physical field force to move the cells. Forces used for cell manipulation have been reviewed by Yun et al. (2013) and include optical, electric, magnetic and acoustic forces. To date acoustic forces have mainly lacked the resolution needed for single-cell manipulation. Most biological materials, including most cells, are diamagnetic and show little to no response to externally applied magnetic fields unless modified by the attachment of (super)paramagnetic particles or suspended in a paramagnetic suspending fluid (Safarik and Safarikova 1999). The response of cells to electrical or optical forces is strong, however, and can be achieved without modification of the cells.
9.4 Cell Manipulation with Electric Fields
Cells can be moved by direct (DC) and alternating currents (AC); DC fields move cells through electrophoresis because cells have a net (negative) charge, and AC fields induce movement of the cells by the interaction between the induced dipole moment and electric field gradient (dielectrophoresis). Cell manipulation with high frequency (>10 kHz) AC fields is generally preferred over cell manipulation with DC fields as it suffers less from interference by local fluid streaming induced by the strong electric fields near the electrodes. Dielectrophoresis has been extensively used to create aggregates of microorganisms for the study of microbial interactions in biofilms, including metabolic interactions, quorum sensing, and resuscitation of dormant cells (Andrews et al. 2006; Mason et al. 2005; Zhu et al. 2010). However, to achieve the resolution needed for single-cell manipulation, structures are required of a size similar to that of a single cell. As a result, the manipulation of single cells with electric fields puts high demands on microfabrication skills. The electric field rapidly declines away from the electrode structures, and the electrode structures therefore have to be close to the cell to be isolated. Electric forces are strongly dependent on the composition of the medium, in particular its conductivity. The use of low salt media is often essential, although this can be to some extent alleviated using high frequency electric fields (Schnelle et al. 1999). Despite all this, various examples of single-cell manipulation and isolation with electric fields can be found in the literature (Graham et al. 2012; Hsiao et al. 2010; Schnelle et al. 1999; Yang et al. 2010; Zhang et al. 2015). On the whole, however, single-cell manipulation is simpler with optical techniques than with electrical techniques, and therefore preferable.
9.5 Optical Manipulation
Optical trapping of dielectric particles from tens of nanometers in diameter to tens of micrometers by a single-beam gradient force trap (also known as optical tweezers) (Ashkin et al. 1986; Ashkin and Dziedzic 1987) uses the phenomenon of optical force, or radiation pressure. A laser beam focused through a high numerical aperture objective lens is tightly focused and results in a three-dimensional gradient of laser intensity due to the Gaussian intensity profile of the laser in the transverse direction and the tight focusing in the longitudinal direction. A cell or any other microscopic, dielectric material with a refractive index greater than that of the surrounding medium will experience an optical pressure from transverse and longitudinal gradient forces that draws the cell towards the region of highest intensity—the laser beam focus. The scattering force also acts on the particle and the net effect can be stable trapping of the cell near the focal point. Optical tweezers are straightforward to implement, compatible with other microscopy techniques, and have been used to trap, position, and manipulate cells and molecules in a variety of experiments (Fig. 15.8). The wavelength of laser light can be selected to minimize photothermal and photochemical damage to cells (Haro-Gonzalez et al. 2013; Liang et al. 1996; Neuman et al. 1999). Cells are trapped at the focal point of a microscope objective lens, therefore working at a distance is possible, and as such experiments can be performed in enclosed, sterile sample chambers. There is no mechanical contact that can introduce risk of contamination, and subcellular organelles or endophytes can be manipulated within the cell by focusing the laser beam through the membrane (Sacconi et al. 2005). In addition, optical tweezers provide the ability to accurately measure small forces in biology, down to the level of piconewtons (Block et al. 1989).

Microfluidics with integrated optics for single-cell isolation. Top schematic of a working device created using ultrafast laser inscription and selective chemical etching and micrographs of the device. The cell sample is hydrodynamically focused using a sheath flow (i). The optical force from the laser beam emanating from the integrated waveguide deflects a single, selected cell (ii), which is collected from a side channel. The single isolated cell can be used to create a pure culture or other cells can be selected and deflected to create a co-culture. A micrograph of the device is shown in panel iii
Optical tweezers have been used for cell sorting (Ericsson et al. 2000) to select single cells and position them in patterns on a microscope slide, immobilize them, and study their viability. There have been several studies reported in the literature on the development of automated optical tweezers for manipulating and precise positioning of microspheres (Banerjee et al. 2010; Ozcan et al. 2006), diatoms (Tanaka et al. 2008), and eukaryotic cells (Grover et al. 2001; Hu and Sun 2011; Wang et al. 2013). In addition to using a tightly focused beam, laser light can be loosely focused, collimated or diverging and can exert a guiding force on a cell to push it in the direction of beam propagation (Arlt et al. 2001; Ashkin et al. 1970; Imasaka et al. 1995).
A novel, microfluidic device with integrated channels and waveguides fabricated using ultrafast laser inscription combined with selective chemical etching (Choudhury et al. 2014) is being developed in order to enable sorting and isolation of biological cells using the optical force of laser light (Fig. 15.8). The complex three-dimensional microfluidic structures within the device allow the injected cell population to focus in a hydrodynamic flow (Keloth et al. 2015; Paie et al. 2014). Continuous wave laser in the near infrared light is coupled into the integrated waveguide in the device. The laser light emerges from the waveguide into the microfluidic channel and is used to exert radiation pressure on the selected cells (Keloth et al. 2015) as these cells in the focused stream flow past the waveguide. The optical scattering force then pushes the cell from the focused stream into the sheath fluid. Thus, individual cells can be controllably deflected from the focused flow to the side channel for downstream analysis or culture.
