
Abstract
Amyloidoses encompass a broad category of cutaneous and systemic disorders with important pathogenic consequences. These derive from the direct deposition of abnormal proteins or indirectly relate to potentially deadly systemic disorders that produce such deposits. Each of amyloidoses can be defined by certain histomorphologic and chemical properties that permit their identification and inclusion into disease categories (J Am Acad Dermatol 18:1, 1988). These designations can be loosely grouped into systemic and cutaneous delimited forms.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Synonyms::
-
S—
N—none
ED—macular and lichenoid
- Etiology::
-
S—plasma-cell-derived light chains
N—plasma-cell-derived light chains
ED—epidermal keratins
- Associations::
-
S—multiple myeloma in 1/3 pts
N—10 % with systemic amyloidosis
ED—none
- Clinical::
-
S—hemorrhagic papules/plaques
N—waxy nodules
ED—hyperpigmented macules and lichenoid papules
- Histology::
-
S—fissured hyaline deposits and perivascular deposits
N—hyaline deposits with plasma cells
ED—hyaline dermal globules
- IHC::
-
S—monoclonal kappa or lambda light chain
N—monoclonal kappa or lambda light chain
ED—keratins
- Staging::
-
N/A
- Prognosis::
-
S—poor in myeloma patients
N—dependent upon systemic status
ED—excellent
- Adverse outcome::
-
S—myeloma and cardiac disease
N—systemic amyloidosis
ED—none
- Treatment::
-
S—chemotherapy
N—excision, laser and cryotherapy
ED—topical retinoids
Amyloidoses encompass a broad category of cutaneous and systemic disorders with important pathogenic consequences. These derive from the direct deposition of abnormal proteins or indirectly relate to potentially deadly systemic disorders that produce such deposits. Each of amyloidoses can be defined by certain histomorphologic and chemical properties that permit their identification and inclusion into disease categories [1]. These designations can be loosely grouped into systemic and cutaneous delimited forms.
The term amyloidosis historically derives from the gross starch-like deposits seen in conjunction with systemic amyloidosis. Despite the appellation, the chemical constituency of amyloid is either protein or glycoprotein. Each of the 16 chemical types possesses similar histomorphologic and chemical properties including a predominantly extracellular location, an amorphous eosinophilic appearance on hematoxylin and eosin staining, and a meshwork of hollow 7.5–10-nm linear nonbranched fibrils on ultrastructural examination [2]. The fibrils often align into a three-dimensional beta-pleated sheet configuration that is held responsible for their birefringence properties on diagnostic polaroscopic examination. Amyloid fibrils often associate with certain disease defining nonamyloid glycoproteins including protein P (systemic disease) and apolipoprotein E (Alzheimer’s disease). Among the various types of amyloid, only a few are commonly observed in the skin and thus merit discussion. The two most important responsible for the bulk of dermatologic disease derive from plasma-cell-produced immunoglobulin kappa or lambda light chains or from epidermal keratinocyte keratins. The former are seen in conjunction with primary or systemic amyloidosis and nodular amyloidosis, whereas the latter are seen in macular and lichenoid amyloidosis. The light chains may be monoclonal and systemically produced from abnormal collections of bone marrow or reticuloendothelial associated plasma cells in multiple myeloma or lymphoma or monoclonal and produced locally within the skin from associated plasma cells. The keratins deposited in macular/lichenoid amyloidosis derive from degenerated epidermal keratinocytes and as such are typically seen as aggregates in the superficial dermis juxtaposed to the overlying epithelium.
Primary and myeloma-associated amyloidosis involves the skin in approximately one-third of patients [3]. Typical patients are in their 60s with a slight male predominance. There is no known ethnic predilection. Although the dermatologic manifestations may be overshadowed by the systemic stigmata of macroglossia, bilateral or unilateral carpal tunnel syndrome, or hepatosplenomegaly, distinctive cutaneous changes are often present. Waxy papules and nodules are typically seen on the flexural folds of the face, trunk, and extremities. The papules may undergo secondary hemorrhage and are rarely ulcerated. Mucocutaneous nodules may assume a warty appearance reminiscent of condyloma or a flattened plaque-like configuration suggestive of xanthoma. Non-vasculitic purpuric macules, petechiae, and ecchymoses are common, particularly in flexural fold areas such as the eyelids. Purpuric lesions may be elicited with trauma (pinch purpura) (Fig. 17.1) or following a precipitous increase in intrathoracic pressure (Valsalva maneuver). Less common manifestations include diffuse involvement of the scalp (cutis verticis gyrata) and alopecia, scleroderma-like induration of the extremities, bullous lesions, nail dystrophy, cutis laxa, and cord-like thickening of the blood vessels. Important systemic signs and complications stemming from visceral organ involvement include the heart, reticuloendothelial system, blood vessels, and peripheral and autonomic nervous system. Cardiac disease is the most important cause of mortality with most cases resulting in cardiomyopathy, arrhythmias, congestive heart failure, or ischemic heart disease [4]. Hepatosplenomegaly is seen in approximately one-half of patients and is an important cause of morbidity. Amyloid infiltration of the blood vessels may produce ischemic claudication of the gastrointestinal tract, extremities, or heart. Peripheral and autonomic neuropathy is common, with the latter often producing orthostatic hypotension. Massive amyloid deposits within the soft tissues of the wrist or when located around the shoulders (providing the shoulder pad sign) may result in carpal tunnel syndrome.

