
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a primary immune system disorder that results from defective cell-to-cell signaling and disruption of inflammatory system homeostasis. This disorder usually affects children, although cases of HLH in adults have been reported (Zhang et al. 2011). HLH usually is initiated by an external stimulus, most commonly by viral infections, but familial cases have also been described (Janka 2006). Epstein-Barr virus is one of the more frequently implicated infections in the pathogenesis of HLH, but many other infections have been associated (Filipovich 2008). There are several inherited gene mutations which have been demonstrated to cause HLH. All of these genes encode proteins that participate in target-cell killing within the immune system and that effect homeostasis of the inflammatory response. Without such a regulatory system, cytokines and other inflammatory mediators are produced unchecked, resulting in fever, multiple organ system failure, pancytopenia, and death in many patients if treatment is not initiated promptly.
✠ Author was deceased at the time of publication
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Serum Ferritin
- Complete Response Rate
- Hematopoietic Cell Transplantation
- Cord Blood Transplantation
- Hemophagocytic Lymphohistiocytosis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a primary immune system disorder that results from defective cell-to-cell signaling and disruption of inflammatory system homeostasis. This disorder usually affects children, although cases of HLH in adults have been reported (Zhang et al. 2011). HLH usually is initiated by an external stimulus, most commonly by viral infections, but familial cases have also been described (Janka 2006). Epstein-Barr virus is one of the more frequently implicated infections in the pathogenesis of HLH, but many other infections have been associated (Filipovich 2008). There are several inherited gene mutations which have been demonstrated to cause HLH. All of these genes encode proteins that participate in target-cell killing within the immune system and that effect homeostasis of the inflammatory response. Without such a regulatory system, cytokines and other inflammatory mediators are produced unchecked, resulting in fever, multiple organ system failure, pancytopenia, and death in many patients if treatment is not initiated promptly.
Recognition of this rare disorder continues to increase as improved diagnostic tools become available to medical centers worldwide. The growth of translational research over the last several years, as well as clinical trials investigating more targeted therapies for HLH, has led to the potential for significant improvement in the diagnosis and treatment of patients with HLH.
Case 1: Review of the Pathogenesis, Diagnosis, and Treatment Options for HLH
A 14-month-old male with a 1-week history of high fevers and recent onset of total-body maculopapular rash presents to the local emergency department. He has no significant medical or family history. On examination, he was found to have widened pulse pressures and difficulty in breathing. He has massive hepatosplenomegaly but no lymphadenopathy. A CBC is performed and shows severe pancytopenia. Serum ferritin is markedly elevated. Plasma fibrinogen is normal, but the serum triglyceride level is elevated. He is transferred to the intensive care unit for further monitoring and evaluation. HLH is strongly suspected.
Question 1.
What other laboratory studies should be performed to support this diagnosis according to internationally accepted criteria?
-
A.
C-reactive protein, hepatic transaminases, MRI brain
-
B.
C-reactive protein, NK cell function, serum soluble IL-2 receptor
-
C.
NK cell function, serum soluble IL-2 receptor, bone marrow aspiration/biopsy
-
D.
NK cell function, C-reactive protein, bone marrow aspiration/biopsy
Expert Perspective
Prompt diagnosis of a patient with HLH is critical for improved survival. It would be reasonable for this patient to be admitted to the intensive care unit, only to be extensively evaluated for a fever of unknown origin and to have consultations with the infectious disease and/or rheumatology services prior to the recognition that this patient may have HLH. The Histiocyte Society uses the following internationally accepted criteria to establish the diagnosis of HLH:
Criterion (absolute) |
Familial disease or known genetic defect |
Criteria (5/8 needed) |
Fever |
Splenomegaly |
Bicytopenia |
Hemoglobin <9 g/dl |
Platelets <100,000/μl |
Neutrophils <1,000/μl |
Hypertriglyceridemia (fasting >265 mg/dl) and/or |
Hypofibrinogenemia (<150 mg/dl) |
Ferritin (>500 ng/dl) |
Soluble IL-2 receptor (> upper limit of normal for reference laboratory) |
Decreased or absent NK cell activity |
Hemophagocytosis in the bone marrow, liver, spleen, lymph nodes, or cerebrospinal fluid |
If a patient has a known mutation in a gene that is associated with HLH, or if a patient has a family history of HLH and symptoms associated with HLH, the diagnosis of HLH can be made. Otherwise, at least five of the eight additional criteria need to be documented for a diagnosis of HLH. The challenge, however, lies in the case of the patient who does not have a family history or documented genetic defect and satisfies only three or four of the stated criteria. These patients must be monitored with extreme vigilance due to the risk of rapid disease progression without treatment. It would be reasonable to perform weekly evaluations of the aforementioned criteria, including bone marrow aspirations, in order to demonstrate a diagnosis of HLH and to proceed with treatment to prevent clinical decompensation of the patient.
