
Abstract
The molecular structures, conformational stabilities, and infrared vibrational wave numbers of 2-formylpyridine, 3-formylpyridine, and 4-formylpyridine have been computed using Becke-3-Lee-Yang-Parr (B3LYP) density functional theory (DFT) method with 6-31+G* basis set. From the computations, 2-formylpyridine and 3-formylpyridine were predicted to exist predominantly in cis conformation both in gas and solution phases. The infrared vibrational wave numbers of the molecules in Cs symmetry were computed and compared with the observed infrared vibrational wave numbers. The effect of solvents on the conformational stability of the molecules in nine different solvents was investigated. The Integral Equation Formalism in the Polarizable Continuum Model (IEF-PCM) was used for all solution phase computations.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Density functional theory
- Conformational stability
- Vibrational wave numbers
- Solvent effect
- Formylpyridine
10.1 Introduction
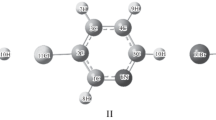
The molecules 2-formylpyridine (picolinaldehyde), 3-formylpyridine (nicotinaldehyde), and 4-formylpyridine (isonicotinaldehyde) are pyridine derivatives that contain an aldehyde (CHO) group, substituted ortho, meta, or para to the nitrogen of a pyridine ring. The rotation of the aldehyde group on the pyridine rings of both 2-formylpyridine and 3-formylpyridine leads to two equilibrium configurations. As such, both 2-formylpyridine and 3-formylpyridine can exist in two asymmetric planar rotational conformers whereby the nitrogen and oxygen can be either ON-cis or ON-trans. On the other hand, 4-formylpyridine has only one conformer as illustrated in Fig. 10.1. Several experimental and theoretical studies examined the conformational preference and vibrational spectra of these molecules [1–4]. A brief summary of the experimental and theoretical studies on conformational preference of the aldehyde group on 2-formylpyridine and 3-formylpyridine has been reported [2, 3].

Structures and atomic numbering of 2-formylpyridine, 3-formylpyridine, and 4-formylpyridine
It is well known that the position of equilibrium between alternative rotational isomers of a particular molecule can be remarkably medium dependent and to the best of our knowledge, computational studies about the effect of solvent on the conformational preference of these molecules have not been reported. The ability to predict the conformational preference and the rotational barrier reliably are of great interest for systems where experimental conformational preference is not clear and is strongly solvent dependent. Such study will contribute to a better understanding of many controversial aspects of their molecular structures, structural stabilities, and vibrational spectra. Thus, the aim of this work is to take advantage of the quantum mechanics to carry out calculations that will aid in clarifying experimental data and contribute to better understanding of the molecular structures and conformational stabilities of the studied molecules. In this study, molecular geometry, potential energy surface, and infrared vibrational spectra of 2-formylpyridine, 3-formylpyridine, and 4-formylpyridine are investigated using the Gaussian 03 program package [5]. Computations were performed at DFT/B3LYP level of theory, which had been previously used successfully in a variety of conformational studies [6–11].
10.2 Computational Methods
GAUSSIAN 03 program package was used to optimize the structures, predict energies, and calculate infrared vibrational wave numbers for 2-formylpyridine, 3-formylpyridine, and 4-formylpyridine in their possible conformations (Fig. 10.1). The trans and cis conformers of the studied molecules are defined by the position of the carbonyl oxygen atom with respect to the adjacent C–C bond in pyridine ring (Fig. 10.1).
The energies of the possible conformers of the molecules were optimized by Becke’s three-parameter exchange functional [12] combined with Lee-Yang-Parr [13] correlation functional (B3LYP) method using the standard 6-31+G* basis set. From the data, the relative conformational stability and the barrier to internal rotation were determined. The infrared vibrational wave numbers were computed and then scaled by 0.955 and 0.967 for wave numbers above 1800 cm−1 and below 1800 cm−1 respectively [14]. The effect of solvents on the conformational stability of 2-formylpyridine and 3-formylpyridine was investigated using solvents with different polarities. Dielectric constants of 1.92, 4.90, 7.58, 10.36, 20.70, 24.55, 32.63, 46.70, and 78.39 were used as the values for heptane, chloroform, tetrahydrofuran, dichloroethane, acetone, ethanol, methanol, dimethylsulfoxide and water, respectively. The integral equation formalism in the Polarizable Continuum Model (IEF-PCM) [15–17] was used for all solution phase computations.
10.3 Results and Discussion
Generation of potential energy functions from the experimental data is a difficult task, since only conformations near the minima are appreciably populated. The alternative is to carry out accurate computation for appropriate numbers of rotational angles. Therefore, to study the different conformations of the molecules, potential energy scan (PES) for the internal rotation of the aldehyde group about the C–C single bond was performed at B3LYP/6-31+G* level of theory. The rotational energy profiles were obtained by optimizing the total energy at a fixed dihedral angle, and the other parameters were relaxed to their equilibrium values. The torsional angle ϕ(OC-CC) was varied in steps of 15ο between ϕ = 0ο (cis position, where C=O bond eclipses the adjacent C–C bond) to ϕ = 180ο (trans position, where the C=O bond is anti to the adjacent C–C bond). The adjacent C–C bond refers to C2–C3, C3–C4, and C4–C5 (atoms numbering are given in Fig. 10.1) in 2-formylpyridine, 3-formylpyridine and 4-formylpyridine , respectively. The cis and trans correspond to ON-trans and ON-cis, respectively. The saddle points were determined, and full geometry optimization was carried out at the transition state. Figure 10.2 shows the potential energy surface of the three molecules as a function of the dihedral angle (ϕ). All the reported minima along the potential energy surface were subjected to full geometry optimization, and the minimum was verified by calculating the vibrational wave numbers that result in the absence of imaginary wave numbers. The lowest energy structure for 2-formylpyridine and 3-formylpyridine was found to be the planar conformer with dihedral angle of zero degree. On the other hand, both conformers of 4-formylpyridine have equal energy due to the symmetry of the molecule. The highest energy structure for the three molecules occurs at dihedral angle close to 90° (referred to as perpendicular structure) which is the transition state with one imaginary infrared vibrational wave number corresponding to the rotation of the aldehyde (CHO) group. The transition states for 2-formylpyridine, 3-formylpyridine, and 4-formylpyridine are located at a dihedral angle (ϕ) of 81.7°, 89.9° and 90.2°, respectively. The trend of the internal rotational barriers is 2-formylpyridine > 3-formylpyridine > 4-formylpyridine. The same trend was observed experimentally [19].