9.6 Single-Cell Culture and Modification of Its Microenvironment
A large variety of methods has been or is currently under development for culture of single cells. In many cases, the work is done not with cells from the marine environment but with model organisms such as Escherichia coli that are easy to culture and readily modified. However, the work described can often translate well to working with (marine) uncultured microorganisms. Working with natural or artificial seawater is in itself not usually an issue when working in miniaturization; the most common materials used such as the photoresist SU8 and the polymer PDMS are highly biocompatible and resistant to seawater. The long incubation times can be an issue as dehydration is a recurrent danger when working with small volumes but can be controlled by ensuring samples remain well isolated and humidity levels are controlled. Also, PDMS is a material that is highly permeable to small molecules.
Prominent amongst single-cell culture methods are those based on micro-droplet formation in microfluidic devices (Eun et al. 2011; Joensson and Andersson Svahn 2012; Liu et al. 2009; Pan et al. 2011). Typically a stream of suspended cells is introduced into a stream on a non-miscible biocompatible fluid such as a mineral or vegetable oil or a fluorocarbon carrier fluid. Droplet formation is controlled by the interaction between hydrodynamic and interfacial forces, although electric fields may also be used to control the timing of droplet formation and droplet size (Link et al. 2006). An alternative is the formation of arrays of individual droplets by interfacial effects, for example at micro-wells in an SU8 surface (Boedicker et al. 2009). Although in each case, whether you have a single cell in a droplet or not is mainly determined by chance. The technique has great merit because it facilitates high-throughput experimentation and also automation is straightforward (Khorshidi et al., 2014). Of particular interest for the study of uncultured microorganisms is also the chemistrode, which allows sampling and formation of nanoliter volume droplets directly from the environment (Liu et al. 2009). Droplet microfluidics has allowed the investigation of the effect of cell number (Boedicker et al. 2009) on cell growth and quorum sensing responses. Confining a cell to a(n) (insulated) micro-droplet is essentially equivalent to increasing the cell density more than a thousand fold compared to having a cell in a Petri dish (Boedicker et al. 2009; Vincent et al. 2010). As a result, when confining cells to micro-droplets, Boedicker et al. (2009) found that the single cells confined in micro-droplets were able to induce a quorum sensing response. This raises the question whether quorum sensing actually is a community response. Such confinement of microorganisms to micro-droplets or micro-chambers, which have no mechanism of exchange of signaling molecules, could help inducing growth of uncultured microorganisms (Ma et al. 2014; Vincent et al. 2010). At the same time, if there is exchange, then it can be fast due the high surface to volume ratio of devices at the microscale (Boitard et al. 2015). This could be used to advantage, for example, to more effectively remove accumulated toxins.
Miniaturized methods that use compartmentalization can eliminate competition among species (Ma et al. 2014). However, as interactions between microorganisms are thought to be an important factor for cell culturability, the ability to control interactions between cells is highly attractive. A micro-droplet-based example is that developed by Park et al. (2011) for parallel co-culture of cells. Kim et al. (2008) developed a system of interconnected micro-chambers in which three of them are located close to each other but communication could only occur by diffusion. When Kim et al. (2008) constructed a community of three different species of wild-type soil bacteria with syntrophic interactions using this device they found that the spatial separation of the different species was essential for the community to survive: if the cells mixed then competition between the different species would cause the community to collapse.
The micromanipulation techniques described in the previous part of this chapter allowed one to select cells from a mixture rather than leaving the choice of cells to be investigated by chance. Yasuda et al. (Umehara et al. 2003; Wakamoto et al. 2003; Yasuda et al. 2013) have taken this approach further and used laser tweezers to move single cells of E. coli into individual chambers in a micro-chamber array (Wakamoto et al. 2003). After a cell had divided, the authors moved one of the two daughter cells to a vacant chamber, allowing differences between generations to be studied. In later sets of experiments, the same group also developed methods for observing the adaptation of single cells, tweezed into individual micro-chambers, to changes in nutrient concentrations (Umehara et al. 2003). The ability to change the response of cells to changes in nutrient concentration is also important for studies of culturability. Also of interest is therefore the work by Eriksson et al. (2007) who used optical tweezers to move single (yeast) cells in a gradient created within a microfluidic device, thus exposing cells to different environments and allowing the detection and analysis of rapid changes.
10 Conclusion
For a long time, microbiologists have judged the presence of microbial growth only by observing changes in the turbidity with the naked eye or measuring it with a spectrophotometer. Low biomass detection methods (microscopy, flow cytometry, ATPmetry, and microplate reader) now allow the observation of microbial growth with a low threshold of 103 cells/ml. Therefore, microbiologists have to get accustomed to working with microbial cultures with no visible turbidity. The high sensitivity of molecular methods permits the extraction of DNA from low biomass and allows one to obtain taxonomic affiliations from 16S rRNA gene amplification and sequencing. Moreover, single-cell genomic DNA can be amplified and sequenced in order to identify the metabolic functions which could help to design artificial media best suited for an optimal growth.
