
Abstract
A 20-month-old boy recently moved to the USA from Thailand with his family. He has a history of thalassemia and has been treated with intermittent transfusions whenever he has symptoms of anemia, according to his parents. He is otherwise active and playful. He is growing well, with height and weight at the 10th and 25th percentiles, respectively. His head circumference is at the 50th percentile. His physical examination is remarkable for pallor, frontal bossing, scleral icterus, and moderate splenomegaly (2–3 cm below left costal margin). His parents report that his hemoglobin (Hb) is usually around 6 g/dL. A complete blood count at your office shows Hb 6.3 g/dL and Hb electrophoresis with HbA 20 %, Hb E 43 %, HbA2 6 %, and HbF 31 %. His serum ferritin was 1150 mg/dL and he has never been on iron chelation therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Case 1: Toddler (Transfusion Regimen and Initiation of Iron Chelation)
A 20-month-old boy recently moved to the USA from Thailand with his family. He has a history of thalassemia and has been treated with intermittent transfusions whenever he has symptoms of anemia, according to his parents. He is otherwise active and playful. He is growing well, with height and weight at the 10th and 25th percentiles, respectively. His head circumference is at the 50th percentile. His physical examination is remarkable for pallor, frontal bossing, scleral icterus, and moderate splenomegaly (2–3 cm below left costal margin). His parents report that his hemoglobin (Hb) is usually around 6 g/dL. A complete blood count at your office shows Hb 6.3 g/dL and Hb electrophoresis with HbA 20 %, Hb E 43 %, HbA2 6 %, and HbF 31 %. His serum ferritin was 1150 mg/dL and he has never been on iron chelation therapy.
Question 1.
What transfusion regimen is most appropriate for him?
-
A.
Intermittent transfusions when symptomatic.
-
B.
Chronic transfusions to maintain his pre-transfusion hemoglobin at 9.5 g/dL.
-
C.
Discontinue transfusions to avoid iron overload.
-
D.
Transfuse to maintain a normal hemoglobin >12 g/dL.
Expert Perspective:
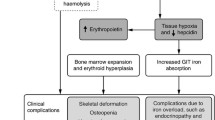
First, it is always important to confirm the exact diagnosis when establishing care for a new thalassemia patient. In this child, the hemoglobin electrophoresis is consistent with HbE-beta thalassemia, a common beta thalassemia variant in Thailand (the presence of normal adult HbA reflects previous transfusions). The combination of chronic hemolysis and ineffective erythropoiesis leads to scleral icterus, bony deformities due to increased bone marrow expansion as well as hepatosplenomegaly (HSM) in patients with thalassemia.
The decision to start regular packed red blood cells (pRBCs) transfusion should take into consideration clinical and laboratory factors. Initiation of a regular pRBCs transfusion regimen is recommended for patients with Hb levels persistently below 7 g/dL or even if >7 g/dL, but with poor growth, facial changes, recurrent fractures, or significant HSM (Cappellini et al. 2014). A typical transfusion regimen is every 2–5 weeks to maintain a pre-transfusion hemoglobin level of 9–10.5 g/dL (Cazzola et al. 1995, 1997). The frequency of transfusions varies based on growth needs, fatigue, and the presence of hypersplenism. Younger children may need a more specific volume calculation:\( \left[\begin{array}{l}\left(\mathrm{Desired}-\mathrm{actual}\ \mathrm{H}\mathrm{b}\right)\times \mathrm{weight}\times 3/\ \\ {}\mathrm{hematocrit}\ \mathrm{of}\ \mathrm{transfused}\ \mathrm{unit}\end{array}\right]=\mathrm{mL} \) to be transfused (Davies et al. 2007), generally 10–15 mL/kg. Each patient needs a detailed record of their transfused blood to be able to monitor their annual transfusion requirements.
Later in life, comorbidities, and lifestyle considerations, such as school and work schedules may become more impactful. For example, patients with heart failure or more clinically significant extramedullary hematopoiesis may require more frequent transfusions to maintain a higher pre-transfusion Hb level of 11–12 g/dL.
Most children with β-thalassemia major (β-TM) will initiate regular transfusions before 1 year of age. Delayed transfusion initiation in early life may seem advantageous, but it comes with an increased risk of alloimmunization in patients who start their regular transfusions after the first few years of life, which can be a challenge for patients with lifelong transfusion needs (Michail-Merianou et al. 1987; Spanos et al. 1990). Also, frontal bossing and relative macrocephaly in this patient suggest marrow expansion to compensate for ineffective erythropoiesis. Early and optimal transfusions can mitigate skeletal deformities.
Question 2.
How would you monitor his iron overload status?
-
A.
Serum ferritin levels every 3–6 months
-
B.
Liver biopsy
-
C.
Magnetic resonance imaging (MRI) R2/R2* or FerriScan every 12 months
-
D.
Superconducting quantum interference device (SQUID)
Expert Perspective
Serum ferritin
In general, ferritin level is an easy and inexpensive test that correlates well with body iron stores in thalassemia and thus is a useful marker to identify trends. Serum ferritin is more sensitive than specific. In other words, down- or up-trending ferritin levels would suggest decreasing or increasing total body iron burden, respectively, but neither is specific because of other possible explanations. In patients with beta thalassemia major (β-TM,), only 57 % of the variability of ferritin levels can explain the variation in body iron stores (Brittenham et al. 1993), which can be due to infections or other inflammatory conditions. Patients with good long-term control of ferritin levels have better outcomes overall with lower risk of cardiac disease and death (Olivieri et al. 1994; Borgna-Pignatti et al. 2004). High serum ferritin levels (>3000 μg/L) are less reliable as predictors of body iron (Davis et al. 2004), and differences in this correlation depend on the type of chelation used (Ang et al. 2010) and duration of chelation therapy (Fischer et al. 2003). Serial ferritin monitoring is essential for determining when to initiate iron chelation therapy and how to adjust the dose.