Computed internal rotational potential energy profiles for 2-formylpyridine, 3-formylpyridine and 4-formylpyridine
Table 10.1 shows the gas phase computed total energies (hartree), relative energy (kcal/mol), rotational barriers (kcal/mol), and relative stabilization energy of the stable conformers of the studied molecules. The cis conformers of 2-formylpyridine and 3-formylpyridine are more stable than their trans conformer but the relative energy between the cis and trans conformer of 2-formylpyridine (4.42 kcal/mol) is higher than the cis-trans relative energy of 3-formylpyridine (0.73 kcal/mol). The energies of the conformers have been used to calculate the relative energy (ΔE = E trans − E cis ). The cis-trans and trans-cis rotational barriers of the three pyridine derivatives are presented in Table 10.1.
The relative stabilization energy which is the energy differences between the stable conformers (cis) shows that 3-formylpyridine has the smallest energy, followed by 4-formylpyridine and then 2-formylpyridine having the highest energy. These relative stabilization energies of the stable conformers (cis) of the studied molecules can be explained in terms of the resonance structures shown in Fig. 10.3.

Conjugation in 2-formylpyridine (a), 3-formylpyridine (b), and 4-formylpyridine (c)
The conjugations of both 2-formylpyridine (Fig. 10.3a) and 4-formylpyridine (Fig. 10.3c) show destabilizing contributions. In addition, steric effect could contribute to the high energy of 2-formylpyridine. In general, the higher energy of 4-formylpyridine could be due to electronic effect and the higher energy of 2-formylpyridine could be due to both electronic and steric effects.
It is well known that the position of equilibrium between alternative rotational isomers of a particular molecule can be remarkably medium dependent. In order to establish the preferred conformations in solution, IEF-PCM model implemented at the B3LYP/6-31+G* level of theory was used to investigate the conformational preference of 2-formylpyridine and 3-formylpyridine in nine different solvents. Figure 10.4 shows the variation of cis-trans relative energy with dielectric constant of solvent obtained from IEF-PCM optimization at B3LYP/6-31+G* level of theory. The relative energy of both molecules decreases with the increase in dielectric constant of solvents. Thus, the cis conformer of both 2-formylpyridine and 3-formylpyridine which are more stable in gas phase remained the more stable comformers in solution, but the stability decreases as the dielectric constant of the solvent increases (Fig. 10.4).