Periorbital purpura seen in systemic amyloidosis.
Other less common forms of systemic amyloidosis that rarely manifest in the skin include secondary or inflammatory-associated amyloidosis and the extremely rare heredofamilial syndromes of familial Mediterranean fever (urticaria, vasculitic purpura, fever, serositis, and renal amyloid) and Muckle-Wells syndrome (urticaria, fever, deafness, and renal amyloidosis) [1].
The lesions of nodular or localized amyloidosis tend to be less numerous and individually larger than systemic amyloidosis. Demographically, most patients are in their 60s and there tends to be a male predominance. Lesions may occur on the extremities as well as the face or trunk. Although most lesions are asymptomatic, they tend to gradually enlarge in time. In recent studies, as low as 7 % of patients are found to have or subsequently develop systemic amyloidosis, which is indicative of an overall good prognosis. Management of these nodules, however, is difficult since there is no consistently effective treatment, likely due to their high rate of recurrence [5].
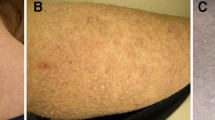
The epithelial-derived forms of cutaneous lichenoid and macular amyloidosis have distinctive demographic and clinical attributes [6]. Both forms may coexist and likely represent a spectrum of disease with similar etiologic origins. Often pruritic, many believe that the lesions follow excoriation and represent the turnover of epithelial-derived keratinocytes. The lesions are more commonly encountered among races of darker skin color including individuals of Asian or Latin-American ancestry and consist of hyperpigmented macules or lichenoid papules. The former tend to occur over the trunk, in particular the interscapular area, with the lichenoid lesions typically seen over the extensor aspects of the lower extremities and penis. Less common forms of cutaneous limited amyloidosis include anosacral amyloidosis, consisting of lichenified plaques located on the buttocks of Chinese persons; familial primary cutaneous amyloidosis, a rare autosomal dominant genodermatoses consisting of amyloid-containing keratotic papules and swirled areas of cutaneous hypo-/hyperpigmentation, encompassing subtypes such as cutis dyschromica; and poikilodermatous amyloidosis, in which typical patients possess a short stature and show photosensitivity and palmoplantar keratoderma [7, 8]. In the latter, amyloid deposition is due to failed DNA repair secondary to sunlight-induced damage [9]. Keratinocyte-derived amyloid may also be seen as part of a degenerative phenomenon permeating and surrounding the stroma of epithelial-derived tumors such as basal cell carcinoma, squamous cell carcinoma, and various adnexal tumors.
Biopsy of skin lesions and blind abdominal fat pad and rectal aspirations are the most reliable means to establish a diagnosis [10]. The latter techniques have a high diagnostic yield in systemic and myeloma-associated forms of the disease only. The histomorphologic features of amyloid are identical regardless of the type examined or clinical context and consist of amorphous aggregates of slightly eosinophilic extracellular material. Larger aggregates may contain internal artifactual clefts or fissures and may be associated with chronic inflammatory cells including plasma cells. The systemic and myeloma-associated forms of disease are more often characterized by larger aggregates of amyloid deposition located throughout the dermis and subcutaneous fat (Figs. 17.2 and 17.3). Furthermore, the deposits are found within and surrounding vascular structures associated with purpura and may similarly outline the cell membranes of adipocytes within the panniculus, producing amyloid rings. Localized forms of the disease are characterized by smaller aggregates of amyloid usually located in the superficial dermis adjacent to the epithelium. The exception to this is the nodular localized form of disease in which large amyloid deposits are observed with abundant associated plasma cells [11]. The deposits are accentuated surrounding the blood vessels and adnexal structures. This infiltration of the reticular dermis and subcutaneous tissue is indistinguishable from systemic amyloidosis but differentiates the nodular amyloidosis from other cutaneous localized types of amyloidosis [12]. In the lichenoid form, the overlying epithelium usually shows slight acanthosis. In both the macular and lichenoid forms, the overlying epithelial keratinocytes are hyperpigmented, and scattered dyskeratotic or apoptotic basilar keratinocytes are observed (Fig. 17.4). Dermal incontinence of melanin and a sparse interface or superficial dermal perivascular lymphocytic infiltrate may be seen.

Low-power photomicrograph depicting superficial dermal eosinophilic (amyloid) deposits.