The dilution-to-extinction method combined with high-throughput cultivation is expected to become a widely used method in the different microbiology laboratories in order to isolate relevant microorganisms. This approach, combined with low biomass detection methods and longtime incubations, has proved to be efficient for the isolation of key microbial players. Even if a majority of the isolates that is brought into culture with this approach is later on found to be difficult to grow to high cell numbers in artificial seawater media or even under the same growth conditions, there generally should be enough cells to obtain the genomic information that may later on be useful for taming these isolates.
Culture chips and immobilization culture approaches (beads, chambers) are also promising, because they allow the maintenance of cell communication, and thus the study of syntrophic relations and interactions between cells. Moreover, many of such culture chip methods use the natural environment as the basis for the “growth medium” with advantages in culturability. It is desirable both to decrease the costs of miniaturized culture devices and automate functions such as targeting desirable micro-colonies, recovery, creating replicates, and integrating culture methods with molecular techniques.
Single-cell approaches require specific pieces of equipment (such as FACS and optical tweezers), which can be expensive and need to be handled by experts. Optical tweezers, and optical forces in general, allow one to target a specific cell in a mixture, to isolate it and study it after having isolated it in a micro-chamber.
The marine environment, the largest continuous habitat on Earth, harbors the greatest biodiversity on earth, with an untapped resource of bioactive compounds. The recent progress in the culture approaches described above could help to target marine microorganisms with high biotechnological potential, such as actinobacteria, that have been found to produce many bioactive compounds.
References
Akselband Y, Cabral C, Castor TP, Chikarmane HM, McGrath P (2006) Enrichment of slow-growing marine microorganisms from mixed cultures using gel microdrop (GMD) growth assay and fluorescence-activated cell sorting. J Exp Mar Biol Ecol 329(2):196–205
Andrews JS, Mason VP, Thompson IP, Stephens GM, Markx GH (2006) Construction of artificially structured microbial consortia (ASMC) using dielectrophoresis: Examining bacterial interactions via metabolic intermediates within environmental biofilms. J Microbiol Methods 64(1):96–106
Aoi Y, Kinoshita T, Hata T, Ohta H, Obokata H, Tsuneda S (2009) Hollow-fiber membrane chamber as a device for in situ environmental cultivation. Appl Environ Microbiol 75(11):3826–3833
Arlt J, Garces-Chavez V, Sibbett W, Dholakia K (2001) Optical micromanipulation using a Bessel light beam. Optics Commun 197:239–245
Ashkin A (1970) Acceleration and Trapping of Particles by Radiation Pressure. Phys Rev Lett 24:156–159
Ashkin A, Dziedzic JM (1987) Optical trapping and manipulation of viruses and bacteria. Science 235(4795):1517–1520
Ashkin A, Dziedzic JM, Bjorkholm JE, Chu S (1986) Observation of a single-beam gradient force optical trap for dielectric particles. Opt Lett 11(5):288–290
Azam A, Laflin K, Jamal M, Fernandes R, Gracias D (2011) Self-folding micropatterned polymeric containers. Biomed Microdevices 13(1):51–58
Balagadde FK, You LC, Hansen CL, Arnold FH, Quake SR (2005) Long-term monitoring of bacteria undergoing programmed population control in a microchemostat. Science 309(5731):137–140
Banerjee AG, Pomerance A, Losert W, Gupta SK (2010) Developing a stochastic dynamic programming framework for optical tweezer-based automated particle transport operations. IEEE Trans Autom Sci Eng 7(2):218–227
Ben-Dov E, Kramarsky-Winter E, Kushmaro A (2009) An in situ method for cultivating microorganisms using a double encapsulation technique. FEMS Microbiol Ecol 68(3):363–371
Blainey PC (2013) The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev 37(3):407–427
Block SM, Blair DF, Berg HC (1989) Compliance of bacterial flagella measured with optical tweezers. Nature 338(6215):514–518
Boedicker JQ, Vincent ME, Ismagilov RF (2009) Microfluidic confinement of single cells of bacteria in small volumes initiates high-density behavior of quorum sensing and growth and reveals its variability. Angewandte Chemie-Int Edn 48(32):5908–5911
Boitard L, Cottinet D, Bremond N, Baudry J, Bibette J (2015) Growing microbes in millifluidic droplets. Eng Life Sci 15(3):318–326
Bustard MT, Burgess JG, Meeyoo V, Wright PC (2000) Novel opportunities for marine hyperthermophiles in emerging biotechnology and engineering industries. J ChemTechnol Biotechnol 75:1095–1109
