
Abstract
The development of synthetic routes to hierarchically organized porous materials containing multiple, discrete sets of pores having disparate length scales is of high interest for a wide range of applications. One possible route toward the formation of multilevel porous architectures relies on the processing of condensable, network-forming precursors (sol-gel processes) in the presence of molecular porogens, lyotropic mesophases, supramolecular architectures, emulsions, organic polymers, or ice. In this review the focus is on sol-gel processing of inorganic and organic precursors with concurrently occurring microscopic and/or macroscopic phase separation for the formation of self-supporting monoliths. The potential and the limitations of the solution-based approaches are presented with special emphasis to recent examples of hierarchically organized silica, metal oxides, and phosphates as well as carbon monoliths.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others

Introduction
Research on complex and hierarchically organized porous materials has seen tremendous progress in the last decades, and the field is still rapidly evolving (Su et al. 2012). As a result huge progress has been made in the development of synthetic approaches toward porous materials that exhibit interconnected pore dimensions on several length scales, from molecular (<2 nm) via nano- (2–100 nm) to macroscopic (>100 nm). Pores smaller than 2 nm are typically termed micropores, pores with sizes between 2 and 50 nm mesopores, and pores larger than 50 nm are macropores (McCusker et al. 2001). Such multilevel porous architectures confer unique properties to materials depending on the combination of pore sizes, e.g., micro- and mesopores impart high surface areas and pore volumes providing size and shape selectivity and large interfacial areas, while larger pores (>50 nm) reduce transport limitations in the material and facilitate mass transport to the active sites. A variety of preparation techniques have already been reported for the preparation of micro-/macroporous, micro-/mesoporous, meso-/macroporous, or micro-/meso-/macroporous materials (Dong et al. 2002; Sen et al. 2003; Kuang et al. 2004; Gawel et al. 2010; Lopez-Orozco et al. 2011; Sun et al. 2011; Triantafillidis et al. 2013; Depardieu et al. 2015) with great potential for applications in the fields of catalysis, sorption, separation, energy storage and conversion, sensing, and biomedicine, i.e., medical diagnostics or therapies (Su et al. 2012). However, the applicability of a material depends not only on its pore sizes and size distributions but also on structural characteristics, such as the total amount of pores, the accessibility of the pores (ratio of closed to open pores), tortuosity and interconnectivity, gradients, etc., and, very importantly, the chemical composition as well as the processability in terms of shaping (films, fibers, monoliths, etc.). Shaping of the material is for many applications an inevitable requirement. To name just some of the advantages of highly porous, macroscopic monolithic materials, they can give lower back pressures, a higher permeability, and better performance in flow-through catalytic or separation systems (Siouffi 2006).
Well-controlled top-down and bottom-up self-assembly techniques providing a high level of structural control have been reported as very elegant approaches for the synthesis of hierarchically organized porous powders, particles, and monoliths (Yuan and Su 2006; Colombo et al. 2010; Su et al. 2012; Inayat et al. 2013). All these methods can in principle roughly be divided in the following categories: (1) posttreatment, starting from porous/nonporous objects and introducing a first, second, or third level of porosity by, e.g., selective leaching processes (Inayat et al. 2013); (2) typical ceramic processing, e.g., starting from powders, including sintering, foaming, and/or leaching (Galassi 2006); (3) synergetic solution-based processes, such as co-assembly of molecular precursors with or without “soft” templates, such as polymers, surfactants, emulsions, etc. (Colombo et al. 2010; Triantafillidis et al. 2013); and (4) transcriptive processes using pre-organized or self-assembled molecular, supramolecular, or solid molds (organic, biological, or inorganic) as templates, also termed “nanocasting” or exotemplating (Schüth 2003; Sanchez et al. 2005; Petkovich and Stein 2013).
Due to the nature of this review, we primarily limit the discussion to solution-based processes toward the preparation of monolithic materials of macroscopic dimensions with well-controlled pore sizes and pore orientations over multiple length scales. As a central topic, the simultaneous processing of condensable precursors in the presence of molecular porogens, lyotropic mesophases, supramolecular architectures, emulsions, organic polymers, ice, etc. is discussed. The possibilities in tailoring such multilevel porous architectures arising from concurrent microscopic and macroscopic phase separation in sol-gel systems with the ongoing competition between the timing of gelation versus phase separation will be covered. With that a detailed description of nanocasting or “hard” templating approaches will be beyond the scope of this Chapter, and only a few examples can be found throughout the text.
The next sections will first briefly highlight the fundamentals of sol-gel processing, phase separation, and other often applied templating schemes, followed by more recent key examples for silica-based materials as well as non-silica oxides or phosphates and carbon-based structures. These examples have been chosen to illustrate the general applicability of the synthetic routes toward these hierarchically organized materials and give an overview over the range of accessible materials. Many more examples can be found in the literature, and the list of materials is extended every day. We hope this chapter will not only be useful for experienced researchers already working in the field but also for encouragement of others to enter this exciting area of research with new ideas.
Fundamentals
Before beginning the exploration of combined phase separation and templating strategies with sol-gel processing, a few remarks on the materials under discussion and the network-forming processes need to be made.
As mentioned above, we will focus the discussion on monolithic materials. As defined by IUPAC, “a monolith is a shaped, fabricated, intractable article with a homogeneous microstructure that does not exhibit any structural components distinguishable by optical microscopy” (McNaught and Wilkinson 1997). In this chapter all materials have an interconnected porosity on multiple length scales within the monolithic structure as a second feature in common. Depending on the synthetic strategies, the pores might be arranged and connected in very different ways as schematically shown in Fig. 1.

Concept of hierarchy in a porous material: Schematics of a hierarchical porous buildup from the micrometer scale (inner blue circle), via the mesoscopic regime (orange circle) to the macroscopic porous dimension (light blue circle) within a monolithic material: In the macroporous regime, the different pore arrangements, such as inverse opal-like structures (a), isolated pores (b), co-continuous porosity (c), and a cellular buildup (d), are shown (in the scheme, the blue part represents the solid network, and the white part is the pore space). For the mesoporous regime, either well-organized pores with monomodal character as shown for a 2D hex structure or disordered arrangements are possible
Sol-Gel Processing to Yield Porous Materials
The sol-gel process is a method to produce a solid material (the gel) from molecular precursors via the formation of colloidal particles (the sol) (Brinker and Scherer 1990). Condensation reactions of hydrolyzable precursors, e.g., metal or semimetal alkoxides, but also salts, induced by the controlled addition of water represent the key steps in the synthesis of monolithic materials. The network evolves via nucleation and growth of nanometer-sized sol particles as well as their aggregation. A large variety of parameters, such as the choice of the precursor, its concentration, pH value, temperature, solvent, etc., can be adjusted to deliberately tailor the final network morphology, the network chemistry, and the pore structure (ranging from polymeric to particulate network buildup of either small or large particles, etc.). With that the homogeneity or even heterogeneity of the network is adjusted. This process is routinely used in the formation of oxidic and even hybrid inorganic–organic oxidic networks (Hench and West 1990; Schubert et al. 1995; Corriu and Leclercq 1996) but can also be found in an analogous manner in purely organic systems, i.e., resorcinol–formaldehyde polymers (Pekala et al. 1992; Al‐Muhtaseb and Ritter 2003; Elkhatat and Al‐Muhtaseb 2011). Figure 2 schematically shows the similarity of the basic chemical reactions for metal alkoxides as well as resorcinol–formaldehyde and the resulting network formation.

Analogy in the condensation-based network formation reactions starting from silicon alkoxides or resorcinol/formaldehyde
Micro- and mesoporosity is an inherent feature of amorphous gels prepared by sol-gel processing (Brinker and Scherer 1990; Al‐Muhtaseb and Ritter 2003; Antonietti et al. 2014). As described above, the network is build up from aggregated particles, whose size, number and density in the given volume are adjusted by the synthetic conditions. The solvent space between the solid network represents the potential pore space after drying. Thus, the critical step determining the porous character of the final dried material is the removal of the solvent, which is especially true when monoliths are prepared. Drying of large monolithic pieces is often difficult, since surface tension and evolution of capillary pressures can result in large shrinkages or even destruction of the whole gel body. One typical procedure to prevent cracking and collapse of the gel body is drying with supercritical fluids (scf), e.g., carbon dioxide or alcohols, since the building up of a gas–liquid interface is avoided; hence no capillary pressures evolve (Kistler 1931; Hüsing and Schubert 1998). This process is routinely used in the preparation of mesoporous materials, such as aerogels of variable composition with porosities as high as 97% and statistically distributed pore sizes in the upper mesoporous range. This process can also be applied for monolithic systems with hierarchical organization of pores. However, scf extraction is expensive and time-consuming and requires high pressures, sometimes even combined with high temperatures. This process can also be applied for monolithic systems with a hierarchical organization of pores. Another approach for drying hierarchically organized porous monoliths has been presented by Mukai et al. via freeze gelation and freeze-drying (Mukai et al. 2004; Nishihara et al. 2006). Freeze-drying can also be applied to purely mesoporous bodies; however in many cases the monolithic structures cannot be fully retained. A very promising procedure for drying large silica gels relying on a simple surface modification treatment with trimethylchlorosilane was presented in the mid-1990s as an alternative to supercritical drying (Smith et al. 1995). The capillary pressure, Pc, generated during drying is a function of the pore fluid–vapor surface tension, γLV, the contact angle, θ, between the fluid–vapor interface and pore wall, and the pore radius, a, as follows: Pc = −(2γLVcosΘ)/a. For a wetting fluid (θ < 90°), Pc is negative, thus indicating that the fluid is in tension. The presence of organic methyl groups on the surface of the silica gel in combination with a proper selection of the final pore fluid, e.g., hexane, allowed to change the contact angle to lower capillary pressures and thus dry monolithic wet silica gels without cracking (Smith et al. 1995). However, due to the requirement of surface silylation, this process is mostly limited to silica-based monoliths.
Phase Separation and Templating Strategies Combined With Sol-Gel Processing
Only when the porogen shows a univocal relationship between its own structure and the final porous structure, it can be termed a template. In most synthetic approaches occurring in solution and relying on phase separation on different length scales, this clear relationship is not given. Even in zeolite synthesis, the porogen typically is not a true template, but more a structure-directing agent. In other words, a template is mostly a “hard” object which does not significantly alter its shape when the solid counterpart is being formed. Because of the strong kinetic control and cooperative nature of the processes that result in mesopore and/or macropore formation via “soft”-templating routes as discussed in this chapter, the term “structure-directing agent” is to be preferred to “template.”
The sol-gel process is a dynamic process, in which the ongoing condensation reactions (cross-linking) of the mostly hydrophilic precursor molecules/oligomers result in solidification of the network. In principle, any kind of structure-directing agent can be added as porogen in this solidification process to induce some kind of phase separation. Already the nucleation and growth of the sol particles can be considered as a type of phase separation forming two heterogeneous phases: a solid network and a solution phase. This can be extended on the microscopic, but also macroscopic length scale by the addition of molecules, polymers, or supramolecular arrangements that enforce demixing. With the progress in understanding the hydrolysis and condensation reactions, the deliberate design of the porous structures by different phase separation strategies advanced more and more, e.g., by increasing the relative volume of the hydrophobic components within the porogens, the characteristic size of the pore dimensions can be increased from less than 1 nm to tens of micrometers and larger. Examples of suitable structure-directing agents/templates include molecular species as used in zeolite synthesis (Cundy and Cox 2003), low-molecular-weight and block copolymer surfactants (Wan and Zhao 2007), emulsions (Studart et al. 2006), and/or solid particles (Petkovich and Stein 2013). For materials comprising multiple levels of pore sizes (bimodal such as micro–meso, meso–macro, or even trimodal such as micro–meso–macro, meso–meso–macro, etc.), in principle, a combination of the above mentioned templating strategies is possible. This would mean that mixtures of, i.e., molecules, polymers, or supramolecular arrays are added to the gelling solution, with the intrinsic difficulty in the preservation of the existing levels of organization upon introducing another one. The major challenges arising in the preparation of such bi- or multimodal micro-/meso-/macroporous materials are: (1) to avoid macroscopic demixing of the components, (2) to avoid the formation of significant proportions of closed pores, (3) to control the different pore sizes independently, and (4) to manage shrinkage of the whole structure while retaining the macroscopic shape.
As one example for such a phase separation process: Monolithic materials with well-defined, co-continuous porous structures on multiple levels can be obtained by combining liquid–liquid phase separation and sol-gel processing. The phase separation process is induced by the presence of a porogen, which is in many cases a water-soluble polymer such as poly(ethylene oxide) or poly(acrylic acid). Nakanishi and Soga were the first to prepare monolithic silica with interconnected macropores and textural mesoporosity by the addition of poly(sodium styrene sulfonate) to a silica sol-gel mixture, and they could clearly show that the formation of different biphasic morphologies (isolated pores, particle aggregates, interconnected continuous pores) is induced by the polycondensation reaction of the network-forming silica species and is finally irreversibly frozen by the sol-gel transition (Nakanishi and Soga 1991). Therefore, all parameters that change the relative rates of phase separation versus gelation will have a profound influence on the architectural properties of the final gel, including mesoporosity, interconnected macroporosity, and the degree of macroscopic phase separation. A very detailed discussion on the topic of phase separation would be beyond the scope of this chapter, but the reader is referred to some excellent review articles, the original work of Cahn and Hilliard or the Handbook of Porous Solids, all providing a profound summary in the context of porous materials (Cahn and Hilliard 1958; Weitkamp et al. 2002; Nakanishi and Tanaka 2007; Kanamori and Nakanishi 2011).
As has been shown by Nakanishi et al., if a phase separation process is occurring concomitantly to the gelation process, a certain structure is irreversibly frozen in (just like a “snapshot” in time of the heterogeneity). Depending on the timing between phase separation and sol-gel transition as well as the stability of the different heterogeneous phases, different structures will be obtained as shown in Fig. 3. As typical for phase separation phenomena relying on spinodal decomposition of a system, the larger the time difference between phase separation and gelation, the coarser the structure will become, sometimes even breaking up into fragmented particles (in this case no monolithic structure is obtained). Thus, all parameters resulting in faster sol-gel transitions, e.g., higher temperatures, pH value changes, water–precursor ratio, etc., influence the final structure.

