
Abstract
Morphea, also called localized scleroderma, is a sclerosing skin disorder that resembles scleroderma (systemic sclerosis) in terms of cutaneous histopathological features, but differs demographically and clinically. Hallmark clinical and serological features of scleroderma (sclerodactyly, Raynaud’s phenomenon, internal organ involvement, and scleroderma-specific antibodies) are absent in morphea. Affecting adults and children equally, morphea is characterized by single or multiple indurated cutaneous plaques that can have variable appearance depending on the subtype and activity of disease. The predominant subtypes are circumscribed or plaque-type, linear, generalized, pansclerotic, and mixed morphea. Treatment of morphea, which may include corticosteroids, methotrexate, and/or phototherapy, is aimed at shutting down inflammation in active morphea and preventing cosmetic/functional impairment that may result from unabated activity.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others



Keywords
- Clinical features of morphea
- Classification of morphea
- Localized forms of scleroderma
- Morbidity in morphea
- Treatment in morphea
- Morphea
Classification and Epidemiology of Morphea
Morphea, also called localized scleroderma, is a chronic autoimmune disease characterized by inflammation and sclerosis of the skin. Morphea, like scleroderma, is characterized as a sclerosing skin disorder, due to characteristic histological findings shared by both disorders, including sclerosis of the dermis and sometimes subcutis in the absence of fibroblast proliferation. However, morphea differs from scleroderma demographically and clinically. In contrast to scleroderma, involvement of internal organs in morphea is unusual and very different from that in scleroderma, and the diagnosis does not carry the same implications in terms of morbidity and increased mortality. Thus, it is the opinion of the authors that the term “localized scleroderma” should be avoided to limit unnecessary evaluation and anxiety for patients and confusion among providers. For the purposes of this chapter, we will exclusively use the term “morphea.”
The clinical features of morphea are single or multiple sclerotic or indurated cutaneous plaques that are often dyspigmented (hypo- or hyperpigmented) and may have an erythematous border, depending on their stage of evolution. These plaques vary in appearance, depending on the subtype of morphea (Table 8.1) and activity of disease (see Assessment of disease activity and Stages of morphea lesions). Notably, cutaneous features of scleroderma, including Raynaud’s phenomenon, mat-like telangiectasias, sclerodactyly, acrosclerosis, decreased oral aperture, and nailfold capillary changes, are not seen in morphea.
The major subtypes of morphea include circumscribed or plaque-type, linear, generalized, pansclerotic, and mixed forms. Clinical images of the morphea subtypes are presented in Fig. 8.1. The classification scheme presented herein was developed by the Committee on Classification Criteria for Juvenile Systemic Sclerosis, composed of members of the Pediatric Rheumatology European society (PRES), the American College of Rheumatology (ACR), and the European League Against Rheumatism (ELAR) [1] (Table 8.1). Although there are many reported classifications of morphea, the authors have found this to be the most clinically relevant. The classification of morphea is based on the morphology of the skin lesions, as histopathology is similar in all the forms of morphea, and there are no known biomarkers for morphea subtypes. Histology is useful, however, in excluding other entities in the differential diagnosis (see Differential diagnosis of morphea).


(a) Plaque-type morphea. Circumscribed hyperpigmented plaques are present on the posterior legs of this patient with morphea. (b) Generalized morphea on the trunk. Hyperpigmented sclerotic plaques are present on the chest and abdomen. (c) Generalized morphea on the extremities. Symmetric, hypopigmented, sclerotic plaques are present on the legs of this patient with generalized morphea. (d) Linear morphea. Linear lesions of both the extremity and trunk are seen in this patient. Note that early linear lesions may not completely coalesce and may be confused with plaque-type morphea if not carefully examined. Note that on the trunk, linear lesions characteristically obey the midline. (e, f) Pansclerotic morphea. Note sheets of contiguous sclerosis, encompassing the majority of the body surface area (e) and characteristically sparing the fingertips, stopping at the metacarpophalangeal joints (f)
Variants and Related Entities
When linear morphea occurs on the upper face, especially the paramedian forehead, it is often called en coup de sabre (ECDS) (Fig. 8.2a, b). When it involves the lower face or produces hemifacial atrophy of deeper tissues, it is called progressive hemifacial atrophy (PHA), also known as Parry-Romberg syndrome (PRS) (Fig. 8.2c–f). Whether these conditions represent an entity different from morphea has been a subject of considerable debate, but recent literature suggests that they are part of the morphea disease spectrum [2] in the sense that PHA represents deep involvement of the facial tissues with resultant residual atrophy.


(a, b) En coup de sabre morphea. Depressed linear plaques are present on the foreheads of these patients. These lesions are often dyspigmented and may have more obvious dermal changes (a) or change predominantly in the subcutis. Interestingly, years after his initial period of activity on the forehead, this patient (b) also developed tenosynovitis. (c–f) Progressive hemifacial atrophy also known as Parry-Romberg Syndrome may be subtle (c), requiring additional exam maneuvers to detect. Asymmetry can sometimes be better appreciated using different facial expressions (d). When progressive, hemifacial atrophy may lead to more obvious lesions, as seen here on the chin, mandible, neck and tongue (e–f)
Overlying lichen sclerosus, changes may be seen in morphea lesions of all subtypes (Fig. 8.3). This observation has led to controversy over whether these represent two independent processes or whether changes similar to lichen sclerosus occur in morphea. Eosinophilic fasciitis (EF) has also been considered by some to be a variant of morphea, as about a third of patients with EF have findings typical of classic plaque-type morphea [3].

Morphea and lichen sclerosus. Sclerotic lesions of morphea with overlying areas of fine, hypopigmented, wrinkled skin are present. This lesion also has features of active inflammatory morphea with an erythematous border
Other entities such as bullous morphea and guttate morphea have been described in case reports and case series [4–13]. Examination of reports of bullous morphea reveals that bullae were largely present in areas of dense sclerosis in dependent areas [7]. This suggests that the bulla is a secondary change related to edema in dependent areas or lymphatic obstruction due to sclerotic changes in the skin, rather than a primary process [7, 11, 14]. Therefore, it is the opinion of the authors that bullae should not warrant a designation as a separate subtype. Similarly, guttate lesions likely represent a variant of circumscribed morphea.
Epidemiology and Clinical Course
Morphea has an estimated incidence of 2.7/10000 with a female to male ratio of 2–3:1 [15]. Existing studies suggest that morphea occurs most frequently in Caucasians, though population-based studies are needed to confirm this finding [16–18]. The reported frequency of different subtypes varies likely due to differing classification systems. However, linear morphea is more common in the pediatric population [15, 19], while the generalized and plaque subtypes predominate in adults. The clinical course is not well described, with periods of disease activity ranging from 3 to 6 years, and reactivation occurring after periods of remission in some patients [20]. Patients with pediatric-onset disease may also experience persistent disease and/or recurrences in adulthood [21, 22]. A retrospective evaluation of long-term outcomes of adults with pediatric-onset morphea from the Morphea in Adults and Children (MAC) cohort revealed that 89 % of patients (24/27) developed new or expanding lesions over time, suggesting that patients may need lifelong evaluation and repeated courses of treatment to prevent morbidity [21] (Fig. 8.4).

Chronic nature of morphea and sequelae. Lesions began on the right leg at age five in this patient. Note muscle atrophy, limb length discrepancy, and pes planus foot deformity (a). In adulthood, the same patient had a recurrence of morphea in the form of active inflammatory lesions of the trunk, seen here on the abdomen (b), as well as a plaque that appeared on the shoulder (c)
Studies examining recurrence rates after treatment with methotrexate and/or systemic steroids report recurrence 6–19 months after discontinuation of therapy in 10–44 % of patients [23–26]. Studies evaluating long-term outcomes after methotrexate therapy vary greatly in methods and duration of follow-up, so comparison of recurrence is difficult [23, 27]. Recurrence has also been described in patients after successful treatment with UVA-1 phototherapy at rates as high as 46 %, which may exceed those rates described with methotrexate. Median time to recurrence ranges from 10 months up to 20–30 months for those treated with UVA-1 and methotrexate, respectively [22, 23, 27–29]. These findings emphasize the need for regular monitoring of patients with morphea for signs of new disease activity even after successful treatment (see Assessment of disease activity in morphea).
Factors associated with recurrence in previous studies have included longer duration of disease before treatment [28], older age of onset in the pediatric population [25], and the presence of linear morphea of the limbs [25, 30]. However, these associations have not always been replicated across studies. Prognostic markers have not yet been identified for recurrent or chronic disease in the form of prospective longitudinal studies, but retrospective reviews and cross-sectional studies have implicated that increased duration of disease before treatment may be associated with increased likelihood of recurrent disease [28]. Preliminary analysis from the prospective MAC cohort has revealed similar recurrence rates between pediatric and adult patients and across subtypes of morphea. The only variable associated with recurrence in this cohort has been disease duration, in that patients with longer disease duration have been more likely to recur.
Etiology and Pathogenesis of Morphea
The etiology and pathogenesis of morphea is not well understood. Most pathological events ascribed to morphea have been extrapolated from research in scleroderma, as the two disorders are assumed to arise from a similar etiology [31]. Morphea, like other autoimmune disorders, likely arises from a genetic background of increased immune disease susceptibility, combined with other causative factors, such as trauma or environmental exposures, which modulate the expression of disease.