Button DK, Schut F, Quang P, Martin R, Robertson BR (1993) Viability and isolation of marine-bacteria by dilution culture theory, procedures, and initial results. Appl Environ Microbiol 59(3):881–891
Button DK, Robertson BR, Lepp PW, Schmidt TM (1998) A small, dilute-cytoplasm, high-affinity, novel bacterium isolated by extinction culture and having kinetic constants compatible with growth at ambient concentrations of dissolved nutrients in seawater. Appl Environ Microbiol 64(11):4467–4476
Cachon R, Lacroix C, Divies C (1997) Mass transfer analysis for immobilized cells of Lactococcus lactis sp. using both simulations and in-situ pH measurements. Biotechnol Tech 11(4):251–255
Carini P, Steindler L, Beszteri S, Giovannoni SJ (2013) Nutrient requirements for growth of the extreme oligotroph ‘Candidatus Pelagibacter ubique’ HTCC1062 on a defined medium. ISME J 7(3):592–602
Carini P, Campbell EO, Morre J, Sanudo-Wilhelmy SA, Thrash JC, Bennett SE, Temperton B, Begley T, Giovannoni SJ (2014) Discovery of a SAR11 growth requirement for thiamin’s pyrimidine precursor and its distribution in the Sargasso Sea. ISME J 8(8):1727–1738
Cassidy MB, Lee H, Trevors JT (1996) Environmental applications of immobilized microbial cells: a review. J Ind Microbiol 16:79–101
Cho JC, Giovannoni SJ (2004) Cultivation and growth characteristics of a diverse group of oligotrophic marine Gammaproteobacteria. Appl Environ Microbiol 70(1):432–440
Choudhury D, Macdonald JR, Kar AK (2014) Ultrafast laser inscription: perspectives on future integrated applications. Laser Photonics Rev 8(6):827–846
Connon SA, Giovannoni SJ (2002) High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol 68(8):3878–3885
Czechowska K, Johnson DR, van der Meer JR (2008) Use of flow cytometric methods for single-cell analysis in environmental microbiology. Curr Opin Microbiol 11(3):205–212
Davey HM, Kell DB (1996) Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analyses. Microbiol Rev 60(4):641
Doleyres Y, Fliss I, Lacroix C (2004) Increased stress tolerance of Bifidobacterium longum and Lactococcus lactis produced during continuous mixed-strain immobilized-cell fermentation. J Appl Microbiol 97(3):527–539
D’Souza SF (2002) Trends in immobilized enzyme and cell technology. Ind J Biotechnol 1:321–338
Duetz WA, Ruedi L, Hermann R, O’Connor K, Buchs J, Witholt B (2000) Methods for intense aeration, growth, storage, and replication of bacterial strains in microtiter plates. Appl Environ Microbiol 66(6):2641–2646
Enger J, Goksor M, Ramser K, Hagberg P, Hanstorp D (2004) Optical tweezers applied to a microfluidic system. Lab Chip 4(3):196–200
Ericsson M, Hanstorp D, Hagberg P, Enger J, Nystrom T (2000) Sorting out bacterial viability with optical tweezers. J Bacteriol 182(19):5551–5555
Eriksson E, Enger J, Nordlander B, Erjavec N, Ramser K, Goksor M, Hohmann S, Nystrom T, Hanstorp D (2007) A microfluidic system in combination with optical tweezers for analyzing rapid and reversible cytological alterations in single cells upon environmental changes. Lab Chip 7(1):71–76
Eun Y-J, Utada AS, Copeland MF, Takeuchi S, Weibel DB (2011) Encapsulating bacteria in agarose microparticles using microfluidics for high-throughput cell analysis and isolation. ACS Chem Biol 6(3):260–266
Ferrari BC, Winsley T, Gillings M, Binnerup S (2008) Cultivating previously uncultured soil bacteria using a soil substrate membrane system. Nat Protoc 3(8):1261–1269
Flickinger MC, Schottel JL, Bond DR, Aksan A, Scriven LE (2007) Painting and printing living bacteria: engineering nanoporous biocatalytic coatings to preserve microbial viability and intensify reactivity. Biotechnol Prog 23(1):2–17
Fröhlich J, König H (1999) Rapid Isolation of single microbial cells from mixed natural and laboratory populations with the aid of a micromanipulator. Syst Appl Microbiol 22(2):249–257
Fröhlich J, König H (2000) New techniques for isolation of single prokaryotic cells1. FEMS Microbiol Rev 24(5):567–572
Fu AY, Chou HP, Spence C, Arnold FH, Quake SR (2002) An integrated microfabricated cell sorter. Anal Chem 74(11):2451–2457
Futra D, Heng LY, Surif S, Ahmad A, Ling TL (2014) Microencapsulated Aliivibrio fischeri in alginate microspheres for monitoring heavy metal toxicity in environmental waters. Sensors (Basel) 14(12):23248–23268
Gan M, Su J, Wang J, Wu H, Chen L (2011) A scalable microfluidic chip for bacterial suspension culture. Lab Chip 11(23):4087–4092. doi:10.1039/c1lc20670b
Gavrish E, Bollmann A, Epstein S, Lewis K (2008) A trap for in situ cultivation of filamentous actinobacteria. J Microbiol Methods 72(3):257–262
