
Abstract
Adipose tissue is considered an important source of mesenchymal stromal cells since it is an abundant tissue with great distribution throughout the body and human adipose-derived stem cells (hASCs) have high proliferation rates in vitro. These adult stromal cells have the ability to differentiate into tissues including bone, cartilage and adipose in vitro, and also display some biological functions such as trophic, paracrine and immunomodulatory effects that may have the greatest therapeutic impact in vivo for diverse pathologies. The considerable therapeutic potential of hASCs turned them of substantial interest in many areas of research. hASCs transplantation is currently being tested in more than 70 clinical trials for many diseases, including bone fractures, graft versus host disease, multiple sclerosis, brain injury, acute respiratory distress syndrome, idiopathic pulmonary fibrosis and diabetes. In this chapter, we will describe the protocols for hASCs isolation and characterization, besides summarizing current reported studies about their potential use.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
Multipotent mesenchymal stromal cells (MSCs) were first identified as non-hematopoietic cells found in the bone marrow, comprising the multipotent precursors of the bone marrow stroma (Friedenstein et al. 1968). More recently, it was demonstrated that MSCs are derived from perivascular cells or pericytes (revised in Caplan and Sorrell 2015). Although mesenchymal stromal cells (MSCs) are partially defined by their ability to differentiate into tissues including bone, cartilage and adipose in vitro, experiments in animal models revealed that it is their trophic, paracrine and immunomodulatory functions that might have the greatest therapeutic impact in vivo (Murphy et al. 2013; Caplan and Sorrell 2015).
According to the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy, these fibroblast-like plastic-adherent cells, regardless of the tissue from which they are isolated, should be termed multipotent mesenchymal stromal cells (MSCs) (Horwitz et al. 2005). The committee also proposes 3 minimum criteria to define a cell population as MSCs: (1) cells must be plastic-adherent when maintained in standard culture conditions, (2) express CD105, CD73 and CD90, and lack expression of CD45, CD34, CD14 or CD11b, CD79 alpha or CD19 and HLA-DR surface molecules, and (3) must differentiate into osteoblasts, adipocytes and chondroblasts in vitro (Dominici et al. 2006).
MSCs from the adipose tissue, human adipose-derived stromal cells (hASCs), were described in 2001 by Zuk and coworkers (2001). In 2002, the same group demonstrated their differentiation capacity and clonogenicity, and reported hASCs as a new adult stromal cell population (Zuk et al. 2002). The first use of hASCs for a therapeutic purpose occurred in 2004 by combining these cells with bone grafts in an attempt to treat extensive craniofacial damage in a 7 year-old girl (Lendeckel et al. 2004).
Adipose tissue is considered an interesting source of MSCs, as it is an abundant tissue with great distribution throughout the body. Moreover, hASCs have high in vitro proliferation rates . These adult cells can be isolated from liposuction samples, a material usually discarded, or from biopsies, in which even a small amount of tissue can provide high yield of cells (Zhang et al. 2014). Since they are obtained from adult tissues, there are no ethical issues associated with their use, unlike other kinds of stem cells, such as embryonic stem cells or neural stem cells obtained from aborted fetuses.
MSCs and hASCs include a mixture of several heterogeneous populations characterized by the presence of specific cell surface markers which may indicate a particular function for that cell population (revised by Sorrell and Caplan 2010; Murphy et al. 2013). Therefore, MSCs may have different profiles according to the relative proportion of different sub-populations, or even due to the presence or absence of a particular population (Crisan et al. 2008; Zimmerlin et al. 2010, 2013; Li et al. 2011).
Similar to other MSCs, hASCs are immune privileged , as they lack expression of class II MHC and co-stimulatory molecules (CD80, CD86 or CD40) (Jacobs et al. 2013). This property allows the treatment of genetic disorders with cells from both healthy autologous or heterologous donors without immunosuppression. Interestingly, it was recently reported that the use of MSCs simultaneously with anti-inflammatories alters the activity of MSCs (Chen et al. 2014), suggesting that they should be applied without concomitant use of steroids.
At first it was believed that, since hASCs have the potential to differentiate into various cellular lineages in vitro, they could regenerate injured tissues in vivo (Vieira et al. 2008b, 2012). However, several studies, including xenogeneic transplants from our group (Valadares et al. 2014), reported functional recovery without significant differentiation of cells, supporting a new hypothesis according to which the beneficial effect of these cells occurs predominantly due to the production of trophic factors, cytokines, and antioxidants with the ability to promote diverse biologic responses, such as modulation of the immune system (Ichim et al. 2010; English et al. 2010; Gharaibeh et al. 2011; Singer and Caplan 2011; da Justa Pinheiro et al. 2012; Caplan and Sorrell 2015).
In short, the considerable therapeutic potential of hASCS turned them of great interest in many areas of research. hASCs transplantation are currently being tested in 74 clinical trials for many diseases and conditions such as ischemic revascularization (NCT01709279), diabetic foot ulcers (NCT02394886), bone/cartilage repair (NCT02307435) and brain injury (NCT01649700), among others (clinical trials.gov). hASCs have also been applied in trials for graft versus host disease (Fang et al. 2007). Here, we will describe the protocols for isolation, characterization and potential uses of hASCs.
3.2 Protocols
3.2.1 hASCs Separation from Adipose Tissue
This protocol was adapted from the original work of Zuk and coworkers (2001).
-
1.
Obtain a sample of adipose tissue (lipoaspirate or biopsy). Sample must be collected in 50 ml tubes containing Dulbecco’s modified Eagle’s medium (DMEM) and 2 % antibiotic/antimycotic;
-
2.
The sample can be processed immediately after collection or stored up to 24 h in the refrigerator at 4 °C in the tube with DMEM and antibiotic, until the processing (see Note 1);
-
3.