Genetics and Autoimmunity in Morphea
Like many autoimmune connective tissue diseases, morphea is likely a complex genetic disease. Familial clustering has been reported, although rarely [18, 32, 33], and morphea is also associated with higher than expected rates of familial autoimmune disorders [18, 21, 34]. In retrospective studies, morphea patients have demonstrated concomitant autoimmune disease, including psoriasis, systemic lupus erythematosus, multiple sclerosis, and vitiligo, at higher than expected frequency when compared with published population-based prevalence estimates [18, 34–36]. A population-based study examining a rheumatoid arthritis population in Sweden reported a higher risk of morphea among these patients with a reported standardized incidence ratio (SIR) of 2.40 [35]. A similar population-based study in Sweden also reported a higher risk of morphea among siblings of patients with multiple sclerosis with an SIR of 1.72 [36]. Morphea has been reported, in case studies, to coexist with other autoimmune diseases, including inflammatory bowel disease [37], autoimmune thyroid disease [38, 39], alopecia areata, type I diabetes mellitus [37], antiphospholipid syndrome [40], and necrotizing vasculitis [68]. Other types of inflammatory skin disorders can also be associated with morphea, such as psoriasis [41] and lichen planopilaris [42].
Of interest, the risk for morphea has been associated with the presence of specific HLA class I and II alleles, further implicating a possible underlying genetic predisposition for the disease. A large case-control association study of patients from the MAC cohort revealed strong associations with specific HLA class I and II alleles, including, most significantly, HLA-B*37, as well as another, DRB1*04:04, in common with a risk allele previously identified for systemic sclerosis [43]. Alleles conferring the greatest risk included HLA DRB1*04:04 and HLAB37 [43]. Risk alleles identified in this case-control study have also been associated with the risk in rheumatoid arthritis and autoimmune thyroid disease [44, 45], implying that there may be a common genetic susceptibility to these disorders.
Increasing evidence supports immune dysregulation as an important pathogenic event early in the course of morphea. Early morphea lesions are characterized by the influx of large amounts of mononuclear lymphocytes (usually activated T-lymphocytes), plasma cells, and eosinophils [31]. This is likely the result of autoimmunity, as there is widespread autoimmune reactivity in morphea patients including elevated ANAs (see Laboratory findings in morphea), cytokines, and adhesion molecules [18, 21, 46]. Vessel damage and upregulation of adhesion molecules (ICAM-1, VCAM 1, and E-selectin) occur related to the inflammatory cell infiltrate in morphea, which facilitates local monocyte recruitment [47]. These adhesion molecules also facilitate the recruitment of T-lymphocytes that are capable of producing pro-fibrotic cytokines (IL-4, IL-6, and TGF-beta) and may contribute to the development to sclerosis [31, 48]. Of note, increased levels of these vascular adhesion molecules have been detected in serum from patients with morphea [49]. These adhesion molecules are upregulated by cytokines classically associated with a Th2 immune response (IL-4, IL-1, and TNFs). Cytokines found in increased concentration in the sera and skin of morphea patients include IL-4, IL-6, and IL-8 [47, 50]. These cytokines, especially IL-4, upregulate TGF-beta, initiating a cascade of events that results in increased production of collagen and other extracellular matrix components via the induction of connective tissue growth factor, platelet-derived growth factor, and matrix metalloproteinases. Chimerism or nonself cells may also play a role in the pathogenesis of morphea by initiating a local inflammatory reaction [51].
In a recent study of 69 pediatric patients with morphea, interferon-gamma-inducible protein 10 (IP-10) levels were significantly elevated in the plasma of morphea patients when compared with healthy controls. Immunohistochemistry staining of IP-10 was also present in the dermal infiltrate of a subset of morphea patients who had available skin biopsies. IP-10 levels were significantly elevated in those with active versus inactive disease and correlated with standardized disease outcome measures of activity, further suggesting that IP-10 may be a potential biomarker for disease activity in morphea [52].
Although large-scale studies examining gene expression profiles in morphea are lacking, Milano et al. [53] have used gene expression array analysis to establish a gene expression signatures for scleroderma skin subsets as compared to unaffected skin of the same patient. In their first study, they included the skin from three patients with morphea. They identified five significantly different gene expression clusters: diffuse proliferation, inflammatory, limited, and normal-like in scleroderma. Gene expression profiles from all three biopsies of morphea skin fell into the inflammatory category, which was characterized by markers for an increase in immune response, response to pathogen, humoral defense, lymphocyte proliferation, chemokine binding, and chemokine receptor activity, and response to virus. These findings provide further evidence for an important role for immune dysregulation in morphea.
Taken together and extrapolating from research in scleroderma, these studies implicate an important role for interferons and Th1-skewed immune responses in early morphea, while Th2 cell lineages may become dominant in later disease as sclerosis predominates. Studies of this sort are necessary to identify pathways relevant to the pathogenesis of morphea and to pave the way for identification of biomarkers and therapeutic targets.
Histopathology of Morphea
Histopathological changes in morphea evolve over the duration of individual lesions. The histopathological changes have been divided into indeterminate (early), inflammatory, mixed inflammatory and sclerotic, and sclerotic (late) stage morphea [54] (Figs. 8.5 and 8.6). Increased numbers of T-cells are present in the inflammatory stage compared with a normal skin. As in scleroderma, the inflammatory stage of morphea is characterized by dermal edema and by lymphocytic and histiocytic inflammatory cell infiltrates with plasma cells in a perivascular, periadnexal, and interstitial pattern. Eosinophils, mast cells, and macrophages may also be present. One finding particularly characteristic of morphea is the presence of an inflammatory cell infiltrate in the junction between the dermis and subcutaneous fat. From there, the inflammatory cell infiltrate may stream down the septa of the subcutaneous fat (Fig. 8.6c, d).


Disease features in morphea. (a) Deep morphea. The areas affected by deep morphea, also known as morphea profunda, may have a cobblestone appearance with subcutaneous atrophy. (b–d) Inflammatory morphea. An active plaque in the inflammatory stage is present on the left leg of this patient (b) note the violaceous border. There are also multiple early lesions present on the right. (c) Early morphea lesions may be subtle, as seen here on the patient’s abdomen, where ill-defined indurated plaques with peripheral erythema indicate active inflammatory morphea. (d) This well-circumscribed plaque on the patient’s breast exemplifies the erythematous border and central sclerosis typical of an early evolving morphea lesion. (e) Sclerotic morphea. This lesion demonstrates exuberant sclerosis centrally with surrounding hypopigmentation and erythema, likely indicating that the lesion is transitioning toward a more inactive state. Also note telangiectasias, which can occur in the surrounding atrophy and should not be mistaken for erythema indicating inflammation. (f) Atrophoderma of Pasini and Pierini. Preadolescent girl with several-year history of a large plaque and small patches of atrophy on the lumbar back. The lesions have a sharp drop-off with a “punched out” appearance and are not indurated clinically. (g) Atrophy. Severe subcutaneous atrophy is present in this patient with linear morphea in the atrophic stage, as well as postinflammatory hyperpigmentation changes and limb length discrepancy. (h, i) Linear morphea of the extremity associated with disabling contractures. Note contractures of the hands in children with long-standing linear morphea. Contractures can be a manifestation of damage occurring most commonly in linear lesions crossing joints, but may also be a component of other subtypes, including generalized morphea, when distributed over the joints

(a) Skin biopsies of morphea have a characteristic “squared-off” shape because of the dense dermal sclerosis. There are patchy perivascular mononuclear cell infiltrates of lymphocytes, histiocytes, and occasional plasma cells, but otherwise the dermis is acellular compared with the normal skin. Flattening and atrophy of the epidermis are associated with underlying sclerosis. Hair follicles and sebaceous glands normally seen in a skin biopsy of hair-bearing skin are absent. (Magnification = 2.5×). (b) This specimen shows a pattern of “bottom-up” sclerosis that can be seen in morphea before the entire dermis is involved. The sclerotic collagen is in the lower half of dermis, while the normal collagen is in the upper half. Dense collagen has replaced normal collagen from the arrow down. The left upper corner of the image also shows the edge of an atrophic hair follicle. (Magnification = 5×). Sclerosis can also occur from the top down or may encompass the entire dermis, even extending into the subcutis and beyond in some cases. (c, d) Biopsies of active inflammatory morphea reveal perivascular, periadnexal, and interstitial inflammatory infiltrate. Also note the inflammatory cell infiltrate in the junction between the dermis and subcutaneous fat, streaming down through the septa of the subcutaneous fat. Though difficult to appreciate at this power, inflammatory infiltrate is typically composed of lymphocytes, plasma cells, and, occasionally, eosinophils. (e, f) Higher-power views of the non-sclerotic (e) and sclerotic (f) dermal areas show that the collagen in the sclerotic area has lost the fine fibrillar texture of normal collagen (Magnification = 40×). (g) Encasement of adnexal structures by dense collagen with loss of the fat around eccrine glands is shown. Eventually, atrophy of hair follicles, sebaceous glands, and eccrine glands (arrow) occurs in long-standing morphea (Magnification = 20×)
Recently, a cross-sectional study of the MAC cohort revealed a predilection for inflammatory cell infiltrate to occur in the border between the subcutis and the dermis. This was particularly true in cases where sclerosis followed a bottom-heavy pattern, characterized by hyalinized collagen bundles in the deep dermis and subcutis, sparing the papillary through the mid-dermis, a pattern which was typical of those with morphea profunda [55] (Fig. 8.6b). A biopsy with such changes, even in the absence of sclerosis, should alert providers to the possibility of early inflammatory morphea profunda. In contrast, in this the same study, a top-heavy pattern, characterized by collagen bundles exclusively in the papillary to superficial reticular dermis, was typical of those with lichen sclerosus overlap. Patterns of sclerosis varied equally in those with linear and plaque-type morphea between top-heavy, bottom-heavy, and full thickness patterns, with thickened collagen bundles throughout the dermis. Interestingly, among patients with generalized morphea, those with isomorphic pattern of distribution in patterns of chronic friction more often had top-heavy patterns of sclerosis, while bottom-heavy patterns predominated among those with symmetric patterns of distribution in generalized morphea. When inflammation was present, it consisted most often of lymphocytes (83/91, 91 %) and plasma cells (68/91, 75 %), but eosinophils were also noted with some frequency (17/91, 19 %).