Giebel H-A, Kalhoefer D, Gahl-Janssen R, Choo Y-J, Lee K, Cho J-C, Tindall BJ, Rhiel E, Beardsley C, Aydogmus O, Voget S, Daniel R, Simon M, Brinkhoff T (2013) Planktomarina temperata gen. nov., sp. nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int J Syst Evolut Microbiol 63(Pt 11):4207–4217
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappé MS, Short JM, Carrington JC, Mathur EJ (2005) Genome streamlining in a cosmopolitan oceanic bacterium. Science 309(5738):1242–1245
Gorlas A, Alain K, Bienvenu N, Geslin C (2013) Thermococcus prieurii sp nov., a hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent. Int J Syst Evol Microbiol 63:2920–2926
Graham DM, Messerli MA, Pethig R (2012) Spatial manipulation of cells and organelles using single electrode dielectrophoresis. Biotechniques 52(1):39–43
Grover SC, Skirtach AG, Gauthier RC, Grover CP (2001) Automated single-cell sorting system based on optical trapping. J Biomed Opt 6(1):14–22
Hahnke RL, Bennke CM, Fuchs BM, Mann AJ, Rhiel E, Teeling H, Amann R, Harder J (2015) Dilution cultivation of marine heterotrophic bacteria abundant after a spring phytoplankton bloom in the North Sea. Environ Microbiol 17:3515–3526
Haro-Gonzalez P, Ramsay WT, Martinez Maestro L, del Rosal B, Santacruz-Gomez K, del Carmen Iglesias-de la Cruz M, Sanz-Rodriguez F, Chooi JY, Rodriguez Sevilla P, Bettinelli M, Choudhury D, Kar AK, Garcia Sole J, Jaque D, Paterson L (2013) Quantum dot-based thermal spectroscopy and imaging of optically trapped microspheres and single cells. Small 9(12):2162–2170
Hsiao AP, Barbee KD, Huang X (2010) Microfluidic device for capture and isolation of single cells. In: Biosensing Iii 7759
Hu S, Sun D (2011) Automatic transportation of biological cells with a robot-tweezer manipulation system. Int J Robot Res 30(14):1681–1694
Hu XY, Bessette PH, Qian JR, Meinhart CD, Daugherty PS, Soh HT (2005) Marker-specific sorting of rare cells using dielectrophoresis. Proc Natl Acad Sci USA 102(44):15757–15761
Imasaka T, Kawabata Y, Kaneta T, Ishidzu Y (1995) OPTICAL CHROMATOGRAPHY. Anal Chem 67:1763–1765
Ingham CJ, van Hylckama JET (2008) MEMS and the microbe. Lab Chip 8(10):1604–1616
Ingham CJ, Sprenkels A, Bomer J, Molenaar D, van den Berg A, van Hylckama Vlieg JET, de Vos WM (2007) The micro-Petri dish, a million-well growth chip for the culture and high-throughput screening of microorganisms. Proc Natl Acad Sci 104(46):18217–18222
Ingham C, Bomer J, Sprenkels A, van den Berg A, de Vos W, van Hylckama Vlieg J (2010) High-resolution microcontact printing and transfer of massive arrays of microorganisms on planar and compartmentalized nanoporous aluminium oxide. Lab Chip 10(11):1410–1416
Ishii S, Tago K, Senoo K (2010) Single-cell analysis and isolation for microbiology and biotechnology: methods and applications. Appl Microbiol Biotechnol 86(5):1281–1292
Ishøy T, Kvist T, Westermann P, Ahring B (2006) An improved method for single cell isolation of prokaryotes from meso-, thermo- and hyperthermophilic environments using micromanipulation. Appl Microbiol Biotechnol 69(5):510–514
Joensson HN, Andersson Svahn H (2012) Droplet microfluidics—a tool for single-cell analysis. Angew Chem Int Ed 51(49):12176–12192
John RP, Tyagi RD, Brar SK, Surampalli RY, Prevost D (2011) Bio-encapsulation of microbial cells for targeted agricultural delivery. Crit Rev Biotechnol 31(3):211–226
Johnstone KI (1969) The isolation and cultivation of single organisms. In: Norris JR, Ribbons DW (eds) Methods in microbiology. Academic Press, New York, vol 1, pp 455–471
Johnstone KI (1973) Micromanipulation of bacteria. The cultivation of bacteria and their spores by the agar gel dissection technique. Churchill Livingstone, Edinburgh
Joint I, Muehling M, Querellou J (2010) Culturing marine bacteria - an essential prerequisite for biodiscovery. Microb Biotechnol 3(5):564–575
Kaeberlein T, Lewis K, Epstein SS (2002) Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296(5570):1127–1129
Kanasawud P, Hjiirleifsdottir S, Hoist O, Mattiasson B (1989) Studies on immobilization of the thermophilic bacterium Thermus aquaticus YT-1 by entrapment in various matrices. Appl Microbiol Biotechnol 31:228–233
Keloth A, Paterson L, Markx GH, Kar AK (2015) Three-dimensional optofluidic device for isolating microbes. In: Microfluidics, biomems, and medical microsystems, vol Xiii, p 9320
Keymer JE, Galajda P, Muldoon C, Park S, Austin RH (2006) Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA 103(46):17290–17295
Khorshidi MA, Rajeswari PKP, Wahlby C, Joensson HN, Andersson Svahn H (2014) Automated analysis of dynamic behavior of single cells in picoliter droplets. Lab Chip 14(5):931–937