Wash sample with PBS (Phosphate Buffered Saline), with 2 % antibiotic/antimycotic. The volume of PBS should be twice the sample volume. Repeat this step two times (this step is important to remove the blood cells) (see Note 2);
-
4.
After the washing step, prepare the digestion solution: 0.075 % collagenase IA. Collagenase is diluted with PBS; the appropriate volume of this solution is approximately half the tissue sample volume;
-
5.
Digest the sample for 30 min at 37 °C with digestion solution;
-
6.
Enzyme activity is neutralized with DMEM containing 10 % FBS (Fetal Bovine Serum) and 1 % antibiotic/antimycotic, and centrifuged at 1200 RCF for 10 min to obtain a high-density pellet;
-
7.
The pellet should be resuspended in DMEM containing 10 % FBS and 1 % antibiotic/antimycotic, plated and incubated overnight at 37 °C and 5 % CO2. (Concentration: 5000 cells/cm2);
-
8.
Following incubation, the plates must be extensively washed with PBS with 2 % antibiotic/antimycotic to remove debris;
-
9.
Incubate cells with growth medium (see Note 3).
The same protocol was used by our group to isolate ASC from other species such as mouse, dog and cat. Adipose samples from healthy dogs and cats are obtained in animals submitted to castration surgery, under anesthesia. The adipose tissue is collected from the subcutaneous region without any harm to these animals.
Note 1: We tested a protocol for freezing samples immediately after collection, in 90 % FBS and 10 % DMSO while other groups reported freezing without cryopreservation or other frozen agents. According to Devitt and coworkers ( 2015 ), long-term storage of adipose tissue at −70 °C without cryopreservative agents can reduce viable cells yield, but these cells can be submitted to extended culturing, irrespective of cryopreservation duration or patient’s age. Since these cells are cryopreserved for future use in plastic and reconstructive surgical procedures or cell therapy, they must be stored for a long period. According to Minonzio and coworkers ( 2014 ), adipose-derived stromal fraction retain 85 % of cell viability, normal proliferative capacity and differentiation potential when stored in liquid nitrogen using serum-free cryopreservation media.
Note 2: Some protocols include an additional step of red blood cell lysing with a blood lysis buffer. When we compared the sample processing of a sample with or without the blood lysis step, we observed that cell viability may be reduced in the first option and thus we suggest removal of red blood cells (suspension cells) with culture passaging.
Note 3: As firstly reported by Zuk and coworkers ( 2001 ), cells were maintained with a growth medium composed by DMEM, 10 % FBS and 1 % antibiotic/antimycotic. To our knowledge, both DMEM low glucose and DMEM high glucose are efficient in promoting cell growth (Vieira et al. 2008a , b , 2012 ). Recently, our group began to use also a medium richer in nutrients (DMEM F12 + glutamax, 20 % FBS, 1 % non-essential aminoacids and 1 % antibiotic/antimycotic).
Protocol variations: Some variability can be found in published protocols such as collagenase type, and/or concentration. Some groups use cell strainers to help in removing debris. We observed that the use of these filters is not critical, particularly with small samples.
3.2.2 Maintenance
3.2.2.1 Growth/Expansion
-
1.
Growth medium is composed of DMEM low glucose supplemented with 10 % FBS and 1 % antibiotic/antimycotic (see Note 3);
-
2.
Growth medium should be changed every 3–4 days until subculture is performed.
3.2.2.2 Subculturing
-
1.
Passage is done when cells reach 80–100 % of confluence;
-
2.
Wash plates two times with PBS (3 ml for every 25 cm2);
-
3.
Incubate cells with trypsin at 37 °C for 5 min (1 ml for every 25 cm2) (see Note 4);
-
4.
Add growth medium to inactivate trypsin (3 ml for every 25 cm2);
-
5.
Count cells;
-
6.
For expansion, cells must be re-plated in concentration of 5000 cells/cm2.
Note 4: Detachment of plate can be done incubating cells for 5 min with porcine derived trypsin-EDTA (in concentrations of 0.25 % or 0.05 %) or using recombinant and xenofree enzymatic products.
3.2.2.3 Freezing
-
1.
Grow cells until they reach 80–100 % of confluence;
-
2.
Wash plates two times with PBS (3 ml for every 25 cm2);
-
3.
Incubate cells with trypsin at 37 °C for 5 min (1 ml for every 25 cm2);
-
4.
Add growth medium to inactivate trypsin (3 ml for every 25 cm2);
-
5.
Count cells;
-
6.
Centrifuge at 500 RCF for 5 min;
-
7.
Discard supernatant;
-
8.
Resuspend in cryopreservative medium containing 10 % DMSO and 90 % FBS (see Note 5) and transfer cells to cryovial. Number of cells/vial may vary;
-
9.
Quickly transfer cryovials to a freezing container, and then place it in −80 °C freezer overnight (see Note 6);
-
10.
Transfer cryovials to liquid nitrogen for long-term storage.
Note 5: Cryopreservative medium is made of 10 % dimethyl sulfoxide (DMSO) and with variable proportions of other components (90 % FBS or 80 % FBS + 10 % growth medium or 20 % FBS + 70 % growth medium).
Note 6: Cells may be frozen by means of a programmable freezer with different controlled-rates or using a freezing container, which contains isopropanol alcohol, providing a 1 °C/min cooling rate in a −80 °C freezer. After overnight storage in a −80 °C freezer, cryotubes are transferred to liquid nitrogen at −196 °C and can be maintained for long-term storage.
3.2.2.4 Thawing
-
1.
Cell thawing requires cryovials immersion in a waterbath at 37 °C for 2–3 min to gently defrost cryotubes (see Note 7);
-
2.