Liver Iron Concentration (LIC)
LIC is the most reliable indicator of total body iron stores, response to chelation therapy, and risk of hepatic and extrahepatic damage due to iron overload (Angelucci et al. 2000). LIC up to 3 mg/g liver dry wt (gldw) is tolerable because of the liver’s capacity for transferrin binding and buffering of iron. Persistently elevated LIC (>15 mg/g dry wt) have been associated with poor prognosis, progressive liver fibrosis (Angelucci et al. 1997), abnormal liver functions (Jensen et al. 2003), and death due to myocardial siderosis (Olivieri et al. 1994). Measurement of LIC can be helpful prior to starting iron chelation, if there are variable responses to chelation or if the current regimen needs to be modified. LIC correlates with total iron burden and ferritin levels (Angelucci et al. 2000), but this relationship is not linear with high ferritin levels (>4000 μg/L) (Cappellini et al. 2014).
Measurement of LIC can be done by biopsy, magnetic resonance imaging (MRI), or magnetic biosusceptometry using superconducting quantum interference device (SQUID). Liver biopsy had been the method of choice to determine LIC, but the risk of the invasive procedure and the propensity for sampling variability has limited its acceptability to patients and providers. There is a good correlation between liver iron evaluated by either MRI R2 and R2* techniques or liver biopsy, and all can give reliable estimates of total body iron and monitor iron balance with adequate long-term control overtime (St Pierre et al. 2005; Wood et al. 2005). SQUID has moderate to strong correlation with serum ferritin but has a very limited use worldwide, as it is available in only a few centers.
Myocardial Iron and Heart Function Assessment
MRI techniques are the most widely used tools for monitoring liver and myocardial iron burden (Kwiatkowski et al. 2012). Myocardial T2* (mT2*) MRI with shortened T2* values (<20 ms) is associated with decreased left ventricular ejection fraction (LVEF) (Anderson et al. 2001). Cardiac MRI T2* values of >20 ms are normal, while values of <10 ms signify severe myocardial iron loading. β-TM patients at risk of developing clinical heart failure can be identified earlier when intensification of iron chelation can be beneficial (Davis et al. 2001, 2004). Moreover, patients with T2* values <10 ms are at a 160-fold increased risk heart failure in the next 12 months (Kirk et al. 2009).
The risk of myocardial iron loading in β-TM increases with age, and it is uncommon to have an abnormal cardiac T2* in the first decade of life. The risk increased to 24 and 36 % in children aged 9.5–15 and 15–18 years old in one retrospective study (Wood et al. 2008). Therefore, in children with β-TM, cardiac T2* surveillance is recommended annually starting at 10 years of age.
Iron assessment in endocrine organs
Although MRI may be helpful to evaluate iron burden in endocrine organs, this has been supported by only small number of studies. Patients with higher pancreatic and pituitary iron levels were associated with an increased risk of glucose dysregulation (Noetzli et al. 2012a) and hypogonadism (Noetzli et al. 2012b), respectively.
Question 3.
When should iron chelation therapy be started?
-
A.
After he has received chronic transfusions for 1–2 years
-
B.
After approximately 20–25 units of pRBCs
-
C.
When the serum ferritin level is >1000 ng/mL for at least two steady-state separate measurements
-
D.
When the liver iron concentrations is >3 mg Fe/g dry wt measured by biopsy or MRI
-
E.
All of the above
Expert Perspective:
Iron chelation therapy should be initiated in children after receiving chronic transfusions for 1–2 years or approximately 20–25 units of pRBCs. Iron chelation initiation is not recommended before age 2 years and there is no evidence of harm for waiting. Elevated serum ferritin level >1000 ng/mL for at least two steady-state separate measurements or with elevated liver iron >3 mg/g dry wt measured by biopsy or MRI has also been used as criteria for chelation initiation (Rachmilewitz and Giardina 2011).
Iron chelation therapy is used to remove accumulated iron from regular transfusions and to minimize ongoing iron loading process (Cohen et al. 2008). Higher chelation doses may be needed with increased transfusional needs. There are differential rates of iron loading and unloading in different organs. While iron tends to accumulate earlier in the liver before the heart, it can be cleared faster from the liver than the heart (Noetzli et al. 2008; Anderson et al. 2001, 2004; Deborah Chirnomas et al. 2008); iron chelators generally remove iron from the liver faster than from the heart, protect against further cardiac iron accumulation and reverse possible iron-related cardiac abnormalities, and improve some endocrine abnormalities with more intensive chelation regimens(Anderson et al. 2004; Farmaki et al. 2010).
The common practice is to maintain the following goals while on chelation therapy: ferritin 500–1500 mg/mL, LIC between 2 and 7 mg/g dry wt, and cardiac T2* >20 ms. Some centers have used more intensive chelation regimens in adult patients with β-TM to target lower serum ferritin levels with improvement in endocrinopathies and other iron-related morbidities (Farmaki et al. 2010, 2011). This approach has not been studied in children and careful chelation dose adjustment would be important to avoid additional toxicities.