Time evolution of phase-separated domains (Reprinted with permission from reference Konishi et al. (2006). Copyright (2006) American Chemical Society)
This phase separation strategy can be complemented by variations on the sol-gel precursor side. Not only alkoxides but also inorganic salts, tailor-made precursor molecules, or even nanoscale building blocks can be used to influence the phase separation tendency in a sol-gel solution. Depending on the desired chemical composition of the final material, an almost unlimited choice of precursors is available and will be discussed in more detail below. To mention just one example, the formation of hierarchical structures involving the assembly of preformed inorganic nanoparticles into materials with higher-order architectures has been applied for oxides derived from highly reactive molecular precursors, e.g., alumina (Davis et al. 2001). These methods are often also found in the literature by using the term “nanotectonics.”
Emulsions and Foams
The basic idea behind emulsion templating is to use sol-gel processing to deposit an inorganic material at the exterior of emulsion droplets. Imhof and Pine were the first to show the applicability of this method in the formation of macroporous oxides (Imhof and Pine 1997).
An emulsion is a two-phase mixture (droplet and continuous) of immiscible fluid phases in which one is dispersed in a second in the form of droplets (Fig. 4). Generally there are two types of emulsion: oil-in-water (O/W) emulsion where the droplet phase is an organic solvent while the continuous phase is water and water-in-oil (W/O) emulsion where water or an aqueous solution is the droplet phase with an organic continuous phase. To form an emulsion, a suitable surfactant (emulsifier) is generally required to stabilize the droplets dispersed in the continuous phase. In order to use emulsions as templates in the preparation of porous oxides, the ceramic precursor sol is added to the continuous phase (mostly aqueous for the synthesis of monoliths), and gelation is induced by, i.e., changing the pH value. Upon polymerization the oxide structure is frozen while the emulsion structure is maintained, and, upon removal of the emulsion phase, a macroporous, mostly closed cell, structure is produced. Mesopores can be generated when a structure-directing agent is co-added to the ceramic precursor sol. In this case, hierarchically organized meso-/macroporous objects are obtained as nicely reviewed recently for organic polymers by Silverstein et al. (Silverstein 2014). Emulsion templating takes advantage of the fact that oil droplets – compared to solid particulate templates – are: (1) highly deformable to allow the inorganic gel to accommodate large shrinkages and thus prevents cracking during drying, (2) can yield architectures on a scale ranging from 5 to 600 μm, and (3) the emulsion droplets are easily removable by evaporation, extraction, or calcination. Compared with the often applied hard-templating approach, the combination of using mesostructure-directing agents together with an emulsion has the advantage that the emulsion droplet size can be adjusted by changing the emulsification conditions, and the use of block copolymer species, which self-assemble to a large extent independently of the emulsion formation, allows tailoring the macro- and mesopore size.

Schematic diagram of emulsions and high internal phase emulsions (HIPEs). (top) Normal oil-in-water (O/W) and the reverse (W/O) emulsion; (bottom) high internal phase emulsion
The number of droplets in an emulsion can be varied, normally expressed as the volume ratio of the droplet phase to the continuous phase or the volume percentage of the droplet phase in the emulsion. The droplet volume fraction can even exceed the close-packing limit of 74%, which corresponds to the most compact arrangement of uniform undistorted spherical droplets; this type of emulsion is called high internal phase emulsion (HIPE) in which the structure consequently consists of deformed and/or polydispersed droplets (Fig. 4). The droplets are separated by a thin continuous phase, and a structure resembling gas–liquid foams is formed. Consequently interconnected macroporous structures can be prepared with HIPE systems. Emulsion templating, especially using HIPEs, is typically situated at the borderline between soft and hard templating. Two cases have to be distinguished: On one hand the purely liquid state, in which the emulsion-forming species are in a dynamic equilibrium, and on the other, emulsions, in which the continuous or droplet phase has been polymerized prior to the actual solidification of the continuous network, thus forming a solid foam (Studart et al. 2006; Alvarez and Fuertes 2007).
Ice Templating
Freeze-casting or ice templating takes advantage of the growth of ice crystals to template molecular, high-molecular-weight precursors, or colloidal suspensions in either a water solution, a suspension, or a hydrogel followed by sublimation of the ice phase (Deville 2008, 2013; Gutiérrez et al. 2008). The formation of crystalline ice causes the substances originally dispersed in the aqueous medium to be expelled to the boundaries between adjacent ice crystals. High-vacuum sublimation of the ice by subsequent freeze-drying gives rise to cryogels (even monolithic ones) with macroporous structures, which are replicas of the original ice structure. When compared with conventional methods, ice templating has many advantages, such as: Flawless components with monolithic shape can be produced; it does not require the addition of special templates which usually lead to high production costs and require severe removal processes (e.g., calcination and chemical etching, using a strong base). Since the solidification is often directional, the porous channels run from the bottom to the top of the samples (Fig. 5). The final porosity content can be tuned by varying the particle content within the slurry, and the size of the pores is affected by the freezing kinetics.

The four processing steps of freeze casting: slurry preparation, solidification, sublimation, and sintering (Reprinted with permission from reference Deville (2008). Copyright (2008) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim)
The variety of materials processed by ice templating suggests that the underlying principles of the technique are not strongly dependent on the materials but rely more on physical rather than chemical interactions.
Hierarchically Organized Porous Materials: Selected Examples
Silica
The formation of silica monoliths comprising either, micro-, meso-, or macropores is well known for several years. The most prominent macroporous example is Vycor glass, which is prepared by phase separation of a melt-quenched metastable glass that is reheated close to its glass-transition temperature followed by a leaching process (Enke et al. 2003). Mesoporous silica monoliths are well known from silica aerogels (Hüsing and Schubert 1998), and zeolite monoliths exhibiting micro- and macropores are accessible by, i.e., pseudomorphic transformation reactions to name only some examples (Sachse et al. 2011).
In the early nineties two pioneering discoveries resulted in a boost in the development of high surface area materials with controlled porosity. On one hand, two groups in the USA and Japan independently published work on porous silica templated with surfactants with a periodic ordering in the mesoscopic regime (Yanagisawa et al. 1990; Kresge et al. 1992), and on the other in 1991 Nakanishi and Soga published the first paper on a meso-/macroporous material prepared via a sol-gel route accompanied by a phase separation process (Nakanishi and Soga 1991). Both processes have seen extensive progress in the last decades, and in many cases combinations of both – soft templating with surfactants or block copolymers and phase separation – in sol-gel system result in the desired pore structure.
Phase Separation by Addition of Polymers
In a typical synthesis, a tetraalkoxysilane is mixed with water in ratios of water/Si > 4 (in a solvent), and a polymeric phase separation agent is added, e.g., poly(ethylene oxide), poly(acrylic acid), etc. In simple terms, this phase separation process – and with that the macroporous structure – is governed by the interaction between the condensing precursor molecules, the polymeric species, and the solvent. This has been well investigated for silica-based systems, showing that polymers not having any specific attractive interactions with silanols, e.g., poly(acrylic acid) or poly(sodium styrene sulfonate), stay in the solvent phase and thus directly relate to the volume fraction of macropores in the dried material (Nakanishi and Soga 1991, 1992). Polymers that strongly interact with the growing silica network by, i.e., hydrogen bonding interactions, such as poly(ethylene oxide) or cationic surfactants, are typically distributed in the gel phase. In this case, the macropore volume fraction is more or less only determined by the amount of solvent, but the domain size and tendency of phase separation are influenced by the polymeric additive (Nakanishi 1997). If the timing and the dynamics driven by the interfacial energy between phase separation and sol-gel transition are chosen properly, bicontinuous gels constituted by two interconnected phases on the micrometer length scale are formed, one being rich in silica, the other one being rich in solvent. After removal of the solvent and drying, structures with macropores resembling the solvent phase and solid architectures comprising textural mesoporosity in the 10–20 nm range and high specific surface areas are obtained.
Keeping in mind that hydrogen bonding results in polymers strongly interacting with the gel phase, a natural extension of the described process is the addition of polyether-based or cationic surfactants/block copolymers to the sol-gel mixture to tailor the mesoporous structure of the materials. Smått et al. (Smått et al. 2003) as well as the group of Nakanishi (Sato et al. 2001; Nakanishi et al. 2003) successfully added different kinds of surfactants (triblock copolymers based on poly(ethylene oxide) and poly(propylene oxide) units or cetyltrimethylammonium bromide) in a double-templating approach. While Smått et al. (2003) relied on a combination of a homopolymeric phase separation agent (PEO) and a cationic surfactant (CTAB) to obtain a hierarchically organized silica monolith with small mesopores (~3 nm) and macropores of 0.5–35 μm, Nakanishi solely used the amphiphilic block copolymer or cationic surfactant as phase separation and supramolecular templating agent. In the latter cases, the mesopores exhibit a certain degree of long-range ordering.
A key problem in the combined sol-gel processing and phase separation/supramolecular templating strategy toward materials with a periodic arrangement of the mesopores lies in the presence of the low-molecular-weight alcohol that is released upon hydrolysis of, e.g., tetraethoxy- or tetramethoxysilanes. Many of the supramolecular arrangement of block copolymers or surfactants are not compatible to higher concentrations of these alcohols as shown by Alexandridis et al. (Ivanova et al. 2000b). Hüsing et al. avoided this problem by applying tetrakis- (2-hydroxyethyl) orthosilicate as the silica source instead of TEOS or TMOS (Hüsing et al. 2003). Here, ethylene glycol is released upon hydrolysis of the silane, which has been proven to be compatible with a variety of lyotropic surfactant phases (Ivanova et al. 2000a, 2001). Processing of this glycol-based silane in the presence of an amphiphilic triblock copolymer surfactant (Pluronic P123®) gave hierarchical macro-/mesoporous silica monoliths with ordered mesopore organization after supercritical extraction with carbon dioxide. This work has been extended to a variety of diol- and polyol-modified silanes and typically networks with macropores between 500 nm to 5 μm, and periodically arranged, uniform mesopores of about 5–10 nm are obtained (Fig. 6; Brandhuber et al. 2005; Hartmann et al. 2007; Triantafillidis et al. 2013). It is again noteworthy to mention that the key point in the preparation of these hierarchically organized silica monoliths lies in the timing of the concurrently occurring phase separation and gel formation processes. Thus, the network structure is not only influenced by the type of diol that has been used to modify the silane (Brandhuber et al. 2005) but also by the choice of acid that is used to start the sol-gel reactions (Flaig et al. 2015) or the amphiphilic molecule that is added as phase separation agent (Hartmann et al. 2014).

