
Abstract
The regulatory T cells are a subset of T cells which suppress or regulate the immune response, protect from autoimmune disease, and maintain tolerance to self-antigens. This chapter describes the nomenclature and function of various types of Treg cells that possess immunosuppressive function. Their major subsets include natural Treg cells, peripheral Treg cells, Tr1 cells, TH3 cells, CD8 Treg cells, Qa-1-restricted CD8+, CD8+ CD28−, and NKT cells. The factors involved in the mechanism of action of each subset are discussed. Finally, the potential of Treg cells as therapeutic targets for diseases including allergic disease, autoimmune diseases, inflammatory diseases, infections, cancer, transplant rejection, and amyotrophic lateral sclerosis is described. The treatment of patients with antigen-specific Treg cells would be an interesting approach if the issue of their propagation at a mass scale is resolved.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- CD25
- CTLA-4
- CD40L
- Treg
- Self-tolerance
- CD+CD25+ T cells
- Tr1 cells
- TH3 cells
- CD8+CD28− T cells
- CD8+CD122+ T cells
- Qa-1-restricted CD8+ T cells
- γ/δ T cells and NKT cells
- Perforin
- TGF-β
- Lag3
- Granzyme B-dependent killing
- IL-10
- IL-12
- FoxP-3
- TGF-β
- Polyendocrinopathy candidiasis ectodermal dystrophy
- Thymic hypoplasia
- T-cell receptor excision circles
- CD45RBLow
- CD62L
- CD103
- CD152
- αЄ β7
- Integrin
- GITR
- JAK1
- JAK3
- PTEN
- PI3K
- Peripheral Treg cells
- Dendritic cells
- CSCR4/CSCL12 signals
- n regulatory T cells
- I regulatory T cells
- NKT cell
- NKG2A
- NKG2B
- CD94
- Including CD45Rbl0
- CD4+
- CD103+
- CD4+CD8+IELS
- TCRαβ+ CD8+IELS
- CD101
- CD103
- Anti-ergotypic Treg cells
- Anti-idiotypic Treg cells
- TCRγ/δ+ anti-erg T cells
- T-bet
- GATA-3
- RORϒt
- Treg and disease
- Allergic disease
- Autoimmune diseases
- Inflammatory diseases
- Infections
- Cancer
- Transplantation
- Cancer immunotherapy
- Amyotrophic lateral sclerosis
- STAT3
- STAT5
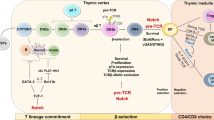
10.1 Introduction
The immune response is designed to protect human and other organisms from disease-causing agents; it also protects from detrimental responses to self. The immune system needs to be strictly regulated because of its ability to produce inflammatory mediators, killer cells, and antibodies, which are synthesized to eliminate the invading organisms but can also harm other normal cells. Consequently, an immune response cannot only produce autoimmunity but it is also capable of producing other diseases due to its ability to attack normal cells that are damaged mainly by the inflammatory cytokines in a collateral damage. A number of regulatory mechanisms keep these harmful effects of the immune response in check.
Immune response is triggered by antigen presentation to the TCR in context of MHC molecules, resulting in the activation and proliferation of CD4+ cells and secretion of cytokines. This activation also causes CD4+ T cells to express a number of cell surface receptors including CD25 (IL-2 receptor), CTLA-4, and CD40 ligand (CD40L). This growth of antigen-activated T cells was suggested to be controlled by suppressor T cells. Niels Jerne proposed that immune response may be inhibited by special lymphocytes, which pointed to the existence of suppressor T cells. The presence of such cells initially was hinted by the observations that injection of polyclonal T lymphoblasts from a parent to F1 hybrid blocked allograft rejection in rats. Similarly, injection with myelin basic protein (MBP)-reactive cloned T-cell line abrogated autoimmune encephalomyelitis specific for this antigen. The studies of the regulation of the anti-MBP response in autoimmune encephalomyelitis provided further foundational work in identifying the regulatory T cells. This model system allowed the understanding of the recognition of TCR peptides by regulatory T cells.
Regulatory T (Treg) cells have been defined as either having suppressor or regulatory functions. The term regulatory has been preferred over suppressor because of questions about I-J-regulated suppressor T cells. Gershon initially suggested that T cells could also have a regulatory function in addition to the role of T cells as helper cells for antibody synthesis. The initial focus was on soluble factors secreted by suppressor T cells as immune regulatory agents, and some of these soluble suppressor factors were characterized as MHC restricted. However, due to a lack of a definite cell surface marker on suppressor T cells, their existence remained controversial. The concept of the presence of suppressor T cells suffered further with the discovery of TH1 and TH2 cell subsets.
Studies by Nishizuka and Sakakura led to the conclusions that the suppression of the disease was due to the actions of thymus-derived lymphocytes and the splenocytes providing protection against the disease also originated in the thymus. However, they did not attempt to isolate the suppressor factor from the thymus. Later studies by other investigators demonstrated that only a small number of Treg cells were required to stop the development of autoimmune disease in mouse models, but the number of cells required to inhibit antibody response was much higher. The splenic T cells from mice with disease were able to transfer autoimmune disease to the newborn or adult nu/nu mice. CD4+ CD8− cells were later identified as the effector and suppressor T-cell population. It was found that the removal of supressor T cells from lymphoid cells will result in the disease and the re-administration of these cells will induce self-tolerance and suppression of autoimmunity. The suppressor T cells are a very small number of cells among CD4+ T lymphocytes that also express CD25 antigen, and selective depletion of CD4+ CD25+ T cells results in multiple manifestations of autoimmune diseases. This is a result of depletion of Treg cells from either the thymus or the peripheral lymphoid tissue.