Case 2: Adolescent (Disease Complications)
A 17-year-old girl with β-TM presents with bone pain and delayed puberty. She has been on chronic transfusions since age of 2 years and she started iron chelation at age 3 years with deferoxamine. She had poor compliance with the nightly subcutaneous deferoxamine infusions and chelation changed to deferasirox 4 years ago. Her serum ferritin levels are elevated (3500–4500 mg/dL range). Over the last year, she has required more frequent transfusions with an annual transfusion volume of 240 mL/kg/year to maintain her pre-transfusion hemoglobin at 9 g/dL. She is active and likes to play soccer. On exam, she is short (<3rd percentile for height) and pale with scleral icterus and has marked splenomegaly and Tanner 2 breast development with scant pubic or axillary hair. She has not had menarche yet.
Question 4.
How would you manage her growth impairment and delayed puberty?
-
A.
Provide reassurance and monitor height and weight annually.
-
B.
Perform testing to identify possible causes.
-
C.
Consider an empiric trial of growth hormone treatment for a year.
-
D.
Start dual iron chelation therapy.
Expert Perspective:
Many children with β-TM will have growth failure, mainly short stature. Growth failure and pubertal delay are multifactorial and can be due to chronic anemia, ineffective erythropoiesis with a hypermetabolic state, nutritional deficiencies (protein-calorie malnutrition, vitamin D and A, zinc and carnitine deficiencies), chelation toxicity (particularly deferoxamine), chronic liver disease, chronic heart failure, and iron-induced endocrinopathies (hypogonadism, hypothyroidism, growth hormone deficiency) (Vogiatzi et al. 2009; De Sanctis et al. 2013).
Careful clinical evaluation is necessary for thalassemia children with growth failure. An important first step is to document measurements of height (cm) and growth velocity every 6 months (cm/year) using national standard growth chart for age and sex, taking into account the height of the parents. Other signs of possible underlying causes of growth failure should be explored as well.
A laboratory work up would include the following (Cappellini et al. 2014):
-
Thyroid function tests (Free T4, thyroid-stimulating hormone)
-
Pituitary-gonadal axis assessment (testosterone, estradiol, luteinizing hormone [LH], follicle-stimulating hormone [FSH])
-
Pituitary growth axis with insulin growth factor-1, insulin growth factor binding protein-3, and growth hormone (GH) stimulation test (clonidine, glucagon, or growth hormone releasing hormone)
-
Calcium homeostasis (calcium, phosphate, alkaline phosphatase, parathormone, and 25-OH vitamin D levels)
-
Oral glucose tolerance tests
Prevention is the best treatment for growth impairment and delayed puberty in children with β-TM. This includes regular pRBCs transfusion (pre-transfusion goal of Hb 9–10 g/dL), proper iron chelation (serum ferritin goal of <1000 mg/dL), and adequate nutrition. Patients with hypothyroidism and diabetes mellitus often need thyroid replacement therapy and insulin, respectively. Hormone replacement therapy may improve manifestations of hypogonadism and help to achieve puberty. Ovarian reserve testing including a pelvic ultrasound to evaluate ovarian and uterine size as well as number of ovarian antral follicle counts may be appropriate if there is no response to hormone replacement. A consultation with an endocrinologist would be beneficial in most cases. Early diagnosis and proper timely management of pubertal and growth delay in children with β-TM are critical to try to achieve normal growth spurt and pubertal maturation.
Question 5.
How would you evaluate her bone health? What is the most appropriate initial intervention for osteopenia in children with β-TM?
-
A.
Sufficient calcium and vitamin D supplements and increased physical activity
-
B.
Adequate iron chelation
-
C.
Growth hormone replacement therapy
-
D.
Bisphosphonates to inhibit the function of osteoclasts
Expert Perspective:
Patients with β-TM are at high risk for osteoporosis, fractures, and skeletal deformities. Osteopenia and osteoporosis are common causes of morbidity in patients with β-TM, seen approximately 40–50 % (Voskaridou and Terpos 2004). Bone pain can be symptom of low bone density and increases with age particularly if patients have been on deferoxamine for iron chelation (Haines et al. 1984). A radiological evaluation of the skeleton should include assessment of bone maturation (bone age) and measuring bone mineral density (DEXA scan) in late childhood and early adolescence. Delayed puberty in girls is more often due to hypogonadotrophic hypogonadism than from ovarian iron deposition and should be suspected if the FSH and the estradiol are both low.
Low bone mineral density (BMD) in children with β-TM is due to an imbalance between bone formation by osteocytes and bone resorption by osteoclasts. This can be secondary to genetic factors as well as iron deposition in the bone with direct toxic effects of iron on osteoblasts, deferoxamine exposure, vitamin C and D deficiencies, lack of physical activity, and iron-related endocrinopathies including hypothyroidism, hypoparathyroidism, diabetes mellitus, IGF-1 and growth hormone deficiencies, and mainly hypogonadism with delayed sexual maturation.
DEXA scan is the gold standard imaging modality for the assessment of BMD. Osteoporosis is defined as 2.5 standard deviations (SD) below the normal mean in BMD, whereas osteopenia occurs with BMD between 1.5 and 2.5 SD below the normal mean. Annual assessment of minerals (calcium and phosphorus), vitamins (C and D), and hormonal levels is crucial for adequate monitoring of bone health in children with β-TM, in addition to an annual DEXA scan starting in early adolescence.
Management of osteopenia and osteoporosis in children β-TM consists of sufficient calcium and vitamin D supplements, adequate iron chelation (better to avoid deferoxamine, if possible), increased physical activity, adequate glycemic control, hormone replacement therapy for different iron-related endocrinopathies, and bisphosphonates to inhibit the function of osteoclasts. The use of bisphosphonates, particularly intravenous formulations (pamidronate, neridronate, or zoledronic acid), has been associated with reduction of bone resorption, increase of BMD, reduction of back pain, and improved quality of life overtime (Voskaridou et al. 2003, 2006; Forni et al. 2012). However, bisphosphonates should not be given concurrently with calcium and vitamin D for longer than 2 years (Cappellini et al. 2014).