Kim HJ, Boedicker JQ, Choi JW, Ismagilov RF (2008) Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA 105(47):18188–18193
Klingeberg M, Vorlop KD, Antranikian G (1990) Immobilization of anaerobic thermophilic bacteria for the production of cell-free thermostable alpha-amylases and pullulanases. Appl Microbiol Biotechnol 33(5):494–500
Konneke M, Bernhard A, de la Torre J, Walker C, Waterbury J, Stahl D (2005) Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437:543–546
Kopf A, Bicak M, Kottmann R, Schnetzer J, Kostadinov I, Lehmann K, Fernandez-Guerra A, Jeanthon C, Rahav E, Ullrich M, Wichels A, Gerdts G, Polymenakou P, Kotoulas G, Siam R, Abdallah RZ, Sonnenschein EC, Cariou T, O’Gara F, Jackson S, Orlic S, Steinke M, Busch J, Duarte B, Cacador I, Canning-Clode J, Bobrova O, Marteinsson V, Reynisson E, Loureiro CM, Luna GM, Quero GM, Loscher CR, Kremp A, DeLorenzo ME, Ovreas L, Tolman J, LaRoche J, Penna A, Frischer M, Davis T, Katherine B, Meyer CP, Ramos S, Magalhaes C, Jude-Lemeilleur F, Aguirre-Macedo ML, Wang S, Poulton N, Jones S, Collin R, Fuhrman JA, Conan P, Alonso C, Stambler N, Goodwin K, Yakimov MM, Baltar F, Bodrossy L, Van De Kamp J, Frampton DM, Ostrowski M, Van Ruth P, Malthouse P, Claus S, Deneudt K, Mortelmans J, Pitois S, Wallom D, Salter I, Costa R, Schroeder DC, Kandil MM, Amaral V, Biancalana F, Santana R, Pedrotti ML, Yoshida T, Ogata H, Ingleton T, Munnik K, Rodriguez-Ezpeleta N, Berteaux-Lecellier V, Wecker P, Cancio I, Vaulot D, Bienhold C, Ghazal H, Chaouni B, Essayeh S, Ettamimi S, Zaid EH, Boukhatem N, Bouali A, Chahboune R, Barrijal S, Timinouni M, El Otmani F, Bennani M, Mea M, Todorova N, Karamfilov V, Ten Hoopen P, Cochrane G, L’Haridon S, Bizsel KC, Vezzi A, Lauro FM, Martin P, Jensen RM, Hinks J, Gebbels S, Rosselli R, De Pascale F, Schiavon R, Dos Santos A, Villar E, Pesant S, Cataletto B, Malfatti F, Edirisinghe R, Silveira JAH, Barbier M, Turk V, Tinta T, Fuller WJ, Salihoglu I, Serakinci N, Ergoren MC, Bresnan E, Iriberri J, Nyhus PAF, Bente E, Karlsen HE, Golyshin PN, Gasol JM, Moncheva S, Dzhembekova N, Johnson Z, Sinigalliano CD, Gidley ML, Zingone A, Danovaro R, Tsiamis G, Clark MS, Costa AC, El Bour M, Martins AM, Collins RE, Ducluzeau A-L, Martinez J, Costello MJ, Amaral-Zettler LA, Gilbert JA, Davies N, Field D, Glockner FO (2015) The ocean sampling day consortium. GigaScience 4:27
Kourkoutas Y, Bekatorou A, Banat IM, Marchant R, Koutinas AA (2004) Immobilization technologies and support materials suitable in alcohol beverages production: a review. Food Microbiol 21(4):377–397
Landenberger B, Hoefemann H, Wadle S, Rohrbach A (2012) Microfluidic sorting of arbitrary cells with dynamic optical tweezers. Lab Chip 12(17):3177–3183
Lederberg J, Lederberg EM (1952) Replica plating and indirect selection of bacterial mutants. J Bacteriol 63(3):399–406
Leong TG, Randall CL, Benson BR, Zarafshar AM, Gracias DH (2008) Self-loading lithographically structured microcontainers: 3D patterned, mobile microwells. Lab Chip 8(10):1621–1624
Liang H, Vu KT, Krishnan P, Trang TC, Shin D, Kimel S, Berns MW (1996) Wavelength dependence of cell cloning efficiency after optical trapping. Biophys J 70(3):1529–1533
Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517(7535):455–459
Link DR, Grasland-Mongrain E, Duri A, Sarrazin F, Cheng ZD, Cristobal G, Marquez M, Weitz DA (2006) Electric control of droplets in microfluidic devices. Angewandte Chemie-Int Edn 45(16):2556–2560
Liu W, Kim HJ, Lucchetta EM, Du W, Ismagilov RF (2009) Isolation, incubation, and parallel functional testing and identification by FISH of rare microbial single-copy cells from multi-species mixtures using the combination of chemistrode and stochastic confinement. Lab Chip 9(15):2153–2162
Lok C (2015) Mining the microbial dark matter. Nature 522(7556):270–273
Lu Z, Moraes C, Ye G, Simmons CA, Sun Y (2010) Single cell deposition and patterning with a robotic system. PLoS ONE 5(10):e13542
Ma L, Datta SS, Karymov MA, Pan Q, Begolo S, Ismagilov RF (2014) Individually addressable arrays of replica microbial cultures enabled by splitting SlipChips. Integr Biol 6(8):796–805
Martens-Habbena W, Sass H (2006) Sensitive determination of microbial growth by nucleic acid staining in aqueous suspension. Appl Environ Microbiol 72(1):87–95. doi:10.1128/aem.72.1.87-95.2006
Mason VP, Markx GH, Thompson IP, Andrews JS, Manefield M (2005) Colonial architecture in mixed species assemblages affects AHL mediated gene expression. FEMS Microbiol Lett 244(1):121–127
Mazard S, Ostrowski M, Holland R, Zubkov MV, Scanlan DJ (2014) Targeted genomics of flow cytometrically sorted cultured and uncultured microbial groups. Methods Mol Biol (Clifton, NJ) 1096:203–212