The later sclerotic stage of morphea is characterized histopathologically by thickened, acellular, homogenized-appearing collagen bundles that may involve all levels of dermis and/or subcutis (where collagen bundles stream downward through the septa) (Fig. 8.6e, f). The adnexal structures (hair, eccrine glands) are surrounded by dense fibrosis with loss of fat around the eccrine glands in chronic disease (Fig. 8.6g). In specimens from morphea profunda, the deep reticular dermis, subcutis, and/or fascia also show sclerotic changes. It is not uncommon for inflammation and sclerosis to coexist (Fig. 8.6a). The atrophic stage is characterized by loss of inflammatory cell infiltrate, decreasing sclerosis, and an absence of appendageal structures. Telangiectasia may be evident.
Triggers and Precipitating Factors
The emergence of morphea following exposure to environmental exposures is intriguing and may provide clues for the pathophysiology of morphea. While there are no definitive associations, the development of morphea lesions has been linked to local tissue trauma including radiation, surgery, insect bites, and intramuscular injections [56]. Although controversial, infectious agents have also been linked to pathogenesis. Studies in Europe had implicated a role for Borrelia infection in the pathogenesis of morphea, but this association has been largely discounted.
Increasing evidence, however, exists for the role of trauma in the development of morphea lesions. In a study of 26 patients with severe juvenile localized scleroderma, four (15 %) reported a history of trauma to the area, and one had a history of dental extraction ipsilateral to the area in which morphea developed [57]. There are many examples in the literature of injection-site morphea [56, 58], including the onset of morphea at the site of vaccinations as well as at the site of an insulin pump placement, observed by the authors. In a cross-sectional analysis of 329 patients in the MAC cohort, 52 (16 %) had trauma-associated lesions at the onset of disease, most commonly chronic friction (isomorphic) and surgery/isotopic triggers [59]. These findings, if confirmed in future studies, might suggest that elective procedures and skin trauma or friction be avoided in morphea patients. However, necessary and lifesaving surgery, radiation, or other procedures should not be avoided merely due to a diagnosis of morphea.
Clinical Features of Morphea
Assessment of Disease Activity in Morphea
The assessment of disease activity in morphea is important to clinical decision-making, as most treatments with proven efficacy are directed at the inflammatory stage of disease and will not be effective in predominantly sclerotic or atrophic lesions. The risk-benefit assessment of pursuing treatment for morphea lesions depends on the potential for functional or cosmetic impairment and symptom burden due to active lesions. It is therefore important to make an accurate assessment of lesion activity in each clinical examination, including clinical photographs to compare lesions to prior visits, since active disease may warrant intervention, while inactive disease may be carefully observed over time (see Fig. 8.10). Though imaging modalities such as ultrasound can be useful for assessing disease activity (see Imaging methods), active morphea lesions can usually be distinguished from inactive lesions by clinical features. Signs of disease activity (which have been established by expert consensus and validated) include new lesions, lesions that have expanded in size from previous visits, the presence of an erythematous or violaceous border around lesions, and possibly increased patient symptomology such as pain or itch at the site of lesions [60, 61].
Stages of Morphea Lesions
Morphea of all subtypes can begin as erythematous patches or plaques and may be preceded by pain or itch. Later, hypopigmentation with skin thickening, which is a manifestation of sclerosis, begins to develop at the center of lesions. These lesions typically have an erythematous/violaceous border indicative of active inflammation and expansion (inflammatory phase) (Fig. 8.5b–d). Sclerosis develops centrally and may lead to a shiny white-yellow color, with surrounding hyperpigmentation, as lesions stop expanding (sclerotic phase) (Fig. 8.5e). The loss of hair follicles can lead to alopecia in areas of morphea.
As activity subsides, these sclerotic plaques will soften over the course of months to years and become atrophic with hyper- or hypopigmentation (atrophic phase). Sclerosis can lead to contractures and limitations in range of motion and may impede growth in the pediatric population, leading to limb length discrepancies. Limitation in range of motion may actually improve as the lesions progress from sclerosis into atrophy.
Atrophy produces varying features depending on the level of skin involvement: cigarette-paper atrophy (papillary dermis), cliff-drop atrophy (dermis), or deep indentations that alter the contour of the affected site (subcutis). Atrophoderma of Pasini and Pierini (Fig. 8.5f) is thought to be the residua of plaque-type morphea involving dermal atrophy, as the borders of these lesions are characterized by the cliff-drop appearance. When morphea affects the subcutis or deeper, long-term sequelae may include limb length discrepancies, limitations in range of motion, and contractures (Fig. 8.5g–i).
Depth of Involvement
Assessment of the level of involvement in morphea is important to recognize potential comorbidities as well as to guide treatment (see Fig. 8.10). Morphea involving the superficial to the mid-dermis may be amenable to topical or phototherapy. Deep morphea, or morphea profunda, on the other hand, which involves the deep dermis, subcutaneous tissue, fascia, and/or muscle, will require systemic therapy to suppress disease activity when lesions are widespread or threaten function or cosmesis.
Deep involvement can occur with any subtype of morphea. Hemifacial atrophy, previously known as Parry-Romberg syndrome, represents deep morphea (morphea profunda) of the face that may or may not be accompanied by a more dermal linear or en coup de sabre (ECDS) lesion. Similar changes are seen in other anatomic sites with deep involvement (morphea profunda). Lesions in which the pathology is predominantly located in the deep dermis/subcutis are poorly circumscribed and may be accompanied by changes such as cobblestoning and altered contour in the sclerotic phase, as the skin becomes tacked down by sclerosis to underlying fascia (Fig. 8.5a). A “groove” sign or depression may be present at the site of tendons and ligaments, and deep tissue loss may result in the atrophic phase. However, as deep involvement can occur without obvious superficial changes, it may be more reliably detected with careful palpation, biopsy, and/or imaging than by visual exam alone (see Figs. 8.9 and 8.10).
Data is lacking on the correlation between depth and involvement and amount of damage in morphea. However, a preliminary analysis of the MAC cohort revealed that the vast majority of patients who had functional abnormalities (including limited range of motion, contractures, limb length discrepancies in morphea-involved areas) also had deep involvement of their disease (80/86, 93 %). These results imply a possible connection between the depth of disease and potential for damage and further emphasize the importance of assessment of the level of involvement as part of the clinical evaluation of the patient with morphea (Fig. 8.10) [62].
Morbidity in Morphea
Quality-of-life assessments show that individuals with morphea have better outcomes than those with disabling severe atopic dermatitis, with the exception of children and adolescents with the more severe forms of morphea affecting the face and limbs [63]. However, large cross-sectional studies of the MAC cohort have demonstrated that morphea patients do experience impairment of health-related quality of life that is greater than those with nonmelanoma skin cancer, vitiligo, and alopecia [64]. Notably, symptoms of pruritus and pain have been significantly associated with impaired quality of life in morphea, contrary to conventional wisdom that morphea is an asymptomatic disorder [64, 65].
A variety of internal disorders are reported to occur in patients with morphea, more frequently in the linear [34] and generalized or mixed subtypes [18], though they are different than the internal organ manifestations of scleroderma. The most commonly associated disorders include musculoskeletal and deep soft tissue abnormalities and neurologic and ophthalmologic problems. Malignancy is a rarely associated morbidity and may include squamous cell carcinoma in long-standing pansclerotic morphea [66]. Few systematic studies have been performed because of the infrequency of morphea and even more infrequent coexisting morbidities, so relatively little is known about the course and treatment of these disorders in the context of morphea [57].
-
Musculoskeletal and soft tissue complications. The most common extracutaneous finding in morphea patients is arthritis/arthralgias, which have been reported in 12 % of pediatric patients with morphea (Fig. 8.7a) [34]. Both articular and soft tissue/bony abnormalities are typically associated with linear morphea. Other musculoskeletal complications include joint swelling, myalgia, and limb contractures [18] (Fig. 8.4a). Individuals with facial linear morphea can have dental abnormalities [66] including gingival involvement (Fig. 8.7b) and even ipsilateral tongue hypoplasia. Musculoskeletal changes including fascial thickening and enhancement, articular synovitis, tenosynovitis, perifascial enhancement, and myositis have been detected using MRI in morphea patients, particularly those with pansclerotic morphea, and have been observed in some cases even when involvement was not expected based on the exam [67]. However, the clinical significance of such findings has yet to be determined, and the authors would not recommend routine imaging in morphea patients without signs or symptoms of musculoskeletal involvement.