Serum ferritin is a useful surrogate marker for inflammation, and patients often can experience hyperferritinemia >50,000 ng/dl at the time of diagnosis. Serum soluble IL-2 receptor is a more sensitive marker of the degree of hypercytokinemia that is experienced by patients with HLH, and levels are often markedly elevated at the time of diagnosis. Of note, both of these serum tests are used to monitor a patient’s disease activity after treatment has been initiated.
Once a diagnosis of HLH is made, care must be taken to document central nervous system (CNS) involvement with a MRI of the brain with and without contrast and a lumbar puncture with histological evaluation of the cerebrospinal fluid (CSF). Patients may present with neurological symptoms (most commonly, seizures or irritability), or they may have an abnormal MRI (white matter lesions). Both clinical symptoms and radiological abnormalities may be associated with abnormal CSF (pleocytosis, increased protein, or demonstration of hemophagocytosis), and at least 50 % of patients have CNS involvement with HLH at the time of diagnosis (Horne et al. 2008).
Question 2.
Laboratory testing for familial causes of HLH reveals a mutation in the PRF1 gene that results in near-absent expression of perforin. What is the role of perforin in the pathogenesis of HLH?
-
A.
Endocytosis of inflammatory cytokines
-
B.
Microtubule formation
-
C.
Direct cytotoxicity
-
D.
Vesicle docking
Expert Perspective
Familial HLH is much more rare an entity than secondary (infection-associated) HLH, but its detection, when possible, is essential for identification of patients who will require hematopoietic stem cell transplant (HSCT) for eventual cure. The following genes can be sequenced for mutations readily and commercially:
Gene | Associated condition | Role in HLH pathogenesis when deficient |
|---|---|---|
AP3B1 | Hermansky-Pudlak syndrome type 2 | Vesicle formation |
BLOC1S6 | Hermansky-Pudlak syndrome type 9 | Vesicle docking and fusion |
CD27 | Lymphoproliferative syndrome type 2 | Cell signaling and apoptosis |
ITK | Lymphoproliferative syndrome type 1 | Differentiation and T-cell receptor signaling |
LYST | Chediak-Higashi syndrome | Size and function of lytic granules |
MAGT1 | XMEN syndrome | Magnesium transport |
PRF1 | Familial hemophagocytic lymphohistiocytosis type 2 | Pore forming for transmembrane channel and target-cell killing |
RAB27A | Griscelli syndrome type 2 | Vesicle docking and granule movement |
SH2D1A | X-linked lymphoproliferative syndrome type 1 | Signal transduction, granule polarization |
SLC7A7 | Lysinuric protein intolerance | Amino acid transport |
STX11 | Familial hemophagocytic lymphohistiocytosis type 4 | Vesicle transport and fusion |
STXBP2 | Familial hemophagocytic lymphohistiocytosis type 5 | Vesicle transport and fusion |
MUNC13-4 | Familial hemophagocytic lymphohistiocytosis type 3 | Vesicle priming |
BIRC4 | X-linked lymphoproliferative syndrome type 2 | Cell signaling and apoptosis |
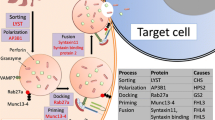
Mutations in any of these genes disrupt any one of a number of steps in the process of cell signaling in an effector cell (e.g., NK cell) that lead to apoptosis of a target cell (e.g., histiocyte). Thus, histiocytes proliferate, remain activated, and cause the symptomatology that was discussed previously in this chapter. The following diagram illustrates the role of these genes along the many steps in this pathway:
Cytolytic granules, including granzyme and perforin (PRF1), are released from the endoplasmic reticulum of the effector cell, possibly with the aid of LYST. The granules and perforin are packaged into vesicles with AP3B1 (a key component of protein trafficking and lysosomal packaging), and the vesicles are prepared for docking with RAB27A. The vesicles are further primed and docked via the actions of MUNC13-4, BLOC1S6, STX11, and STXBP2. The granules are polarized in a SH2D1A-dependent manner, and the entire complex causes the vesicle to undergo exocytosis and release the granules and perforin. Perforin is a pore-forming protein that creates a transmembrane channel in the target cell that allows the cytolytic granules to enter. Via cell signaling and the actions of CD27, ITK, and BIRC4, the target cell undergoes apoptosis. The actions of the transport genes MAGT1 and SLC7A7 are less clear. If any of these genes have mutations, the normal pathways that lead to apoptosis of the target cell are disrupted, and increased inflammatory mediators and cytokine storm ensue as a result. These actions can lead to the development of HLH.
Question 3.
What are considered to be the optimal treatment options for patients with HLH?
-
A.
Dexamethasone and etoposide
-
B.
Alemtuzumab
-
C.
Antithymocyte globulin and methylprednisolone
-
D.
Allogeneic hematopoietic stem cell transplantation
-
E.
All of the above
Expert Perspective
The first trial, HLH-94, was championed by the Histiocyte Society and enrolled 113 patients (Henter et al. 2002; Trottestam et al. 2011). The treatment regimen for this trial employed an initial 8-week phase of dexamethasone and etoposide, followed by a continuation phase (for those patients who require hematopoietic stem cell transplantation, HSCT) consisting of dexamethasone alternating weekly with etoposide, with daily cyclosporine. A second and institutional study enrolled 38 patients (Mahlaoui et al. 2007). This study used a combi and methylprednisolone to induce remission. A maintenance phase (for patients who required HSCT) included cyclosporine. Both trials used multiple doses of intrathecal hydrocortisone and methotrexate to treat CNS involvement.
The HLH-94 trial demonstrated a complete response (CR) rate of 50 %, overall survival (OS) of 78 %, and relapse rate (RR) of 13 %. The Mahlaoui study showed an improved CR rate compared to the HLH-94 trial (74 %), but the overall survival was similar (79 %). This trial also had an increased RR of 29 %. These data indicate that the combination of ATG and methylprednisolone may be very effective in inducing remission, but these patients were more likely to experience reactivation of HLH. In contrast, patients enrolled in the HLH-94 trial had a poorer CR rate than those patients in the Mahlaoui study, but the RR was lower, which indicates that the clinical responses to treatment with HLH-94 therapy were sustained more often. These results provided the rationale for the HIT-HLH (hybrid immunotherapy) clinical trial, which is in process at the time of this writing. This study combines the use of ATG (as in the Mahlaoui study) with a backbone of dexamethasone and etoposide (as in the HLH-94 trial). The hypothesis is to capitalize on the superior CR while providing a favorable RR by combining aspects of both treatment regimens. Of note, the favorable results of the HLH-94 trial laid the groundwork for the HLH-2004 clinical trial that was administered by the Histiocyte Society and is currently completed (Henter et al. 2007). The main difference between HLH-94 and HLH-2004 was that cyclosporine was used in the 8-week induction phase, in addition to dexamethasone and etoposide. Data from this trial have not yet been published. The results of these clinical trials will likely serve as a new baseline for future clinical studies administered by the Histiocyte Society and others to treat HLH.