Variation of relative energy with dielectric constant of the solvent
The decrease in relative energy of the conformers with solvent dielectric constant may be due to the increase in stability of the trans conformer in different solvents, because conformers of higher dipole moment are usually more favored in media of high dielectric constant [19]. The dipole moments of the trans conformers of both 2-formylpyridine and 3-formylpyridine are generally higher than the dipole moments of the cis conformers and the dipole moments of the cis and the trans conformers increase with the increase in dielectric constant of the solvent. The variations of the dipole moment as a function of solvent dielectric constant for the cis and trans conformers of 2-formylpyridine and 3-formylpyridine are presented in Fig. 10.5.

Variation of dipole moment with dielectric constant of the solvent
The computed relative energies (kcal/mol) of 2-formylpyridine and 3-formylpyridine in gas and solution phases are provided in Table 10.2. It is interesting to note that the gas phase relative energy for all the molecules are in good agreement with literature values computed at HF/6-311++G(d,p) and B3LYP/6-311++G(d,p) levels of theory [2]. The percentage of the cis conformers at 298.15 K for 2-formylpyridine and 3-formylpyridine are given in Table 10.2.
Some of the geometry parameters optimized at DFT/B3LYP level of theory for the cis 2-formylpyridine in gas phase and different solvents are presented in Table 10.3. There are no systematic and significant changes in the structures of both 2-formylpyridine and 3-formylpyridine in different solvents. The mean absolute deviations (MAD) from the gas phase for the nine solvents are 0.0028 Å for the bond lengths and 0.160° for the bond angles. The individual MAD values for the nine solvents are given in Table 10.3.
The optimized structural parameters were used to compute the infrared vibrational wave numbers and the resulting scaled infrared vibrational wave numbers and their intensities are given in Tables 10.4, 10.5 and 10.6. Complete assignments of the vibrational modes of the studied molecules have been reported [2, 3, 20]. Both conformers of the studied molecules belong to Cs symmetry and the 33 vibration modes are accounted by the irreducible representations Γvib = 23 A′ + 10 A″ of Cs point group. As seen in Tables 10.4 and 10.5, the experimental vibrations are in good agreement with the calculated values of both the cis and trans conformers of 2-formylpyridine and 3-formylpyridine. The cis/trans mean absolute deviation (MAD) between the experimental infrared vibrational wave numbers and the corresponding scaled vibrational wave numbers are found to be 9.63 cm−1/13.68 cm−1 and 11.47 cm−1/13.62 cm−1 for the 2-formylpyridine and 3-formylpyridine respectively. The vibrational deviations for the cis conformers are better than those of trans conformers. Thus, the preferential conformers for the 2-formylpyridine and 3-formylpyridine are the cis conformers which are in agreement with the calculated relative energies. For 4-formylpyridine, both conformers are equally energetic due to the symmetry of the molecule and the observed vibrations correspond to the average of the conformers. The MAD value of calculated infrared vibrational wave numbers from the experimental wave numbers for 4-formylpyridine is about 11.66 cm−1.
The correlation graphs between computed vibrational [3] and observed wave numbers of the most stable conformers (cis) of the studied molecules are presented in Figs. 10.6, 10.7, and 10.8. The correlations reflect a high level of conformity between the harmonic wave numbers obtained from DFT/B3LYP computations [3] and the observed fundamental wave numbers.

Comparison between the observed and computed infrared vibrational wave numbers of cis 2-formylpyridine

Comparison between the observed and computed infrared vibrational wave numbers of cis 3-formylpyridine