CD4+ T cells mediate suppressor function independent of CD8+ T cells, and these cells classified as Treg cells maintain self-tolerance and suppress responses to foreign antigens. Although various types of Treg cells have been found, the most attention has been paid to CD4+CD25+ T cells that predominantly originate in the thymus and their centralized production is referred to as “the third function of the thymus.” They are mature T cells with a distinct function, and humans lacking CD+CD25+ T cells exhibit severe defects in controlling autoimmune response and have abnormal immune regulation, resulting in autoimmune and allergic diseases.
Based on the immunosuppressive activity, the many different types of Treg cells include natural CD4+ CD25+ T cells, peripheral Treg cells, IL-10-secreting Tr1 cells, TGF-β-secreting TH3 cells, CD8+CD28− T cells, CD8+CD122+ T cells, Qa-1-restricted CD8+ T cells, γ/δ T cells, and NKT cells. The production of different types of Treg cells is distinct; some are produced as a part of innate immune response while others are produced in response to an antigen as the acquired immune response develops with a selective participation of cytokines. Their function varies and includes regulation of autoimmunity, allergic responses, infection, inflammation, and transplant tolerance. The mechanisms of action of Treg cells are also diverse and range from cell–cell interaction involving CTLA-4, perforin, TGF-β, Lag3, granzyme B-dependent killing, regulation of dendritic cells, IL-10-mediated suppression, and Treg cell-mediated IL-12 consumption.
10.2 Types of Regulatory T Cells
10.2.1 Naturally Occurring Treg Cells
The Treg cells have been classified on the basis of their site of origin or mechanism of action (Table 10.1). The site of origin is for the naturally occurring Treg cells is the thymus, and the main mechanism of action is via cell–cell interaction. These Treg cells constitutively express CD4, CD25 (which is the α chain of IL-2 receptors), FoxP-3 (forkhead-winged-helix) transcription factor, and surface CD152. A defect in FoxP3 gene results in the hyperactivation of CD4+ T cells. Foxp3 is also expressed on CD4+CD25+ peripheral T cells and CD4+CD8−CD25+ thymocytes but is not expressed on other thymocytes, T cells, and B cells. Naturally occurring Treg cells do not require antigen exposure for their suppressive effector function; however, their generation and some of their activity may require TGF-β. A number of costimulatory signals and cytokines are also involved in the generation of Treg cells that may include B7, TNF family molecules, CD40L, PD-1, IL-2, TGF-β, or TNF- α, and these mechanisms are independent of the avidity of TCR. This results in induction of Treg cells through a genetic program with concomitant expression of CD25 and FOXP3+ or negative selection of thymocytes. Natural Treg cells possess a broad T-cell receptor repertoire that has high affinity than other T cells for the MHC Class II self-peptide ligands, which have selected them positively in the thymus. They do not produce inflammatory cytokines and they inhibit the activation, proliferation, and differentiation of a number of cell types including CD4+ cells, CD8+ cells, B cells, NK cells, NKT cells, and dendritic cells.
If natural Treg cells cannot be generated in the thymus or have a deficient function, this results in a number of autoimmune diseases. Impaired Treg cell generation is observed in children with thymic hypoplasia resulting from 22q-2 deletion syndrome. Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) is due to mutation in a gene called transcription factor autoimmune regulator (AIRE), and important self antigens on thymic medullary epithelial cells are regulated by AIRE. In the absence of AIRE, T cells that recognize self-antigens do not undergo negative selection and consequently are not deleted. The patients suffering from rheumatoid arthritis and multiple sclerosis exhibiting a reduced number of T-cell receptor excision circles (Trec) have suppressed activity of the thymus and its output. In juvenile rheumatoid arthritis, there are reduced numbers of Trec, suggesting premature aging of the thymus. Patients with autoimmune disease have early aged thymus, which results in poor development of Treg cells and an escape of non-Treg cells with an autoreactive TCR.
Regulatory T cells exhibit different developmental stages: one group (CD4+CD25+) expresses high CD62L and CCR7 levels and inhibits inflammation after binding to antigen-draining lymph nodes; another subgroup (CD4+CD25+ or CD4+CD25−) expresses αЄ β7 integrin and suppresses local immune reactions after homing to nonlymphogenic tissues at sites of inflammation. The expression of CD25 on natural Treg cells varies from none, low, intermediate, to high, suggesting a shift of expression based on the degree of injury or inflammation.
Various other cell surface markers are also expressed on CD4+ Treg cells including CD45RBLow, CD62L, CD103, CD152 (cytotoxic T-lymphocyte antigen-4 or CTLA-4), and GITR (glucocorticoid-induced TNF receptor family-related gene). The phenotype markers expressed on naturally occurring Treg cells are shown in Table 10.2. Many of these markers are associated with activated/memory cells, and it appears that naturally occurring Treg cells may be similar to memory T cells and usually are in an antigen-primed state. The Treg cells exhibit broad antigen specificity and have enhanced ability to recognize self-antigen rather than other T-cell subsets, and their production, maintenance, and function are IL-2 dependent. The development of Treg cells in the thymus is blocked if there is a defect in Foxp3/FOXP3 gene which controls the production of these cells. They suppress a variety of immune cells involved in both the innate and acquired immune responses; IL-2 and TCR stimulation is required to express their suppressive effects on helper T cell proliferation and IL-2 production but the subsequent immune suppression is not antigen specific. Interestingly, the normal T-cell inducers do not cause the proliferation of naturally arising CD4+CD25+ Treg cells or IL-2 secretion. However, they respond to very high doses of IL-2, mature dendritic cells as antigen-presenting cells, or anti-CD28 and, as a result, proliferate and secrete IL-2.
The cell–cell interaction is the critical mechanism of suppression by the natural Treg cells where CTLA-4, GITR, and PD-110 play a role in the contact-dependent suppression. A number of molecules expressed on natural Treg cells including CTLA-4, CD80, CD86, and CD223 are inhibitory molecules. For cell–cell interaction, a competition for antigen-presenting cells and specific MHC/peptide antigenic complexes is required.