There are other novel agents for osteoporosis that are under investigation but their effects in β-TM-induced osteoporosis remain to be proven (Cappellini et al. 2014). Denosumab is a monoclonal antibody against RANKL that has been recently licensed by the US FDA for the treatment of postmenopausal osteoporosis (Miller 2011). Antibodies against Dkk-1 or against sclerostin may be future agents for the effective management of osteoporosis in patients with β-TM. Additionally, teriparatide, a recombinant peptide fragment of parathyroid hormone, and strontium ranelate, a second anabolic agent, are other agents that may help to prevent osteoporosis-related fractures in postmenopausal women. Furthermore, luspatercept, a chimeric protein containing the extracellular domain of the activin receptor 2A (ActRIIA), inhibits activin-A, which increased both BMD and Hb levels in β-TM animal models (Suragani et al. 2014), and a phase II study had recently started in patients with β-TM and thalassemia intermedia.
Question 6.
What are the indications for splenectomy in patients with β-TM?
-
A.
Increased blood requirement (more than 200–220 mL/kg/year)
-
B.
Hypersplenism (leukopenia, thrombocytopenia, and worsening anemia)
-
C.
Symptomatic splenomegaly with left upper quadrant abdominal pain or early satiety
-
D.
All of the above
Expert Perspective:
Splenomegaly is not uncommon in patients with β-TM and is usually due to excess destruction of defective red blood cells and extramedullary hematopoiesis in the spleen. The ultimate goal of splenectomy in patients with β-TM is to reduce iron overload by reducing their transfusion requirement (Rachmilewitz and Giardina 2011). The incidence of splenomegaly and the need for splenectomy has dramatically decreased over time (Piga et al. 2011; Thompson et al. 2011), particularly with the use of regular transfusion regimens maintaining adequate pre-transfusion Hb of 9–10.5 g/dL.
Patients undergoing splenectomy are at increased risk of overwhelming infections (Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis), venous thromboembolism, and pulmonary hypertension. Therefore, splenectomy should be avoided in children with β-TM, particularly those <5 years of age, and should be considered if the patient has increased blood requirement that prevents adequate control with iron chelation therapy (usually >200–220 mL/kg/year), hypersplenism (leukopenia, thrombocytopenia, and worsening anemia), or symptomatic splenomegaly with left upper quadrant abdominal pain or early satiety (Cappellini et al. 2014).
Total splenectomy is the most commonly used approach, either open or laparoscopic. Laparoscopic splenectomy has superior outcomes with shorter hospital stay and more significant reduction in 30-day postoperative mortality and other postoperative complications, such as wound and infection complications (Musallam et al. 2013a). Partial splenectomy is another less commonly used approach, because the splenic volume that needs to be preserved for adequate immune function and the risk of splenic regrowth remains unknown. Additionally, there have been no comparative effectiveness studies comparing total versus partial splenectomy (Rice et al. 2012).
Regardless of the splenectomy approach, patients should receive pneumococcal, meningococcal, and Haemophilus influenza vaccines at least 2 weeks before splenectomy with repeat doses in 3–5 years afterward and should also take lifelong penicillin or other antibiotic alternatives (erythromycin or trimethoprim sulfamethoxazole) for prophylaxis.
Question 7.
What are the assessments that you would consider for her comprehensive care?
-
A.
Liver iron measurement (R2* MRI or FerriScan) every 12 months
-
B.
Serum ferritin every month
-
C.
Annual serologies for blood-borne pathogens
-
D.
All of the above
Expert Perspective:
Children and adults with β-TM require close monitoring of their growth and regular follow-up with physical examination and laboratory assessments by hematologists who are familiar with the management of thalassemia, preferably and adults in a comprehensive thalassemia center. Monthly and annual assessments are recommended (Table 1) (Martin and Thompson 2013).
Case 3: Young Adult (Pregnancy, SCT, and Gene Therapy)
A 27-year-old female with beta thalassemia major presented to your office for routine assessment. She has been doing well since her last visit with no acute illness, emergency room visits, or hospitalizations. She continued to receive chronic transfusions every 4 weeks with an average of pre-Hb level of 9–10 g/dL. She has been on deferasirox (25 mg/kg) for the last 3 years but she is experiencing nausea and bloating. Her serum ferritin has been 2500–3500 mg/dL. She has a history of type I diabetes that is well controlled on insulin. She earned a degree in computer sciences and works as a software programmer. She was married a year ago and is interested in pregnancy in the near future. She has questions about thalassemia recommendations for pregnancy preparation and availability of definitive treatment options including stem cell transplantation and gene therapy.
Question 8.
What, if any, changes should she make in her iron chelation strategy?
-
A.
Increase her deferasirox dose.
-
B.
Add an additional chelating agent.
-
C.
Stop all chelation now.
-
D.
Reduce the frequency of her transfusions.
Expert Perspective:
Three iron-chelating agents, deferoxamine (DFO), deferiprone (DFP), and deferasirox, are FDA-approved commercially available. The dose, route, side effects, and practical considerations for the three agents are summarized in Table 2. Each agent has its own advantages, disadvantages, side-effect profile, and monitoring requirements; therefore, the choice of the chelating agent should be tailored for each individual patient with carefully weighted risks and benefits.
