
Abstract
This chapter has reviewed various complement pathways, physiologic activities, biologic properties of complement fragments and receptors related to allergic diseases, and immunomodulation related to intravenous gamma globulin. It has also listed various autoimmune and inflammatory diseases benefiting from immune globulin and discussed autoimmune urticarial and selected diseases and syndromes associated with excessive complement activation and clinical associations and therapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Complement pathways
- Physiologic activities
- Fragments
- Receptors
- Immunomodulation and mechanisms
- Autoimmunity
- Therapies
The Complement System
Pathways and Physiologic Activities
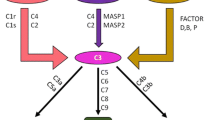
The complement system consists of more than 30 plasma and cell membrane proteins both first discovered classic (C1–C9), activated by antigen–antibody complexes, alternative pathway components (properdin, factors B and D), inhibitors (C1, factor 1, etc.), microbial cell walls, and regulatory proteins (C4 binding, factor H, S protein (Fig. 23.1)). Complement is part of the innate immune system and is an important effector mechanism of humoral immunity. The main physiologic activities are listed which illustrates host defense against infection, bridging innate and adaptive immunity. The removal of immune complexes and inflammatory products is performed by C1q and covalently bound fragments of C3 and C4. Initiators of activation pathways for the classical pathway include apoptotic cells, viruses, gram-negative bacteria, and C-reactive protein in addition to immune complexes. The early steps of complement activation and classical pathways are illustrated in Fig. 23.2. The mannose-binding lectin or collectin, homologous to C1q, is initiated by organisms with terminal mannose groups, and decreased levels have been noted in children with recurrent infections. The late steps of complement activation and the membrane attack complex (MAC) are shown in Fig. 23.3.

Schematic representation of the classical complement cascade showing the initiation of the pathway by binding of the C1q component of C1 binding to the Fc region of an IgG antibody molecule bound to an antigen on the surface of a target cell. The numbers in bold indicated the sequential steps which are involved in the activation of each of the components leading to the final lytic event carried out by the MAC. Some sources now use a revised nomenclature for the fragment of C2, in which C2 is the small fragment diffuses away and C2b is the larger fragment that binds with C4b and acts in the convertase, C4b2b (Source: Immunology IV Text, Editor Bellanti, Berger)

Schematic representation of classical pathway activation. (a) Upon binding of C1 to the Fc region of IgG or IgM, C1r and C1s are activated. C1s cleaves C4 and C2, and the C4b binds covalently to the surface of a target cell and C2a subsequently binds to the C4b forming C4b2a complex, the C3 convertase. (b) Following the binding of C3 to the C4b2a complex, it is cleaved by the C2a component of the complex releasing C3a, which diffuses away, and C3b, which adheres to the C4b2a complex forming C4b2a3b, the C5 convertase; note that C4b2a may deposit at a distance from the initial C1 site and that many molecules of C3b may be deposited. Only a few will join with C4b2a to form the C5 convertase. (c) Cleavage of C5 by C2a releases C5a and allows C5b to bind with C6. The C5b6 complex can insert into the plasma membrane; C5b6 can also insert at a distance from the convertase that cleaved the C5. (d) C7 and C8 can bind with C5b6, forming a complex causing C9 molecules to unfold, polymerize, and insert into the membrane, to a protein-linked pore (Source: Immunology IV Text, Editor Bellanti, Berger)

Schematic representation of the alternative pathway showing the initiation of the pathway when C3b, which may be formed by one of the other pathways or by nonspecific proteolytic cleavage of C3, binds with Factor B, which is analogous to C2. When bound to C3b, B acquires a conformation that allows it to be cleaved of C3, binds with Factor B, which is analogous to C2. When bound to C3b, B acquires a conformation that allows it to be cleaved by the protease D following which the subsequent steps of the pathway are similar to those of the other two pathways (Source: Immunology IV Text, Editor Bellanti, Berger)
Biologic Properties of Complement Fragments Related to Allergic Diseases
Complement cascade activation leads to generation of biologically active fragments (Table 23.1). The products of C3 and C5 are small polypeptide anaphylatoxins that have a variety of biologic properties; C3a, C4a, and C5a release inflammatory mediators from mast cells, induce smooth muscle contraction, promote vascular permeability, and induce adhesion molecules on endothelial cells. C3a can also lead to mucus secretion by goblet cells, and C3a and C3a des arg can modulate synthesis of tumor necrosis factor alpha (TNFα) and interleukin 1 beta (IL-1β) by mononuclear cells to focus the production of proinflammatory cytokines that contribute to the pathophysiology of asthmatic inflammation. Anaphylatoxic peptides can trigger a variety of responses which contribute to allergic and inflammatory reactions. Anaphylaxis is an immediate systemic reaction due to rapid, IgE-mediated release of potent mediators from tissue mast cells and peripheral blood basophils. Anaphylactoid reactions are immediate systemic reactions that mimic anaphylaxis, but are not caused by IgE-mediated immune responses. Mast cell and basophil mediators may play a role in anaphylaxis and anaphylactoid reactions through tryptase which may activate complement by cleavage of C3.