Treg cells express all three chains (α, β, γ) of the high-affinity IL-2 receptor. Signaling for IL-2 receptor is mediated through induction of JAK1 and JAK3, which results in the phosphorylation and activation of STAT3 and STAT5. The translocation of these activated transcriptional factors to the nucleus results in the functional effects mediated by IL-2. Stimulation of IL-2 receptors also results in the activation of other pathways including mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K). IL-2 regulates self-tolerance through its involvement in the development and homeostasis of CD4+CD25+ Treg cells. Treg cells have a distinct IL-2R-mediated signal transduction pathway where, while the JAK-STAT-dependent transduction pathways are not altered, downstream signaling of PI3K is not observed. This difference in transduction pathways is associated with expression of PTEN (phosphatase and tensin homolog deleted on chromosome 10) and is correlated with the hypoproliferative response of Treg cells. PTEN is a lipid phosphatase, which is a catalyst for the reverse reaction of PI3K. Consequently, PTEN negatively activates the induction of downstream signal transduction pathway. In Treg cells, the expression of PTEN is unaltered as opposed to activated T cells where it undergoes downregulation. IL-2-induced T-cell proliferation is dependent on PI3K-mediated signal transduction. PTEN deletion allows the expansion of Treg cells in the presence of IL-2 without compromising their regulatory activity in maintaining homeostasis and self-tolerance.
IL-2 signaling directly targets the Foxp3 gene in Treg cells. This is accomplished as a result of binding between a specific site present in the first intron of the FOXP3 gene and STAT3 and STAT5 proteins. This signaling pathway is specific for CD4+CD25+ Treg cells since an IL-2-induced expression of FOXP3 gene is not seen in CD4+CD25− cells. This lack of effect is not due to the absence of IL-2Rα because conditions which do not require the presence of IL-2Rα such as stimulation with CD3 and treatment with high concentrations of IL-2 do not alter the effects of IL-2 on Foxp3 gene expression in CD4+CD25− cells. Consequently, IL-2 affects CD4+CD25+ Treg cells in a unique manner, which is mediated via expression of Foxp-3 gene (Fig. 10.1).

Development of CD4+ regulatory T cells: Natural CD4+CD25+ Treg cells develop in the thymus as a result of positive selection between TCR and host antigens. The thymus-derived Treg cells are specific for antigens seen in the thymus. The autoreactive T cells undergo negative selection and are depleted by apoptosis. The acquired Treg cells develop in the periphery from naïve precursors and their specificity lie in antigens other than the ones which come in contact with the thymus. Tr1 cells are induced in the periphery when naïve T cells are exposed to an antigen in the presence of IL-10. They are not identified by a particular cell surface marker. Th3 cells are generated from naive CD4+ T cells as a result of low doses of antigen via the oral route and they secrete TGF-ß
10.2.2 Peripheral (Adaptive) Treg Cells
The peripheral (adaptive) Treg cells develop in the periphery, and the stimulus for their generation is either an ongoing immune response or exposure to tolerogenic dendritic cells. Adaptive Treg cells develop from naïve precursors or mature T cells, and their specificities lie in antigens other than the ones which come in contact in the thymus, such as food antigens, pathogens, parasites, and bacterial flora. Their mechanism of action is mediated via suppressive effects of cytokines (IL-10 or TGF-β). The peripheral (adaptive) Treg cells are not generated in the thymus and are specific for both foreign antigens and self-antigens. This is in contrast to the thymus-derived Treg cells, which are specific for antigens seen in the thymus. Two models have been suggested for the generation of peripheral Treg cells. According to the linear model, after antigen recognition, naïve T cells are activated and differentiate into effector and Treg cells. Alternatively, a parallel model suggests that after activation, naïve T cells remain uncommitted and their development into effector and Treg cells is in parallel. As a result, the development of effector cells is faster than peripheral Treg cells. IL-2 is required for the development and differentiation of both types of cells. Non-Treg cells can differentiate into CD4+CD25+ Treg cells in the periphery and can function like natural Treg cells, that is, suppression of T-lymphocyte proliferation by cell–cell interaction independent of cytokines. The only main requirement for the generation of these natural Treg-like cells in periphery is activation of CD4+ T cells, and their production can be achieved by exposure to oral, intravenous, or subcutaneous antigens or continued exposure to superantigen. The development of natural Treg-like cells is possible from peripheral CD4+CD25+ T cells under conditions requiring either TGF-β or both TGF-β and TCR activation, which results in CD4+CD25− cells expressing Treg cell function and FOXP3. The treatment of human peripheral blood lymphocytes with allogeneic dendritic cells in the presence of IL-10 also results in the development of natural Treg-like cells in the periphery, as is the case with the treatment of CD4+CD25− T cells with anti-CD3 and anti-CD28. The induction of FOXP3 is independent of TGF-β, and peripheral Treg cells look identical to natural Treg cells in phenotype, function, and gene expression. They only differ from Treg cells in their requirement for TCR and CD28 for induction. The induction of peripheral Treg cells requires immunogenic antigen exposure and the combination of an antigen directed at dendritic cells and anti-CD40 antibody. Immune response is limited against foreign antigens, and collateral damage to healthy tissue is avoided as a result of early development of FOXP3 Treg cells. The reduction in the number of effector and Treg cells may result from apoptosis and peripheral migration. However, in addition to these short-term Treg cells, long persisting Treg cells can also be induced. This induction is a result of exposure to low levels of antigen, which could be achieved without inflammation. The generation of these Treg cells may be less efficient in the beginning, but they may have prolonged presence and consequently play a role in tolerance to autoantigens.