High-power photomicrograph depicting amyloid deposits within the wall of the dermal blood vessels and perivascular stroma.

High-power photomicrograph depicting fissured amyloid deposits at the dermoepidermal junction. Note additional changes of interface dermatitis.
Several diagnostic adjuncts may be employed to confirm the diagnosis and establish the subtype of amyloidosis. Special stains may be employed that metachromatically stain the deposits (crystal or methyl violet) or that when exposed to polarized light produce birefringence (green with Congo red and yellow with thioflavine T). It is important to realize that all forms of amyloid stain, as do nonamyloid deposits including elastin, colloid milium, and the deposits of lipoid proteinosis. Specificity of the staining can be improved with the addition of immunohistochemical antibodies including amyloid P protein, immunoglobulin light chains, and keratins [12, 13]. Immunolabeling for P protein is found in all forms of amyloidosis. Monoclonal light chain restriction is typically observed within the plasma-cell-associated forms of nodular and systemic/myeloma amyloidosis. Keratin antibodies are positive in the cutaneous derived forms of amyloidosis, most notably CK5, and are negative in the remainder [13–15]. The specific types of amyloid may be determined through mass spectroscopy or amino acid sequencing. Electron microscopy can also be employed to identify the characteristic filaments.
The treatment is type dependent [6]. Patients with systemic and myeloma-associated forms of the disorder are usually administered systemic chemotherapy with or without autologous bone marrow transplantation. Organ transplantation of severely affected organs, most often the liver, heart, or kidney, can be performed, but amyloid may reaccumulate in the transplanted organ. Localized nodular amyloidosis may be removed surgically or ablated with laser or cryotherapy. Lichenoid/macular amyloidosis is difficult to treat, although some have reported variable success with topical retinoids, calcipotriol, and dermabrasion.
The prognosis is similarly impacted by the type and stage of disease [1]. The mean survival of patients with myeloma-associated disease is 5 months with most patients succumbing to complications stemming from cardiac or renal failure. The prognosis is slightly better for patients with systemic non-myeloma-associated disease with a mean survival of 2 years. Response to chemotherapy and single-organ limited forms of the disease (i.e., neuropathy) fare better than most patients. Cardiac disease usually indicates a very poor overall prognosis.
References
Breathnach S. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1.
Tan S, Pepys M. The amyloidosis. Histopathology. 1994;25:403.
Wright J. Clinico-pathologic differentiation of common amyloid syndromes. Medicine. 1981;69:429.
Palecek T, Fikrle M, Nemecek E, Baueroca L, Kuchynka P, Louch WE, Sicka I, Rysava R. Contemporary treatment of amyloid heart disease. Curr Pharm Des. 2014;21:4.
Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145(1):105–9.
Brownstein M, Helwig E. The cutaneous amyloidoses: localized forms. Arch Dermatol. 1970;102:8.
Qiao J, Fang H, Yao H. Amyloidosis cutis dyschromica. Orphanet J Rare Dis. 2012;7:95.
Ratz J, Bailin P. Cutaneous amyloidosis. J Am Acad Dermatol. 1981;4:21.
Unni M, Ankad B, Naidu V, Sudakar RK. A familial poikiloderma-like cutaneous amyloidosis. Indian J Dermatol. 2014;59(6):633.
Hashimoto K. Diseases of amyloid, colloid, and hyaline. J Cutan Pathol. 1985;12:322.
Lee D, Huang C, Wong C. Dermatopathologic findings in 20 cases of systemic amyloidosis. Am J Dermatopathol. 1998;20:438.
Noren P, Westermark P, Cornwell G, et al. Immunofluorescence and histochemical studies of localized cutaneous amyloidosis. Br J Dermatol. 1983;108:277.
Breathnach S, Bhogal B, Dyck R, et al. Immunohistochemical demonstration of amyloid P component in skin of normal subjects and patients with cutaneous amyloidosis. Br J Dermatol. 1981;105:115.
Chang YT, Liu HN, Wang WJ, Lee DD, Tsai SF. A study of cytokeratin profiles in localized cutaneous amyloids. Arch Dermatol Res. 2004;296(2):83–8.
Kobayashi H, Hashimoto K. Amyloidogenesis in organ-limited cutaneous amyloidosis: an antigenic identity between epidermal keratin and skin amyloid. J Invest Dermatol. 1983;80:66.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Vesely, N., Castillo, B., Morgan, M.B. (2016). Amyloidosis: Systemic, Nodular, and Epidermal Derived. In: Crowe, D., Morgan, M., Somach, S., Trapp, K. (eds) Deadly Dermatologic Diseases. Springer, Cham. https://doi.org/10.1007/978-3-319-31566-9_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-31566-9_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31564-5
Online ISBN: 978-3-319-31566-9
eBook Packages: MedicineMedicine (R0)