Fig. 8.7 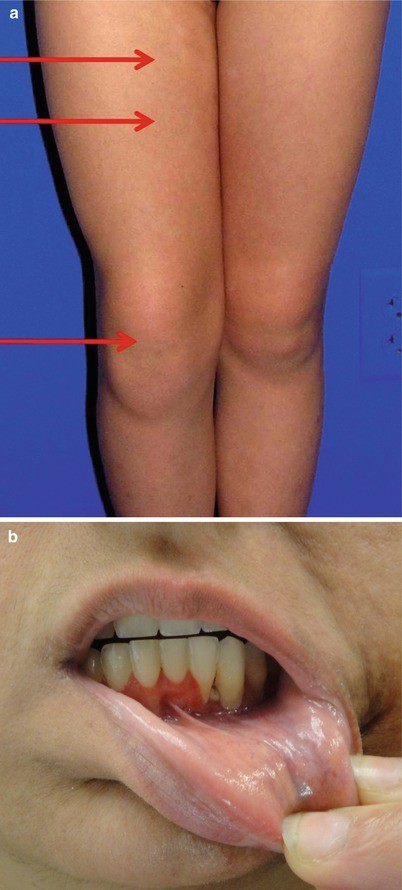
Morbidity in morphea. (a) Arthritis in morphea. A swollen right knee with effusion (lowest arrow) is present in this patient with morphea (upper areas). Notably, this patient also had spondyloarthropathy. (b) Gingival changes in morphea. Gingival changes, in the form of a kind of destructive gingivitis, are seen in this patient with linear morphea of the face. Referral to oral maxillofacial surgery may be warranted to determine need for intervention once lesions are inactive
-
Neurologic complications. Neurologic involvement was reported in 4 % of pediatric patients with morphea [34] and is more common in patients with linear morphea of the head. Complications include seizures, headache, and peripheral neuropathy among others. Intractable partial seizures [68], epileptic encephalopathy [69], status migrainosus [70], and central nervous system vasculitis [71, 72] have all been reported in association with morphea. Kistger et al. reviewed 54 patients with craniofacial scleroderma who also had neuroimaging (head CT or MRI) for neurologic symptoms. They found some common atypical features on imaging including atrophy, calcifications, and T2 hyperintensities. Others have described abnormal MRI results in patients with Parry-Romberg syndrome [73]. The clinical significance of such abnormal findings has yet to be determined, and at the current time, there is not sufficient evidence to recommend imaging in the morphea population in the absence of neurologic manifestations by history or physical examination.
-
Ophthalmologic complications. Ocular involvement was reported in 2 % of pediatric patients with morphea [34]. Ocular involvement in morphea is also more common in patients with linear morphea that affects the face, although it has also been reported to occur in patients without facial lesions [74]. Associated ophthalmologic abnormalities may include enophthalmos, anterior uveitis, episcleritis, glaucoma, xerophthalmia, keratitis, and strabismus [74].
-
Other manifestations. Genital involvement has been reported in association with morphea, particularly the generalized subtype, though to date, the frequency of such changes is not well described. Patients with genital involvement will often complain of pruritus, as well as dyspareunia, dysuria, and pain with defecation. Importantly, patients will not typically volunteer these symptoms without specific inquiry by providers [75, 76]. From the literature, the most common changes in the genital area include lichen sclerosus changes of porcelain-like polygonal macules, as well as more typical classical plaque-type morphea lesions with areas of waxy induration, and even labial fusion [75–78]. Patients with reported genital involvement have typically been postmenopausal women with the generalized subtype of morphea, though lichen sclerosus changes have also been reported with some frequency in plaque-type morphea [75, 78]. Review of the MAC cohort database revealed genital changes were present in 3.5 % of all participants (14/433), though this represented almost 10 % of patients with the generalized subtype of morphea (13/149, 8.7 %). Similar to reports in the literature, those affected were all women and were typically postmenopausal (13/14, 92.8 %). Similar to previous reports, genital involvement in the MAC cohort was primarily seen in patients with the generalized subtype of morphea, typically with features of lichen sclerosus overlap (13/14, 92.8 %).
-
Malignancy. Rare cases of malignancy are reported in morphea lesions, such as squamous cell carcinoma of the lip that developed in an individual with pansclerotic disabling morphea [66]. The etiology is likely similar to carcinoma developing in chronic venous insufficiency ulcerations and burn scars. Providers should take care to exclude cutaneous metastasis in lesions of morphea involving the breast (see Differential diagnosis of morphea).

Laboratory Findings in Morphea
Individuals with all subtypes of morphea, particularly those with deep involvement, may have peripheral eosinophilia and presence of markers of inflammation and autoimmunity (though this is relatively uncommon). The most common of these markers are positive antinuclear antibody (ANA) and/or rheumatoid factor and presence of anti-ssDNA and antihistone antibodies (AHAs), which suggest a predisposition to autoimmunity, but do not reliably correlate with disease activity or severity [16, 37, 38]. In one nested case-control study, morphea patients had a higher prevalence of ANA (63/187, 34 %) and AHA positivity (22/187, 22 %) as compared to matched healthy controls. There was a similar prevalence of ssDNA antibodies in cases (15/187, 8 %) and controls (10/149, 7 %). Of those morphea patients with positive ANA, most had speckled pattern (52/63, 81 %) at high titer (>1:1,280), though few patients with ANA positivity had extractable nuclear antigen (ENA) antibodies (7/63, 11 %), implying the possible presence of an unidentified antigen in morphea. The presence of these autoantibodies, however, has not reliably correlated with any measures of clinical activity. Thus ANAs and AHAs have limited clinical utility except in linear morphea, where their presence may be associated with greater disease burden and functional impairment [79].
There are no laboratory tests that confirm the diagnosis of morphea, which is made on clinical features with confirmatory histopathology. Biomarkers predictive of activity or prognosis are also lacking. Thus clinical assessment remains the mainstay of diagnosis and evaluation.
Differential Diagnosis of Morphea
Many morphea patients experience delay in diagnosis and treatment because providers fail to recognize this relatively rare disease [80]. The development of morphea can be insidious and subtle, and initial misdiagnosis, particularly by non-dermatologists, occurs frequently [80]. Morphea can mimic other cutaneous diseases, including atrophic conditions, vascular lesions, dyspigmenting disorders, and even more common skin lesions such as bruises and scars (see Table 8.2) [81–83]. Early morphea lesions have been periodically confused with port-wine stains in the pediatric population [83, 84], because early lesions may present as erythematous linear patches with minimal sclerosis. Histology can be useful to distinguish these entities, and lesions should also be monitored for changes over time, as induration, atrophy, and sclerosis may develop over time and reveal the correct diagnosis [83].
Providers should consider common entities in their differential diagnosis, including lipodermatosclerosis and trauma-induced fat necrosis. Steroid atrophy can mimic morphea, as subcutaneous atrophy may result at the site of steroid and other intramuscular injections, and injection of the scalp to treat other skin conditions can actually mimic linear morphea of the scalp. These entities can usually be differentiated from morphea by a careful history and physical examination, and, when necessary, a biopsy. Providers should also make sure to exclude more dangerous entities such as metastatic carcinoma in the appropriate settings (for instance, of lesions on the breast, overlying the site of known prior carcinoma). Post-radiation morphea can mimic radiation dermatitis, infection, or recurrent breast carcinoma (Fig. 8.8), and differentiating morphea from radiation dermatitis will depend on both clinical and histopathological features [85]. Special care should also be taken to distinguish morphea from scleroderma based on clinical features already described. Patients will often confuse the two entities, and misplaced concern over systemic disease damage to internal organs may impact negatively on patient quality of life [64].

Postirradiation morphea on the breast. Morpheaform changes occurred after breast reconstruction and radiation therapy in this patient with a history of breast cancer. Note the importance of a wider skin examination to differentiate this process from radiation dermatitis or recurrent carcinoma, as multiple morphea lesions are present on the trunk as well. Biopsy would be warranted in this case to rule out malignancy. Though postirradiation morphea has been reported, patients should not avoid necessary radiation or other lifesaving treatments due to a diagnosis of morphea
The diagnosis of morphea is made clinically, but biopsy and histological examination may be useful to aid decision-making based on the depth of involvement (see Fig. 8.9). Biopsy should also be done to exclude potential malignancy and in cases in which history and physical examination findings are not definitive, and pathology is needed to exclude disorders other than squamous cell carcinoma. Given the lack of accepted biomarkers, laboratory-based tests are not currently recommended for the evaluation of morphea in the absence of specific signs or symptoms that would otherwise prompt them (see Laboratory findings in morphea). Imaging is not recommended routinely for diagnosis, but may be useful to evaluate for depth of involvement or to monitor disease activity (see Imaging methods and Fig. 8.9).