Lymphopenia promotes expansion of Treg cells. Lymphocyte activation is regulated by competition with general lymphocyte population. In the absence of such competition, both regulatory and effector cells develop sequentially as a result of weak signals. In lymphopenia the appearance of Treg cells parallel the recovery, and as soon as the accumulation of cells seizes, the Treg cells are expressed. FOXP3+ cells may contribute to the development of homeostasis, and after the cell number reaches equilibrium, the generation of Treg cells begins. In periphery, generation of antigen-specific Treg cells could be rapid when other T cells are present, resulting in IL-2 secretion and the production of Treg cells. Peripheral Treg cells play an important role in tolerance, tumor immunity, and microbial defense. The suppressive effects of these cells are mediated via production of immunosuppressive cytokines, IL-10 and TGF-β. However, some peripherally induced Treg cells which express FOXP3 also act by cell–cell interaction. Treg cells suppress immune response during infection to avoid tissue damage but this may prolong the infection. Tumor-infiltrating CD4+CD25+ Treg cells are suppressive in nature and are found in increased numbers in human cancers.
Generation of peripheral Treg cells may result in the establishment of homeostasis after its disruption. The examples include infection, autoimmune diseases, certain forms of cancers, and immunodeficiency syndrome. The mechanisms by which peripheral Treg cells induce self-tolerance and homeostasis may involve cytokines or cell–cell interaction. The cytokines IL-10 and TGF-β and molecules such as CTLA-4 are involved in the effector mechanisms of Treg cells. The indirect effects of Treg cells may be mediated via antigen-presenting cells or natural killer cells. More specifically, the assembly of immunologic synapse between antigen-presenting cells and effector cells is modulated by Treg cells, which may be mediated via direct or indirect mechanisms.
The bone marrow is also a significant reservoir for Treg cells. Treg cells enter in the bone marrow and are retained through CSCR4/CSCL12 signals. Functional stromal-derived factor (CXCL12) is strongly expressed in the bone marrow and is the ligand for CXCR4. The human bone marrow CXCL12 expression is suppressed by G-CSF, and this causes the migration of Treg cells from the bone marrow to the peripheral blood. This also explains improvement in autoimmune diseases and graft-versus-host response after treatment with G-CSF. Figure 10.2 depicts the activation and differentiation of Treg cells.

Activation of Treg cells and differentiation. Treg cells expressing FOXP3+ migrate from the thymus and enter the secondary lymphoid tissues where they recirculate as central Treg cells. They differentiate into effector Treg cells after they are induced by TCR ligation along with costimulation with CD28 and proliferation resulting from exposure to IL-2. This effect is mediated via induction of expression of interferon regulatory factor 4. The differentiation of Treg cells also is dependent on activation of B-lymphocyte-I-induced maturation protein 1 and suppression of BTB and CNC homolog 2. There is also induction of transcription factors which along with FOXP3 induce chemokine and homing receptors, causing the polarization of Treg cells. These events are responsible for the recruitment of Treg cells to tissues or site of injury/inflammation (Source: Reproduced with permission. Liston A, Gray HD (2014) Homeostatic control of regulatory T cell diversity. Reviews Immunology. 14: 154–165. Nature Publishing Group)
10.2.3 Tr1 Cells
CD4+ type 1 regulatory T (Tr1) cells were initially characterized after isolation of the peripheral blood lymphocytes from patients who suffered from combined immunodeficiency and have received successful HLA-mismatched bone marrow transplant, resulting in their generation from naïve CD4+ cells following the development of antigen-specific immune response. Tr1 cells can exist naturally or may be induced. These cells are induced in the periphery when naïve T cells are exposed to an antigen in the presence of IL-10. Tr1 cells are generated from naïve CD4+ T cells after they are activated by TCR and CD28. IL-10-producing Tr1 cells are also produced by treatment with a combination of anti-IL-4 and anti-IL-12 antibodies, dexamethasone, and active vitamin D3 (Table 10.3). Furthermore, IL-10-secreting Tr1 cells are generated when naïve CD4+ T cells are treated with immature dendritic cells, IFN-α, or immunosuppressive agents. Tr1 cells are not identified on the basis of any particular surface marker and also do not constitutively express FoxP3; however, they may express markers associated with TH2 cells and repressor of GATA (ROG). They do express high levels of surface CD152 as is the case with natural Treg cells. Tr1 cells could be either CD4+ or CD8+; proliferate poorly and migrate to the inflamed tissue; and secrete high amounts of IL-10, TGF-β, and IL-5 and low concentrations of IL-2 and IFN-γ; but do not secrete IL-4. In response to signaling through TCR, IL-2R, and CD28, they exhibit anergy and suppress antigen-specific proliferation of naïve CD4+ cells. The immune suppression is mediated via cytokines and not cell–cell interaction. In animal models, Tr1 cells regulate mucosal tolerance, diabetes, responses to transplant antigens, infectious agents, and allergens, after they migrate to the inflamed or injured site. Their role is pivotal in the maintenance of peripheral tolerance. A member of the TNF superfamily, OX40 ligand, inhibits the generation of Tr1 cells, and as a consequence, OX40L augments immunity and abrogates tolerance.
10.2.4 ILT3+ Regulatory T-Cell Subpopulation
The Treg cells protect the mucosal surfaces in the lung from the allergens as they get activated in response to their exposure. The defect in the regulatory mechanism results in the development of the immune response against the allergen and subsequently the development of the atopic state, including allergic asthma. The Notch ligand, Jagged1, expressed on DCs, is responsible for TH2 differentiation by enhancing the expression of GATA-3 and IL-4 in T cells in vivo. Furthermore, the stimulus for TH2 cell-derived immune responses depends on the expression of the transcription factor IRF4 in DCs. This regulates the maturation of a PDL2-expressing DC subset 23. IRF4 also induces the expression of IL-33 and IL-10 by DCs, which is essential for the induction of TH2 differentiation. The number of PD-L2+IRF4+ DCs is substantially greater in Csnk2bfl/flFoxp3-Cre mice than in Csnk2bfl/fl mice, implying that CK2β controls the ability of Treg cells to regulate PD-L2+IRF4+ DCs and TH2 immune responses.