Mix defrosted cells with growth medium in a conical tube (5 ml of growth media for every 1 ml of defrosted cells);
-
3.
Centrifuge at 500 RCF for 5 min;
-
4.
Discard the supernatant;
-
5.
Resuspend cell pellet in growth medium and plate cells in concentration of 5000 cells/cm2.
Note 7: Cell thawing must be performed quickly and gently to avoid cell death caused by DMSO toxicity.
3.2.3 Characterization
3.2.3.1 In Vitro Differentiation
One of the minimum criteria proposed for defining a multipotent stromal cell population is their ability to differentiate in vitro into chondroblasts, osteoblasts and adipocytes (Fig. 3.1). Differentiation medium are commercially available, or may be home-made prepared. All differentiation media, both commercial and home-made, have subtle variations, including different compounds or concentrations. Here, we summarize medium composition to induce in vitro differentiation as described by Zuk and coworkers (2002).

In vitro differentiation of hASC. Left and right columns represent evaluation performed with hASCs maintained for 21 days with growth media and with differentiation media, respectively. (a, b) chondrogenic differentiation visualized by toluidine blue staining; (c, d) osteogenic differentiation by Von Kossa staining; and (e, f) adipogenic differentiation by Oil Red staining
Besides quantitative methods specified below, RNA expression analysis through qRT-PCR may be also performed. The in vivo differentiation potential should be tested in a specific animal model that represents a disease, or in conditions that highlight the cell capacity of tissue regeneration. However, in practice, MSCs are not always tested for their in vivo differentiation potential.
3.2.3.1.1 Chondrogenic Differentiation
Cell Preparation
-
1.
Grow cells until they reach 80–100 % of confluence;
-
2.
Wash plates two times with PBS (3 ml for every 25 cm2);
-
3.
Incubate cells with trypsin at 37 °C for 5 min (1 ml for every 25 cm2);
-
4.
Add growth medium to inactivate trypsin (3 ml for every 25 cm2);
-
5.
Count cells;
-
6.
Centrifuge at 500 RCF for 5 min;
-
7.
Discard supernatant;
-
8.
Resuspend cells to achieve droplets with high cell concentration (ranging from 1 × 105 to 1 × 107 cells per drop of 5 μl) (see Note 8);
-
9.
Drip every high cell concentration droplet in a 24-well plate, keeping the droplets spaced from each other;
-
10.
Incubate at 37 °C for 2 h for cell adhesion;
-
11.
Gently add growth medium in control wells and differentiation medium in experimental wells (see Note 9);
-
12.
Medium must be changed every 3–4 days until achieving 21 days of differentiation.
Note 8: Instead of high cell concentration droplets, one can also maintain a cell pellet (ranging from 1 × 10 5 to 1 × 10 7 cells) in a conic tube during differentiation. After 21 days, pellet must be collected and analyzed by histology.
Note 9: Chondrogenic medium is composed of DMEM-LG supplemented with 1 % FSB, 6.25 μg/ml insulin, 10 ng/ml TGF-beta1, 50 nM ascorbate-2-phosphate, 1 % antibiotic/antimycotic.
Chondrogenic differentiation is confirmed on day 21 by analysing presence of extracellular matrix mucopolysaccharides with toluidine blue staining.
Staining with Toluidine Blue
-
1.
Prepare toluidine blue at 1 %;
-
2.
Wash cells two times with PBS;
-
3.
Fix cells with paraformaldehyde 4 % diluted in PBS for 30 min;
-
4.
Discard solution;
-
5.
Incubate with toluidine blue 1 % for 30 min;
-
6.
Wash 3 times with PBS.
3.2.3.1.2 Osteogenic Differentiation
Cell Preparation
-
1.
Grow cells until they reach 80 % of confluence in 24-well plate or larger;
-
2.
Wash plates two times with PBS;
-
3.
Add growth medium in control wells and differentiation medium in tested wells (see Note 10);
-
4.
Medium must be changed every 3–4 days until achieving 21 days of differentiation.
Note 10: Osteogenic differentiation medium is composed of DMEM-LG supplemented with 10 % FBS, 50 μM ascorbate-2-phosphate, 10 mM B-glycerophosphate and 0.1 μM dexamethasone.
Osteogenesis is demonstrated by accumulation of mineralized calcium phosphate assessed by Von Kossa or Alizarin Red S staining. Quantitative analysis of osteogenic differentiation may be performed with alkaline phosphatase enzyme activity at 7 days of differentiation and at 14 or 21 days of differentiation with alizarin discoloration. Quantitative analysis should take into consideration the number of cells per well and the basal expression of alkaline phosphatase of non-differentiated cells in order to normalize results for comparison among samples.
Alkaline Phosphatase Activity
-
1.
Prepare substrate buffer and substrate solutions (see Note 11);
-
2.
For each well, prepare a 1.5 ml tube;
-
3.
Add a volume of 1 M NaOH equal to the volume of substrate that will be used;
-
4.
Rinse cells twice with PBS;
-
5.
Add an appropriate volume of substrate solution to each well size (0.5 ml per well of 12-well plate);
-
6.
Incubate for 15 min;
-
7.
Transfer the substrate solution to the NaOH-containing tubes;
-
8.
Rinse cells twice with PBS and add differentiation medium to continue differentiation;
-
9.
Set up a standard curve of p-Nitrophenol (see Table 3.1).
Table 3.1 Components of the p-nitrophenol standard curve
Note 11
-
Substrate buffer: Dissolve 188 g of glycine and 0.1017 of MgCl 2 in 500 ml water. Adjust pH to 10.5 with 1 N NaOH.
-
Substrate solution: Dissolve 1 tablet (5 mg) of phosphatase per 5 ml of substrate buffer.