C5a also plays an important role in recruiting phagocytic cells to sites of immune complex deposition in the lung leading to enhanced oxidative and lipoxygenase activity with leukotriene B4 (LTβ4) production. LTβ4 and other leukotriene mediators are known to play important roles in asthma allergic rhinitis and cystic fibrosis. The presence of C3a and C5a in the lung can also induce respiratory distress through contraction of smooth muscle walls in bronchioles and pulmonary arteries. Animal studies have demonstrated the expression of C3aR and C5aR by cells in the lung suggesting a role for these receptors during lung inflammation both in sepsis and asthma (Table 23.2).
Cellular Receptors and Regulators
Receptors for complement components are expressed on many cells with important functions as listed in Table 23.3a. However, unlike most cellular receptors, some of the complement receptors also act as control molecules and interact with the molecule they bind to allow for further degradation of the bound fragment. Various inhibitors and regulators of complement activation and actions are listed in Table 23.3b. C5a also acts as potent chemoattractant for LFA integrins (CD11a/CD18) to enhance leukocyte movement into tissues at the site of infection. There are four cell membrane receptors for bound C3 or CR1, CR2, Cr3, and Cr4 that are within two gene families. CR1 or CD35 is found on mononuclear cells, neutrophils, mast cells, basophils, eosinophils, B and T lymphocytes, and kidney podocytes. It functions in phagocytosis and clearance of immune complexes. CR2 or CD35 expressed on B cell and follicular dendritic cells in addition to immature epithelial cells is utilized by Epstein–Barr viruses (EBVs) as a cellular receptor to promote cell entry.
Clinical Associations
Clinical effects of hereditary complement deficiencies related to infection, glomerulonephritis, angioedema, hemolysis, and systemic lupus erythematous (SLE) have been reported by Walport in articles on Complement. First and second of two parts N Engl J Med. 2001 Apr 5;344(14):1058–66 and N Engl J Med. 2001 Apr 12;344(15):1140–4. Complement is part of the innate immune system and underlies one of the main effector mechanisms of antibody-mediated immunity. It has three overarching physiologic activities defending against pyogenic bacterial infection, bridging innate and adaptive immunity, and disposing of immune complexes and the products of inflammatory injury. In this review, each of these activities will be placed in a clinical context. The pathways leading to the cleavage of C3 are triggered enzyme cascades, analogous to the coagulation, fibrinolysis, and kinin pathways. The terminal complement pathway, leading to the formation of the membrane attack complex, is a unique system that builds up a lipophilic complex in cell membranes from several plasma proteins.
SLE can be associated with allergic disease such as urticaria and can masquerade as atopy.
Complement deficiency can lead to increased susceptibility to pyogenic infections such as Haemophilus influenzae and Streptococcus pneumoniae, abnormality of function of the mannose-binding lectin, defective regulation of C3 associated with membranoproliferative glomerulonephritis, or compromise of the lytic activity increasing neisserial infections. C3b and iC3b which are covalently bound cleavage fragments of C3 are the most significant opsonins for bacterial host defense. Mannose-binding lectin as previously mentioned is low in recurrent infections but also involved in tissue inflammation and necrosis. The mechanism of entry used by various organisms involving complement is discussed in Walport’s article as three pathways of activation of the complement system: the classical, mannose-binding lectin, and alternative pathways (Tables 23.4a and 23.4b).
Three types of complement deficiency can cause increased susceptibility to pyogenic infections: a deficiency of the opsonic activities of the complement system, which causes a general susceptibility to pyogenic organisms; any deficiency that compromises the lytic activity of complement, which can increase the susceptibility to neisserial infections; and deficient function of the mannose-binding lectin pathway.
EBV uses glycoprotein 350/20, measles and picornaviruses employ hemagglutinin and capsid, and M. tuberculosis uses C3 fragments.
Hereditary angioedema (HAE), an autosomal dominant disease, is a deficiency of the C1 inhibitor with loss of regulation and failure to activate kallikrein. This disorder can lead to severe illness when it involves the intestinal submucosa or obstruction of the upper airway leading to death by suffocation. Symptoms usually begin in adolescence, and edema of the gastrointestinal tract results in severe colicky abdominal pain, nausea, and vomiting. Urticaria is not part of the syndrome, and swelling can be triggered by trauma, psychological stress with increased frequency with angiotensin inhibitors. Over 100 mutations in the C1-INH gene have been described. Type 1 HAE is due to a mutation which prevents the transcription of the abnormal allele, whereas type 2 variant is due to a point mutation in the gene abolishing its activity as a serine protease inhibitor. Patients with the type 2 variant have normal or elevated antigenic levels but synthesize a dysfunctional protein with reduced or absent C1-INH function.