A Treg cell subpopulation identified by the presence of immunoglobulin-like transcript 3 (ILT3) provides inhibition of the main function of Treg cells. Thus the activity of this subpopulation of Treg cells can be regulated. Protein kinase CK2 is involved in the suppression of excessive TH2 response in the lung upon exposure to allergen by Treg cells. CK2 is a highly conserved serine–threonine kinase and takes part in many signal transduction pathways, including the NF-κB, PI (3)K, and Wnt pathways. By using Treg cell-specific gene targeting, it is observed that the inhibition of allergic immune response by TH2 cells is protein kinase CK2 dependent. There is proliferation of ILT3+ Treg cells as a result of genetic ablation of the β-subunit of CK2 in Treg cells. ILT3+ Treg cells subpopulation does not inhibit the development of IRF4+PD-L2+ dendritic cells. These dendritic cells play a major role in the development of TH2-mediated immune responses. A different explanation for the TH1/TH2 imbalance favoring TH2 cells is that during inflammation CK2β deficiency results in the reprogramming of Treg cells into TH2 cells.
ILT3 has been identified as a CK2-controlled protein that regulates Treg cells and modulates TH2 cell-induced inflammation. Negative signals are transduced by ILT3, which impair TCR-induced pathway. This causes activation of tyrosine phosphatases (SHP-1 and SHP-2) and as a result dephosphorylating of Zap70. The TCR signaling is much lower in ILT3+ Treg cells than ILT3− Treg cells. The expression of all three receptors of the Nr4a family is lower in ILT3+ Treg cells than ILT3− Treg cells. Deletion of Nr4a family receptors results in defective thymic Treg cell development and in a TH2 cell-induced inflammation, which is fatal in animal models. In contrast, the attenuation of suppression or deletion of SHP-1 increases the inhibitory function of Treg cells. The polymorphisms in the LILRB4 locus are associated with high IgE levels in the serum of children with asthma. There appears to be a close association between ILT3-dependent signaling and Treg cell-induced suppression of inflammatory responses caused by TH2 cells. CK2β deficiency leads to increased expression of ILT3 and inhibition of the activity of Treg cells; this results in the proliferation the TH2 cell-inducing IRF4+PD-L2+ DC population. As a result there is an induction of TH2 cells that leads to allergy.
10.2.5 TH3 Cells
Antigen-specific Treg cells, TH3, are generated from naïve CD4+ T cells as a result of low doses of antigen via the oral route. This phenomenon is just opposite to hyporesponsiveness resulting from anergy or deletion, resulting following exposure to high antigen doses. Oral tolerance results from such exposure as a result of interaction of dietary antigens with GI immune apparatus. TH3 can also be induced in periphery. They are TGF-β-producing cells, which may also express FOXP3. TH3 cells suppress antigen-specific responses and can transfer tolerance. Their mechanism of action is mediated via TGF-β, and defects in TH3 cells may be associated with the development of autoimmune disease.
10.2.6 n Regulatory T Cells (nTreg) and i Regulatory T Cells (iTreg)
nTreg are derived in the thymus from Foxp3−CD25+CD4+ cells. iTreg develops outside the thymus from FOXP3+ CD25− CD4− cells, in chronically inflamed tissues such as spleen, lymph node, and gut-associated lymphoid tissue, from naïve T cells. Both cell types become FOXP3+ CD25+ CD4+. The activation of nTreg requires CD28 for costimulation, and the activation of iTreg requires CTLA-4 for costimulation. Both are inhibitors of the autoreactive T-cell signaling and cell–cell interaction dependent as well as independent signaling.
10.2.7 Natural Killer T (NKT) Cells
Since NKT expresses a TCR, they are defined as T lymphocytes and are distinct from NK cells, although they both share CD161 or NKR-P1. They also differ from T lymphocytes and other Treg cells because they lack the ability to interact with peptide antigens in classical MHC Class I or Class II restricted manner. Instead, they recognize antigens in context of a glycolipid, which is a nonclassical antigen-presenting molecule called CD1d. NKT cells have two main subsets, CD4+ and CD4−, but some of the NKT cells are also CD8+. The subsets of NKT cells are present along with other lymphocytes, but their numbers are tissue dependent. In mice, their numbers are most prevalent in the liver and with lower frequencies in the bone marrow, spleen, thymus, blood, and lymph nodes.
They recognize a class of antigens that is not recognized by T lymphocytes. After recognition of the antigen, they are activated within 1–2 h of TCR ligation and produce both TH1 (IFN-γ, TNF-α) and TH2 (IL-4, IL-13) cytokines. Many glycolipids including glycophosphatidylinositol, gangliosides, and phosphoethanolamine can activate NKT cells. Although their cytokine production pattern is TH0-like, they can produce either a TH1 or TH2 immune response.
NKT cells express receptors both for the NK lineage and for T cells (TCR αβ) and thus are a unique population of lymphocytes. Tumor cells expressing lipid antigens are recognized and killed by NKT cells. These lipid antigens related to the glycolipid α-galactosylceramide are presented to NKT cells in context of MHC (Ib, CD1d) molecules. In addition to their ability to kill tumor cells, they also regulate autoimmune diseases. Their effector function as natural killer cells and the ability to secrete a number of cytokines including INF-α, TGF β, IL-4, and IL-10 are enhanced following antigen recognition by the TCR in context of MHC Class Ib molecules. On the basis of secretion of cytokines, it appears that NKT cells may be involved in modulating both innate- and TH2-dependent acquired immune response. In animal models, NKT cells have been shown to inhibit the onset of type 1 diabetes mellitus and multiple sclerosis, and their depletion accelerates the development of the disease. In humans, there is also an association of NKT cells and autoimmune disease. A decrease in the frequency of Vα24-J αQNKT cells is associated with relapse in patients with multiple sclerosis, and the patients with diabetes also have lower expression of Vα24-JαQ NKT cells.