-
p-Nitrophenol for standard curve: Prepare 100 nmol/ml solution of p-nitrophenol by combining 100 μl of 10 μmol/ml p-nitrophenol standard solution with 9.90 ml 0.02 N NaOH. Prepare further dilutions as described in Table 3.1 . As blank, use p-Nitrophenol and substrate solution. Read in spectrophotometer at 405 nm.
Staining with Alizarin Red S
-
1.
Prepare Alizarin Red S at 2 % and filtrate solution;
-
2.
Wash cells two times with PBS;
-
3.
Fix cells with ethanol 70 % diluted in PBS for 30 min;
-
4.
Discard solution;
-
5.
Wash once with Milli-Q water;
-
6.
Incubate with alizarin red for 30 min;
-
7.
Wash three times with PBS;
-
8.
After microscopic analysis, quantitative evaluation may be performed (see Note 12).
-
1.
Note 12: To perform alizarin red discoloration, an alcoholic solution (20 % methanol, 10 % acetic acid, 70 % PBS) is used for 15 min incubation. The resultant solution can be quantified through spectrophotometer assay (at 450 nm).
Staining with Von Kossa
-
1.
Carefully, wash cells with distilled water;
-
2.
Add silver nitrate at 1 %;
-
3.
Incubate under UV light for 40 min;
-
4.
Carefully, wash cells with distilled water;
-
5.
Remove silver nitrate excess using sodium thiosulfate 3 % or safranin O 1 % for 5 min;
-
6.
Wash cells carefully, with distilled water;
-
7.
Stain with Van Gieson solution (3.75 ml fuchsin 1 %, 46.25 ml saturated picric acid, 50 ml water) for 5 min;
-
8.
Wash carefully with 100 % ethanol.
3.2.3.1.3 Adipogenic Differentiation
Cell Preparation
-
1.
Grow cells until they reach 80 % of confluence in a 24-well plate or larger;
-
2.
Wash plates two times with PBS;
-
3.
Add growth medium in control wells and differentiation medium in tested wells (see Note 13);
-
4.
Medium must be changed every 3–4 days until achieving 21 days of differentiation.
Note 13: Adipogenic differentiation medium is composed of DMEM-LG supplemented with 10 % FBS, 1 μM dexamethasone, 500 μM 3-isobutyl-1-methylxanthine (IBMX), 200 μM indomethacin, 10 μM insulin and 1 % antibiotic/antimycotic.
Adipogenic differentiation is confirmed on day 21 by intracellular accumulation of lipid-rich vacuoles stainable with Oil Red O.
Staining with Oil Red O
-
1.
Prepare oil red stock solution diluting 0.15 g in 50 ml of isopropyl alcohol;
-
2.
Prepare oil red work solution diluting 8.2 ml of stock solution in 6.8 ml of distilled water;
-
3.
Wash cells two times with PBS;
-
4.
Fix cells with para paraformaldehyde at 4 % diluted in PBS for 30 min;
-
5.
Discard solution;
-
6.
Incubate with oil red work solution for 30 min;
-
7.
Wash three times with PBS;
-
8.
After microscopic analysis, quantitative evaluation may be performed (see Note 14).
Note 14: Quantitative analysis may be performed using a commercial colorimetric assay (AdipoRed, Lonza), where lipid droplets are stained and quantitatively measured in a fluorescent spectrophotometer, according to manufacturer’s instructions.
3.2.3.2 Immunophenotyping
Cell surface proteins are used as markers for cell characterization. Mesenchymal stromal cells are positively marked for CD105, CD73 and CD90 (adhesion molecules) and lack the presence of CD45, CD34, CD14 or CD11b, CD79a or CD19 and HLA-DR. Immunophenotyping is performed using specific antibodies for these proteins, and presence or absence of antibody ligation is verified when cells are read in flow cytometer (Fig. 3.2).

Exemplification of hASC immunophenotyping . In this assay, cells were evaluated in the 4th passage. Coloured peaks represent labeled population of hASCs, with respective antibodies. Comparison was performed with the same population of cells without any step of antibody incubation (white peaks)
-
1.
Grow cells until they reach 80–100 % of confluence;
-
2.
Wash plates two times with PBS (3 ml for every 25 cm2);
-
3.
Incubate cells with trypsin at 37 °C for 5 min;
-
4.
Add growth medium to inactivate trypsin (6 ml for every 25 cm2);
-
5.
Count cells;
-
6.
Resuspend with PBS in a concentration required in antibody manufacturer’s instructions;
-
7.
Incubate with antibody in dark room at 4 °C for 45 min, according to manufacturer’s instructions (see Note 15);
-
8.
Wash with 500 μl PBS;
-
9.
Centrifuge at 500 RCF for 5 min;
-
10.
Discard supernatant;
-
11.
Resuspend cells in 200 μl PBS (see Notes 15 and 16);
-
12.
Place cells in 96-wells plate with round bottom;
-
13.
Perform reading in flow cytometer.
Note 15: If secondary antibody is required, incubate for another 15 min with secondary antibody after step 11. Then, repeat steps 8–11 and proceed to steps 12–13.
Note 16: It is possible to fix cells after antibody labeling, allowing the analysis to be performed in the flow cytometer 2 days after labeling. To fix cells, instead of resuspending cells with PBS in step 11, resuspend with PFA at 4 % for 15 min, then repeat steps 9–10 and proceed to the next steps.
3.2.3.3 Karyotype
Karyotype analysis is crucial when cells are being considered for therapy. It has been reported that after long-term culture, cells may show chomosomal abnormalities (Roemeling-van Rhijn et al. 2013; Pan et al. 2014). Therefore, karyotype analysis should be performed in the same passage in which cells will be used for treatment.