Close monitoring is needed for patients with transfusional iron overload to optimize their chelation therapy. Routine monitoring may vary with different iron chelators and at minimum should include serum ferritin levels (every 3 months) and measurements of cardiac and liver iron burden with MRI scans (annually). Modification of chelation regimen may be warranted in patients with rising serum ferritin levels despite adherence to chelation therapy, cardiac T2* <20 ms, or LIC >7 mg/gldw.
Combined chelation therapy should be considered in patients with severe transfusional iron overload (LIC >15 mg/gldw or cardiac T2* <10 ms), dose-limiting toxicity of a single chelator used, and evidence of iron-induced cardiac dysfunction (arrhythmias, reduced left ventricular ejection fraction). There has been accumulating evidence to support the effectiveness of a combined chelation with both DFP and DFO in reducing serum ferritin levels and LIC (Tanner et al. 2007; Kattamis et al. 2006). This combination is advantageous, compared to deferoxamine alone, in patients with severe cardiac iron overload in improving their cardiac T2* and left ventricular ejection fraction (Tanner et al. 2007).
The combination of deferoxamine and deferasirox in patients with β-TM has showed significant improvement in serum ferritin levels, LIC, and cardiac T2* with satisfactory safety profile in two different studies over 12-month period (Lal et al. 2013; Cassinerio et al. 2014). Similarly, the use of combined deferasirox and deferiprone for 1 year was associated with significant reduction in serum ferritin levels and LIC with comparable reversible side effects to either drug alone (Farmaki et al. 2011; Totadri et al. 2015). Furthermore, a recent randomized trial of DFP/DFO versus DFP/DFX has shown a significant comparable reduction of serum ferritin and LIC as well as improvement in quality of life in both study arms with tolerable side effects. However, DFP/DFX arm was associated with more significant improvement in cardiac T2* and higher reported satisfaction and adherence rates (Elalfy et al. 2015).
Question 9.
How would you counsel her about pregnancy and other precautions during pregnancy?
-
A.
Ovarian reserve testing should include anti-müllerian hormone levels and antral follicle counts.
-
B.
No testing, since all women with TM are infertile.
-
C.
Continue taking her current chelation during pregnancy.
Expert Perspective:
Some women with thalassemia may have spontaneous puberty, normal menstrual function with a preserved hypothalamic-pituitary-gonadal axis, but they may later experience premature ovarian failure. Many adult patients with β-TM are subfertile due to hypogonadotrophic hypogonadism (HH) secondary to iron overload (Skordis et al. 1998). Other endocrinopathies, such as hypothyroidism and diabetes, may affect reproductive capacity. Ovarian reserve testing should include serum levels of anti-müllerian hormone (AMH) and ultrasound to assess the number of antral follicles. Patients with low ovarian reserve have lower chances of spontaneous pregnancy and poor response to hormonal stimulation, particularly with aging (Singer et al. 2011). AMH is produced by the pre-antral and early antral follicles with small inter- and intra-cycle variability. Low ovarian reserve is considered predictive for low chances of spontaneous pregnancy and for poor ovarian response to hormonal stimulation.
Pregnancy planning is critical for patients with β-TM because of the high risk for serious complications for both mother and baby. A multidisciplinary team of hematologist, geneticist, reproductive medicine specialist, cardiologist, obstetrician, and nurse specialist is needed. Prior to pregnancy, screening echocardiogram, electrocardiogram, testing for acquired red cell antibodies, and testing of partner for hemoglobinopathies should be performed. Assisted reproductive techniques are viable options for successful pregnancies in women with thalassemia.
During pregnancy, some patients will need increased transfusion support to maintain Hb level >10 g/dL. For better outcomes, patients should have a cardiac MRI with T2* more than 20 ms, echocardiography with left ventricular ejection fraction >65 % and fractional shortening >30 %, and electrocardiogram with no significant arrhythmias (Cappellini et al. 2014) prior to attempting a pregnancy. Most patients should discontinue all iron chelation as soon as they become pregnant. While there is limited data on potential teratogenicity of deferasirox or deferiprone, deferoxamine has been used safely after the first trimester. Bisphosphonates have long half-life and, therefore, should be stopped about 6 months prior to conception and during breastfeeding. Interferon, ribavirin, and hydroxyurea should also be discontinued at least 6 months before conception. Patients should be advised about healthy lifestyle and the need for supplementation with calcium, vitamin D, and folic acid. Breastfeeding should be recommended unless the mother is HIV positive.
Question 10.
What option for allogeneic hematopoietic cell transplantation is associated with the best outcomes in patients with β-TM?
-
A.
Unrelated umbilical cord blood donor
-
B.
Matched sibling donors
-
C.
Matched unrelated donors
-
D.
Haploidentical related donors
Expert Perspective:
Hematopoietic cell transplantation (HCT) is the only available curative treatment option for patients with β-TM. With more than 3000 HCTs performed worldwide (Angelucci 2010), outcomes continue to improve, with transplant-related mortality of ≤5 % and a cure rate of 80–90 % (Angelucci 2010), compared to 73 % in earlier reports (Angelucci and Baronciani 2008). This is due to the availability of more effective graft-versus-host disease prophylaxis, better treatment options for cytomegalovirus infection, broad spectrum systemic antibiotic therapy, higher resolution and better HLA typing, and the wide adoption of aseptic techniques.
One common risk stratification tool for HCT in thalassemia is the Pesaro classification, which incorporates the presence of portal fibrosis, hepatomegaly, or history of inadequate chelation for the more accurate prediction of overall survival, disease-free survival, and risk for graft rejection. Three-year probabilities for overall survival and thalassemia-free survival varied among classes; class 1 (no risk factors) had 94 % and 94 %, class 2 (one risk factor) had 80 % and 77 %, and class 3 (both risk factors) had 61 % and 53 %, respectively (Lucarelli et al. 1990, 1993). Patients with β-TM are also at higher risk for mortality if they were ≥7 years old and had hepatomegaly at the time of or prior to HCT (Sabloff et al. 2011).