NKT cells also play a role in allograft tolerance, which involves NKT cell-dependent allospecific regulatory T-cell generation. They induce cardiac allograft tolerance and inhibit graft-versus-host disease. NKT cells may also play a role in tumor rejection and are also required for IL-12-mediated cancer therapy. A-GalCer, a glycolipid recognized by NKT cells, causes rejection of a number of tumor cell lines via activating NKT cells.
NKT cells are unique regulatory cells although they also act as effector cells. Their regulatory role is reflected by the pattern of cytokines they secrete, interaction with dendritic cells, and their small numbers. Interaction with dendritic cells suggests their role in acquired immune response. It seems that the phenotype of these cells may be distinct at different locations such as the thymus versus the liver, which may be a deciding factor in their ability to promote or suppress the immune response. The human CD4+ NKT cells produce a high TH2/TH1 cytokine ratio with distinct expression of cytotoxic and chemokine receptors as opposed to CD4− NKT cells. The type of signal that NKT cells receive may also determine whether they will produce pro- or anti-inflammatory response. The examples include the production of INF-γ when cross-linked with IL-12 or anti-NK1.1 and IL-4 production after cross-linking with IL-7; therefore, the cytokines secreted by NKT cells may be dependent on the type of TCR stimulation. The OCH analog of A-GalCer induces TH2 responses and C-glycoside induces TH1 responses. Alternatively, the products produced by NKT cells may be the same, but different physiologic or pathologic circumstances may interpret them differently and thus NKT cells are not responsible for the final impact. The example includes the immediate production of both INF-γ and IL-4 following a-GalCer stimulation, which are the products of preformed mRNA, but IL-4 secretion stops within a few hours while NKT cells continue to secrete IFN-γ for another 2–3 days. This suggests that this differential response may be attributed to the temporal nature of the interactions of NKT cells with other immune cells.
CD4+CD25+ T cells and NKT cells exist as natural suppressor cells from the early fetal life before antigen exposure and play an important role in immune regulation during innate and/or primary immune responses. The NKT cells recognize glycoproteins via Vα chain of TCRαβ, which is expressed by tumor cells, pathogens, injured apoptotic cells, and blast cells. The primary immune response balance of TH1 and TH2 cells is modulated by NKT cells via their secretion of IL-4 and IL-10. Naturally occurring CD4+CD25+ T-cell subset is also present in the peripheral lymphoid system and without antigen stimulation can affect the primary immune response. Their effect is mediated via cell–cell interaction as well as the secretion of TGF-β. Antigen recognition activates NKT cells, which respond by secreting IL-13. IL-13 receptors are expressed on certain myeloid cells, and these cells, as a result of IL-13 binding and signal transduction, produce TGF-β and suppress the CD8+ CTLs. This results in the suppression of tumor immunity since CD8+ CTLs kill tumor cells.
10.2.8 Regulatory CD8+ T Cells
Most of the information about Treg cells involve CD4+ sublineage with regulatory activity. Following the identification of CD4+ Treg cells, a subpopulation of CD8+ T cells was identified, which suppressed helper T cell and B cell responses in an MHC-dependent manner, requiring the expression of HLA Class Ib MHC molecule Qa-1 on target cells. CD8+ Treg cells in mice are divided at least into two groups: Qa-1 restricted and Qa-1 nonrestricted; Qa-1 is an equivalent of human HLA-E.
CD8+ Treg cells are activated by autologous CD4+ T cells after their induction during the primary immune response, differentiating into functional suppressor T cells. Their effector function is prominent during the secondary immune response as well as memory-based immune responses. CD8+ Treg cells may be responsible for producing suppression to autoimmunity after the patient recovers from the first episode of the disease and consequently will resist to a relapse and may decrease the severity of the symptoms in future episodes of the same disease. Qa-1-restricted CD8+ Treg cells recognize their target through their TCRαβ in an MHC-restricted manner. Some Qa-1 self-peptide-expressing activated T cells are downregulated by these cells, but this is not the case for all activated T cells. Both types of Qa-1 receptors, TCR and CD94/NKG2, can be expressed on CD8+ Treg cells. CD94/NKG2 is a C-type lectin receptor present on NK and CD8+ cells. The TCR can recognize Qa-1 complex on induced CD4+ cells, resulting in suppressor activity, and CD94/NKG2 recognize Qa-1/Qdm ligands. NKG2 receptors play a dual role as they can either enhance suppression in response to NKG2C, E, or H or inhibit suppression in response to NKG2A or NKG2B. A non-Qa-1-restricted CD8+CD28− Treg cell subset has been identified, which mediate suppression via antigen-presenting cells.
Human CD8+ Treg cells express CD25, CD69, CTLA-4, and FOXP3. They secrete IL-4, IL-5, IL-13, and TGF-β but do not secrete IFN-γ and contribute to immunoregulation. Naïve CD8+CD25− cells are considered to differentiate into CD8+ Treg cells when presented with an antigen. CD8+CD28− Treg cells are induced in the presence of IL-10. IL-10 may be involved in the downregulation of dendritic cell costimulation as well as in the upregulation of ILT-3 (immunoglobulin-like transcript 3) and ILT-4. An additional human Treg subset has been identified, which includes CD8+, LAG-3+ (lymphocyte activation gene 3, an MHC class II-binding CD4 homolog), CD25+, FOXP3+, CCL4+, and it suppresses T-cell responses via secretion of chemokine CC chemokine ligand 4.