-
1.
Grow cells in a 75 cm2 flask until they reach 70–80 % of confluence;
-
2.
Incubate with colchicine for 4 h;
-
3.
Before trypsinization, prepare a conical tube to save all the material that would normally be discarded during the procedure (such as the growth media and the PBS used to rinse cells);
-
4.
Wash plates two times with PBS (9 ml each time);
-
5.
Incubate cells with trypsin at 37 °C for 5 min (3 ml);
-
6.
Add growth medium to inactivate trypsin (12 ml);
-
7.
Centrifuge at 250 RCF for 10 min;
-
8.
Resuspend cell pellet in KCl hypotonic solution and incubate them for 20 min (see Note 17);
-
9.
Centrifuge at 250 RCF for 10 min;
-
10.
Resuspend with ice cold fixation solution and incubate for 1 min (see Note 18);
-
11.
Centrifuge at 250 RCF for 10 min;
-
12.
Repeat steps 10 and 11 for three times;
-
13.
Drop resuspended cells in slides;
-
14.
Stain slides with giemsa or band for microscopic analysis (see Note 19);
Note 17: KCl hypotonic solution (KCl 0,075 M): 5,6 g KCl diluted in 1 L of distilled water.
Note 18: Fixation solution: Mix 1 volume of glacial acetic acid with 3 volumes of methanol.
Note 19: Giemsa/Banding: See manufacturer’s instructions.
3.3 Potential Use
Historically, the therapeutic use of MSCs aimed to explore its regenerative potential into diverse tissues/organs such as liver (Theise et al. 2000) and heart (Zimmet and Hare 2005). A recent study reported that a proliferative population of myogenic progenitors was efficiently derived from ASCs. This population had characteristics similar to muscle satellite cells and was capable of terminal differentiation into myotubes. When transplanted into the mdx mice, the murine model for Duchenne muscular dystrophy, these progenitor cells successfully engrafted in skeletal muscle for up to 12 weeks, generating new muscle fibers and restoring dystrophin expression (Zhang et al. 2015).
Beside their regenerative potential, MSCs are capable of secreting paracrine factors with diverse biological functions such as immunomodulation, anti-scarring, chemoattraction, angiogenesis support and anti-apoptotic effect (reviewed in Meirelles et al. 2009). According to Caplan and Sorrell, the most notable immunomodulatory capacity of MSCs is that they can be used in allogenic and xenogenic transplants, in a variety of diseases (Caplan and Sorrell 2015).
MSCs can promote immunomodulation through several mechanisms. Proliferation of naïve T lymphocytes and memory lymphocytes can be suppressed by MSCs in the presence of alloantigens, mitogens, CD3 or CD28. Suppression was observed with autologous and allogeneic co-cultures and thus seems to be independent of MHC interaction (Di Nicola et al. 2002; Krampera et al. 2003; Le Blanc et al. 2003; Tse et al. 2003; Le Blanc et al. 2004; Meisel et al. 2004; Aggarwal and Pittenger 2005; Klyushnenkova et al. 2005). Additionally, the immunosuppressive property is maintained when MSCs and lymphocytes are co-cultured separated by a semi-permeable membrane, pointing out the involvement of soluble factors (Tse et al. 2003; Meisel et al. 2004; Glennie et al. 2005; Maccario et al. 2005; Mellor 2005; Plumas et al. 2005; Rasmusson et al. 2005; Selmani et al. 2009; Sioud et al. 2011).
In comparison to bone marrow and umbilical cord derived MSCs (BM-MSC and UC-MSC, respectively), hASCs display a more potent suppressor effect in dendritic cells differentiation and NK cell activation. Moreover, hASCs were shown to block the T cell activation process in an earlier phase than BM or UC-MSCs, yielding a greater proportion of T cells in the non-activated state. Concerning B cells and NK cells, hASCs and BM displayed an inhibitory effect while UC-MSCs did not influence their activation kinetics (Puissant et al. 2005; Ribeiro et al. 2013).
Immunosuppression is a desirable function to treat some inflammatory diseases, such as kidney diseases and graft versus host disease (GvHD). Animal models for severe acute renal failure have shown that administration of MSCs improves renal structure and function through paracrine effects that inhibit pro-inflammatory cytokines and stimulate anti-inflammatory cytokines (Tögel et al. 2005). A phase I clinical trial for treating idiopathic Focal Segmental Glumero Sclerosis (FSGS) with allogenic adipose derived MSCs (NCT02382874) was recently created and will start recruiting patients. Moreover, for patients diagnosed with GvHD, hASCs have been used in a number of Phase I and II trials with success (Herrmann and Sturm 2014).
In addition to immunomodulatory properties, the anti-fibrotic potential of hASCs was also demonstrated. Administration of hASCs on irradiated rats (model for radiation-induced pulmonary fibrosis), preserved the architecture of lungs without marked activation of fibroblasts or collagen deposition within the injured sites. The effect was mediated via host secretions of hepatocyte growth factor (HGF) and prostaglandin E2 (PGE2) (Dong et al. 2015). Based on this effect, a multicentric Phase I/II Clinical Trial with hASCs transplantation is currently recruiting participants in order to evaluate the safety and efficacy of autologous treatment of patients with diagnosis of idiopathic pulmonary fibrosis (NCT02135380).
Cell therapy is also being considered to treat some neurodegenerative diseases such as Parkinson’s disease (as reviewed in Nanette et al. 2010) and Amyotrophic Lateral Sclerosis (as revised in Coatti et al. 2015). Although transdifferentiation of MSCs (mesodermal origin) into neuronal cells (ectodermal origin) is a still very debated issue, it was observed that in addition to the paracrine factors mentioned above, some MSCs subpopulations can also secrete neuroregulatory factors such as brain derived neurotrophic factor (BNDF) and nerve growth factor (NGF) (Crigler et al. 2006). In this scenario, a multi-center phase I trial (NCT02326935) is currently recruiting patients for an autologous transplantation of hASCs in patients diagnosed with multiple sclerosis, an incurable neurodegenerative disease.