TEM image (left), SEM image (middle), and photograph of a silica monolith prepared from EGMS in the presence of a nonionic block copolymer (P123) at pH = 1
Other precursor systems that have been applied in the synthesis of silica monoliths with bimodal pore systems are based on triethanolamine solution with the corresponding silatrane silane derivatives (El Haskouri et al. 2002). Monoliths with 4 nm mesopores and a second level of pores with sizes of 30–60 nm are obtained in the presence of the cationic surfactant CTAB. Instead of liquid precursors, preformed, high surface area, mesoporous silica nanoparticles could also be used to prepare hierarchically organized materials (Huerta et al. 2007). Many more examples can be found by applying combinations of supramolecular arrangements and hard templates to achieve a hierarchical organization (Petkovich and Stein 2013).
In principle, dual or even multiple micellar-templating approaches based on two or more different structure-directing agents, such as surfactants, block copolymers, or ionic liquids, could be used in the formation of monoliths with multimodal pore sizes (Sel et al. 2006). However, the different structure-directing agents will show a rather complex mixing behavior, thus becoming very difficult to control, and the delicate interaction behavior between the different species will determine whether hierarchical structures are formed or not. So far, the formation of monolithic silica has not explicitly be mentioned; however, the process allows for the formation of different morphologies as presented by Yuan et al. (Yuan et al. 2010).
Not only purely silica-based monoliths can be prepared by the abovementioned approaches but also inorganic–organic hybrid materials are accessible (Hüsing et al. 2006; Hartmann et al. 2007; Kanamori and Nakanishi 2011). Co-condensation reactions of tetraalkoxysilane or tetrakis(2-hydroxyethyl) orthosilicate with organo-functional trialkoxysilanes, such as methyl-, phenyl-, vinyl-, aminopropyl, methacryloxypropyl-, or glycidoxypropyl-derivatized ones to name only a few, are routinely used in the preparation of functionalized silica-based materials. However, one has to keep in mind that changes in polarity in the sol might result in completely different phase separation behaviors, thus resulting in different porous network structures. A very detailed investigation on the formation of hybrid chloroalkyl-modified, meso-/macroporous silica monoliths and the structural changes observed due to the presence of the organofunctional silane and its organic chain, as well as due to post-synthetic processes, such as azide–alkyne Click reactions, has been presented by Keppeler et al. (Keppeler et al. 2011, 2015; Keppeler and Hüsing 2011).
In addition to co-condensation reactions, pure silsesquioxane-based hybrids are accessible via condensation reactions of the sole organotrialkoxysilane or bis-(trialkoxysilyl) precursors, such as 1,4-bis[tris(2-hydroxyethoxy)silyl]benzene or the corresponding alkoxy derivatives (Brandhuber et al. 2006; Kanamori and Nakanishi 2011). Even dendrimeric silanes as well as cyclic preceramic precursors, such as a glycol-modified 1,3,5-trisilacyclohexane carbosilane, have been used in the formation of meso-/macroporous hybrid monoliths (Weinberger et al. 2008, 2010).
Emulsions and Foams
Dual meso–macroporous silica monoliths from polymer foams have been presented by the group of Chmelka in 2003 (Maekawa et al. 2003). However, this is an example, in which styrene first was prepolymerized in an emulsion to give a foam that was soaked in a second step with an acidic silane-based sol-gel solution containing amphiphilic block copolymer species as structure-directing agents. Silica monoliths with cellular macropores (0.3–2 μm) comprising 0.2–0.5 μm cell windows and highly ordered mesopores (5.1 nm) were obtained.
A true emulsion-based approach was used by Sen et al., who reported the formation of mesoporous silica, meso-cellular silica foams (MCFs), macro-cellular silica foams (UMCFs) and ordered macroporous silica in a one-pot synthesis at room temperature. However, no comment on the macroscopic morphology of the material was made. At very low oil concentration with slow stirring mesoporous silica was obtained, whereas meso-cellular silica foams were formed with faster stirring. Syntheses using intermediate-to-high oil concentration produced macroporous solids with various pore sizes and wall thicknesses. Upon increasing the pH from acidic to neutral to basic, the macroporous structure starts to disappear, and a mesoporous solid is formed (Sen et al. 2005).
The group of Backov applied concentrated emulsions, so-called HIPE systems prepared from dodecane in the presence of an aqueous tetradecyltrimethylammonium bromide (TTAB)–tetraethoxysilane mixture for the preparation of highly porous silica monoliths (Fig. 7; Carn et al. 2004a). TTAB serves as a mesoscopic texturing agent. They obtained hierarchically organized materials with very low densities, vermicular-type mesoporosity, and macropore sizes in the range of 1–100 μm that they labeled as Si (HIPE). The macroscopic void space could be varied by varying the starting oil volume fraction of the O/W concentrated emulsion; however the texture always resembled hollow spheres.

SEM visualization of the inorganic monolith-type material macrostructure. (a) and (b) 1Si-HIPE, (c) and (d) 2Si-HIPE, (e) and (f) 3Si-HIPE (Image taken from reference Carn et al. 2004a)
Even air–liquid interfaces in foam structures have been applied in the formation of monolithic materials comprising multiple levels of pore sizes. Carn et al. (2004b) could show that foams give a high level of control over macropore characteristics, such as size, topology (open versus closed cell), and morphology (spherical versus polygonal cell structures). The foaming solution consisted of colloidal silica that has been prepared via the Stöber method and a cationic surfactant in an aqueous medium (pH = 9); the foam was generated by continuous bubbling of perfluorohexane-saturated nitrogen through a porous glass disk.
Another unusual approach toward meso-/macroporous silica monoliths has been presented by Vuong et al., who used the release of oxygen gas from hydrogen peroxide decomposition through a silica gel with low viscosity that has been prepared in the presence of a nonionic surfactant. The escape of the oxygen bubbles results in a significant and rapid expansion of the gel body and a spongelike silica monolith exhibiting periodically ordered mesopores within grains of 10–20 μm in diameter, and wall thicknesses of 0.5 μm was obtained (Fig. 8; Vuong et al. 2008).

Scanning electron micrographs (SEMs) of meso-/macroporous silica spongelike networks as well as a photograph of the spongelike network formed in the presence of hydrogen peroxide (With kind permission of Springer Science+Business Media; reference Vuong et al. 2008)
Ice Templating
As described above, ice crystals can be used as macrotemplates, as demonstrated by Nishihara et al. who prepared ordered macroporous materials with micro–mesoporosity by thermally induced phase separation (Nishihara et al. 2005). By this method, it is possible to precisely control the macroporosity, wall thickness, and micro-/mesoporosity of silica materials via very simple procedures.
Tamon and co-workers successfully produced silica monoliths that were not only macroporous (the cell size of the micro-honeycomb structure ranges between 10 and 15 μm) but also meso- and microporous (the BET surface area ranges between 400 and 700 m2/g). Faster immersion rates of the gel in the cold bath produces smaller macropores. It is noted that the microporosity is simply a consequence of the voids left between silica colloids packaged at the boundaries of adjacent ice crystals (Fig. 9) and can be adjusted by the pH of the parent silica sol.

Morphology and structure of silica monoliths exhibiting a micro-honeycomb structure. Upper: (a) An overall image. SEM images of (b) cross section, (c) microchannel structure, and (d) longitudinal section. Lower (a) Detail of a cross section. Nitrogen isotherms (b) are also represented. The inset shows the mesopore size distribution in the desorption branches (Reprinted with permission from reference Mukai et al. 2004. Copyright (2004) The Royal Society of Chemistry)
Non-siliceous Monoliths
The methods for preparing non-siliceous materials are in principle similar to those for preparing silica monoliths. However, a one-to-one transfer is not possible due to the typically higher reaction rates of the metal precursors (alkoxides and metal salts) and the tendency of metal oxides to crystallize at relatively low temperatures. The first mesoporous non-siliceous materials were reported by Ying et al. in 1995 (Antonelli and Ying 1995). Since then, the synthesis of mesoporous non-siliceous metal oxides and mixed oxides has seen major progress. The most common synthetic procedure to these materials is based on hard templating (nanocasting) or colloidal crystal templating by using preformed silica or carbon materials. While some excellent reviews and papers concerning these synthetic approaches are available (Lu and Schüth 2005; Smått et al. 2006, 2012; Ren et al. 2012; Petkovich and Stein 2013), only few publications focusing on the preparation of non-siliceous monoliths with hierarchical porosity via sol-gel processing directly from solution can be found. In this chapter we give an overview of the progress in this topic in the last years and introduce a few recent examples.
The main difficulties in the synthesis of porous, non-siliceous, hierarchically organized monoliths are: (i) to control the high reactivity of the metal precursors, e.g., metal alkoxides or salts, (ii) to preserve the monolithic form during calcination processes, and (iii) to simultaneously control the resulting crystallinity and porosity. With respect to the first point, metal alkoxides are stronger Lewis acids than silicon alkoxides and thus facilitate the nucleophilic attack of water or other molecules. Furthermore, most metals have several stable coordination numbers and are therefore often present with a coordinatively unsaturated valence state. Both effects dramatically increase the reactivity and thus make it difficult to control homogeneous gelation. As a result, precipitates instead of monolithic materials are often obtained.
Today, various approaches to moderate the reactivity of metal alkoxides have been reported. One possibility is to add chelating agents, e.g., acetylacetone (Dutoit et al. 1995) or carboxylic acids (Takenaka et al. 2000) to attenuate the reactivity of the titanium precursor by replacing part of the alkoxy groups. Therefore, the coordination state of titanium is stabilized, and the reactivity toward nucleophilic attacks is lowered. Another possibility to tackle this problem is the addition of strong acids. At low pH, the particle surfaces are positively charged, and condensation is slowed down due to electrostatic repulsion between equally charged particles. If the pH is raised gradually, e.g., by the addition of formamide, the electrostatic repulsion is reduced, and the particles can aggregate until the formation of a gel (Konishi et al. 2006a).
Gash and co-workers developed a similar approach for the synthesis of stable non-siliceous aerogel monoliths without the need of strong acids (Gash et al. 2001). In their alkoxide-free sol-gel approach, they raised the pH gradually by the addition of epoxides to aqueous and/or ethanolic solutions of metal salts. The effect of the epoxide as an acid scavenger is illustrated in Fig. 10. First, the oxygen atom of an epoxide, e.g., propylene oxide, is protonated by an acid and subsequently undergoes a ring-opening reaction by a nucleophilic attack of an acid anion. This epoxide method allows the formation of various metal oxides in different morphologies, e.g., monoliths, powders, or thin films.

Propylene oxide as acid scavenger (Adapted with permission from reference Kido et al. (2012). Copyright (2012) American Chemical Society)
The amount of water in the sol represents another possibility to influence the hydrolysis rate of the precursor molecules. Water can either be directly added to the system or it can be released during the reaction, e.g., by esterification reactions. A summary of different possibilities to control the reaction rate of metal precursors is shown in Fig. 11.