CD8+ Treg cells are generated in neonatal life when T lymphocytes enter into nonlymphoid tissue and maintain tolerance during adulthood. The thymus does not contribute to this antigen-specific tolerance, and the continued presence of the antigen is necessary to maintain tolerance. Antibody-mediated inhibition of T-cell migration abrogates this tolerance. TGF-β1 may play a role in the upregulation of this TCR+CD8+ subset-mediated tolerance. Furthermore, granzyme B is activated in this TCR+CD8+ Treg cell subset, which has been implicated in the induction of cell death of effector T cells by CD4+CD25+ Treg cells. This naturally occurring TCR+CD8+ Treg cell subset is induced by self-antigens that are expressed in neonatal mice on parenchymal cells. They maintain tolerance during adult life as a result of the downregulating effector function of T cells. This mechanism is independent of CD4+ T cells.
10.3 Regulatory T Cells in the Mucosal System
Tolerance is an important goal of the immune response in the gastrointestinal tract. Harmful pathogens are recognized by the mucosa-associated lymphoid tissue to protect the epithelial layer from their deleterious effects. Moreover, the mucosa-associated lymphoid tissue develops tolerance against dietary and bacterial antigens. In normal individuals, a number of regulatory cell types control inflammatory response when pathogenic bacteria and viruses attack the intestinal mucosa. A lack of appropriate regulatory responses, which limit inflammation in the gut results in the development of inflammatory bowel disease. A number of Treg cell subsets may be involved in mucosal immunity including CD45Rbl0, CD4+CD25+, CD4+, CD103+, CD4+Tr1, CD4+CD8+IELS, TCRαβ+ CD8+IELS, and CD8+CD28− cells. CD4+Tr1 are present in intestinal mucosa, and their immunosuppressive effects are mediated via IL-10, which they produce in large amounts. They are induced by IL-10, secreted by intestinal epithelial cells and other Treg cells in intestinal mucosa. The generation of mucosal CD4+ Tr1 cells is negatively modulated by a subset of dendritic cells. The role of these cells in the prevention of human inflammatory bowel disease has not yet been established. Another subset of Treg cells associated with mucosal immunity is CD4+TH3. These cells are present in the human intestinal mucosa and play a role in controlling inflammation in the gut.
The intestinal epithelial cells play an important role in the generation of these Treg cells that maintain tolerance in the mucosal immune system because of their ability to serve as antigen-presenting cells. The intestinal epithelial cells process and present antigenic fragments in context of the MHC molecules to the TCR. Several regulatory subsets, which include both CD4+ and CD8+ T cells, are involved in oral tolerance, and most of the immune suppression, which they cause is mediated via IL-4, IL-10, and TGF-β. The mechanism of tolerance resulting from antigen exposure in the lamina propria is different from oral tolerance. This tolerance is not dependent on perforin, and these cells act like CD4+, CD25+, or CD8+ and CD28− Treg cells and express CD8, CD101, and CD103. These CD8+ Treg cells are not present in patients with inflammatory bowel disease, which may be due to the epithelial cell glycoprotein gp180, a molecule expressed on all normal intestinal epithelial cells. The interaction of the gp180/CD1d complex on intestinal epithelial cells with a subset of CD8+ Treg cells results in oligoclonal expansion of CD8+ Treg cells in the intestinal mucosa.
10.4 T-Cell Vaccination and Regulatory T Cells
Anti-idiotypic and anti-ergotypic Treg cells are activated after T-cell vaccination and are regulators of the immune response. The regulatory T cells induced by activated T-cell vaccines, which are not anti-idiotypic, are called anti-ergotypic. They proliferate in response to autologous T cells after their activation, and their presence does not require T-cell vaccination or prevalence of an autoimmune disease. They are widely distributed in the thymus, spleen, and lymph nodes in naïve rats, and they do not need antigen exposure. Anti-erg T cells include both TCR α/β+ and TCRγ/δ+ T cells, and CD8+ markers are present on naive anti-erg T cells. IFN-γ and TNF-α are secreted by TCRγ/δ+ anti-erg T cells following activation of T cells. No detectable levels of cytokines are produced by TCR α/β+ anti-erg T cells as they proliferate in response to T-cell activation. Cell–cell interaction is involved for interaction between anti-erg T cells and activated stimulator T cells. Anti-erg T cells can also recognize ergotype on the cell surface of macrophages and other antigen-presenting cells, but this results in a much weaker response as opposed to the activated stimulator T cells. The naïve TCR α/β+ CD8+ and TCRγ/δ+ anti-ergotypic T cells respond in a classical MHC Class I restricted manner, and the B7 and CD28 molecules are involved in this recognition process. CD4+CD25+ Treg cells do not play a role in anti-ergotypic response.
A regulatory cell needs to meet two conditions in order for it to be called an ergotype. It must be expressed and presented by the activated and not the resting cells, resulting in the activation of anti-erg T cells; TCR, CD25, and HSP60 epitopes are some examples of ergotopes. Only activated T cells can present ergotypic TCR peptides to anti-erg T cells, despite the expression of TCR on resting T cells.
Anti-id Treg cells utilize unique TCR CDR3 peptides on the cell surface of effector cells. Anti-idiotypic T cells generated after T-cell vaccination are cytolytic T cells, which are CD8+ that kill after interaction with target T-cell receptors in context with MHC Class I molecules. This CD8+ anti-idiotypic T-cell response is responsible for the depletion of circulating autoreactive T cells. Furthermore, CD4+ regulatory T-cell responses also occur in response to T-cell vaccination in addition to the generation of CD8+ anti-idiotypic T cells. The production of CD4+ Treg cells in anti-idiotypic response may be responsible for the production of T-cell vaccination-induced clinical effects. The precise mechanisms relating to the involvement of CD4+ Treg cells in these processes have not yet been established. However, interaction with ergotypes including IL-2 receptors and heat shock protein 60 may result in the induction of CD4+ Treg cell responses.