Algorithm for the evaluation of patients with morphea (Reproduced with permission from: Jacobe H. Treatment of morphea (localized scleroderma) in adults. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (Accessed on 19 Dec 2014.) Copyright © 2014 UpToDate, Inc. For more information visit www.uptodate.com)
Approach to the Evaluation of the Patient with Morphea
Approach to the patient with morphea depends on a number of factors, including disease subtype, disease activity, and depth of involvement, as well as patient symptomatology and the potential for cosmetic or functional impairment. Patients may benefit from a multidisciplinary approach to address functional and cosmetic issues as well as comorbidities when present (Fig. 8.9). As noted earlier, there are no widely accepted biomarkers for morphea, so laboratory-based tests are not routinely recommended for the evaluation of morphea in the absence of specific signs or symptoms that would otherwise warrant them. Depending on features found on clinical examination or on patient complaints (muscle pain, limited range of motion, neurologic symptoms or deficits, etc.), imaging may be warranted to evaluate the depth of lesions and involvement of underlying structures (see Imaging methods).
Imaging Methods
-
Ultrasound [86–89]. The most reliable assessment uses 20–25 MHz ultrasound (US). Ultrasound at 10–15 MHz – which is more readily available in the United States – is also useful [86, 87]. This modality has been more extensively used in making the diagnosis of morphea, but has been used more recently for measuring disease activity based on increased cutaneous blood flow and increased subcutaneous tissue echogenicity, and has been found to be a sensitive tool for monitoring localized scleroderma in the pediatric population [86, 90, 91]. However, ultrasound assessment is operator dependent and lacks standardization.
-
Magnetic resonance imaging (MRI) [67, 92]. Although the MRI findings in morphea can overlap with other soft tissue abnormalities such as fibromatoses and myofibromatoses, the experienced radiologist can distinguish features of morphea including thickening of the dermis and increased signal intensity during the inflammatory stages, as well as changes in signal in the bone marrow with deeper involvement. MRI is particularly useful for evaluation of the depth of infiltration in morphea and sequential analysis of disease activity [92]. MRI has also been used to detect musculoskeletal involvement, including fascial thickening and enhancement, articular synovitis, and tenosynovitis, in patients with morphea, even some in whom musculoskeletal involvement was not suspected based on the history and exam [67] However, the clinical significance of such findings has yet to be determined, and at this point there is no evidence for pan imaging in the patient diagnosed with morphea without signs or symptoms concerning for musculoskeletal involvement. Imaging is best done after conferring with a radiologist to optimize the methods in which the patient is imaged and to ensure attention to possible features of morphea.
Therapeutic Options for Morphea
A majority of morphea patients experience delay in diagnosis of greater than 6 months, as well as quite varied treatment that appears to depend more on provider specialty than on disease characteristics or evidence for treatment modalities [80]. However, there are a number of therapeutic options with proven efficacy in morphea (Table 8.3). Each of these may be considered depending on disease subtype, activity, depth of involvement, and other patient considerations. The authors recommend using the evidence-based algorithm below when considering therapeutic options for patients with morphea (Fig. 8.10).

Therapeutic algorithm for morphea based on existing evidence. Histological examination and/or MRI are encouraged to evaluate lesions for the depth of involvement and, likewise, determine appropriate treatment as well as evaluation of therapeutic efficacy. Superficial involvement is defined by histological evidence of papillary dermal involvement. Deep involvement is defined as sclerosis or inflammation of the deep dermis, subcutis, fascia, or muscle (Reproduced with permission from: Jacobe H. Treatment of morphea (localized scleroderma) in adults. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (Accessed on [19 Dec 2014].) Copyright © 2014 UpToDate, Inc. For more information visit www.uptodate.com). *There is very little evidence for any therapy addressing disease damage in morphea. The risk of disease reactivation is also unknown, but possible with the use of invasive procedures. Therefore, surgery and the like should only be undertaken after prolonged inactivity of disease. + There is no evidence for efficacy in the literature
It is important to note that, contrary to conventional wisdom, morphea can be successfully treated with proper therapy and patient selection. However, providers should remember, and emphasize to patients, that the therapeutic end point in morphea is not the complete resolution of lesions but rather the loss of features of activity (reduction of erythema, halting progression of lesions or development of new lesions). Disease damage may actually increase for a time with treatment as lesions transition from inflammatory to atrophic or sclerotic stages (though skin thickening is likely to soften over time, even after treatment cessation). Providers should make sure to distinguish features of damage, such as dyspigmentation or atrophy, which can result in more visible blood vessels and apparent erythema, from true erythema mediated by inflammation (this may be accomplished by examining areas of erythema with magnification and looking for the induration that accompanies inflammation).
The approach to the patient with morphea depends on the assessment of disease activity, depth of involvement, and the presence of other sequelae, which is typically based on clinical examination. Careful clinical photographs should be taken at each visit to monitor patient’s lesions, as subtle changes indicative of disease activity may otherwise be missed. Clinical scoring measures, such as the LOSCAT, may also be useful for quantifying and monitoring progression of disease or efficacy of therapy [60, 93]. Histological examination may aid in initial therapeutic decision-making, as it can be difficult to determine the depth of involvement by clinical exam alone. In these cases, biopsy of the advancing edge of a lesion can be undertaken to provide insight into both activity and depth of morphea (see Histopathology of morphea).
Phototherapy
There is substantial evidence for the efficacy of phototherapy in morphea, particularly for broadband UVA, narrowband UVB, and UVA-1 [94–99] (Table 8.3). UVB is more appropriate for lesions restricted to the superficial dermis, which are relatively thin on palpation and, on biopsy, have sclerosis and inflammation in the papillary and superficial reticular dermis. UVA-based treatments can penetrate to a greater depth and are therefore more effective for deeper dermal lesions. UVA-1, in particular, has been shown to normalize dermal collagen and reduce inflammation in morphea and thus may be effective for disease in either inflammatory or sclerotic phase [95]. There is also evidence in the literature for the efficacy of broadband UVA phototherapy, which can be an appropriate alternative when UVA-1 is not available [94]. Improvement in disease, noted by the halting of lesion progression and the reduction of erythema, should be seen after 10–20 treatments. A trial can usually be stopped after 20–30 treatments if improvement is not seen in that period. Treatments are usually given three to five times weekly for a period of several weeks until this number of total treatments has been reached. Evidence suggests that patients may continue to improve even after the cessation of therapy, leading some to suggest an even greater number of treatments (30–50) for further therapeutic benefit. Optimum doses and regimen for UVA-1 phototherapy have yet to be determined; however, one randomized controlled trial has suggested that both low-dose (20 J per square centimeter) UVA-1 and medium-dose (50 J per square centimeter) UVA-1 are equally effective, while medium-dose UVA-1 is more effective than narrowband UVB [96]. Phototherapy is not likely to be effective for lesions with deep involvement of the subcutis, fascia, or muscle and therefore should not be considered a primary therapy for such disease.
Vitamin D Derivatives
Only one study provides level-1 evidence on the effect of vitamin D derivatives in morphea and actually showed no difference between oral calcitriol and placebo, with both groups improving equally [100] (Table 8.3). The authors also point out that this study was underpowered, making definitive conclusions about the efficacy of oral calcitriol difficult. Various uncontrolled trials and case reports have shown improvement in patients using topical vitamin D derivatives applied under occlusion. However, this improvement took place over a period of several months, during which time lesions might be expected to improve independently.
Immunomodulators
-
Methotrexate with or without corticosteroids. The use of methotrexate monotherapy and methotrexate combined with systemic corticosteroids has been shown to be effective based on multiple prospective trials [2, 26, 29, 101–103] (Table 8.3). One double-blind randomized controlled trial has also shown that pediatric morphea patients treated with 15–20 mg weekly of methotrexate for 12 months, after induction with 1 mg/kg/day oral prednisone for 3 months, had lower clinical disease scores, decreased lesion temperature, and lower rates of relapse than those treated with prednisone induction alone (plus placebo) [29]. Relapse occurred within 12 months in 15 patients treated with methotrexate in addition to corticosteroids (32.6 %) versus 17 of those treated with placebo only after steroids (70.8 %). More specifically, new lesions developed in three methotrexate-treated patients (6.5 %) versus four placebo-treated patients (16.7 %) within 3–9 months. As the primary end point in the study was response to treatment within 12 months, it is difficult to determine the actual time for effect of this medication regimen. Similarly, optimum dose, route, and indications for the additions of corticosteroids to methotrexate, as well as duration of therapy, have not been definitively established. In most studies with combined therapy, including the randomized trial described, corticosteroids are used for induction therapy either orally or via intravenous pulse (IV methylprednisolone 30 mg/kg/day for 3 days per month or 1 mg/kg/day prednisone) over the first 3 months. Methotrexate is used as a steroid-sparing agent and initiated simultaneously (0.6 mg/kg/week in children or 15–25 mg/week for adults), then maintained for a prolonged period (1–2 years) and gradually tapered. Consensus treatment plans for pediatric morphea have recently been established that detail these monotherapy and combined therapy approaches [61]. As evidence to date has not suggested a difference in disease processes between pediatric and adult morphea, it is likely that adults will respond similarly to this regimen. From the literature, most patients will respond in a mean of 2–5 months with this approach, with patients early in their disease course typically responding best. Importantly, relapse has been noted frequently after cessation of therapy (see Epidemiology and clinical course), indicating that therapy suppresses disease activity but is not curative [27].