Most patients with β-TM lack a HLA-matched sibling donor. Alternative options include the use of matched unrelated marrow donor, matched unrelated cord blood, and mismatch related donor, which remain experimental (Cappellini et al. 2014). Allogeneic HCT with reduced intensity conditioning regimen has proven safety and effectiveness with a 5-year probability of survival and disease-free survival of 93 % and 84 %, respectively, but at this time is not recommended outside of a clinical trial (Bernardo et al. 2012). HCT may be cost effective considering the cost of lifelong regular transfusions, iron chelation therapy, and management of other disease complications and comorbidities.
Question 11.
What about the use of other alternative or novel drugs might help her?
-
A.
Hydroxyurea to induce fetal hemoglobin
-
B.
Short-chain fatty acids
-
C.
New agents to induce erythroid maturation
-
D.
All of the above
Expert Perspective:
Hemoglobin-F (HbF) inducers may reduce transfusion frequency and volumes. The evidence of the effectiveness of these agents varied in previous studies that included different patient populations and considered different end points (Musallam et al. 2013b). The most commonly studied drug is hydroxyurea. Observational studies of HU in patients with different forms of β-thalassemia have shown some benefits with higher hemoglobin levels and decreased transfusion requirements (Musallam et al. 2013b). Additionally, the use of HU combined with recombinant erythropoietin has shown higher hemoglobin levels than using HU alone in patients with non-transfusion-dependent thalassemia, but not β-TM (Loukopoulos et al. 1998).
Demethylating agents have been also used to increase HbF levels in patients with β-TM. Earlier studies included 5-azacytidine with remarkable increase in Hb level and HbF percent; however, there have been concerns regarding its safety profile and long-term side effects with possible mutagenicity (Musallam et al. 2013b). More recently, a small pilot study of decitabine in patients with β-thalassemia intermedia has shown an increase in both total Hb and HbF levels and improvement in markers of hemolysis with tolerable side effects (Olivieri et al. 2011).
Short-chain fatty acids include sodium phenylbutyrate and isobutyramide, and both can increase HbF levels as well. The use of oral sodium phenylbutyrate in patients with homozygous β-thalassemia was associated with higher total Hb and HbF levels and an improvement in markers of ineffective erythropoiesis (Collins et al. 1995). In contrast, isobutyramide had shown variable responses of HbF in patients with non-transfusion-dependent thalassemia (Taher et al. 2012).
Activin receptor antagonist (ACE-536) is another novel agent that has shown promising results in patients with β-thalassemia. ACE-536 works by promoting late erythrocyte differentiation independent of erythropoietin. Recently, a phase 2 study of ACE-536 in adults with β-thalassemia demonstrated more than 50 % and 12–60 % reduction in transfusion requirements and ferritin levels in patients with β-TM from their baseline, respectively, with increased Hb levels in patients with non-transfusion-dependent thalassemia (Piga et al. 2014). ACE-536 had satisfactory safety profile with no reported serious side effects.
Question 12.
What do we know about gene therapy in thalassemia so far?
-
A.
It has proven efficacy and should be offered to all patients.
-
B.
It should be considered to control iron overload.
-
C.
Early data on safety and efficacy are promising.
-
D.
The risk of leukemia is high with gene therapy and should be avoided.
Expert Perspective:
Efforts to evaluate the role of gene therapy in thalassemia started in early 2000 with promising results in mouse models (May et al. 2002; Rivella et al. 2003). The principles of gene therapy involve autologous stem cell collection ex vivo globin gene transduction with a lentiviral vector coding for normal β-globin gene and followed by reinfusion of the transduced stem cells (Cavazzana-Calvo et al. 2010; Chandrakasan and Malik 2014). The ultimate goal is to increase patients’ ability to produce normal adult Hb to become transfusion independent or reduce their transfusion requirement to a minimum. Risks of gene therapy include conditioning toxicity, transfer of replication-competent virus, clonal dominance with insertional oncogenesis, and infertility. Early results of gene therapy have been very promising in terms of safety and efficacy, but follow-up data is limited.
Answers
-
Question 1. B
-
Question 2. C
-
Question 3. E
-
Question 4. B
-
Question 5. A
-
Question 6. E
-
Question 7. D
-
Question 8. A
-
Question 9. A
-
Question 10. B
-
Question 11. D
-
Question 12. C
References
Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22(23):2171–9.
Anderson LJ, Westwood MA, Holden S, et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. Br J Haematol. 2004;127(3):348–55.
Ang AL, Shah F,BD, et al. Deferiprone is associated with lower Serum Ferritin (SF) relative to Liver Iron Concentration (LIC) than deferoxamine and deferasirox-implications for clinical practice. Blood. 2010;116:4246.
Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematol Am Soc Hematol Educ Program. 2010;2010:456–62.
Angelucci E, Baronciani D. Allogeneic stem cell transplantation for thalassemia major. Haematologica. 2008;93(12):1780–4.
Angelucci E, Giovagnoni A, Valeri G, et al. Limitations of magnetic resonance imaging in measurement of hepatic iron. Blood. 1997;90(12):4736–42.
Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med. 2000;343(5):327–31.
Bernardo ME, Piras E, Vacca A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood. 2012;120(2):473–6.
Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–93.