Comparison between the observed and computed vibrational wave numbers of cis 4-formylpyridine
10.4 Conclusions
The B3LYP/6-31G* computations indicate that the internal rotation potential energy profiles of 2-formylpyridine, 3-formylpyridine, and 4-formylpyridine have the same skeleton and the trend of the internal rotational barriers is 2-formylpyridine > 3-formylpyridine > 4-formylpyridine. From the computations, 2-formylpyridine and 3-formylpyridine were predicted to exist predominantly in cis conformation with the cis-trans rotational barrier of 9.38, 8.55, and 7.18 kcal/mol for 2-formylpyridine, 3-formylpyridine and 4-formylpyridine, respectively. The computations of solvent effects were performed over nine different values of dielectric constant to illustrate the effects of varying dielectric constant using IEF-PCM methods. The cis conformer of both 2-formylpyridine and 3-formylpyridine which are more stable in gas phase remain the more stable conformers in solution but the stability decreases as the dielectric constant of the solvent increases. The computed vibrational wave numbers at B3LYP/6-31G* give a reasonable agreement with the observed vibrational wave numbers.
References
Taurian OE, De Kowalewski DG, Pérez JE, Contreras RH (2005) NMR J(13C,13C) spin–spin coupling constants in pyridine-carboxaldehydes: experimental and DFT-B3LYP studies. J Mol Struct 754:1–9
Saglam A, Ucun F, Guclu V (2007) Molecular structures and vibrational frequencies of 2-, 3- and 4-pyridine carboxaldehydes by ab initio Hartree-Fock and density functional theory calculations. Spectrochim Acta A 67:465–471
Umar Y (2009) Density functional theory calculations of the internal rotations and vibrational spectra of 2-, 3-, and 4-formylpyridine. Spectrochim Acta A 71:1907–1913
John IG, Ritchie GLD, Radom L (1977) Conformations of furan-, pyrrole-, and pyridine-carbaldehydes: an ab initio molecular orbital study. J Chem Soc Perkin Trans 2:1601–1607
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven JT, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Peterson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian, Inc., Wallingford CT Gaussian 03, Revision C.02
Ramasami P (2009) Theoretical study of 2-selenophenecarboaldehyde in the gas and solution phases: rotational barrier, energy difference and thermodynamic parameters. J Mol Struct (Theochem) 907:57–61
Ashish H, Ramasami P (2008) Rotational barrier and thermodynamic parameters of furfural, thiofurfural, and selenofurfural in the gas and solution phases: theoretical study based on density functional theory method. Mol Phys 106:175–185
Umar Y, Morsy MA (2007) Ab initio and DFT studies of the molecular structures and vibrational spectra of succinonitrile. Spectrochim Acta A 66:1133–1140
Umar Y (2006) Theoretical investigation of the structure and vibrational spectra of carbamoylazide. Spectrochim Acta A 64:568–573
Umar Y, Jimoh T, Morsy AM (2005) Ab initio and density functional calculations of the structures and vibrational spectra of formaldoxime. J Mol Struct (Theochem) 725:157–161
Balci K, Akyuz S (2008) A vibrational spectroscopic investigation on benzocaine molecule. Vib Spectrosc 48:215–228
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5653
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Tursun M, Kesan G, Parlak C, Senyel M (2013) Vibrational spectroscopic investigation and conformational analysis of 1-heptylamine: a comparative density functional study. Spectrochim Acta A 114:668–680
Cancès E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041
Mennucci B, Tomasi J (1997) Continuum solvation models: a new approach to the problem of solute’s charge distribution and cavity boundaries. J Chem Phys 106:5151–5158
Tomasi J, Mennucci B, Cancès E (1999) The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J Mol Struct (Theochem) 464:211–226
Drakenberg T (1976) 13C Nuclear magnetic studies on aromatic aldehydes torsional barriers and conformational equilibria in pyridine carboxaldehydes. J Chem Soc Perkin II:147–149
Baldridge KK, Jonas V (2000) Ground state gas and solution phase conformational dynamics of polar processes: furfural systems. J Chem Phys 113:7519–7529
Jose SP, Mohan S (2006) FT-IR and FT-RAMAN investigations of nicotinaldehyde. Spectrochim Acta A 33:75–79
Green JHS, Harrison DJ (1977) Vibrational spectra of cyano-, fomyl- and halogeno-pyridines. Spectrochim Acta A 64:205–209
Acknowledgment
Facilities provided by Jubail Industrial College of Royal Commission for Jubail and Yanbu are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
Umar, Y., Tijani, J. (2016). Theoretical Investigation of the Conformational Stabilities, Internal Rotations, and Vibrational Infrared Spectra of 2-Formylpyridine, 3-Formylpyridine, and 4-Formylpyridine. In: Ramasami, P., Gupta Bhowon, M., Jhaumeer Laulloo, S., Li Kam Wah, H. (eds) Crystallizing Ideas – The Role of Chemistry. Springer, Cham. https://doi.org/10.1007/978-3-319-31759-5_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-31759-5_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31758-8
Online ISBN: 978-3-319-31759-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)