To the present, more than 70 human clinical trials are listed in the clinical trial database (www.clinicaltrial.gov), for pathologies such as graft versus host disease (NCT01222039), severe brain injury (NCT01649700) and osteoarthitis (NCT01809769). Banking of hASCs is being performed by various companies such as the American Bank Celletex.
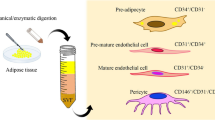
The classical steps to obtain a MSC lineage results in a heterogeneous population. Attempts to isolate specific subpopulations with known biological function from the stromal fraction are ongoing. Zimmerlin and coworkers (2013) reported different subpopulations of cells in human adipose stromal fractions, according to immunophenotyping verified by immunostaining and flow cytometry. Thee major population were described in this fraction: pericytes (CD45−, CD31−, CD146+), endothelial progenitor cells (CD45−, CD31+, CD34+), and supra adventitial adipose stromal cells (CD45−, CD31-, CD146-, CD34+) (Zimmerlin et al. 2013).
The therapeutic potential of hASCs and pericytes were investigated by our group in murine and canine models for muscular dystrophy, without immunosuppression. In the SJL mouse, the murine model for dysferlinopathy (or LGMD2B muscular dystrophy), hASCs transplantation revealed a functional benefit in transplanted animals when compared to untreated controls (Vieira et al. 2008b). The clinical effect as well as the safety of repeated hASCs injections from different donors was also tested in Golden Retriever Muscular Dystrophy (GRMD) dogs (Vieira et al. 2012). More recently, we compared the clinical effect of human pericytes obtained from different sources (adipose tissue, fallopian tube, muscle and endometrium) from a single female donor, in a double knockout mouse model (mdx/utr-) for Duchenne Muscular Dystrophy. It was observed that only pericytes obtained from adipose tissue, but not from other sources, were able to significantly increase the lifespan of treated mice (Valadares et al. 2014).
Cell therapy is also being considered for treating some pets’ diseases. In a recent study, ten lame dogs with severe hip osteoarthitis (OA) and a control group of five dogs received intra-articular transplantation of autologous ASCs. Mean values of diseases scores were significantly improved within the first 3 months post-treatment in the OA group (Vilar et al. 2014). In humans, intra-articular injection of 1 × 108 hASCs into the osteoarthitic knee improved function and pain of the knee joint without causing adverse events, and reduced cartilage defects by regeneration of hyaline-like articular cartilage (Jo et al. 2014).
Besides its therapeutic use, hASCs can be used for disease modeling. Dossena and coworkers proposed the use of Spinal and Bulbar Muscular Atrophy (SBMA) patients’ adipose derived MSCs as a new human in vitro model. They found that hASCs from SBMA patients showed less growth potential and differentiated only into adipocytes. Moreover, lower expression of key receptors was also found. The authors propose the use of hASCs from SMBA patients to test novel therapeutic approaches (Dossena et al. 2014).
In short, MSCs and hASCs have multiple biological functions as well as regenerative, anti-fibrotic and immunomodulatory properties. Such plasticity, in association with the facility to obtain and handle them, makes hASCs an important tool for future use in medicine. To achieve this goal, scientific experiments should be carefully designed and implemented.
Reagent specification (See Table 3.2)
References
Aggarwal S, Pittenger MF (2005) Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105:1815–1822
Caplan AI, Sorrell JM (2015) The MSC curtain that stops the immune system. Immunol Lett Euro Fed Immunol Societ 168:136–9. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0165247815001030.
Chen X, Gan Y, Li W, Su J, Zhang Y, Huang Y, Roberts AI, Han Y, Li J, Wang Y, Shi Y (2014) The interaction between mesenchymal stem cells and steroids during inflammation. Cell Death Dis 5:e1009
Coatti GC, Beccari MS, Olávio TR, Mitne-Neto M, Okamoto OK, Zatz M (2015) Stem cells for amyotrophic lateral sclerosis modeling and therapy: myth or fact? Cytom Part A 87:197–211
Crigler L, Robey RC, Asawachaicharn A, Gaupp D, Phinney DG (2006) Human mesenchymal stem cell subpopulations express a variety of neuro-regulatory molecules and promote neuronal cell survival and neuritogenesis. Exp Neurol 198:54–64
Crisan M, Yap S, Casteilla L, Chen C-W, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng P-N, Traas J, Schugar R, Deasy BM, Badylak S, Buhing H-J, Giacobino J-P, Lazzari L, Huard J, Péault B (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313
Da Justa Pinheiro CH, de Queiroz JCF, Guimarães-Ferreira L, Vitzel KF, Nachbar RT, de Sousa LGO, de Souza-Jr AL, Nunes MT, Curi R (2012) Local injections of adipose-derived mesenchymal stem cells modulate inflammation and increase angiogenesis ameliorating the dystrophic phenotype in dystrophin-deficient skeletal muscle. Stem Cell Rev 8:363–374
Devitt SM, Carter CM, Dierov R, Weiss S, Gersch RP, Percec I (2015) Successful isolation of viable adipose-derived stem cells from human adipose tissue subject to long-term cryopreservation: positive implications for adult stem cell-based therapeutics in patients of advanced age. Stem Cells Int 2015:1–11
Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, Grisanti S, Gianni AM (2002) Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 99:3838–3843
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Dong L-H, Jiang Y-Y, Liu Y-J, Cui S, Xia C-C, Qu C, Jiang X, Qu Y-Q, Chang P-Y, Liu F (2015) The anti-fibrotic effects of mesenchymal stem cells on irradiated lungs via stimulating endogenous secretion of HGF and PGE2. Sci Rep 5:8713
Dossena M, Bedini G, Rusmini P, Giorgetti E, Canazza A, Tosetti V, Salsano E, Sagnelli A, Mariotti C, Gellera C, Navone SE, Marfia G, Alessandri G, Corsi F, Parati EA, Pareyson D, Poletti A (2014) Human adipose-derived mesenchymal stem cells as a new model of spinal and bulbar muscular atrophy. PLoS One 9:e112746
English K, French A, Wood KJ (2010) Mesenchymal stromal cells: facilitators of successful transplantation? Cell Stem Cell 7:431–442
Fang B, Song Y, Zhao RC, Han Q, Lin Q (2007) Using human adipose tissue-derived mesenchymal stem cells as salvage therapy for hepatic graft-versus-host disease resembling acute hepatitis. Transplant Proc 39:1710–1713
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP (1968) Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 6:230–247
Gharaibeh B, Lavasani M, Cummins JH, Huard J (2011) Terminal differentiation is not a major determinant for the success of stem cell therapy—cross-talk between muscle-derived stem cells and host cells. Stem Cell Res Ther 2:31
Glennie S, Soeiro I, Dyson PJ, Lam EW-F, Dazzi F (2005) Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 105:2821–2827
Herrmann R, Sturm M (2014) Adult human mesenchymal stromal cells and the treatment of graft versus host disease. Stem Cells Cloning Adv Appl 7:45–52
Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, Deans RJ, Krause DS, Keating A (2005) Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 7:393–395
Ichim TE, Alexandrescu DT, Solano F, Lara F, Campion RDN, Paris E, Woods EJ, Murphy MP, Dasanu CA, Patel AN, Marleau AM, Leal A, Riordan NH (2010) Mesenchymal stem cells as anti-inflammatories: implications for treatment of Duchenne muscular dystrophy. Cell Immunol 260:75–82
Jacobs SA, Roobrouck VD, Verfaillie CM, Van Gool SW (2013) Immunological characteristics of human mesenchymal stem cells and multipotent adult progenitor cells. Immunol Cell Biol 91:32–39
Jo CH, Lee YG, Shin WH, Kim H, Chai JW, Jeong EC, Kim JE, Shim H, Shin JS, Shin IS, Ra JC, Oh S, Yoon KS (2014) Intra-articular injection of mesenchymal stem cells for the treatment of osteoarthitis of the knee: a proof-of-concept clinical trial. Stem Cells 32:1254–1266
Klyushnenkova E, Mosca JD, Zernetkina V, Majumdar MK, Beggs KJ, Simonetti DW, Deans RJ, McIntosh KR (2005) T cell responses to allogeneic human mesenchymal stem cells: immunogenicity, tolerance, and suppression. J Biomed Sci 12:47–57
Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, Dazzi F (2003) Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 101:3722–3729
Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringdén O (2003) Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol 57:11–20
Le Blanc K, Rasmusson I, Götherström C, Seidel C, Sundberg B, Sundin M, Rosendahl K, Tammik C, Ringdén O (2004) Mesenchymal stem cells inhibit the expression of CD25 (interleukin-2 receptor) and CD38 on phytohaemagglutinin-activated lymphocytes. Scand J Immunol 60:307–315
Lendeckel S, Jödicke A, Chistophis P, Heidinger K, Wolff J, Fraser JK, Hedrick MH, Berthold L, Howaldt H-P (2004) Autologous stem cells (adipose) and fibrin glue used to treat widespread traumatic calvarial defects: case report. J Cranio-Maxillofacial Surg 32:370–373
Li H, Zimmerlin L, Marra KG, Donnenberg VS, Donnenberg AD, Rubin JP (2011) Adipogenic potential of adipose stem cell subpopulations. Plast Reconstr Surg 128:663–672
Maccario R, Podestà M, Moretta A, Cometa A, Comoli P, Montagna D, Daudt L, Ibatici A, Piaggio G, Pozzi S, Frassoni F, Locatelli F (2005) Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of CD4 + T-cell subsets expressing a regulatory/suppressive phenotype. Haematologica 90:516–525
Meirelles LDS, Fontes AM, Covas DT, Caplan AI (2009) Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev 20:419–427
Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D (2004) Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 103:4619–4621
Mellor A (2005) Indoleamine 2,3 dioxygenase and regulation of T cell immunity. Biochem Biophys Res Commun 338:20–24
Minonzio G, Corazza M, Mariotta L, Gola M, Zanzi M, Gandolfi E, De Fazio D, Soldati G (2014) Frozen adipose-derived mesenchymal stem cells maintain high capability to grow and differentiate. Cryobiology 69:211–216
Murphy MB, Moncivais K, Caplan AI (2013) Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med 45:e54
Nanette J, Geralyn A, Louisa W, Scott O, Gerhard B, Nolta J, Joyce N, Annett G, Wirthlin L, Olson S, Bauer G, Nolta A (2010) Mesenchymal stem cells for the treatment of neurodegenerative disease. Regen Med 5:933–946
Pan Q, Fouraschen SM, Ruiter PED, Dinjens WN, Kwekkeboom J, Tilanus HW, Laan LJVD (2014) Detection of spontaneous tumorigenic transformation during culture expansion of human mesenchymal stromal cells. Exp Biol Med 239:105–115