Methods to control the reaction rate of metal precursors
These sol-gel techniques are often combined with a polymerization-induced phase separation process that has already been described in the previous section for siliceous materials. In these cases, typical bimodal porosities are obtained with micro- and mesopores that often result from interstices between metal oxide crystallites. The size and shape of the macropores, however, can mostly be adjusted by the concentration of the polymer, which is used as phase separator and/or the gelation time of the sol. Table 1 gives an overview over the syntheses known to date toward non-siliceous monolithic materials with hierarchically organized pore sizes prepared via sol-gel processing. In Table 2 selected follow-up reactions, e.g., carbothermal reduction or solvothermal treatment, of the monolithic compounds of Table 1 are summarized.
A selection of some recent key examples is presented below in more detail. Particular focus is given to meso-/macroporous titania monoliths since several of the underlying principles have been developed for this class of non-siliceous monoliths. Commonly, metal alkoxides, metal salts, or even preformed metal oxide particles are used as precursor molecules, and the wet gels are typically dried at 30–60 °C in an oven and calcined at higher temperatures. This and further details for each synthesis are described in the individual examples.
Phase Separation by Adjusting the Sol-Gel Processing Conditions
The interest in hierarchically organized titania monoliths in diverse application areas, e.g., for separation science is significant. Despite the difficulty to decrease and control the reaction rates of the precursor molecules, a variety of approaches toward monolithic materials with bicontinuous porosity has been published. Lindén et al. reported a template-free synthetic approach toward hierarchically macro-/mesoporous anatase monoliths based on the sol-gel reaction of titanium isopropoxide in the presence of two different acids, namely, hydrochloric and acetic acids (Backlund et al. 2007). Hydrochloric acid as a strong acid enhances hydrolysis, but decreases the reaction rate of condensation. Acetic acid, however, slows down both hydrolysis and condensation rates, by acting as a chelating agent. Thus, the prepared wet gels were first dried at 60 °C for 24 h prior to calcination at 300 °C. After thermal treatment the material consists of fully crystalline anatase particles with crystallite sizes between 10 and 15 nm. Aggregation of these near to spherical particles leads to the formation of an interconnected, three-dimensional macroporosity. The macropore size can be controlled by the careful adjustment of the sol-gel processing conditions. Above a molar ratio of HAc/Ti(OiPr)4 2:1, small amounts of water are released by an esterification reaction between acetic acid and isopropanol. This in turn causes an increase of the condensation rate of the titanium alkoxide and further as well as of the solvent volume. Both contribute to an increase in the macropore diameter.
A slightly modified form of this approach was used by Wei et al. for the synthesis of anatase monoliths for chromatographic applications (Wei et al. 2013). They successfully synthesized a material that comprises several properties which are prerequisites for the use in chromatographic applications, e.g., high specific surface areas and large-pore diameters.
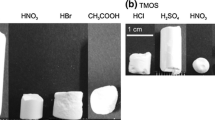
A further possibility to generate multiscale porous titania monoliths in template-free conditions was reported by Konishi et al. and is based on sol-gel processing of Ti(OnPr)4, hydrochloric acid, formamide, and water (Konishi et al. 2006a). Formamide is known to react with strong acids, thereby producing ammonia and thus increases the pH value gradually, e.g., from below 0 to 5 after 24 h aging time. This increase in pH can promote condensation reactions and induce sol-gel transitions. At the same time, the number of OH groups and thus the polarity of the gel phase are lowered with progressive degree of condensation.
Since the polarity of the solvent remains high, phase separation between the condensed and the liquid phase occurs, and macropores are formed after drying of the sample at 60 °C. In addition, the material crystallizes during drying and small anatase crystallites are formed. The authors further show that the macropore size of the crystalline network can be controlled via the composition of the starting solution and/or the temporal relationship between phase separation and gelation time (Fig. 12). In this context, high amounts of water delay the onset of phase separation, resulting in a finer bicontinuous structure of the monoliths, whereas, with small amounts, mainly spherical particles are formed.

SEM images of dried TiO2 gels with increasing ratios of water/TiO2 (a–e). Digital picture of monolithic TiO2 gels prepared in Teflon tubes and a coin (f) (Figure adapted with permission from ref Konishi et al. (2006a). Copyright (2006) American Chemical Society)
Polymers as Phase Separation Agents
TiO2
Konishi et al. extended their studies to the application of the abovementioned monoliths as chromatographic separation media (Konishi et al. 2009). The need for high mechanical strength was addressed by increasing the titanium precursor content in the starting sol. On one hand this indeed strengthens the network, whereas on the other the reactivity of the precursor solution in such concentrated systems is dramatically increased resulting in a loss of control over phase separation. To regain better control, the ammonium source formamide was replaced by N-methyl formamide (NFA), which acts as acid scavenger and hydrolyzes much slower than formamide. Therefore, the pH is gently increased enabling a better control of the sol-gel transition even at high precursor concentrations. As a consequence of the high precursor concentration, the amount of propanol in the system is relatively high and almost no phase separation was observed. To improve the phase separation tendencies, the authors added the water-soluble polymer poly(ethylene oxide) PEO to the system. The polymer adsorbs to TiO2 oligomers via hydrogen bonding and is therefore able to reduce the solubility of the oligomers in the solvent. The careful choice of the Ti(OnPr)4–NFA–PEO composition finally enables the production of TiO2 monoliths with controllable porous morphology. Several similar approaches in which polymers are used as phase separation agents are listed in Table 1.
The influence of mineral salts on the sol-gel processing of TiO2 oligomers and the role of strong acid anions as blocking agent to prevent titanium atoms from nucleophilic reactions were reported by Hasegawa et al. (2010b, c). The authors reported the synthesis of monolithic titania with multiscale porosity by utilizing PEO as phase separator, ethyl acetoacetate as chelating agent, and ammonium nitrate as mineral salt; the latter one is reported to further stabilize the chelated species and decrease the hydrolysis rate. In order to retain the monolithic shape of the wet gels during calcination, the authors removed the employed chelating agent by hydrolysis in EtOH/H2O, followed by decarbonation into acetone and carbon dioxide (Fig. 13). After crystallization of the amorphous gel skeleton in warm water, macroporous TiO2 monoliths with well-defined mesopores attributed to interstices between anatase crystallites were obtained.

Ethyl acetoacetate converts to acetoacetic acid by hydrolysis. The generated acetoacetic acid immediately decomposes into acetone and carbon dioxide
A biocompatible approach toward meso-/macroporous titania monoliths was described by Brook and his co-workers (Chen et al. 2006). They used glycerol to slow down the reaction rate of hydrolysis by transesterification of Ti(OiPr)4 (Fig. 14) and to accomplish the sol-gel reaction at neutral pH without the need for the addition of any catalyst. With the addition of PEO, they obtained bimodal meso- and macroporous amorphous structures that could be crystallized to anatase monoliths at temperatures above 600 °C.

Glycerol-modified titanium precursors (Figure adapted with permission from reference Chen et al. 2006. Copyright (2006) American Chemical Society)
In addition to the fully oxidized phases, reduced materials with tailored pore structures are known. Hasegawa and co-workers reported a novel synthesis toward reduced titanium oxide monoliths with well-defined hierarchical pore structure by the use of an ethylenediamine-modified titanium precursor (Hasegawa et al. 2013). The alkoxy groups of the titanium precursor Ti(OiPr)4 were substituted by ethane-1,2-diamine, and the authors obtained inorganic–organic gels with Ti–N linkages (Fig. 15). Heating of the samples in an argon atmosphere initially resulted in the formation of anatase or rutile that converted toTi4O7 and Ti3O5 at 800–900 °C by carbothermal reduction. The temperatures required for these reactions are exceptionally low, since the reduction reactions of anatase and rutile phases to Ti4O7 by H2 gas (Kolbrecka and Przyluski 1994), metals (Hauf et al. 1999; Kitada et al. 2012), or carbons (White et al. 1992) known to date require temperatures of more than 1000 °C. The authors explain this low-temperature reduction with: (i) the small size of the anatase and rutile crystallites, (ii) the carbon coating, and (iii) the N-doping which distorts the Ti-O lattice and decrease the stability against reduction. With increasing temperature, the amount of micro- and mesopores increases, and specific surface areas of up to 200 m2g−1 have been obtained. Further reduction of Ti3O5 to Ti2O3 initially results in the loss of micropores and the formation of mesopores, whereas the formation of TiOxNy at 1400 °C is accompanied with the loss of micro- and mesoporosity. Further studies on the effect of calcination conditions on the micro- and mesoporosity of these samples as well as on the electric conductivity have been reported recently (Hasegawa et al. 2015).

Scheme of sol-gel processing of titanium alkoxides in the presence of ethane-1,2-diamine and the development of the crystalline character of the resulting TiO2 monoliths upon treatment at different temperatures
Fe3O4, Fe
Similar approaches were reported for various iron- (Fe3O4, iron, and Fe2O4) and chromium-based (CrN and Cr3C2) crystalline monoliths (Kido et al. 2012, 2014). The main difficulty in preparing iron(III) oxide monoliths lies in the tendency of the precursor sol to form precipitates of iron(III) hydroxide. Nakanishi and his group reported a synthetic route toward iron-based monoliths from an aqueous solution of iron(III) chloride hexahydrate (Kido et al. 2012). Upon adjusting the solvent composition, polymer, and epoxide content, the authors were able to control the morphology and the gel formation of iron(III) hydroxide. As a simultaneous phase separation inducing and network-forming agent poly(acrylamide) was added. The conversion of the amorphous iron(III) hydroxide structure into crystalline hematite (α-Fe2O3) by calcination of the material in air was accompanied with collapse of the monolithic form, whereas it can be preserved by calcination in an inert argon gas flow (Fig. 16).

Scheme of sol-gel processing of titanium alkoxides in the presence of ethane-1,2-diamine and the development of the crystalline character of the resulting TiO2 monoliths upon treatment at different temperatures
In nonoxidizing atmospheres, the organic species are converted to carbon, which in turn acts as a reducing agent to yield crystalline Fe3O4, iron, and Fe3C from iron(III) species. With this process, the monolithic form as well as the macrostructure can be retained; however, the pore size and pore volume decrease with increasing temperature. The mesopore size decreases from 5 to 6 nm for the as-dried gels to 3–4 nm for the heat-treated samples. For the samples heated above 400 °C, micropores appeared due to the combustion of carbon in the skeleton. Since the specific surface area of the heat-treated samples mainly depends on the proportion of micropores, the BET values increased from 5 m2g−1 at 300 °C, through 224 m2g−1 at 700 °C to 262 m2g−1 at 1000 °C. This approach can be transferred to the synthesis of nickel–carbon composite monoliths from rigid nickel hydroxide-based xerogels (Kido et al. 2013).
For the preparation of chromium-based monoliths, the authors combined the “urea glass route” in which urea is employed as the nitrogen and/or carbon source with the epoxide-mediated sol-gel route (Kido et al. 2014). Subsequent heat treatment under an inert atmosphere led to the formation of crystalline chromium nitride and chromium carbide.
Al2O3
Porous alumina (Al2O3) is another class of oxidic material that attracts considerable attention due to its high thermal stability and moderate Lewis acidity. The first report on macroporous Al2O3 monoliths prepared by combining the phase separation with sol-gel processing was reported by Tokudome et al. in 2007 (Tokudome et al. 2007b). Their recipe of success was the use of: (i) aluminum salts instead of aluminum alkoxides to decrease the hydrolysis rate of the precursor, (ii) propylene oxide to start gelation by gradually increasing the pH value of the sol, and (iii) PEO to induce the formation of phase-separating structures. With variation of the PEO concentration, the authors were able to change the gel morphology from nonporous through bicontinuous to particle aggregates while the macropore size can simultaneously be controlled in the range of 400 nm to 1.8 μm. Additionally, the authors were able to influence the degree of agglomeration grade and size of primary particles via the drying process (Tokudome et al. 2009). Hartmann et al. extended this work and mixed the aluminum precursor with the PEO/EtOH/H2O solution under ice-cooled conditions prior to the addition of propylene oxide (PO) at 25 °C (Hartmann et al. 2012). With this method, they were able to perfectly control condensation and phase separation. Gawel et al. reported a method in which an aluminum nitrate precursor was hydrolyzed in the presence of boehmite (Gaweł et al. 2012). The interactions between the hydrolyzed aluminum molecules and the wetted surface of boehmite particles are formed leading to a three-dimensional network by condensation reactions (Fig. 17). Crystalline alumina with boehmite crystal structure is formed at temperatures up to 300 °C, whereas at temperatures higher than 400 °C, the crystalline phase is transformed into γ-alumina. The authors confirmed the hierarchical structure of the alumina samples with nitrogen sorption and mercury porosimetry measurements revealing the existence of meso- and macropores. Both originate from the aggregation of plate crystallites or inter-aggregate voids formed during the drying and calcination steps.