-
Other immunomodulators. Level 2 evidence supports the use of topical tacrolimus 0.1 % ointment under occlusion for active, inflammatory superficial plaque-type morphea [104] (Table 8.3). Recent case series using oral mycophenolate mofetil indicate possible efficacy in patients who are refractory to methotrexate or have intolerable side effects [105]. Mycophenolate mofetil also may be effective as a treatment adjunct in children with morphea; in one retrospective analysis, ten patients with severe or methotrexate-resistant pediatric morphea experienced clinical improvement with mycophenolate mofetil that resulted in withdrawal or reduction of doses of corticosteroids and methotrexate [106]. Retrospective case reports on the use of oral cyclosporine, bosentan, infliximab, and topical imiquimod have reported some efficacy, but definitive studies are lacking (Table 8.3).
Antimicrobials
Despite the widespread use of antimicrobials in morphea, including antibiotics and antimalarials, no published clinical trials of these agents exist [107]. Literature supporting the use of antimalarials is limited to a case series in which two patients improved with hydroxychloroquine, while simultaneously receiving methotrexate and corticosteroids [108]. In one retrospective review, 7 out of 11 patients had persistently active disease 3–153 months after initiating therapy with hydroxychloroquine [16]. At this time, the use of these agents in severe morphea is not indicated, pending more definitive evidence for their efficacy (Table 8.3).
Treatments Not Supported by Current Evidence
The efficacy of the most commonly used treatment for morphea, topical corticosteroids, has never been formally evaluated [62, 80]. There have also been no studies investigating the use of intralesional steroids. In the authors’ experience, intralesional steroids have been effective in treating circumscribed plaques of morphea or as an adjuvant for recalcitrant areas in patients receiving phototherapy or systemic treatment. Current evidence does not support the use of interferon-gamma in morphea [109], and the risks of penicillamine, including toxicities, outweigh its potential benefits, which have not been well established. At this time, there is no sufficient evidence to support the use of these agents, especially in moderate-to-severe morphea, when other agents have proven efficacy (unless contraindications exist to their use) (Table 8.3).
Adjunctive Therapy
A significant number of patients with morphea suffer irreversible sequelae that persist even after disease activity has subsided. Patients should be carefully evaluated for limitations in range of motion, contractures, limb length discrepancies, or other functional impairments. Treatment modalities may include physical therapy techniques for improving range of motion of affected limbs, stretching exercises for contractures, orthotics or shoe inserts to adjust for limb length discrepancies and to compensate for loss of subcutaneous fat, or even tendon release in some cases [16]. Referral to specialists, such as rheumatologists, physical therapists, orthopedists, oral maxillofacial surgeons, or plastic surgeons, may be helpful to maximize cosmesis and function and to minimize further damage. Importantly, studies to date are completely lacking in addressing this type of damage due to morphea.
Surgical Therapies
There is limited evidence to support the use of surgical therapies for morphea. Importantly, these therapies should only be considered when morphea has been inactive, and a patient has been off treatment for a number of years. Patients should be monitored closely with serial examinations for reactivation, and collaboration with an experienced plastic surgeon is vital.
Summary
Morphea is a relatively uncommon idiopathic inflammatory disorder that leads to the development of sclerotic plaques in the skin. Though morphea may share common histopathological features with scleroderma, the disease differs demographically and clinically. Involvement of internal organs in morphea is unusual and very different from that in scleroderma. Morphea occurs in adults and children and preferentially affects females. The pathogenesis of morphea is not well understood, but is likely to involve autoimmunity, as well as genetic and environmental factors. Lesions typically begin as inflammatory patches that evolve into firm, sclerotic plaques. Involvement may be limited to the dermis or may extend to underlying subcutaneous fat, muscle, or bone. Atrophic changes often remain after resolution of lesion activity.
Morphea has a variety of clinical presentations. Circumscribed and generalized morphea occurs more frequently in adults, while linear morphea predominates in children. The identification of characteristic clinical findings often is sufficient for the diagnosis of morphea, but biopsy may be performed to exclude other entities or to obtain information on the depth of disease. Antinuclear antibody (ANA) levels are elevated in some patients with morphea; however, routine testing for autoantibodies is not indicated. Magnetic resonance imaging (MRI) or ultrasound can be used to assess the extent of involvement, in those patients for whom clinical examination suggests involvement deeper than the dermis, and may be useful for following disease activity during treatment.
The assessment of disease activity and depth of involvement guides therapeutic decision-making in morphea. Because of the self-limited nature of morphea, patients with limited plaque disease may elect to defer therapy. For patients with superficial (dermal) circumscribed disease who desire treatment, but do not have access to or prefer to avoid the frequent visits required for phototherapy, treatment options include high-potency topical corticosteroids, intralesional corticosteroids, a topical vitamin D analogue, topical tacrolimus, or imiquimod. For patients with superficial (dermal) forms of morphea who are able to receive phototherapy, treatment with UVA-1 is preferred; alternatives include broadband UVA, narrowband UVB, or PUVA. Phototherapy is unlikely to be effective for morphea involving the subcutaneous tissue, muscle, or bone. Rapidly progressive, severe, disabling disease requires systemic therapy, involving a combination of methotrexate and systemic corticosteroids. Morphea may cause joint contractures and other functional impairments secondary to deep tissue sclerosis. All patients should be clinically assessed for the development of these findings. Physical therapy is essential for patients who are at risk for or who show evidence for functional impairments.
References
Zulian F, Woo P, Athreya BH, Laxer RM, Medsger Jr TA, Lehman TJ, et al. The Pediatric Rheumatology European Society/American College of Rheumatology/European League against Rheumatism provisional classification criteria for juvenile systemic sclerosis. Arthritis Rheum. 2007;57(2):203–12.
Tollefson MM, Witman PM. En coup de sabre morphea and Parry-Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol. 2007;56(2):257–63.
Pinal-Fernandez I, Selva-O’Callaghan A, Grau JM. Diagnosis and classification of eosinophilic fasciitis. Autoimmun Rev. 2014;13(4–5):379–82.
Su WP, Greene SL. Bullous morphea profunda. Am J Dermatopathol. 1986;8(2):144–7.
Rencic A, Goyal S, Mofid M, Wigley F, Nousari HC. Bullous lesions in scleroderma. Int J Dermatol. 2002;41(6):335–9.
Trattner A, David M, Sandbank M. Bullous morphea: a distinct entity? Am J Dermatopathol. 1994;16(4):414–7.
Daoud MS, Su WP, Leiferman KM, Perniciaro C. Bullous morphea: clinical, pathologic, and immunopathologic evaluation of thirteen cases. J Am Acad Dermatol. 1994;30(6):937–43.
Kobayasi T, Willeberg A, Serup J, Ullman S. Generalized morphea with blisters. A case report. Acta Derm Venereol. 1990;70(5):454–6.
Blaya B, Gardeazabal J, de Lagran ZM, Diaz-Perez JL. Patient with generalized guttate morphea and lichen sclerosus et atrophicus. Actas Dermosifiliogr. 2008;99(10):808–11.
Yamanaka M, Ishikawa O. Guttate morphea in a scleroderma spectrum disorder patient with anticentromere antibody. Eur J Dermatol: EJD. 2009;19(6):630–1.
Synkowski DR, Lobitz Jr WC, Provost TT. Bullous scleroderma. Arch Dermatol. 1981;117(3):135–7.
Rencic A, Brinster N, Nousari CH. Keloid morphea and nodular scleroderma: two distinct clinical variants of scleroderma? J Cutan Med Surg. 2003;7(1):20–4.
Fernandez-Flores A, Gatica-Torres M, Tinoco-Fragoso F, Garcia-Hidalgo L, Monroy E, Saeb-Lima M. Three cases of bullous morphea: histopathologic findings with implications regarding pathogenesis. J Cutan Pathol. 2015;42(2):144–9.
Kavala M, Zindanci I, Demirkesen C, Beyhan EK, Turkoglu Z. Intertriginous bullous morphea: a clue for the pathogenesis? Indian J Dermatol Venereol Leprol. 2007;73(4):262–4.
Peterson LS, Nelson AM, Su WP, Mason T, O’Fallon WM, Gabriel SE. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960–1993. J Rheumatol. 1997;24(1):73–80.
Christen-Zaech S, Hakim MD, Afsar FS, Paller AS. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol. 2008;59(3):385–96.
Sehgal VN, Srivastava G, Aggarwal AK, Behl PN, Choudhary M, Bajaj P. Localized scleroderma/morphea. Int J Dermatol. 2002;41(8):467–75.
Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe HT. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol. 2009;145(5):545–50.
Vierra E, Cunningham BB. Morphea and localized scleroderma in children. Semin Cutan Med Surg. 1999;18(3):210–25.
Marzano AV, Menni S, Parodi A, Borghi A, Fuligni A, Fabbri P, et al. Localized scleroderma in adults and children. Clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13(2):171–6.
Saxton-Daniels S, Jacobe HT. An evaluation of long-term outcomes in adults with pediatric-onset morphea. Arch Dermatol. 2010;146(9):1044–5.
Condie D, Grabell D, Jacobe H. Comparison of outcomes in adults with pediatric-onset morphea and those with adult-onset morphea: a cross-sectional study from the morphea in adults and children cohort. Arthritis Rheum. 2014;66(12):3496–504.
Kroft EB, Creemers MC, van den Hoogen FH, Boezeman JB, de Jong EM. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study. Br J Dermatol. 2009;160(5):1075–82.
Cox D, OR G, Collins S, Byrne A, Irvine A, Watson R. Juvenile localised scleroderma: a retrospective review of response to systemic treatment. Ir J Med Sci. 2008;177(4):343–6.