Plumas J, Chaperot L, Richard M-J, Molens J-P, Bensa J-C, Favrot M-C (2005) Mesenchymal stem cells induce apoptosis of activated T cells. Leukemia 19:1597–1604
Puissant B, Barreau C, Bourin P, Clavel C, Corre J, Bousquet C, Taureau C, Cousin B, Abbal M, Laharrague P, Penicaud L, Casteilla L, Blancher A (2005) Immunomodulatory effect of human adipose tissue-derived adult stem cells: comparison with bone marrow mesenchymal stem cells. Br J Haematol 129:118–129
Rasmusson I, Ringdén O, Sundberg B, Le Blanc K (2005) Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res 305:33–41
Ribeiro A, Laranjeira P, Mendes S, Velada I, Leite C, Andrade P, Santos F, Henriques A, Grãos M, Cardoso CMP, Martinho A, Ml P, da Silva C, Cabral J, Trindade H, Paiva A (2013) Mesenchymal stem cells from umbilical cord matrix, adipose tissue and bone marrow exhibit different capability to suppress peripheral blood B, natural killer and T cells. Stem Cell Res Ther 4:125
Roemeling-van Rhijn M, de Klein A, Douben H, Pan Q, van der Laan LJW, Ijzermans JNM, Betjes MGH, Baan CC, Weimar W, Hoogduijn MJ (2013) Culture expansion induces non-tumorigenic aneuploidy in adipose tissue-derived mesenchymal stromal cells. Cytotherapy 15:1352–1361
Selmani Z, Naji A, Gaiffe E, Obert L, Tiberghien P, Rouas-Freiss N, Carosella ED, Deschaseaux F (2009) HLA-G is a crucial immunosuppressive molecule secreted by adult human mesenchymal stem cells. Transplantation 87:S62–S66
Singer NG, Caplan AI (2011) Mesenchymal stem cells: mechanisms of inflammation. Annu Rev Pathol Mech Dis 6:457–478
Sioud M, Mobergslien A, Boudabous A, Fløisand Y (2011) Mesenchymal stem cell-mediated T cell suppression occurs through secreted galectins. Int J Oncol 38:385–390
Sorrell JM, Caplan AI (2010) Topical delivery of mesenchymal stem cells and their function in wounds. Stem Cell Res Ther 1:30
Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Teperman L, Henegariu O, Krause DS (2000) Liver from bone marrow in humans. Hepatology 32:11–16
Tögel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C (2005) Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol 289:F31–F42
Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC (2003) Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation 75:389–397
Valadares MC, Gomes JP, Castello G, Assoni A, Pellati M, Bueno C, Corselli M, Silva H, Bartolini P, Vainzof M, Margarido PF, Baracat E, Péault B, Zatz M (2014) Human adipose tissue derived pericytes increase life span in Utrn (tm1Ked) Dmd (mdx)/J mice. Stem Cell Rev 10(6):830–840
Vieira NM, Brandalise V, Zucconi E, Jazedje T, Secco M, Nunes VA, Strauss BE, Vainzof M, Zatz M (2008a) Human multipotent adipose-derived stem cells restore dystrophin expression of Duchenne skeletal-muscle cells in vitro. Biol Cell 100:231–241
Vieira NM, Bueno CR, Brandalise V, Moraes LV, Zucconi E, Secco M, Suzuki MF, Camargo MM, Bartolini P, Brum PC, Vainzof M, Zatz M (2008b) SJL dystrophic mice express a significant amount of human muscle proteins following systemic delivery of human adipose-derived stromal cells without immunosuppression. Stem Cells 26:2391–2398
Vieira NM, Valadares M, Zucconi E, Secco M, Bueno CR, Brandalise V, Assoni A, Gomes J, Landini V, Andrade T, Caetano HVA, Vainzof M, Zatz M, Bueno Junior CR, Brandalise V, Assoni A, Gomes J, Landini V, Andrade T, Caetano HVA, Vainzof M, Zatz M (2012) Human adipose-derived mesenchymal stromal cells injected systemically into GRMD dogs without immunosuppression are able to reach the host muscle and express human dystrophin. Cell Transplant 21:1407–1417
Vilar JM, Batista M, Morales M, Santana A, Cuervo B, Rubio M, Cugat R, Sopena J, Carrillo JM (2014) Assessment of the effect of intraarticular injection of autologous adipose-derived mesenchymal stem cells in osteoarthitic dogs using a double blinded force platform analysis. BMC Vet Res 10:143
Zhang N, Dietrich MA, Lopez MJ (2014) Therapeutic doses of multipotent stromal cells from minimal adipose tissue. Stem Cell Rev 10:600–611
Zhang Y, Zhu Y, Li Y, Cao J, Zhang H, Chen M, Wang L, Zhang C (2015) Long-term engraftment of myogenic progenitors from adipose-derived stem cells and muscle regeneration in dystrophic mice. Hum Mol Genet 24(21):6029–6040
Zimmerlin L, Donnenberg VS, Pfeifer ME, Meyer EM, Péault B, Rubin JP, Donnenberg AD (2010) Stromal vascular progenitors in adult human adipose tissue. Cytom Part A 72:22–30
Zimmerlin L, Donnenberg VS, Rubin JP, Donnenberg AD (2013) Mesenchymal markers on human adipose stem/progenitor cells. Cytom Part A 83A:134–140
Zimmet JM, Hare JM (2005) Emerging role for bone marrow derived mesenchymal stem cells in myocardial regenerative therapy. Basic Res Cardiol 100:471–481
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH (2001) Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7:211–228
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279–4295
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Assoni, A.F., Coatti, G.C., Gomes, J.P.A., Pelatti, M.V., Zatz, M. (2016). Adipose-Derived Mesenchymal Stromal Cells. In: Working with Stem Cells. Springer, Cham. https://doi.org/10.1007/978-3-319-30582-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-30582-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30580-6
Online ISBN: 978-3-319-30582-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)