Formation of a three-dimensional network by condensation of boehmite particles with hydrolyzed aluminum molecules
CuO
Recently, Fukumoto et al. reported the possibility to convert hierarchically organized copper hydroxide-based monolithic xerogels into copper oxides (CuO and Cu2O) under preservation of the monolithic form and macrostructure (Fukumoto et al. 2015). A mixture of copper(II) chloride dihydrate, water, ethanol, glycerol, and PAAm was used as starting solution. In their approach, the presence of glycerol had a significant influence on the crystal growth with lower amounts leading to crystalline precipitates and higher amounts suppressing crystallization and allowing for preservation of the monolithic shape. In contrast, the added PAAm in the starting solution had no influence on the crystallization process, but the morphology was strongly influenced due to the incorporation of PAAm in the gel skeletons. The obtained morphologies varied from co-continuous structures consisting of similar-sized globular units for low PAAm concentrations to isolated macropores for excessive amounts. Therefore, it not only controls phase separation but also physically supports the network. The latter is particularly evident in the calcination process from copper(II) hydroxide to copper(II) oxide. Calcination in air resulted in the formation of crystalline copper(II) oxide, but also in collapse of the monolithic shape. This was improved by a pre-calcination step in argon at 800 °C, followed by calcination in air at 400 °C. Although, the monolith was retained, calcination in argon at such high temperatures is accompanied by the collapse of the mesopores. Simply a solvothermal treatment resulted in meso-/macroporous monoliths. However, the crystalline phase consisted of a mixture of copper(0)/PAAm and copper(I)/PAAm.
The positive effect of solvothermal treatment in tailoring the meso- and crystal structure without destroying the macroporous structure was also reported by Hirao et al. and Guo et al. for the synthesis of meso-/macroporous zirconia (Konishi et al. 2008; Guo et al. 2015). With the solvothermal treatment, the authors obtained a high density of mesopores in the crystallized material via Ostwald ripening and high specific surface areas of up to 584 m2g−1.
Carbon Monoliths With Hierarchical Pore Structure
Porous carbon materials have remarkable physicochemical properties, such as hydrophobicity, high corrosion resistance, good thermal stability, easy handling, and in many cases rather low costs in manufacturing, resulting in materials suitable for many areas of applications, such as energy storage or conversion, e.g., as battery electrodes or supercapacitors, in capacitive desalination, chemical catalysis, and electrocatalysis, to mention only a few examples (Zhai et al. 2011; Roberts et al. 2014a). As in the previous sections discussed, pore structure control is of major importance for carbon materials as well, not only to increase the surface area but also to adjust the accessibility of the active sites and to deliberately tailor the material for the specific application. Hierarchically organized, porous carbons are of special interest, for the reasons mentioned above. Numerous reports on the preparation of porous carbon monoliths via hard-templating approaches (sometimes even in combination with soft templating) can be found in the literature, and many excellent articles have been published (Yang and Zhao 2005; Ungureanu et al. 2015). As mentioned above, we will limit ourselves to “soft-templating” routes in combination with solution-based processes relying on polycondensation reactions as the network-forming mechanism due to the nature of this chapter. In addition, only hierarchical pore structures within a macroscopic monolithic shape will be covered. One major difference to the materials discussed in the previous sections is the high chemical similarity of the organic precursor and network-forming species to the majority of structure-directing agents used. Thus, chemical interactions during network formation are very likely, and removal of the structure-directing agent is expected to be more difficult in some cases. An overview of different approaches to monolithic carbon materials with hierarchical pore structure is shown in Table 3.
Polymerization of Organic Monomers
Acid-catalyzed sol-gel polymerization of resorcinol with formaldehyde (Moreno et al. 2013) is a widely used method toward monolithic carbon materials with large-pore volumes, micro- and mesopores, and specific surface areas up to 3000 m2 g−1 after carbonization of the as-synthesized material (Baumann et al. 2008). It was found that, e.g., shape, size, and arrangement of the primary particulate network, can be influenced by the amount and type of the catalyst (Roberts et al. 2014a). The general procedure comprises four main steps: (1) dissolution of the precursors, e.g., resorcinol and FA in water or a water–alcohol mixture with a catalyst, (2) gelation of the precursor sol, (3) drying to remove the solvent, and (4) carbonization and thermal treatment of the dried polymeric monolith. Additional porosity can be generated by an activation or etching process (acid based or by CO2 treatment) resulting in the formation of micropores. The reaction with CO2 (at ~950 °C) is based on the Boudouard equilibrium, by selectively reacting carbon with carbon dioxide to carbon monoxide at that temperature (Biener et al. 2012).
Drying of a porous monolith (step 3) is known to need particular care because capillary forces might destroy pores; cracks can evolve and even shrinkage can take place as discussed before. Besides supercritical extraction of the organic gels, freeze-drying processes to yield cryogels have been developed to avoid collapse of the network. For this purpose, the gelled monolith is frozen, and the frozen solvent crystals, which simultaneously act as templates, are removed by freeze-drying or solvent exchange (Candelaria et al. 2012).
Typical monomers, used for the synthesis of organic gels and after pyrolysis, carbon monoliths, are shown in Fig. 18. The important factors for the selection of the most appropriate monomers are the condensation rate and the solubility in the respective solvent. After the organic polycondensation reactions and drying of the gels, carbonization in inert atmosphere allows to obtain monoliths with carbon contents of up to 95%. In addition to the pores resulting from polycondensation, pores can be generated by subsequent treatments, e.g., drying or etching of the sample (see etching with carbon dioxide).

Various organic monomers used in polymerization reactions to build up porous carbon networks
In principle, organic polymers can also be formed by other polymerization techniques. As one example of gel formation not relying on polycondensation reactions, a living radical polymerization resulting in macroporous poly(divinylbenzene (PDVB)) monoliths has been published by Hasegawa et al. (Hasegawa et al. 2010a). These gels were stabilized against shrinkage by sulfonation and subsequently carbonized under inert atmosphere to give carbon materials with open and statistically distributed pores of three different levels (macro, meso, and micro) and a high specific surface area of 2360 m2g−1.
Phase Separation
From a thermodynamic point, demixing of a system can be achieved by physical- or chemical cooling (Triantafillidis et al. 2013). In particular, polymerization or condensation, e.g., during sol-gel processing, causes chemical cooling and therefore phase separation. Many organic polymers (e.g., poly(acrylamides), polystyrene/divinylbenzene) can be polymerized in the presence of a poor cosolvent resulting in macroporous, phase separated structures, typically exhibiting ill-defined pore structures with a broad pore size distribution. Microporosity can easily be introduced by etching with, e.g., carbon dioxide (Nakanishi 2011).
Phase separation between mesophase pitch (MP) and polystyrene (PS as phase separation agent) can be induced by evaporation of tetrahydrofuran (THF) to synthesize carbon monoliths with meso- and macroporosity as demonstrated by Adelhelm at al. (2007). FeCl3 as catalyst to accelerate the carbonization is added to a homogeneous solution of MP and PS (MW 35000) in the volatile solvent (THF). With continuing evaporation of THF, the separation into MP-rich and polymer-rich phases starts (spinodal decomposition) and can be extended to the nanometer scale by a heat treatment with temperatures just below the decomposition regime of the polymer (<300 °C). These temperatures (including annealing) induce a better chemical connectivity between the MP molecules, thus strengthening the network. As a result, the spongelike structure can be retained even during the carbonization step at 600 °C. The resulting monolith comprises macropores with 100 μm and mesopores between 10 and 100 nm in diameter, which are not long range ordered but connected in an open-pore manner (see Fig. 19). Worthwile mentioning is the high amount of graphene stacks, which can be attributed to the carbonization of MP. Other carbon sources, such as phenol, resorcinol, or sugars (“hard carbons”), result in carbon structures with less graphitic but more amorphous carbon content (Adelhelm et al. 2012).

Images of a hierarchically structured monolith from MP (a) Photograph of a PS-templated monolith (b) and (c) SEM images of a sample carbonized at 340 °C (inset with higher magnification) and 600 °C (d) Hg porosimetry data of the sample, carbonized at 340 °C (Figure taken from reference Adelhelm et al. 2007)
A dual-phase separation approach (Liang and Dai 2009) to hierarchically structured carbon simultaneously uses microphase separation and spinodal decomposition induced by polymerization and subsequent carbonization resulting in a porous network with pores in the nanometer and micrometer range. The polymerization of phloroglucinol with FA is conducted with two structure-directing agents acting on two length scales: first the microphase separation is induced by Pluronic F127, a triblock poly(EO-PO-EO) copolymer, and afterward phase separation in the μm scale to form macropores results from adding glycolic solvents (EG, DEG, or TEG). The final carbon structure is depicted in Fig. 20. It consists of a bicontinuous, open polymeric network with macropore sizes of 3 μm, skeletal sizes of 1 μm, and mesopores of 8 nm in diameter, which are generated after the pyrolysis step with decomposition of the block copolymer template. The method allows tunability of the macropore size, while the size of the mesopores remains constant. Hasegawa et al. (2012) prepared a carbon–silica composite material via a sol-gel approach followed by pyrolysis in inert atmosphere. As precursors for the formation of the porous network, phenylene- or biphenylene-bridged alkoxysilanes, (BTEB), or (BTEBP), respectively, were used in the presence of a phase separation agent to yield a macroporous structure. After calcination in inert atmosphere, a carbon–silica composite was obtained from which the nano-silica phases could be extracted with NaOH to form micropores. The resulting monolithic carbon exhibits a high specific surface area of >1500 m2g−1. In the case of BTEB as precursor, a mesopore diameter of 4.9 nm is obtained, while the pore diameter can be increased to 10.7 nm for the phenylene-bridged source. Overall, a trimodal porous carbon material with interconnected, non-ordered pores is obtained following this synthetic pathway. In Fig. 21 a SEM picture revealing the open macroporous network is shown, in addition to the schematic processing pathway (Hasegawa et al. 2012).

Schematic drawing of hierarchically structured porous carbon by the dual-phase separation approach with glycolic solvents and F127 as structure-directing agents (Figure used with permission from reference Liang and Dai 2009)

Synthetic scheme to hierarchically porous carbon monoliths over a carbon–silica composite material with structural investigations of the final carbon material by a SEM image (Figure adapted from reference Hasegawa et al. 2012)
Emulsion Templating
A specific type of emulsions in which the droplets exceed 74% of the volume are called high internal phase emulsions (HIPEs) (Brun et al. 2011; Kimmins and Cameron 2011). These closed-packed droplets can also be applied as soft templates to form porous carbon materials. The organic monomers and initiators are first dissolved in the continuous phase, while the emulsion should be stabilized by a suitable surfactant. After polymerization of the monomers (known as polyHIPEs), the solvents from both phases, droplet and continuous phases, are removed, and the remaining polymer is carbonized to obtain the porous carbon structure (c.f. Fig. 22). Recent progress shows the large variability of the emulsion-templating approach: Several monomers, such as glycidyl methacrylate and 2-hydroxyethyl methacrylate of even biomaterials, such as gelatin or dextran, have been successfully used (Kimmins and Cameron 2011). The emulsion system itself can consist of a water-in-oil (W/O) or oil-in-water (O/W) type, and the droplet concentration (HIPE or low concentration) has influence on the resulting porous carbon structure – highly interconnected or closed, isolated pores. Due to the variety of droplet sizes, the resulting macropores are not equal in size. Cohen et al. (Cohen and Silverstein 2011) showed the synthesis of porous carbon monoliths with macro- and mesoporosity from acrylonitrile (AN) copolymerized with divinylbenzene (DVB). The miscibility of AN with water causes difficulties in preparing polyacrylonitrile (PAN)-based polyHIPEs, which was overcome by the stabilization of the HIPE system with a polyglycerol surfactant. Photoinduced polymerization can reduce the polymerization time to some seconds; therefore, no need for longtime stable emulsions is required.

Emulsion-templating approach to porous carbon materials: (a) carbon monomers dissolved in the continuous phase surround the droplet templates (b) porous carbon structure after polymerization and carbonization
A foamlike carbon material was presented by Asfaw et al. (2013), consisting of cellular pores of ~25 μm and meso- and macropores in the foam walls. Their HIPE approach is described as a W/O emulsion system with styrene/DVB/vinylbenzylchloride/span 80 in the oil phase and stabilizing/initializing salts in the aqueous phase, forming a polyHIPE structure. Its pyrolysis results in a final open 3D structure with hierarchical porosity and high surface area of 433 m2 g−1 and is suggested to be used in microbatteries.
Colloidal Crystal Templating
Polymer particles, and in general colloidal particles, can be arranged in closed packing of spheres and are widely used as colloidal crystal templates to produce macroporous monolithic carbon materials (c.f. Fig. 23) (Stein et al. 2013). Among them, poly(methylmethacrylate) (PMMA) and polystyrene (PS) particles have the advantage to decompose in volatile species during carbonization to form spherical voids with sizes corresponding to the particle size. In comparison to silica particles that can only be extracted by treatment with strong bases or HF, polymer-based templates can be extracted from the structure by suitable solvents, such as toluene or THF. If these closed-packed colloidal PMMA templates are infiltrated with a resorcinol–formaldehyde solution, a purely carbonaceous material with hierarchically organized pores remains after polymerization at 85 °C and carbonization at 900 °C (Wang et al. 2008). The macropores are well ordered, while the mesopores are organized in domains of ~150 nm with a cubic arrangement.