Morono Y, Terada T, Kallmeyer J, Inagaki F (2013) An improved cell separation technique for marine subsurface sediments: applications for high-throughput analysis using flow cytometry and cell sorting. Environ Microbiol 15(10):2841–2849
Nagai M, Oohara K, Kato K, Kawashima T, Shibata T (2015) Development and characterization of hollow microprobe array as a potential tool for versatile and massively parallel manipulation of single cells. Biomed Microdevices 17(2). doi:10.1007/s10544-015-9943-z
Neuman KC, Chadd EH, Liou GF, Bergman K, Block SM (1999) Characterization of photodamage to Escherichia coli in optical traps. Biophys J 77(5):2856–2863
Nichols D, Cahoon N, Trakhtenberg EM, Pham L, Mehta A, Belanger A, Kanigan T, Lewis K, Epstein SS (2010) Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl Environ Microbiol 76(8):2445–2450
Norton S, Lacroix C (2000) Gellan gum gel as entrapment matrix for high temperature fermentation processes: a rheological study. Biotechnol Tech 4(5):351–356
Nussinovitch A (2010) Bead formation, strengthening, and modification. Polymer macro- and micro-gel beads: fundamentals and applications. Springer, New York, pp 27–52
Ozcan M, Onal C, Akatay A (2006) A compact, automated and long working distance optical tweezer system. J Mod Opt 53(3):357–364
Paie P, Bragheri F, Vazquez RM, Osellame R (2014) Straightforward 3D hydrodynamic focusing in femtosecond laser fabricated microfluidic channels. Lab Chip 14(11):1826–1833
Pan J, Stephenson AL, Kazamia E, Huck WTS, Dennis JS, Smith AG, Abell C (2011) Quantitative tracking of the growth of individual algal cells in microdroplet compartments. Integrative Biology 3(10):1043–1051
Park J, Kerner A, Burns MA, Lin XN (2011) Microdroplet-enabled highly parallel co-cultivation of microbial communities. PLoS ONE 6(2):e17019
Rappe MS, Connon SA, Vergin KL, Giovannoni SJ (2002) Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418(6898):630–633
Rathore S, Desai PM, Liew CV, Chan LW, Lieng PWS (2013) Microencapsulation of microbial cells. J Food Eng 116(2):369–381
Robert D, Pamme N, Conjeaud H, Gazeau F, Iles A, Wilhelm C (2011) Cell sorting by endocytotic capacity in a microfluidic magnetophoresis device. Lab Chip 11(11):1902–1910
Roy D (2015) Novel bioreactors for culturing marine organisms. In: Kim S-K (ed) Springer handbook of marine biotechnology. Springer, Berlin, pp 353–358
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers Y-H, Falcon LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC (2007) The sorcerer II global ocean sampling expedition: northwest atlantic through eastern tropical pacific. PLoS Biol 5(3):398–431
Sacconi L, Tolic-Norrelykke IM, Stringari C, Antolini R, Pavone FS (2005) Optical micromanipulations inside yeast cells. Appl Opt 44(11):2001–2007
Safarik I, Safarikova M (1999) Use of magnetic techniques for the isolation of cells. J Chromatogr B 722(1–2):33–53
Schnelle T, Muller T, Hagedorn R, Voigt A, Fuhr G (1999) Single micro electrode dielectrophoretic tweezers for manipulation of suspended cells and particles. Biochimica Et Biophysica Acta-Gen Subj 1428(1):99–105
Schut F, Devries EJ, Gottschal JC, Robertson BR, Harder W, Prins RA, Button DK (1993) Isolation of typical marine-bacteria by dilution culture growth-growth, maintenance and characteristics of isolates under laboratory conditions. Appl Environ Microbiol 59(7):2150–2160
Sharpe AN, Michaud GL (1974) Hydrophobic grid-membrane filters: new approach to microbiological enumeration. Appl Microbiol 28(2):223–225
Song J, Oh H-M, Cho J-C (2009) Improved culturability of SAR11 strains in dilution-to-extinction culturing from the East Sea. West Pac Ocean. FEMS Microbiol Lett 295(2):141–147
Staley JT, Konopka A (1985) Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol 39:321–346
Stepanauskas R (2012) Single cell genomics: an individual look at microbes. Curr Opin Microbiol 15(5):613–620
Stingl U, Tripp HJ, Giovannoni SJ (2007) Improvements of high-throughput culturing yielded novel SAR11 strains and other abundant marine bacteria from the Oregon coast and the Bermuda Atlantic Time Series study site. ISME J 1(4):361–371
Stumpf F, Schoendube J, Gross A, Rath C, Niekrawietz S, Koltay R, Roth G (2015) Single-cell PCR of genomic DNA enabled by automated single-cell printing for cell isolation. Biosens Bioelectron 69:301–306
Tanaka Y, Kawada H, Hirano K, Ishikawa M, Kitajima H (2008) Automated manipulation of non-spherical micro-objects using optical tweezers combined with image processing techniques. Opt Express 16(19):15115–15122