A third type in women has clinical findings but normal C1-INH levels and function. Acquired angioedema in older patients with lymphoproliferative or monoclonal gammopathies has consumption of C1q. Laboratory features of HAE are decreased C1-INH, C2, and C4 levels.
A review by Khan was performed of historical and current literature of HAE. HAE I and II are related to insufficient production of C1-esterase inhibitor (C1-INH) or production of a dysfunctional C1-INH protein, respectively. HAE III is not related to C1-INH deficiency and the pathogenesis is unknown. Bradykinin appears to be the main mediator responsible for angioedema in patients with C1-INH deficiencies. Angioedema of the extremities, face, and upper airway along with gastrointestinal angioedema is the most common clinical feature in HAE. The laboratory tests that are most commonly used in the diagnosis of HAE include C4 and C1-INH concentration and C1-INH function. Advances in our understanding of the pathogenesis of HAE have led to several advances in the therapy of this disease. Despite our more thorough understanding of the genetics and pathophysiology of HAE, many questions remain unanswered.
Table 23.5 summarizes the complement profiles of the major forms of recurrent angioedema. As can be appreciated from this table, it is relatively easy to distinguish HAE with normal C1-INH from HAE due to C1-INH deficiency. The major challenge is distinguishing HAE with normal C1-INH from unknown or sporadic angioedema.
Treatment with infusion of C1 inhibitor can be lifesaving for HAE as listed in Walport’s article. Patients with complement deficiencies are also associated with various rheumatic diseases such as SLE, anaphylactoid purpura, dermatomyositis, and vasculitis (Table 23.6).
Paroxysmal nocturnal hemoglobinuria is a rare disease characterized by intravascular hemolysis, hemoglobinuria, and venous thrombosis due to the absence of decay accelerating factor (CD55) and inhibitor of the MAC (CD59).
Hemolytic uremic syndrome is due to factor H and I deficiency. Total deficiency of C3 and factor H mutations is associated with membranoproliferative glomerulonephritis. These patients have a complement consuming antibody called nephritic factor also found in partial lipodystrophy as listed in Walport’s article. Apoptosis has been linked with autoimmune diseases associated with complement deficiencies. C1q can bind to cells undergoing apoptosis with facilitation of elimination. Clearance of apoptotic cells has occurred through reactivity with collectin receptors or phagocytic cells that interact with C1q and mannose-binding lectin.
Immunomodulation of Autoimmunity with Intravenous Immune Globulin and Mechanisms of Immunomodulation
The mode of action of immune globulin involves modulation of the expression and function of Fc receptors with complement activation and the cytokine network. The immunoregulatory effects of immune globulin which involve complement include blockade of Fc receptors on macrophages and other cell inhibitions of the Fcγ receptor IIB. The effect on inflammation includes the decrease of complement-mediated damage and immune complex-mediated inflammation, induction of anti-inflammatory cytokines, inhibition of endothelial cell activation, neutralization of bacterial toxins, and reduction in requirements of corticosteroids. The effects on B cells and antibody production, T cells and cell growth illustrated in immunomodulatory mechanisms, and agents for the treatment of autoimmune diseases include antigen-specific tolerance using intravenous or mucosal antigen application, altered peptides, or vaccines. In addition to immunoglobulin treatment, immunomodulation may also involve a change in the cytokine balance, administration of agents that suppress regulatory cytokines such as IL-10 and TGFß which can occur in allergen immunotherapy, and administration of agents that antagonize, TNFα or stem cell transplantation.
Various autoimmune and inflammatory diseases benefiting from immune globulin and the immunomodulatory effects of immune globulin on B and T cells are illustrated in Walport’s article (Tables 23.7 and 23.8).
Autoimmune Urticaria
Patients with SLE also can present with chronic urticaria, and a subpopulation of patients with chronic urticaria also possess IgG antibody directed to the α-subunit of high-affinity type I IgE receptor. IgG can activate basophils, which is dependent on or augmented by complement. SLE, a prototype of immune complex disease, and other autoimmune diseases are caused by a breakdown of tolerance and other factors. Factors that influence the pathogenesis of T-cell-mediated autoimmune diseases are due to genetic susceptibility, activation of autoreactive T cells or infiltration of target organs by T cells, and damage to target organs by T-cell effector molecules or other cell populations. Breakdown to tolerance can occur in SLE, autoimmune diabetes, and multiple sclerosis.