Emulsion-templating approach to porous carbon materials: first step (a–c) carbon monomers dissolved in the continuous phase surround the droplet templates; second step (d)) porous carbon structure after polymerization and carbonization
An extension of this approach toward carbon monoliths with ordered macro- and mesopores was reported by Wang et al. (Wang and Stein 2008). The authors added Pluronic F127 to a mixture of monodispersed silica colloidal crystals and resols as carbon source to produce a composite material consisting of carbon and silica (Deng et al. 2007). Silica was removed by washing with a HF solution, and a hierarchically organized porous carbon material with macropores of 312–438 nm and mesoporous walls (mesopore diameter ~12 nm) was obtained.
Ice Templating
The method of “ice templating” (Deville 2013; Nardecchia et al. 2013; Roberts et al. 2014b) has widely been used to prepare highly interconnected macroporous structures (Estevez et al. 2011). Starting with an aqueous solution, suspension, or emulsion, the water is frozen in a controlled fashion (temperature gradient, freezing velocity), forming ice crystals that act as template for the formation of macropores as discussed above. The latter ones are obtained after removal of the ice by freeze-drying (also called lyophilization). A schematic of the preparation is shown in Fig. 24. Nucleation and growth of the ice crystals have a significant impact on the orientation and shape/size of the resulting pore structure. Smaller sizes of the ice crystals are achieved by increasing the temperature gradient and the freezing rate. The ice-templating method has been in use for various materials, such as silica or alumina (Gutiérrez et al. 2008), and can also be utilized for porous carbons (Roberts et al. 2014b). In the work of Estevez et al. (Estevez et al. 2013), ice templating was applied in combination with silica colloidal assemblies and glucose as precursor to generate hierarchically porous monoliths with pores covering three orders of magnitude (macro, meso, and micro). Within the monolith, the macropores are oriented in the growth direction of the former ice crystals and show a pore size distribution mostly in the range from 3 to 12 μm. The size of the mesopores can be tuned by using different silica particles, which are removed in a final step by etching with HF. Additional microporosity was obtained by thermal treatment of the material with carbon dioxide. As a result, carbon monoliths with an ultrahigh specific pore volume of ~11.4 cm3g−1 and a specific surface area of up to 2100 m2g−1 were obtained.