Mirsky L, Chakkittakandiyil A, Laxer RM, O’Brien C, Pope E. Relapse after systemic treatment in paediatric morphoea. Br J Dermatol. 2012;166(2):443–5.
Weibel L, Sampaio MC, Visentin MT, Howell KJ, Woo P, Harper JI. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155(5):1013–20.
Zulian F, Vallongo C, Patrizi A, Belloni-Fortina A, Cutrone M, Alessio M, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J Am Acad Dermatol. 2012;67(6):1151–6.
Vasquez R, Jabbar A, Khan F, Buethe D, Ahn C, Jacobe H. Recurrence of morphea after successful ultraviolet A1 phototherapy: a cohort study. J Am Acad Dermatol. 2014;70(3):481–8.
Zulian F, Martini G, Vallongo C, Vittadello F, Falcini F, Patrizi A, et al. Methotrexate treatment in juvenile localized scleroderma: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2011;63(7):1998–2006.
Mertens JS, Seyger MM, Kievit W, Hoppenreijs EP, Jansen TL, van de Kerkhof PC, et al. Disease recurrence in localized scleroderma: a retrospective analysis of 344 patients with paediatric- or adult-onset disease. Br J Dermatol. 2015;172(3):722–8.
Badea I, Taylor M, Rosenberg A, Foldvari M. Pathogenesis and therapeutic approaches for improved topical treatment in localized scleroderma and systemic sclerosis. Rheumatology. 2009;48(3):213–21.
Wadud MA, Bose BK, Al Nasir T. Familial localised scleroderma from Bangladesh: two case reports. Bangladesh Med Res Counc Bull. 1989;15(1):15–9.
Rees RB, Bennett J. Localized scleroderma in father and daughter. AMA Arch Dermatol Syphilol. 1953;68(3):360.
Zulian F, Vallongo C, Woo P, Russo R, Ruperto N, Harper J, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52(9):2873–81.
Hemminki K, Li X, Sundquist J, Sundquist K. Familial associations of rheumatoid arthritis with autoimmune diseases and related conditions. Arthritis Rheum. 2009;60(3):661–8.
Hemminki K, Li X, Sundquist J, Hillert J, Sundquist K. Risk for multiple sclerosis in relatives and spouses of patients diagnosed with autoimmune and related conditions. Neurogenetics. 2009;10(1):5–11.
Firoz EF, Kamino H, Lehman TJ, Orlow SJ. Morphea, diabetes mellitus type I, and celiac disease: case report and review of the literature. Pediatr Dermatol. 2010;27(1):48–52.
Dervis E, Acbay O, Barut G, Karaoglu A, Ersoy L. Association of vitiligo, morphea, and Hashimoto’s thyroiditis. Int J Dermatol. 2004;43(3):236–7.
Hiremath NC, Madan Mohan NT, Srinivas C, Sangolli PM, Srinivas K, Vrushali VD. Juvenile localized scleroderma with autoimmune thyroid disorder. Indian J Dermatol. 2010;55(3):308–9.
Hasegawa M, Fujimoto M, Hayakawa I, Matsushita T, Nishijima C, Yamazaki M, et al. Anti-phosphatidylserine-prothrombin complex antibodies in patients with localized scleroderma. Clin Exp Rheumatol. 2006;24(1):19–24.
Slimani S, Hounas F, Ladjouze-Rezig A. Multiple linear sclerodermas with a diffuse Parry-Romberg syndrome. Joint Bone Spine: Rev Rhum. 2009;76(1):114–6.
Saleh Z, Arayssi T, Saleh Z, Ghosn S. Superficial morphea: 20-year follow up in a patient with concomitant psoriasis vulgaris. J Cutan Pathol. 2009;36(10):1105–8.
Jacobe H, Ahn C, Arnett FC, Reveille JD. Major histocompatibility complex class I and class II alleles may confer susceptibility to or protection against morphea: findings from the morphea in adults and children cohort. Arthritis Rheum. 2014;66(11):3170–7.
Simmonds MJ, Gough SC. Unravelling the genetic complexity of autoimmune thyroid disease: HLA, CTLA-4 and beyond. Clin Exp Immunol. 2004;136(1):1–10.
Weyand CM, Hicok KC, Conn DL, Goronzy JJ. The influence of HLA-DRB1 genes on disease severity in rheumatoid arthritis. Ann Intern Med. 1992;117(10):801–6.
Takehara K, Moroi Y, Nakabayashi Y, Ishibashi Y. Antinuclear antibodies in localized scleroderma. Arthritis Rheum. 1983;26(5):612–6.
Gruschwitz MS, Hornstein OP, von Den Driesch P. Correlation of soluble adhesion molecules in the peripheral blood of scleroderma patients with their in situ expression and with disease activity. Arthritis Rheum. 1995;38(2):184–9.
Fett N, Werth VP. Update on morphea: part I. Epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64(2):217–28. quiz 29–30.
Yamane K, Ihn H, Kubo M, Yazawa N, Kikuchi K, Soma Y, et al. Increased serum levels of soluble vascular cell adhesion molecule 1 and E-selectin in patients with localized scleroderma. J Am Acad Dermatol. 2000;42(1 Pt 1):64–9.
Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch Dermatol Res. 1995;287(2):193–7.
McNallan KT, Aponte C, El-Azhary R, Mason T, Nelson AM, Paat JJ, et al. Immunophenotyping of chimeric cells in localized scleroderma. Rheumatology. 2007;46(3):398–402.
Magee KE, Kelsey CE, Kurzinski KL, Ho J, Mlakar LR, Feghali-Bostwick CA, et al. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther. 2013;15(6):R188.
Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE. 2008;3(7):e2696.
Uziel Y, Feldman BM, Krafchik BR, Laxer RM, Yeung RS. Increased serum levels of TGFbeta1 in children with localized scleroderma. Pediatr Rheumatol Online J. 2007;5:22.
Walker D SJ, Karai L, Currimbhoy S, Jacobe H. Histopathological changes in morphea and their clinical correlates: results from the Morphea in Adults and Children Cohort (MAC) V. In preparation. 2015.
Torrelo A, Suarez J, Colmenero I, Azorin D, Perera A, Zambrano A. Deep morphea after vaccination in two young children. Pediatr Dermatol. 2006;23(5):484–7.
Beltramelli M, Vercellesi P, Frasin A, Gelmetti C, Corona F. Localized severe scleroderma: a retrospective study of 26 pediatric patients. Pediatr Dermatol. 2010;27(5):476–80.
Ueda T, Niiyama S, Amoh Y, Katsuoka K. Linear scleroderma after contusion and injection of mepivacaine hydrochloride. Dermatol Online J. 2010;16(5):11.
Grabell D, Hsieh C, Andrew R, Martires K, Kim A, Vasquez R, et al. The role of skin trauma in the distribution of morphea lesions: a cross-sectional survey of the Morphea in Adults and Children cohort IV. J Am Acad Dermatol. 2014;71(3):493–8.
Arkachaisri T, Vilaiyuk S, Torok KS, Medsger Jr TA. Development and initial validation of the localized scleroderma skin damage index and physician global assessment of disease damage: a proof-of-concept study. Rheumatology. 2010;49(2):373–81.
Li SC, Torok KS, Pope E, Dedeoglu F, Hong S, Jacobe HT, et al. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res. 2012;64(8):1175–85.
Strickland NSA, Fett N, Connolly MK, Hansen C, Jacobe H. Current practices in the evaluation and treatment of morphea. in preparation. 2016.
Orzechowski NM, Davis DM, Mason 3rd TG, Crowson CS, Reed AM. Health-related quality of life in children and adolescents with juvenile localized scleroderma. Rheumatology. 2009;48(6):670–2.
Klimas NK, Shedd AD, Bernstein IH, Jacobe H. Health-related quality of life in morphea. Br J Dermatol. 2015;172(5):1329–37.
Das S, Bernstein I, Jacobe H. Correlates of self-reported quality of life in adults and children with morphea. J Am Acad Dermatol. 2014;70(5):904–10.
Petrov I, Gantcheva M, Miteva L, Vassileva S, Pramatarov K. Lower lip squamous cell carcinoma in disabling pansclerotic morphea of childhood. Pediatr Dermatol. 2009;26(1):59–61.
Schanz S, Fierlbeck G, Ulmer A, Schmalzing M, Kummerle-Deschner J, Claussen CD, et al. Localized scleroderma: MR findings and clinical features. Radiology. 2011;260(3):817–24.
Chiang KL, Chang KP, Wong TT, Hsu TR. Linear scleroderma “en coup de sabre”: initial presentation as intractable partial seizures in a child. Pediatr Neonatol. 2009;50(6):294–8.
Rigante D, Battaglia D, Contaldo I, La Torraca I, Avallone L, Gaspari S, et al. Longstanding epileptic encephalopathy and linear localized scleroderma: two distinct pathologic processes in an adolescent. Rheumatol Int. 2008;28(9):925–9.
Menascu S, Padeh S, Hoffman C, Ben-Zeev B. Parry-Romberg syndrome presenting as status migrainosus. Pediatr Neurol. 2009;40(4):321–3.
Holl-Wieden A, Klink T, Klink J, Warmuth-Metz M, Girschick HJ. Linear scleroderma ‘en coup de sabre’ associated with cerebral and ocular vasculitis. Scand J Rheumatol. 2006;35(5):402–4.