Brittenham GM, Cohen AR, McLaren CE, et al. Hepatic iron stores and plasma ferritin concentration in patients with sickle cell anemia and thalassemia major. Am J Hematol. 1993;42(1):81–5.
Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V, editors. Guidelines for the management of transfusion dependent thalassaemia (TDT). 3rd ed. Nicosia: Thalassaemia International Federation; 2014.
Cassinerio E, Orofino N, Roghi A, et al. Combination of deferasirox and deferoxamine in clinical practice: an alternative scheme of chelation in thalassemia major patients. Blood Cells Mol Dis. 2014;53(3):164–7.
Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467(7313):318–22.
Cazzola M, De Stefano P, Ponchio L, et al. Relationship between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol. 1995;89(3):473–8.
Cazzola M, Borgna-Pignatti C, Locatelli F, Ponchio L, Beguin Y, De Stefano P. A moderate transfusion regimen may reduce iron loading in beta-thalassemia major without producing excessive expansion of erythropoiesis. Transfusion. 1997;37(2):135–40.
Chandrakasan S, Malik P. Gene therapy for hemoglobinopathies: the state of the field and the future. Hematol Oncol Clin N Am. 2014;28(2):199–216.
Cohen AR, Glimm E, Porter JB. Effect of transfusional iron intake on response to chelation therapy in beta-thalassemia major. Blood. 2008;111(2):583–7.
Collins AF, Pearson HA, Giardina P, McDonagh KT, Brusilow SW, Dover GJ. Oral sodium phenylbutyrate therapy in homozygous beta thalassemia: a clinical trial. Blood. 1995;85(1):43–9.
Davies P, Robertson S, Hegde S, Greenwood R, Massey E, Davis P. Calculating the required transfusion volume in children. Transfusion. 2007;47(2):212–6.
Davis B, O’Sullivan C, Porter J. Value of LVEF monitoring in the long-term management of beta-thalassaemia. 8th International Conference on Thalassemia and the hemoglobinopathies (Athens). 2001:147. Abstract 056.
Davis BA, O’Sullivan C, Jarritt PH, Porter JB. Value of sequential monitoring of left ventricular ejection fraction in the management of thalassemia major. Blood. 2004;104(1):263–9.
De Sanctis V, Soliman AT, Elsedfy H, et al. Growth and endocrine disorders in thalassemia: the international network on endocrine complications in thalassemia (I-CET) position statement and guidelines. Indian J Endocrinol Metab. 2013;17(1):8–18.
Deborah Chirnomas S, Geukes-Foppen M, Barry K, et al. Practical implications of liver and heart iron load assessment by T2*-MRI in children and adults with transfusion-dependent anemias. Am J Hematol. 2008;83(10):781–3.
Elalfy MS, Adly AM, Wali Y, Tony S, Samir A, Elhenawy YI. Efficacy and safety of a novel combination of two oral chelators deferasirox/deferiprone over deferoxamine/deferiprone in severely iron overloaded young beta thalassemia major patients. Eur J Haematol. 2015;95(5):411–20.
Farmaki K, Tzoumari I, Pappa C, Chouliaras G, Berdoukas V. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. Br J Haematol. 2010;148(3):466–75.
Farmaki K, Tzoumari I, Pappa C. Oral chelators in transfusion-dependent thalassemia major patients may prevent or reverse iron overload complications. Blood Cells Mol Dis. 2011;47(1):33–40.
Fischer R, Longo F, Nielsen P, Engelhardt R, Hider RC, Piga A. Monitoring long-term efficacy of iron chelation therapy by deferiprone and desferrioxamine in patients with beta-thalassaemia major: application of SQUID biomagnetic liver susceptometry. Br J Haematol. 2003;121(6):938–48.
Forni GL, Perrotta S, Giusti A, et al. Neridronate improves bone mineral density and reduces back pain in beta-thalassaemia patients with osteoporosis: results from a phase 2, randomized, parallel-arm, open-label study. Br J Haematol. 2012;158(2):274–82.
Haines D, Martin M, Carson S, et al. Pain in thalassemia: the effects of age on pain frequency and severity. Clin Haematol. 1984;13(1):151–65.
Jensen PD, Jensen FT, Christensen T, Eiskjaer H, Baandrup U, Nielsen JL. Evaluation of myocardial iron by magnetic resonance imaging during iron chelation therapy with desferrioxamine: indication of close relation between myocardial iron content and chelatable iron pool. Blood. 2003;101(11):4632–9.
Kattamis A, Ladis V, Berdousi H, et al. Iron chelation treatment with combined therapy with deferiprone and deferoxamine: a 12-month trial. Blood Cells Mol Dis. 2006;36(1):21–5.
Kirk P, Roughton M, Porter JB, et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Circulation. 2009;120(20):1961–8.
Kwiatkowski JL, Kim HY, Thompson AA, et al. Chelation use and iron burden in North American and British thalassemia patients: a report from the Thalassemia Longitudinal Cohort. Blood. 2012;119(12):2746–53.
Lal A, Porter J, Sweeters N, et al. Combined chelation therapy with deferasirox and deferoxamine in thalassemia. Blood Cells Mol Dis. 2013;50(2):99–104.
Loukopoulos D, Voskaridou E, Stamoulakatou A, et al. Hydroxyurea therapy in thalassemia. Ann N Y Acad Sci. 1998;850:120–8.
Lucarelli G, Galimberti M, Polchi P, et al. Bone marrow transplantation in patients with thalassemia. N Engl J Med. 1990;322(7):417–21.
Lucarelli G, Galimberti M, Polchi P, et al. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med. 1993;329(12):840–4.