Schematic steps to yield porous carbons by the ice-templating method (Figure taken with permission from reference Estevez et al. 2013)
Conclusions
Hierarchically organized, porous materials prepared via the sol-gel reaction combined with templating or phase separation strategies have been reviewed. The formation of micro-, meso-, and macropores in sol-gel-derived monoliths has been extensively explored in the last 20 years, and many novel synthetic pathways as well as a broad range of chemical compositions are accessible nowadays. While micropores are in many cases an inherent feature of structures formed by sol-gel processing, meso- and macropores are generated via the application of structure-directing agents. These structure-directing agents can be single molecules, oligomers or polymers, or supramolecular arrangements of molecules, but also emulsions or ice crystals that induce phase separation processes on different length scales. The common feature of all these approaches is that they start from a homogeneous solution, in which phase separation is induced by different techniques. In this chapter, the applicability of the different phase separation schemes to various chemical compositions, such as silica, metal oxides or organic polymers, and carbons, has been summarized.
As discussed, for silica-based systems, many approaches are quite well developed. Here, precursor chemistry combined with the simultaneous application of structure-directing agents can be deliberately controlled to obtain the desired meso- and macroporous networks. However, even for the well-estabished techniques available for silica, much more research is required to gain a better control. Especially techniques relying on liquid–liquid phase separation need to be understood in much more detail. This can easily be seen when simple organotrialkoxysilanes are added to the sol-gel solution of a tetraalkoxysilane. Even small amount of organic groups changes the polarity of the system; hence phase separation is strongly influenced and all parameters need to be adjusted again – in many cases on the basis of trial and error experiments.
The situation gets even more complex, when transition metal oxide networks shall be formed. Here, a higher reactivity of the precursors toward hydrolysis combined with higher coordination numbers, and a strong tendency to form crystalline structures requires an even more careful control of the reaction parameters. In many cases, the sol-gel processing conditions have been adjusted in a way that the pH value is slowly changed, e.g., by addition of epoxides that act as an acid scavenger and result in a slow increase of the pH. However, for transition metal networks many possibilities can be found to change the monoliths chemistry by post-synthetic reactions. This chapter gives an overview of examples found in the recent literature, in which these reactions were performed in a way that the hierarchically organized network structure is only slightly compromised.
Many approaches have been published for the formation of hierarchically organized carbon self-supporting structures. This is probably due to the manifold applications that can be envisioned for these materials. The polycondensation reactions of, e.g., resorcinol and formaldehyde, show a rather strong similarity to the classical sol-gel reactions as known for inorganic materials. Therefore, in principle the same underlying concepts as presented for silica and transition metal oxides can be applied for carbon-based materials and have been investigated in great detail.
Although much research has already been devoted to this exciting area, many more ideas can be followed, and we hope that more progress will be made in the future by excellent research contribution.
References
Adelhelm P, Hu YS, Chuenchom L, Antonietti M, Smarsly BM, Maier J. Generation of hierarchical meso‐ and macroporous carbon from mesophase pitch by spinodal decomposition using polymer templates. Adv Mater. 2007;19:4012–7.
Adelhelm P, Cabrera K, Smarsly BM. On the use of mesophase pitch for the preparation of hierarchical porous carbon monoliths by nanocasting. Sci Technol Adv Mater. 2012;13:015010.
Al‐Muhtaseb SA, Ritter JA. Preparation and properties of resorcinol–formaldehyde organic and carbon gels. Adv Mater. 2003;15:101–14.
Alvarez S, Fuertes AB. Synthesis of macro/mesoporous silica and carbon monoliths by using a commercial polyurethane foam as sacrificial template. Mater Lett. 2007;61:2378–81.
Antonelli DM, Ying JY. Synthesis of hexagonally packed mesoporous TiO2 by a modified sol–gel method. Angew Chem Int Ed. 1995;34:2014–7.
Antonietti M, Fechler N, Fellinger TP. Carbon aerogels and monoliths: control of porosity and nanoarchitecture via sol–gel routes. Chem Mater. 2014;26:196–210.
Asfaw HD, Roberts M, Younesi R, Edstrom K. Emulsion-templated bicontinuous carbon network electrodes for use in 3D microstructured batteries. J Mater Chem A. 2013;1:13750–8.
Backlund S, Smått JH, Rosenholm JB, Lindén M. Template-free sol–gel synthesis of hierarchically macro- and mesoporous monolithic TiO2. J Dispers Sci Technol. 2007;28:115–9.
Baumann TF, Worsley MA, Han TY-J, Satcher Jr JH. High surface area carbon aerogel monoliths with hierarchical porosity. J Non Cryst Solids. 2008;354:3513–5.
Biener J, Dasgupta S, Shao L, Wang D, Worsley MA, Wittstock A, Lee JRI, Biener MM, Orme CA, Kucheyev SO, Wood BC, Willey TM, Hamza AV, Weismueller J, Hahn H, Baumann TF. Macroscopic 3D nanographene with dynamically tunable bulk properties. Adv Mater. 2012;24:5083–7.
Brandhuber D, Torma V, Raab C, Peterlik H, Kulak A, Hüsing N. Glycol-modified silanes in the synthesis of mesoscopically organized silica monoliths with hierarchical porosity. Chem Mater. 2005;17:4262–71.
Brandhuber D, Peterlik H, Hüsing N. Facile self‐assembly processes to phenylene‐bridged silica monoliths with four levels of hierarchy. Small. 2006;2:503–6.
Brinker CJ, Scherer GW. Sol–gel sciences: the processing and the chemistry of sol–gel processing. Boston: Academic; 1990.
Brun N, Ungureanu S, Deleuze H, Backov R. Hybrid foams, colloids and beyond: from design to applications. Chem Soc Rev. 2011;40:771–88.
Cahn JW, Hilliard JE. Free energy of a nonuniform system. I. Interfacial free energy. J Chem Phys. 1958;28:258–67.
Candelaria SL, Chen R, Jeong Y-H, Cao G. Highly porous chemically modified carbon cryogels and their coherent nanocomposites for energy applications. Energ Environ Sci. 2012;5:5619–37.
Carn F, Colin A, Achard M-F, Deleuze H, Sellier E, Birot M, Backov R. Inorganic monoliths hierarchically textured via concentrated direct emulsion and micellar templates. J Mater Chem. 2004a;14:1370–6.
Carn F, Colin A, Achard MF, Deleuze H, Saadi Z, Backov R. Rational design of macrocellular silica scaffolds obtained by a tunable sol–gel foaming process. Adv Mater. 2004b;16:140–4.
Chen Y, Yi Y, Brennan JD, Brook MA. Development of macroporous titania monoliths using a biocompatible method. Part 1: material fabrication and characterization. Chem Mater. 2006;18:5326–35.
Cohen N, Silverstein MS. Synthesis of emulsion-templated porous polyacrylonitrile and its pyrolysis to porous carbon monoliths. Polymer. 2011;52:282–7.
Colombo P, Vakifahmetoglu C, Costacurta S. Fabrication of ceramic components with hierarchical porosity. J Mater Sci. 2010;45:5425–55.
Corriu RJ, Leclercq D. Recent developments of molecular chemistry for sol–gel processes. Angew Chem Int Ed. 1996;35:1420–36.
Cundy CS, Cox PA. The hydrothermal synthesis of zeolites: history and development from the earliest days to the present time. Chem Rev. 2003;103:663–702.
Davis SA, Breulmann M, Rhodes KH, Zhang B, Mann S. Template-directed assembly using nanoparticle building blocks: a nanotectonic approach to organized materials. Chem Mater. 2001;13:3218–26.
Deng Y, Liu C, Yu T, Liu F, Zhang F, Wan Y, Zhang L, Wang C, Tu B, Webley PA, Wang H, Zhao D. Facile synthesis of hierarchically porous carbons from dual colloidal crystal/block copolymer template approach. Chem Mater. 2007;19:3271–7.
Depardieu M, Kinadjian N, Backov R. Integrative chemistry: advanced functional cellular materials bearing multiscale porosity. Eur Phys J Spl Topics. 2015;224:1655–68.
Deville S. Freeze‐casting of porous ceramics: a review of current achievements and issues. Adv Eng Mater. 2008;10:155–69.
Deville S. Ice-templating, freeze casting: beyond materials processing. J Mater Res. 2013;28:2202–19.
Dong AA, Wang YJ, Tang Y, Zhang YH, Ren N, Gao Z. Mechanically stable zeolite monoliths with three-dimensional ordered macropores by the transformation of mesoporous silica spheres. Adv Mater. 2002;14:1506–10.
Dutoit D, Schneider M, Baiker A. Titania-silica mixed oxides: I. Influence of sol–gel and drying conditions on structural properties. J Catal. 1995;153:165–76.
El Haskouri J, de Zárate DO, Guillem C, Latorre J, Caldés M, Beltrán A, Beltrán D, Descalzo AB, Rodríguez-López G, Martínez-Máñez R. Silica-based powders and monoliths with bimodal pore systems. Chem Commun. 2002;4:330–1.
Elkhatat AM, Al‐Muhtaseb SA. Advances in tailoring resorcinol‐formaldehyde organic and carbon gels. Adv Mater. 2011;23:2887–903.
Enke D, Janowski F, Schwieger W. Porous glasses in the 21st century – a short review. Micropor Mesopor Mater. 2003;60:19–30.
Estevez L, Kelarakis A, Gong Q, Da’as EH, Giannelis EP. Multifunctional graphene/platinum/nafion hybrids via ice templating. J Am Chem Soc. 2011;133:6122–5.
Estevez L, Dua R, Bhandari N, Ramanujapuram A, Wang P, Giannelis EP. A facile approach for the synthesis of monolithic hierarchical porous carbons – high performance materials for amine based CO2 capture and supercapacitor electrode. Energ Environ Sci. 2013;6:1785–90.
Flaig S, Akbarzadeh J, Peterlik H, Hüsing N. Hierarchically organized silica monoliths: influence of different acids on macro-and mesoporous formation. J Sol–gel Sci Technol. 2015;73:103–11.
Fujita K, Konishi J, Nakanishi K, Hirao K. Strong light scattering in macroporous TiO2 monoliths induced by phase separation. Appl Phys Lett. 2004;85:5595–7.
Fujita K, Konishi J, Nakanishi K, Hirao K. Morphological control and strong light scattering in macroporous TiO2 monoliths prepared via a colloid-derived sol–gel route. Sci Technol Adv Mater. 2006;7:511–8.
Fukumoto S, Nakanishi K, Kanamori K. Direct preparation and conversion of copper hydroxide-based monolithic xerogels with hierarchical pores. New J Chem. 2015;39:6771–7.
Galassi C. Processing of porous ceramics: Piezoelectric materials. J Eur Ceram Soc. 2006;26:2951–8.
Gash AE, Tillotson TM, Satcher JH, Poco JF, Hrubesh LW, Simpson RL. Use of epoxides in the sol–gel synthesis of porous iron (III) oxide monoliths from Fe (III) salts. Chem Mater. 2001;13:999–1007.
Gawel B, Gawel K, Oye G. Sol–gel synthesis of non-silica monolithic materials. Materials. 2010;3:2815–33.
Gaweł B, Gaweł K, Hobæk TC, Yasuda M, Øye G. A simple semi sol–gel method for preparation of alumina monoliths with hierarchical pore structures. Mater Chem Phys. 2012;137:414–20.
Guo X, Song J, Lvlin Y, Nakanishi K, Kanamori K, Yang H. Preparation of macroporous zirconia monoliths from ionic precursors via an epoxide-mediated sol–gel process accompanied by phase separation. Sci Technol Adv Mater. 2015;16:025003.
Gutiérrez MC, Ferrer ML, del Monte F. Ice-templated materials: sophisticated structures exhibiting enhanced functionalities obtained after unidirectional freezing and ice-segregation-induced self-assembly. Chem Mater. 2008;20:634–48.
Hartmann S, Brandhuber D, Hüsing N. Glycol-modified silanes: novel possibilities for the synthesis of hierarchically organized (hybrid) porous materials. Acc Chem Res. 2007;40:885–94.
Hartmann S, Sachse A, Galarneau A. Challenges and strategies in the synthesis of mesoporous alumina powders and hierarchical alumina monoliths. Materials. 2012;5:336–49.
Hartmann S, Elsaesser MS, Hüsing N. Polysiloxane‐based block copolymers as structure‐directing agents in the synthesis of hierarchically organized silica monoliths. Zeitschrift für Anorganische und Allgemeine Chemie. 2014;640:641–8.
Hasegawa G, Kanamori K, Nakanishi K, Hanada T. Fabrication of activated carbons with well-defined macropores derived from sulfonated poly(divinylbenzene) networks. Carbon. 2010a;48:1757–66.
Hasegawa G, Kanamori K, Nakanishi K, Hanada T. Facile preparation of hierarchically porous TiO2 monoliths. J Am Ceram Soc. 2010b;93:3110–5.
Hasegawa G, Kanamori K, Nakanishi K, Hanada T. Facile preparation of transparent monolithic titania gels utilizing a chelating ligand and mineral salts. J Sol–gel Sci Technol. 2010c;53:59–66.
Hasegawa G, Ishihara Y, Kanamori K, Miyazaki K, Yamada Y, Nakanishi K, Abe T. Facile preparation of monolithic LiFePO4/carbon composites with well-defined macropores for a lithium-ion battery. Chem Mater. 2011;23:5208–16.
Hasegawa G, Kanamori K, Nakanishi K. Pore properties of hierarchically porous carbon monoliths with high surface area obtained from bridged polysilsesquioxanes. Micropor Mesopor Mater. 2012;155:265–73.
Hasegawa G, Sato T, Kanamori K, Nakano K, Yajima T, Kobayashi Y, Kageyama H, Abe T, Nakanishi K. Hierarchically porous monoliths based on N-doped reduced titanium oxides and their electric and electrochemical properties. Chem Mater. 2013;25:3504–12.
Hasegawa G, Sato T, Kanamori K, Sun C-J, Ren Y, Kobayashi Y, Kageyama H, Abe T, Nakanishi K. Effect of calcination conditions on porous reduced titanium oxides and oxynitrides via a preceramic polymer route. Inorg Chem. 2015;54:2802–8.
Hauf C, Kniep R, Pfaff G. Preparation of various titanium suboxide powders by reduction of TiO2 with silicon. J Mater Sci. 1999;34:1287–92.
Hench LL, West JK. The sol–gel process. Chem Rev. 1990;90:33–72.
Huerta L, El Haskouri J, Vie D, Comes M, Latorre J, Guillem C, Marcos MD, Martinez-Manez R, Beltran A, Beltran D, Amoros P. Nanosized mesoporous silica coatings on ceramic foams: new hierarchical rigid monoliths. Chem Mater. 2007;19:1082–8.
Hüsing N, Schubert U. Aerogels airy materials: chemistry, structure, and properties. Angew Chem Int Ed. 1998;37:23–45.
Hüsing N, Raab C, Torma V, Roig A, Peterlik H. Periodically mesostructured silica monoliths from diol-modified silanes. Chem Mater. 2003;15:2690–2.
Hüsing N, Brandhuber D, Kaiser P. Glycol-modified organosilanes in the synthesis of inorganic–organic silsesquioxane and silica monoliths. J Sol–gel Sci Technol. 2006;40:131–9.
Imhof A, Pine DJ. Ordered macroporous materials by emulsion templating. Nature. 1997;389:948–51.
Inayat A, Reinhardt B, Uhlig H, Einicke WD, Enke D. Silica monoliths with hierarchical porosity obtained from porous glasses. Chem Soc Rev. 2013;42:3753–64.
Ivanova R, Lindman B, Alexandridis P. Effect of glycols on the self-assembly of amphiphilic block copolymers in water. 1. Phase diagrams and structure identification. Langmuir. 2000a;16:3660–75.
Ivanova R, Lindman B, Alexandridis P. Evolution in structural polymorphism of pluronic F127 poly (ethylene oxide)-poly (propylene oxide) block copolymer in ternary systems with water and pharmaceutically acceptable organic solvents: from “glycols” to “oils”. Langmuir. 2000b;16:9058–69.
Ivanova R, Alexandridis P, Lindman B. Interaction of poloxamer block copolymers with cosolvents and surfactants. Colloids Surf A Physicochem Eng Asp. 2001;183:41–53.
Kanamori K, Nakanishi K. Controlled pore formation in organotrialkoxysilane-derived hybrids: from aerogels to hierarchically porous monoliths. Chem Soc Rev. 2011;40:754–70.
Keppeler M, Hüsing N. Space-confined click reactions in hierarchically organized silica monoliths. New J Chem. 2011;35:681–90.