Bonilla-Abadia F, Munoz-Buitron E, Ochoa CD, Carrascal E, Canas CA. A rare association of localized scleroderma type morphea, vitiligo, autoimmune hypothyroidism, pneumonitis, autoimmune thrombocytopenic purpura and central nervous system vasculitis. Case report. BMC Res Notes. 2012;5:689.
Moseley BD, Burrus TM, Mason TG, Shin C. Neurological picture. Contralateral cutaneous and MRI findings in a patient with Parry-Romberg syndrome. J Neurol Neurosurg Psychiatry. 2010;81(12):1400–1.
Zannin ME, Martini G, Athreya BH, Russo R, Higgins G, Vittadello F, et al. Ocular involvement in children with localised scleroderma: a multi-centre study. Br J Ophthalmol. 2007;91(10):1311–4.
Lutz V, Frances C, Bessis D, Cosnes A, Kluger N, Godet J, et al. High frequency of genital lichen sclerosus in a prospective series of 76 patients with morphea: toward a better understanding of the spectrum of morphea. Arch Dermatol. 2012;148(1):24–8.
Schlosser BJ. Practice gaps. Missing genital lichen sclerosus in patients with morphea: don’t ask? Don’t tell?: comment on “High frequency of genital lichen sclerosus in a prospective series of 76 patients with morphea”. Arch Dermatol. 2012;148(1):28–9.
Farrell AM, Marren PM, Wojnarowska F. Genital lichen sclerosus associated with morphoea or systemic sclerosis: clinical and HLA characteristics. Br J Dermatol. 2000;143(3):598–603.
Kreuter A, Wischnewski J, Terras S, Altmeyer P, Stucker M, Gambichler T. Coexistence of lichen sclerosus and morphea: a retrospective analysis of 472 patients with localized scleroderma from a German tertiary referral center. J Am Acad Dermatol. 2012;67(6):1157–62.
Dharamsi JW, Victor S, Aguwa N, Ahn C, Arnett F, Mayes MD, et al. Morphea in adults and children cohort III: nested case-control study – the clinical significance of autoantibodies in morphea. JAMA Dermatol. 2013;149(10):1159–65.
Johnson W, Jacobe H. Morphea in adults and children cohort II: patients with morphea experience delay in diagnosis and large variation in treatment. J Am Acad Dermatol. 2012;67(5):881–9.
Sung JJ, Chen TS, Gilliam AC, McCalmont TH, Gilliam AE. Clinicohistopathological correlations in juvenile localized scleroderma: studies on a subset of children with hypopigmented juvenile localized scleroderma due to loss of epidermal melanocytes. J Am Acad Dermatol. 2011;65(2):364–73.
Kakimoto CV, Victor Ross E, Uebelhoer NS. En coup de sabre presenting as a port-wine stain previously treated with pulsed dye laser. Dermatol Surg Off Publ Am Soc Dermatol Surg. 2009;35(1):165–7.
Nijhawan RI, Bard S, Blyumin M, Smidt AC, Chamlin SL, Connelly EA. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64(4):779–82.
Kim HS, Lee JY, Kim HO, Park YM. En coup de sabre presenting as a port-wine stain initially treated with a pulsed dye laser. J Dermatol. 2011;38(2):209–10.
Schaffer JV, Carroll C, Dvoretsky I, Huether MJ, Girardi M. Postirradiation morphea of the breast presentation of two cases and review of the literature. Dermatology. 2000;200(1):67–71.
Cosnes A, Anglade MC, Revuz J, Radier C. Thirteen-megahertz ultrasound probe: its role in diagnosing localized scleroderma. Br J Dermatol. 2003;148(4):724–9.
Bendeck SE, Jacobe HT. Ultrasound as an outcome measure to assess disease activity in disorders of skin thickening: an example of the use of radiologic techniques to assess skin disease. Dermatol Ther. 2007;20(2):86–92.
Li SC, Liebling MS. The use of Doppler ultrasound to evaluate lesions of localized scleroderma. Curr Rheumatol Rep. 2009;11(3):205–11.
Li SC, Feldman BM, Higgins GC, Haines KA, Punaro MG, O’Neil KM. Treatment of pediatric localized scleroderma: results of a survey of North American pediatric rheumatologists. J Rheumatol. 2010;37(1):175–81.
Wortsman X, Wortsman J, Sazunic I, Carreno L. Activity assessment in morphea using color Doppler ultrasound. J Am Acad Dermatol. 2011;65(5):942–8.
Li SC, Liebling MS, Haines KA. Ultrasonography is a sensitive tool for monitoring localized scleroderma. Rheumatology. 2007;46(8):1316–9.
Horger M, Fierlbeck G, Kuemmerle-Deschner J, Tzaribachev N, Wehrmann M, Claussen CD, et al. MRI findings in deep and generalized morphea (localized scleroderma). AJR Am J Roentgenol. 2008;190(1):32–9.
Kelsey CE, Torok KS. The localized scleroderma cutaneous assessment tool: responsiveness to change in a pediatric clinical population. J Am Acad Dermatol. 2013;69(2):214–20.
El-Mofty M, Mostafa W, Esmat S, Youssef R, Bousseila M, Nagi N, et al. Suggested mechanisms of action of UVA phototherapy in morphea: a molecular study. Photodermatol Photoimmunol Photomed. 2004;20(2):93–100.
El-Mofty M, Zaher H, Bosseila M, Yousef R, Saad B. Low-dose broad-band UVA in morphea using a new method for evaluation. Photodermatol Photoimmunol Photomed. 2000;16(2):43–9.
Kreuter A, Hyun J, Stucker M, Sommer A, Altmeyer P, Gambichler T. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma. J Am Acad Dermatol. 2006;54(3):440–7.
de Rie MA, Enomoto DN, de Vries HJ, Bos JD. Evaluation of medium-dose UVA1 phototherapy in localized scleroderma with the cutometer and fast Fourier transform method. Dermatology. 2003;207(3):298–301.
Kerscher M, Meurer M, Sander C, Volkenandt M, Lehmann P, Plewig G, et al. PUVA bath photochemotherapy for localized scleroderma. Evaluation of 17 consecutive patients. Arch Dermatol. 1996;132(11):1280–2.
Grundmann-Kollmann M, Behrens S, Gruss C, Gottlober P, Peter RU, Kerscher M. Chronic sclerodermic graft-versus-host disease refractory to immunosuppressive treatment responds to UVA1 phototherapy. J Am Acad Dermatol. 2000;42(1 Pt 1):134–6.
Hulshof MM, Bouwes Bavinck JN, Bergman W, Masclee AA, Heickendorff L, Breedveld FC, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43(6):1017–23.
Uziel Y, Feldman BM, Krafchik BR, Yeung RS, Laxer RM. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. 2000;136(1):91–5.
Seyger MM, van den Hoogen FH, de Boo T, de Jong EM. Low-dose methotrexate in the treatment of widespread morphea. J Am Acad Dermatol. 1998;39(2 Pt 1):220–5.
Kreuter A, Gambichler T, Breuckmann F, Rotterdam S, Freitag M, Stuecker M, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141(7):847–52.
Mancuso G, Berdondini RM. Localized scleroderma: response to occlusive treatment with tacrolimus ointment. J Am Acad Dermatol. 2005;152(1):180–2.
Schlaak M, Friedlein H, Kauer F, Renner R, Rogalski C, Simon JC. Successful therapy of a patient with therapy recalcitrant generalized bullous scleroderma by extracorporeal photopheresis and mycophenolate mofetil. J Eur Acad Dermatol Venereol. 2008;22(5):631–3.
Martini G, Ramanan AV, Falcini F, Girschick H, Goldsmith DP, Zulian F. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil. Rheumatology. 2009;48(11):1410–3.
Mohrenschlager M, Jung C, Ring J, Abeck D. Effect of penicillin G on corium thickness in linear morphea of childhood: an analysis using ultrasound technique. Pediatr Dermatol. 1999;16(4):314–6.
Maragh SH, Davis MD, Bruce AJ, Nelson AM. Disabling pansclerotic morphea: clinical presentation in two adults. J Am Acad Dermatol. 2005;53(2 Suppl 1):S115–9.
Hunzelmann N, Anders S, Fierlbeck G, Hein R, Herrmann K, Albrecht M, et al. Double-blind, placebo-controlled study of intralesional interferon gamma for the treatment of localized scleroderma. J Am Acad Dermatol. 1997;36(3 Pt 1):433–5.
Laxer R, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18:606–3.
Saxton-Daniels S, Jacobe H. Morphea. In: Goldsmith L et al., editors. Fitzpatrick’s dermatology in general medicine. 8th ed. Chicago: McGraw-Hill; 2012.
Acknowledgment
The authors wish to thank Rose Ann Cannon for her support in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media New York
About this chapter
Cite this chapter
Teske, N.M., Jacobe, H.T. (2017). Morphea (Localized Scleroderma). In: Varga, J., Denton, C., Wigley, F., Allanore, Y., Kuwana, M. (eds) Scleroderma. Springer, Cham. https://doi.org/10.1007/978-3-319-31407-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-31407-5_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31405-1
Online ISBN: 978-3-319-31407-5
eBook Packages: MedicineMedicine (R0)