Martin A, Thompson AA. Thalassemias. Pediatr Clin N Am. 2013;60(6):1383–91.
May C, Rivella S, Chadburn A, Sadelain M. Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood. 2002;99(6):1902–8.
Michail-Merianou V, Pamphili-Panousopoulou L, Piperi-Lowes L, Pelegrinis E, Karaklis A. Alloimmunization to red cell antigens in thalassemia: comparative study of usual versus better-match transfusion programmes. Vox Sang. 1987;52(1–2):95–8.
Miller PD. A review of the efficacy and safety of denosumab in postmenopausal women with osteoporosis. Ther Adv Musculoskelet Dis. 2011;3(6):271–82.
Musallam KM, Angastiniotis M, Eleftheriou A, Porter JB. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematol. 2013a;130(2):64–73.
Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood. 2013b;121(12):2199–212; quiz 2372.
Noetzli LJ, Carson SM, Nord AS, Coates TD, Wood JC. Longitudinal analysis of heart and liver iron in thalassemia major. Blood. 2008;112(7):2973–8.
Noetzli LJ, Mittelman SD, Watanabe RM, Coates TD, Wood JC. Pancreatic iron and glucose dysregulation in thalassemia major. Am J Hematol. 2012a;87(2):155–60.
Noetzli LJ, Panigrahy A, Mittelman SD, et al. Pituitary iron and volume predict hypogonadism in transfusional iron overload. Am J Hematol. 2012b;87(2):167–71.
Olivieri NF, Nathan DG, MacMillan JH, et al. Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med. 1994;331(9):574–8.
Olivieri NF, Saunthararajah Y, Thayalasuthan V, et al. A pilot study of subcutaneous decitabine in beta-thalassemia intermedia. Blood. 2011;118(10):2708–11.
Piga A, Serra M, Longo F, et al. Changing patterns of splenectomy in transfusion-dependent thalassemia patients. Am J Hematol. 2011;86(9):808–10.
Piga AG, Perrotta S, Melpignano A, et al. ACE-536 increases hemoglobin and decreases transfusion burden and serum ferritin in adults with beta-thalassemia: preliminary results from a phase 2 study. Blood. 2014;124(21):53.
Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479–88.
Rice HE, Crary SE, Langer JC, Kemper AR, Splenectomy in Congenital Hemolytic Anemia C. Comparative effectiveness of different types of splenectomy for children with congenital hemolytic anemias. J Pediatr. 2012;160(4):684–9.e13.
Rivella S, May C, Chadburn A, Riviere I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101(8):2932–9.
Sabloff M, Chandy M, Wang Z, et al. HLA-matched sibling bone marrow transplantation for beta-thalassemia major. Blood. 2011;117(5):1745–50.
Singer ST, Vichinsky EP, Gildengorin G, van Disseldorp J, Rosen M, Cedars MI. Reproductive capacity in iron overloaded women with thalassemia major. Blood. 2011;118(10):2878–81.
Skordis N, Christou S, Koliou M, Pavlides N, Angastiniotis M. Fertility in female patients with thalassemia. J Pediatr Endocrinol Metab. 1998;11 Suppl 3:935–43.
Spanos T, Karageorga M, Ladis V, Peristeri J, Hatziliami A, Kattamis C. Red cell alloantibodies in patients with thalassemia. Vox Sang. 1990;58(1):50–5.
St Pierre TG, Clark PR, Chua-anusorn W, et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105(2):855–61.
Suragani RN, Cawley SM, Li R, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood. 2014;123(25):3864–72.
Taher AT, Musallam KM, Karimi M, Cappellini MD. Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Rev. 2012;26 Suppl 1:S24–7.
Tanner MA, Galanello R, Dessi C, et al. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation. 2007;115(14):1876–84.
Thompson AA, Cunningham MJ, Singer ST, et al. Red cell alloimmunization in a diverse population of transfused patients with thalassaemia. Br J Haematol. 2011;153(1):121–8.
Totadri S, Bansal D, Bhatia P, Attri SV, Trehan A, Marwaha RK. The deferiprone and deferasirox combination is efficacious in iron overloaded patients with beta-thalassemia major: a prospective, single center, open-label study. Pediatr Blood Cancer. 2015;62:1592–6.
Vogiatzi MG, Macklin EA, Trachtenberg FL, et al. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol. 2009;146(5):546–56.
Voskaridou E, Terpos E. New insights into the pathophysiology and management of osteoporosis in patients with beta thalassaemia. Br J Haematol. 2004;127(2):127–39.
Voskaridou E, Terpos E, Spina G, et al. Pamidronate is an effective treatment for osteoporosis in patients with beta-thalassaemia. Br J Haematol. 2003;123(4):730–7.
Voskaridou E, Anagnostopoulos A, Konstantopoulos K, et al. Zoledronic acid for the treatment of osteoporosis in patients with beta-thalassemia: results from a single-center, randomized, placebo-controlled trial. Haematologica. 2006;91(9):1193–202.
Wood JC, Enriquez C, Ghugre N, et al. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood. 2005;106(4):1460–5.
Wood JC, Origa R, Agus A, Matta G, Coates TD, Galanello R. Onset of cardiac iron loading in pediatric patients with thalassemia major. Haematologica. 2008;93(6):917–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Badawy, S.M., Thompson, A.A. (2016). Management of Thalassemias. In: Abutalib, S., Connors, J., Ragni, M. (eds) Nonmalignant Hematology. Springer, Cham. https://doi.org/10.1007/978-3-319-30352-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-30352-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30350-5
Online ISBN: 978-3-319-30352-9
eBook Packages: MedicineMedicine (R0)