Keppeler M, Holzbock J, Akbarzadeh J, Peterlik H, Hüsing N. Inorganic–organic hybrid materials through post-synthesis modification: impact of the treatment with azides on the mesopore structure. Beilstein J Nanotechnol. 2011;2:486–98.
Keppeler M, Holzbock J, Akbarzadeh J, Peterlik H, Hüsing N. Nucleophilic substitution on silica surfaces: comparison of the reactivity of α-versus γ-chlorosubstituted silanes in the reaction with sodium azide. J Ceram Soc Jpn. 2015;123:764–9.
Kido Y, Nakanishi K, Miyasaka A, Kanamori K. Synthesis of monolithic hierarchically porous iron-based xerogels from iron (III) Salts via an epoxide-mediated sol–gel process. Chem Mater. 2012;24:2071–7.
Kido Y, Nakanishi K, Okumura N, Kanamori K. Hierarchically porous nickel/carbon composite monoliths prepared by sol–gel method from an ionic precursor. Micropor Mesopor Mater. 2013;176:64–70.
Kido Y, Hasegawa G, Kanamori K, Nakanishi K. Porous chromium-based ceramic monoliths: oxides (Cr2O3), nitrides (CrN), and carbides (Cr3C2). J Mater Chem A. 2014;2:745–52.
Kimmins SD, Cameron NR. Functional porous polymers by emulsion templating: recent advances. Adv Funct Mater. 2011;21:211–25.
Kistler SS. Coherent expanded aerogels and jellies. Nature. 1931;127:741.
Kitada A, Hasegawa G, Kobayashi Y, Kanamori K, Nakanishi K, Kageyama H. Selective preparation of macroporous monoliths of conductive titanium oxides TinO2n–1 (n= 2, 3, 4, 6). J Am Chem Soc. 2012;134:10894–8.
Kolbrecka K, Przyluski J. Sub-stoichiometric titanium oxides as ceramic electrodes for oxygen evolution – structural aspects of the voltammetric behaviour of TinO2n−1. Electrochim Acta. 1994;39:1591–5.
Konishi J, Fujita K, Nakanishi K, Hirao K. Monolithic TiO2 with controlled multiscale porosity via a template-free sol–gel process accompanied by phase separation. Chem Mater. 2006a;18:6069–74.
Konishi J, Fujita K, Nakanishi K, Hirao K. Phase-separation-induced titania monoliths with well-defined macropores and mesostructured framework from colloid-derived sol–gel systems. Chem Mater. 2006b;18:864–6.
Konishi J, Fujita K, Oiwa S, Nakanishi K, Hirao K. Crystalline ZrO2 monoliths with well-defined macropores and mesostructured skeletons prepared by combining the alkoxy-derived sol–gel process accompanied by phase separation and the solvothermal process. Chem Mater. 2008;20:2165–73.
Konishi J, Fujita K, Nakanishi K, Hirao K, Morisato K, Miyazaki S, Ohira M. Sol–gel synthesis of macro–mesoporous titania monoliths and their applications to chromatographic separation media for organophosphate compounds. J Chromatogr A. 2009;1216:7375–83.
Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC, Beck JS. Ordered mesoporous molecular-sieves synthesized by a liquid-crystal template mechanism. Nature. 1992;359:710–2.
Kuang DB, Brezesinski T, Smarsly B. Hierarchical porous silica materials with a trimodal pore system using surfactant templates. J Am Chem Soc. 2004;126:10534–5.
Li W, Guo X, Zhu Y, Hui Y, Kanamori K, Nakanishi K. Sol–gel synthesis of macroporous TiO2 from ionic precursors via phase separation route. J Sol–gel Sci Technol. 2013a;67:639–45.
Li W, Zhu Y, Guo X, Nakanishi K, Kanamori K, Yang H. Preparation of a hierarchically porous AlPO4 monolith via an epoxide-mediated sol–gel process accompanied by phase separation. Sci Technol Adv Mater. 2013b;14:045007.
Liang C, Dai S. Dual phase separation for synthesis of bimodal meso-/macroporous carbon monoliths. Chem Mater. 2009;21:2115–24.
Lopez-Orozco S, Inayat A, Schwab A, Selvam T, Schwieger W. Zeolitic materials with hierarchical porous structures. Adv Mater. 2011;23:2602–15.
Lu A-H, Schüth F. Nanocasting pathways to create ordered mesoporous solids. Comptes Rendus Chimie. 2005;8:609–20.
Maekawa H, Esquena J, Bishop S, Solans C, Chmelka BF. Meso/macroporous inorganic oxide monoliths from polymer foams. Adv Mater. 2003;15:591–6.
McCusker LB, Liebau F, Engelhardt G. Nomenclature of structural and compositional characteristics of ordered microporous and mesoporous materials with inorganic hosts – (IUPAC recommendations 2001). Pure Appl Chem. 2001;73:381–94.
McNaught A, Wilkinson A. IUPAC. Oxford: Blackwell Scientific Publications; 1997.
Moreno AH, Arenillas A, Calvo EG, Bermúdez JM, Menéndez JA. Carbonisation of resorcinol–formaldehyde organic xerogels: effect of temperature, particle size and heating rate on the porosity of carbon xerogels. J Anal Appl Pyrolysis. 2013;100:111–6.
Mukai SR, Nishihara H, Tamon H. Formation of monolithic silica gel microhoneycombs (SMHs) using pseudosteady state growth of microstructural ice crystals. Chem Commun. 2004;7:874–5.
Naikoo GA, Dar RA, Khan F. Hierarchically macro/mesostructured porous copper oxide: facile synthesis, characterization, catalytic performance and electrochemical study of mesoporous copper oxide monoliths. J Mater Chem A. 2014;2:11792–8.
Nakanishi K. Pore structure control of silica gels based on phase separation. J Porous Mater. 1997;4:67–112.
Nakanishi K. Hierarchically porous materials by phase separation: monoliths. In: Su B-L, Sanchez C, Yan X-Y, editors. Hierarchically structured porous materials. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2011. p. 241–67.
Nakanishi K, Soga N. Phase-separation in gelling silica organic polymer-solution – systems containing poly(sodium styrenesulfonate). J Am Ceram Soc. 1991;74:2518–30.
Nakanishi K, Soga N. Phase separation in silica sol–gel system containing polyacrylic acid I. Gel formation behavior and effect of solvent composition. J Non Cryst Solids. 1992;139:1–13.
Nakanishi K, Tanaka N. Sol–gel with phase separation. Hierarchically porous materials optimized for high-performance liquid chromatography separations. Acc Chem Res. 2007;40:863–73.
Nakanishi K, Sato Y, Ruyat Y, Hirao K. Supramolecular templating of mesopores in phase-separating silica sol-gels incorporated with cationic surfactant. J Sol–gel Sci Technol. 2003;26:567–70.
Nardecchia S, Carriazo D, Ferrer ML, Gutierrez MC, del Monte F. Three dimensional macroporous architectures and aerogels built of carbon nanotubes and/or graphene: synthesis and applications. Chem Soc Rev. 2013;42:794–830.
Nishihara H, Mukai SR, Yamashita D, Tamon H. Ordered macroporous silica by ice templating. Chem Mater. 2005;17:683–9.
Nishihara H, Mukai SR, Fujii Y, Tago T, Masuda T, Tamon H. Preparation of monolithic SiO2-Al2O3 cryogels with inter-connected macropores through ice templating. J Mater Chem. 2006;16:3231–6.
Pekala RW, Alviso CT, Kong FM, Hulsey SS. Aerogels derived from multifunctional organic monomers. J Non Cryst Solids. 1992;145:90–8.
Petkovich ND, Stein A. Controlling macro-and mesostructures with hierarchical porosity through combined hard and soft templating. Chem Soc Rev. 2013;42:3721–39.
Ren Y, Ma Z, Bruce PG. Ordered mesoporous metal oxides: synthesis and applications. Chem Soc Rev. 2012;41:4909–27.
Roberts AD, Li X, Zhang H. Porous carbon spheres and monoliths: morphology control, pore size tuning and their applications as Li-ion battery anode materials. Chem Soc Rev. 2014a;43:4341–56.
Roberts AD, Wang S, Li X, Zhang H. Hierarchical porous nitrogen-rich carbon monoliths via ice-templating: high capacity and high-rate performance as lithium-ion battery anode materials. J Mater Chem A. 2014b;2:17787–96.
Ruzimuradov O, Hasegawa G, Kanamori K, Nakanishi K. Preparation of hierarchically porous nanocrystalline CaTiO3, SrTiO3 and BaTiO3 perovskite monoliths. J Am Ceram Soc. 2011;94:3335–9.
Sachse A, Galarneau A, Fajula F, Di Renzo F, Creux P, Coq B. Functional silica monoliths with hierarchical uniform porosity as continuous flow catalytic reactors. Micropor Mesopor Mater. 2011;140:58–68.
Sanchez C, Arribart H, Giraud Guille MM. Biomimetism and bioinspiration as tools for the design of innovative materials and systems. Nat Mater. 2005;4:277–88.
Sato Y, Nakanishi K, Hirao K, Jinnai H, Shibayama M, Melnichenko YB, Wignall GD. Formation of ordered macropores and templated nanopores in silica sol–gel system incorporated with EO–PO–EO triblock copolymer. Colloids Surf A Physicochem Eng Asp. 2001;187:117–22.
Schubert U, Hüsing N, Lorenz A. Hybrid inorganic–organic materials by sol–gel processing of organofunctional metal alkoxides. Chem Mater. 1995;7:2010–27.
Schüth F. Endo‐and exotemplating to create high‐surface‐area inorganic materials. Angew Chem Int Ed. 2003;42:3604–22.
Sel O, Kuang D, Thommes M, Smarsly B. Principles of hierarchical meso-and macropore architectures by liquid crystalline and polymer colloid templating. Langmuir. 2006;22:2311–22.
Sen T, Tiddy G, Casci J, Anderson M. One-pot synthesis of hierarchically ordered porous-silica materials with three orders of length scale. Angew Chem Int Ed. 2003;42:4649–53.
Sen T, Tiddy G, Casci J, Anderson M. Meso-cellular silica foams, macro-cellular silica foams and mesoporous solids: a study of emulsion-mediated synthesis. Micropor Mesopor Mater. 2005;78:255–63.
Silverstein MS. PolyHIPEs: recent advances in emulsion-templated porous polymers. Prog Polym Sci. 2014;39:199–234.
Siouffi AM. Special issue – advances in monoliths – foreword. J Chromatogr A. 2006;1109:1–1.
Smått J-H, Schunk S, Lindén M. Versatile double-templating synthesis route to silica monoliths exhibiting a multimodal hierarchical porosity. Chem Mater. 2003;15:2354–61.
Smått JH, Weidenthaler C, Rosenholm JB, Lindén M. Hierarchically porous metal oxide monoliths prepared by the nanocasting route. Chem Mater. 2006;18:1443–50.
Smått JH, Sayler FM, Grano AJ, Bakker MG. Formation of hierarchically porous metal oxide and metal monoliths by nanocasting into silica monoliths. Adv Eng Mater. 2012;14:1059–73.
Smith DM, Stein D, Anderson JM, Ackerman W. Preparation of low-density xerogels at ambient pressure. J Non Cryst Solids. 1995;186:104–12.
Stein A, Wilson BE, Rudisill SG. Design and functionality of colloidal-crystal-templated materials-chemical applications of inverse opals. Chem Soc Rev. 2013;42:2763–803.
Studart AR, Gonzenbach UT, Tervoort E, Gauckler LJ. Processing routes to macroporous ceramics: a review. J Am Ceram Soc. 2006;89:1771–89.
Su B-L, Sanchez C, Yang X-Y. Hierarchically structured porous materials: from nanoscience to catalysis, separation, optics, energy, and life science. Weinheim: Wiley; 2012.
Sun ZK, Deng YH, Wei J, Gu D, Tu B, Zhao DY. Hierarchically ordered macro-/mesoporous silica monolith: tuning macropore entrance size for size-selective adsorption of proteins. Chem Mater. 2011;23:2176–84.
Takenaka S, Takahashi R, Sato S, Sodesawa T. Structural study of mesoporous titania prepared from titanium alkoxide and carboxylic acids. J Sol–gel Sci Technol. 2000;19:711–4.
Tokudome Y, Fujita K, Nakanishi K, Kanamori K, Miura K, Hirao K, Hanada T. Sol–gel synthesis of macroporous YAG from ionic precursors via phase separation route. J Ceram Soc Jpn. 2007a;115:925–8.
Tokudome Y, Fujita K, Nakanishi K, Miura K, Hirao K. Synthesis of monolithic Al2O3 with well-defined macropores and mesostructured skeletons via the sol–gel process accompanied by phase separation. Chem Mater. 2007b;19:3393–8.
Tokudome Y, Nakanishi K, Kanamori K, Fujita K, Akamatsu H, Hanada T. Structural characterization of hierarchically porous alumina aerogel and xerogel monoliths. J Colloid Interface Sci. 2009;338:506–13.
Tokudome Y, Miyasaka A, Nakanishi K, Hanada T. Synthesis of hierarchical macro/mesoporous dicalcium phosphate monolith via epoxide-mediated sol–gel reaction from ionic precursors. J of Sol–gel Scilogy. 2011;57:269–78.
Triantafillidis C, Elsaesser MS, Husing N. Chemical phase separation strategies towards silica monoliths with hierarchical porosity. Chem Soc Rev. 2013;42:3833–46.
Ungureanu S, Birot M, Deleuze H, Schmitt V, Mano N, Backov R. Triple hierarchical micro–meso–macroporous carbonaceous foams bearing highly monodisperse macroporosity. Carbon. 2015;91:311–20.
Vuong G-T, Kaliaguine S, Do T-O. A strategy towards macroporous sponge-like networks of metal oxide-surfactant mesophases and bulk metal oxides. J Porous Mater. 2008;15:679–83.
Wan Y, Zhao D. On the controllable soft-templating approach to mesoporous silicates. Chem Rev. 2007;107:2821–60.
Wang Z, Stein A. Morphology control of carbon, silica, and carbon/silica nanocomposites: from 3D Ordered macro-/mesoporous monoliths to shaped mesoporous particles. Chem Mater. 2008;20:1029–40.
Wang Z, Kiesel ER, Stein A. Silica-free syntheses of hierarchically ordered macroporous polymer and carbon monoliths with controllable mesoporosity. J Mater Chem. 2008;18:2194–200.
Wei J, Jiang Z-T, Jiang S, Li R, Tan J. Sol–gel synthesis and characterization of macro-mesoporous titania monolith and its application in chromatographic separation of carboxylates. J Liquid Chromatgr Relat Technol. 2013;36:1616–30.
Weinberger M, Puchegger S, Rentenberger C, Puchberger M, Hüsing N, Peterlik H. Mesoporous dendrimer silica monoliths studied by small-angle X-ray scattering. J Mater Chem. 2008;18:4783–9.
Weinberger M, Puchegger S, Fröschl T, Babonneau F, Peterlik H, Hüsing N. Sol− Gel processing of a glycolated cyclic organosilane and its pyrolysis to silicon oxycarbide monoliths with multiscale porosity and large surface areas. Chem Mater. 2010;22:1509–20.
Weitkamp J, Sing KSW, Schüth F. Handbook of porous solids. Weinheim: Wiley-VCH; 2002.
White G, Mackenzie K, Johnston J. Carbothermal synthesis of titanium nitride. J Mater Sci. 1992;27:4287–93.
Yanagisawa T, Shimizu T, Kuroda K, Kato C. The preparation of alkyltrimethylammonium-kanemite complexes and their conversion to microporous materials. Bull Chem Soc Jpn. 1990;63:988–92.
Yang H, Zhao D. Synthesis of replica mesostructures by the nanocasting strategy. J Mater Chem. 2005;15:1217–31.
Yuan Z-Y, Su B-L. Insights into hierarchically meso–macroporous structured materials. J Mater Chem. 2006;16:663–77.
Yuan J, Bai X, Zhao M, Zheng L. C12mimBr ionic liquid/SDS vesicle formation and use as template for the synthesis of hollow silica spheres. Langmuir. 2010;26:11726–31.
Zhai Y, Dou Y, Zhao D, Fulvio PF, Mayes RT, Dai S. Carbon materials for chemical capacitive energy storage. Adv Mater. 2011;23:4828–50.
Zhao J, Jiang Z-T, Tan J, Li R. Sol–gel synthesis and characterization of titania monolith with bimodal porosity. J Sol–gel Sci Technol. 2011;58:436–41.
Zhu Y, Shimizu T, Kitajima T, Morisato K, Moitra N, Brun N, Kiyomura T, Kanamori K, Takeda K, Kurata H. Synthesis of robust hierarchically porous zirconium phosphate monolith for efficient ion adsorption. New J Chem. 2015;39:2444–50.
Acknowledgments
This chapter is reproduced from http://dx.doi.org/10.1039/C5CS00710K with permission from the Royal Society of Chemistry.
For parts of the work highlighted in this chapter, we thank the Deutsche Forschungsgemeinschaft within the Priority Programme 1570 “Poröse Medien mit definierter Porenstruktur in der Verfahrenstechnik – Modellierung, Anwendungen, Synthese” (Hu 1427/6-1) and the Austrian Science Foundation FWF (Project I 1605-N20).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this entry

Cite this entry
Feinle, A., Elsaesser, M.S., Hüsing, N. (2018). Hierarchical Organization in Monolithic Sol-Gel Materials. In: Klein, L., Aparicio, M., Jitianu, A. (eds) Handbook of Sol-Gel Science and Technology. Springer, Cham. https://doi.org/10.1007/978-3-319-32101-1_127
Download citation
DOI: https://doi.org/10.1007/978-3-319-32101-1_127
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-32099-1
Online ISBN: 978-3-319-32101-1
eBook Packages: Chemistry and Materials ScienceReference Module Physical and Materials ScienceReference Module Chemistry, Materials and Physics