Evidenced-Based Medicine
Complement Therapeutics in Clinical Practice
Treatment of patients with congenital complement deficiencies focuses on the underlying problems of infection and autoimmunity. Recombinant complement components for a completely deficient patient are possible, and blood transfusion to replace missing components has been tried with some success in two SLE patients with C2 deficiency and several patients with factor H deficiency. Renal transplantation might be a viable therapy specifically for atypical HUS patients with an MCP mutation.
The success of animal studies led to clinical trials, with sCR1 being used for treatment of acute respiratory distress syndrome, myocardial infarction, and lung transplantation and post-cardiopulmonary bypass syndrome and anti-C5 mAb in multicentered trials for myocardial infarction, post-cardiopulmonary bypass syndrome, rheumatoid arthritis, membranous nephropathy, and lupus nephritis.
The complement system as part of innate immunity provides an important effector system for host defense, clearance of immune complexes, and regulation or acquired immune reactions. The future of complement therapy may include targeted gene therapy or replacement with recombinant proteins for patients with complement deficiencies.
Therapeutic complement inhibitor approaches have been considered for treatment of bulbous pemphigus, rejection of transplanted tissues, Alzheimer’s disease since plaques contain high levels of classical and alternative pathway components as well as MAC components and immune-based fetal loss. As listed in an article by Tichaczek-Goska D on deficiencies and excessive human complement system activation in disorders of multifarious etiology in Adv Clin Exp Med. 2012 Jan–Feb;21(1):105–14 who described, selected diseases and syndromes are associated with excessive complement activation HIV and a great many other serious medical conditions. Other disorders that interface importantly with the complement system besides SLE include rheumatoid arthritis and related arthritides including cryoglobulinemia.
Complement also can be an important factor in tissue necrosis after ischemia. In addition, myocardial infarction and stroke are associated with complement activation in the area of tissue infarction. Complement participates in internal homeostasis by removing damaged, neoplastic, or infected cells. Thus, complement science is no longer thought to be just protein pathways involved in esoteric diseases but can be related to both autoimmune and cerebrovascular and myocardial disease. Deficiencies of the C3 and other complement components contribute to the emergence of recurrent bacterial, viral, and fungal infections and autoimmune diseases such as rheumatoid arthritis. The excessive activation of complement proteins is often discovered to be the reason for many diseases that include Alzheimer’s syndrome, schizophrenia, atypical hemolytic uremic syndrome, angioedema, macular degeneration, and Crohn’s disease that was also described by Tichaczek-Goska D etiology in Adv Clin Exp Med. 2012 Jan–Feb;21(1):105–14.
Bibliography
Bellanti J, Chapter 4, Complement Author: Melvin Berger, I Care Press, 2012;101–129.
Walport MJ. Complement, first and second of two parts. N Engl J Med. 2001;344:1140–4; 1058–1066.
Frieri M. Complement mediated diseases. Allergy Asthma Proc. 2002;23:319–24.
Frank MM. Complement disorders and hereditary angioedema. J Allergy Clin Immunol. 2010;125(2, Suppl 2):S262–71.
Frieri M. Asthma concepts in the new millennium-update in asthma pathophysiology. Allergy Asthma Proc. 2005;26:83–8.
Greenberger PA, Lieberman PJ. Idiopathic anaphylaxis. J Allergy Clin Immunol Pract. 2014;2(3):243–50.
Frieri M. Identification of masqueraders of autoimmune disease in the office. Allergy Asthma Proc. 2003;24(6):421–9.
Wen L, Atkinson JP, Giclas PC. Clinical and laboratory evaluation of complement deficiency. J Allergy Clin Immunol. 2004;113:585–93.
Kikuchi Y, Kaplan AP. Mechanisms of autoimmune activating basophils in chronic urticaria. J Allergy Clin Immunol. 2001;107:1056–62.
Kannradt T, Mitchison NA. Tolerance and autoimmunity. N Engl J Med. 2001;344:655–64.
Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747–55.
Khan D. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1–10.
Tichaczek-Goska D. Deficiencies and excessive human complement system activation in disorders of multifarious etiology. Adv Clin Exp Med. 2012;21(1):105–14.
Berger M. Complement. In: Bellanti JA, editor. Immunology IV. Clinical applications in health and disease. Bethesda: I Care Press; 2012. p. 101–29.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Frieri, M. (2016). Complement Systems and Allergy Diseases. In: Mahmoudi, M. (eds) Allergy and Asthma. Springer, Cham. https://doi.org/10.1007/978-3-319-30835-7_23
Download citation
DOI: https://doi.org/10.1007/978-3-319-30835-7_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30833-3
Online ISBN: 978-3-319-30835-7
eBook Packages: MedicineMedicine (R0)