Recurrent or refractory HLH is a challenging entity, mostly because there are very few data that exist to provide clinical evidence for an optimal approach to treatment. Responses using a variety of salvage therapies for recurrent/refractory HLH have been reported, but only in small case series. These therapies include (among others) daclizumab, (anti-CD25 antibody; Tomaske et al. 2002), infliximab (tumor necrosis factor alpha antibody; Henzan et al. 2005), tocilizumab (anti-IL-6 antibody; Rios-Fernández et al. 2015), and anakinra (interleukin-1 receptor antagonist; Bruck et al. 2011). In addition, a clinical trial investigating the treatment of HLH with an inhibitory monoclonal antibody directed against gamma interferon is currently underway. One promising approach is alemtuzumab (monoclonal antibody to the lymphocyte, NK cell, and dendritic-cell marker CD52). A retrospective analysis of 22 adults and children who were treated with alemtuzumab for refractory/recurrent HLH demonstrated effectiveness and safety of alemtuzumab in this setting (Marsh et al. 2012). This author gives alemtuzumab 0.2 mg/kg every 6 weeks with a small dose of continuous oral dexamethasone to be a safe and effective bridge to HSCT while keeping a patient’s HLH in remission. It is clear that clinical trials will be needed to further elucidate the safety and effectiveness of alemtuzumab for refractory/recurrent HLH.
Hematopoietic stem cell transplantation is a treatment modality that is employed for the first-line treatment of asymptomatic patients with familial HLH or as consolidative therapy for patients with refractory/recurrent HLH.
Question 4.
When you search for possible infectious etiologies for this patient’s HLH, you discover that his plasma contains greater than one million copies per milliliter of Epstein-Barr virus (EBV), which is also the likely cause for the patient’s rash. What is the most effective option to treat the EBV?
-
A
Ganciclovir
-
B.
Rituximab
-
C.
Valganciclovir
-
D.
Plasmapheresis
Expert Perspective
EBV is a common trigger for the pathogenesis of HLH. Since EBV usually is found in B-lymphocytes, rituximab, a monoclonal antibody to the B-lymphocyte marker CD20, becomes an excellent therapeutic choice. Evidence for the use of rituximab in patients with HLH is supported from a small case series that first demonstrated its efficacy (Balamuth et al. 2007) and a larger, multi-institutional, retrospective study that investigated the outcome of patients who had received rituximab for EBV-associated HLH (Chellapandian et al. 2013). Both studies reported a rapid decrease in plasma EBV load after the administration of rituximab. Because EBV often drives HLH disease activity, the reduction in viral load was followed quickly by a decrease in serum ferritin and AST, as well as an increase in platelet count (Chellapandian et al. 2013). Decreased EBV load to <1,500 copies/ml and ferritin to ≤1,000 were also associated with increased long-term disease-free survival. The accepted regimen of rituximab is to give one to four weekly doses at a dose of 375 mg/m2, and rituximab can be given concurrently with the HLH therapy consisting of corticosteroids and etoposide. If the EBV load decreases to undetectable levels prior to the fourth weekly dose, the rituximab can be discontinued. While there may appear rationale to utilize antiviral agents in such cases, there is lack of rigorous evidence that ganciclovir or valganciclovir are effective alone in changing the course of EBV-related.
Question 5.
In which of the following scenarios is hematopoietic stem cell transplantation NOT indicated for a patient with HLH?
-
A.
No evidence of current or familial HLH after initial 8 weeks of therapy
-
B.
No evidence of familial HLH, but persistent pancytopenia and fever after initial therapy
-
C.
Reactivation of HLH after initial therapy
-
D.