Tanaka T, Kawasaki K, Daimon S, Kitagawa W, Yamamoto K, Tamaki H, Tanaka M, Nakatsu CH, Kamagata Y (2014) A hidden pitfall in the preparation of agar media undermines microorganism cultivability. Appl Environ Microbiol 80(24):7659–7666
Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ (2008) SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452(7188):741–744
Umehara S, Wakamoto Y, Inoue I, Yasuda K (2003) On-chip single-cell microcultivation assay for monitoring environmental effects on isolated cells. Biochem Biophys Res Commun 305(3):534–540
Vancanneyt M, Schut F, Snauwaert C, Goris J, Swings J, Gottschal JC (2001) Sphingomonas alaskensis sp nov., a dominant bacterium from a marine oligotrophic environment. Int J Syst Evol Microbiol 51:73–80
Venter J, Remington K, Heidelberg J, Halpern A, Rusch D, Eisen J, Wu D, Paulsen I, Nelson K, Nelson W (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66–74
Vincent ME, Liu W, Haney EB, Ismagilov RF (2010) Microfluidic stochastic confinement enhances analysis of rare cells by isolating cells and creating high density environments for control of diffusible signals. Chem Soc Rev 39(3):974–984
Wakamoto Y, Umehara S, Matsumura K, Inoue I, Yasuda K (2003) Development of non-destructive, non-contact single-cell based differential cell assay using on-chip microcultivation and optical tweezers. Sens Actuators B-Chem 96(3):693–700
Wang X, Gou X, Chen S, Yan X, Sun D (2013) Cell manipulation tool with combined microwell array and optical tweezers for cell isolation and deposition. J Micromechan Microeng 23(7):075006
Watve M, Shejval V, Sonawane C, Rahalkar M, Matapurkar A, Shouche Y, Patole M, Phadnis N, Champhenkar A, Damle K, Karandikar S, Kshirsagar V, Jog M (2000) The 'K' selected oligophilic bacteria: A key to uncultured diversity? Current Science 78:1535–1542
Weibel DB, DiLuzio WR, Whitesides GM (2007) Microfabrication meets microbiology. Nat Rev Microbiol 5(3):209–218
Winson MK, Davey HM (2000) Flow cytometric analysis of microorganisms. Method Companion Method Enzymol 21(3):231–240
Wyatt Shields Iv C, Reyes CD, Lopez GP (2015) Microfluidic cell sorting: a review of the advances in the separation of cells from debulking to rare cell isolation. Lab Chip 15(5):1230–1249
Yang S-M, Yu T-M, Huang H-P, Ku M-Y, Hsu L, Liu C-H (2010) Dynamic manipulation and patterning of microparticles and cells by using TiOPc-based optoelectronic dielectrophoresis. Opt Lett 35(12):1959–1961
Yasuda K, Hattori A, Kim H, Terazono H, Hayashi M, Takei H, Kaneko T, Nomura F (2013) Non-destructive on-chip imaging flow cell-sorting system for on-chip cellomics. Microfluid Nanofluid 14(6):907–931
Yun H, Kim K, Lee WG (2013) Cell manipulation in microfluidics. Biofabrication 5(2):022001
Yusof A, Keegan H, Spillane CD, Sheils OM, Martin CM, O’Leary JJ, Zengerle R, Koltay P (2011) Inkjet-like printing of single-cells. Lab Chip 11(14):2447–2454
Zengler K, Toledo G, Rappe M, Elkins J, Mathur EJ, Short JM, Keller M (2002) Cultivating the uncultured. Proc Natl Acad Sci U S A 99(24):15681–15686
Zengler K, Walcher M, Clark G, Haller I, Toledo G, Holland T, Mathur EJ, Woodnutt G, Short JM, Keller M (2005) High-throughput cultivation of microorganisms using microcapsules. Methods Enzymol 397:124–130
Zhang X, Leung C, Lu Z, Esfandiari N, Casper RF, Sun Y (2012) Controlled aspiration and positioning of biological cells in a micropipette. IEEE Trans Biomed Eng 59(4):1032–1040
Zhang P, Ren L, Zhang X, Shan Y, Wang Y, Ji Y, Yin H, Huang WE, Xu J, Ma B (2015) Raman-activated cell sorting based on dielectrophoretic single-cell trap and release. Anal Chem 87(4):2282–2289
Zhu K, Kaprelyants AS, Salina EG, Schuler M, Markx GH (2010) Construction by dielectrophoresis of microbial aggregates for the study of bacterial cell dormancy. Biomicrofluidics 4(2):022810
Acknowledgments
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 311975. This publication reflects the views only of the author, and the European Union cannot be held responsible for any use which may be made of the information contained therein. We wish to thank Jochen Schuster and Anusha Keloth for useful discussions and their help with some of the illustrations. Research was supported by UBO, CNRS, and IFREMER.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
L’Haridon, S., Markx, G.H., Ingham, C.J., Paterson, L., Duthoit, F., Le Blay, G. (2016). New Approaches for Bringing the Uncultured into Culture. In: Stal, L., Cretoiu, M. (eds) The Marine Microbiome. Springer, Cham. https://doi.org/10.1007/978-3-319-33000-6_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-33000-6_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-32998-7
Online ISBN: 978-3-319-33000-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)