Evidence of familial HLH
Expert Perspective
In order to be considered to be “in remission” at the end of the initial 8 weeks of therapy for HLH, patients must have no evidence of disease activity and have no identifiable mutation of genes that are known to be associated with HLH. If this scenario occurs, patients are monitored closely for signs or symptoms of recurrence. If a patient has refractory or recurrent HLH (based on clinical examination or laboratory results), the patient would proceed to maintenance therapy (previously discussed in this chapter) until an adequate HSCT donor is identified. If the patient were found to have a mutation of a gene known to be associated with HLH during the initial 8 weeks of treatment, the patient would proceed to maintenance therapy by default, followed by HCT. HCT would be the best treatment option for an asymptomatic sibling of a patient with a known genetic mutation that is the same as an affected patient; it would be best to perform HCT in the asymptomatic sibling prior to the onset of HLH symptoms due to the high rate of morbidity and mortality that accompanies HLH, for which the timing of onset cannot be predicted.
Allogeneic hematopoietic cell transplantation (HCT) is the only currently known curative approach to treating genetically inherited HLH or recurrent HLH. The outcomes using reduced intensity conditioning (RIC) are superior compared to myeloablative regimen (MAC) for allo-HCT (92 % for RIC 3-year overall survival compared to 43 % for MAC) (Marsh et al. 2010). A similar retrospective analysis from the Japanese transplant registry found that the overall survival of patients who received RIC followed by cord blood transplantation (CBT) was similar to that of MAC followed by CBT (Sawada et al. 2013). Larger, prospective trials are needed to better determine the efficacy of these approaches. In addition, several studies using gene therapy for specific gene-mutated HLH are beginning.
Controversies
-
Clinical/laboratory diagnostic testing for HLH
-
Genetic mutations and their role in the pathogenesis of HLH
-
Optimal treatment for patients with HLH
-
Treatment of EBV-associated HLH
-
Role of allogeneic HCT in the treatment of HLH
Answers
-
Question 1: D
-
Question 2: C
-
Question 3: E
-
Question 4: B
-
Question 5: A
References
Balamuth NJ, Nichols KE, Paessler M, et al. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29(8):569–73.
Bruck N, Suttorp M, Kabus M, et al. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol. 2011;17(1):23–7.
Chellapandian D, Das R, Zelley K, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013;162(3):376–82.
Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008;28(2):293–313.
Henter J-I, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–73.
Henter J-I, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Henzan T, Nagafuji K, Tsukamoto H, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol. 2005;81(1):59–61.
Horne A, Trottestam H, Arico M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;140(3):327–35.
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2006;166(2):95–109.
Mahlaoui N, Ouachée-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–8.
Marsh RA, Vaughn G, Kim M-O, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116(26):5824–31.
Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2012;60(1):101–9.
Marsh RA, Kim M-O, Liu C, et al. An intermediate Alemtuzumab schedule reduces the incidence of mixed chimerism following reduced-intensity conditioning hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Biol Blood Marrow Transplant. 2013;19(11):1625–31.
Rios-Fernández R, Callejas-Rubio J-L, García-Rodríguez S, Sancho J, Zubiaur M, Ortego-Centeno N. Tocilizumab as an adjuvant therapy for hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. Am J Ther. 2015;1–4.
Sawada A, Ohga S, Ishii E, et al. Feasibility of reduced-intensity conditioning followed by unrelated cord blood transplantation for primary hemophagocytic lymphohistiocytosis: a nationwide retrospective analysis in Japan. Int J Hematol. 2013;98(2):223–30.
Tomaske M, Amon O, Bosk A, et al. α-CD25 antibody treatment in a child with hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 2002;38(2):141–2.
Trottestam H, Horne A, Arico M, et al. Chemoimm-unotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–84.
Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial hemophagocytic lymphohistiocytosis. Blood. 2011;118(22):5794–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Henry, M.M., Arceci, R.J. (2016). Hemophagocytic Lymphohistiocytosis: Diagnosis and Management Challenges. In: Abutalib, S., Connors, J., Ragni, M. (eds) Nonmalignant Hematology. Springer, Cham. https://doi.org/10.1007/978-3-319-30352-9_52
Download citation
DOI: https://doi.org/10.1007/978-3-319-30352-9_52
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30350-5
Online ISBN: 978-3-319-30352-9
eBook Packages: MedicineMedicine (R0)