In clinical trials of multiple sclerosis, the therapeutic effects of T-cell vaccination involve CD4+ Treg cells, which are produced as a result of repeated immunization with irradiated autologous T cells selected for autoantigens. The MS patients receiving T-cell vaccination produce two different populations of Treg cells that differ in their expression of FOXP3 gene and cytokine production and may have different mechanisms of action. Most of the cells have an abundant expression of FOXP3 gene and produce IL-10 and INF-γ, while a small number of cells produce only IL-10 and have very low levels of FOXP3 gene. After T-cell vaccination, Treg cells expressing CD4CD25FOXP3 may be derived from the naturally occurring CD4+CD25+ Treg cells. The T-cell vaccination results in the upregulation and proliferation of CD4+ CD25+ Treg cells, the numbers of which are below normal in patients with MS. The other subset of Treg cells, CD4+ CD25+FOXP3−, produces high levels of IL-10, resulting in the suppression of activated T cells. This inhibition by IL-10 is reversed by IL-10 antagonist or monoclonal antibody to IL-10. The CD4+ CD25+FOXP3+ Treg cells recognize an epitope corresponding to 61–73 residues of α chain of IL-2 receptors. This may be clinically relevant in finding new treatments for autoimmune diseases.
10.5 Regulatory T Cells and Antibody Production
The autoantibody production in autoimmune diseases may be attributed to the inability of Treg cells to control their synthesis. In an autoimmune model, T cells regulate the mechanisms through which B cells that were autoreactive to self-antigens do not produce autoantibodies, suggesting a role for suppressor T cells. The administration of irradiation, thymocytes, lymph nodes, or spleen cells inhibits the production of autoantibodies, which is attributed to the suppressor T cells.
10.6 Mechanisms of Induction of Treg Cells
Treg cells are induced by low doses of oral antigen, but high antigen doses result in anergy. CD4+ and CD8+ Treg cells can be produced in autoimmune murine models when self-antigen is administered by the oral route. These antigen-specific Treg cells are of the TH3 subgroup. Antigen-specific Treg cells can also be induced when nonobese diabetic mice are administered human insulin orally or by aerosol. This causes these animals to be hyporesponsive to human insulin when an immunostimulatory route is used. CD8+ Treg cells are produced in kidney grafts in rats after oral exposure to alloantigen, and transfer of these cells to naïve animals will prolong graft survival. Allogeneic cardiac graft survival is prolonged following intratracheal delivery of allogeneic peptides. This is mediated via production of IL-4- and IL-10- secreting Treg cells.
The antigen recognition by TCR involves activation of multiple signal pathways involving additional ligands. CD40 ligand (CD154), expressed on CD4+ T cells, is important in initiating costimulatory signals after it binds to CD40 on antigen-presenting cells. After activation of CD40, other molecules including CD80 and CD86 are upregulated, resulting in the proliferation of T cells and generation of an immune response. CD80 and CD86 also serve as receptors for TCRs, CD28, and CTLA-4. CD28 is an activator of the immune response via IL-2 secretion and induction of T-cell proliferation, whereas CTLA-4 inhibits T-cell responses. Consequently, CTLA-4 is involved in inducing immune tolerance by inhibiting the signals which are responsible for T-cell activation in response to an antigen and result in the induction of Treg cells.
Alloantigen-specific Treg cells can also be generated by a number of other ligands. Administration of anti-CD40LmAb to antagonize CD40-CD40L pathway results in the generation of alloreactive T-cell responses by cell–cell interaction. The antagonism of downstream signal transduction such as inhibition of nuclear factor ĸB results in the generation of Treg cells. Agonist-like signals can also be used to generate Treg cells. For example, LFA-3- or CD58- mediated engagement of CD2 on naïve CD4+ T cells results in the differentiation of Treg cells that are HLA specific. Similarly, the induction of inducible costimulatory molecule on T cells and Notch during antigen presentation will result in the generation of Treg cells. But the most potent positive signals for their generation are provided by cytokines, specifically IL-10 and TGF-β. IL-10 downregulates expression of CD40, CD80, and CD86, resulting in inhibition of generation of CD4+ and CD8+ cells. This environment is optimal for the induction of Treg cells. Other proteins and soluble peptides could also produce antigen-specific Treg cells.
10.7 Antigen Specificity of Regulatory T Cells and Mechanisms of Suppression
Antigen-specific Treg cells function through antigen presentation, activation, and recognition of target cells. The antigen presentation is achieved in context of MHC molecules and in association with costimulatory and regulatory signals. After induction, coming in contact with the same antigen renders functional activity in Treg cells, which is followed by antigen-specific recognition of target cells by Treg cells.
Treg cell-induced suppression is mediated by several different mechanisms. The downregulation of CD40, CD80, and CD86 molecules results in a lack of T-cell activity when inhibited by CD8+CD28− Treg cells. Another mechanism is mediated via cytokines, IL-10 and TGF-β, secreted by antigen-activated Treg cells. IL-10 downregulates CD80 and CD86 molecules via activation of JAK/STAT pathways and inhibits NFĸB activation. As a result, T-cell activation and IL-12 production are affected. The effects of TGF-β are mediated via Smad complex. Another proposed mechanism includes the killing of the effector CD8+ T cells, which kills the graft by Treg cells that involves Fas–Fas ligand pathway.
10.8 FOXP3 Expression and Regulatory T-Cell Activity
The function of Treg cells is controlled by FOXP3 gene, an X-chromosome-linked factor, in a binary function, resulting in the maintenance of immune tolerance. The immune-suppressive activities of T cells are regulated by FOXP3 gene. As a consequence, most attention has focused on equating abnormalities in Treg cells with immunologic diseases. The effector function of Treg cells is as important as their numbers in regulating the immune response. For example, in diabetic NOD mice, there are lower levels of FOXP3 in Treg cells of intra-islet as opposed to other peripheral lymphoid organs, but the frequency of Treg cells expressing this gene in different parts of the body is not different. The differences in the level of expression in diabetic NOD mice are not found in other mice strains, which are not susceptible to diabetes. One of the regulatory mechanisms may be the degree of gene switching, which determines expression levels of FOXP3. Consequently, immune disease may be a result of decreased FoxP-3 expression. It seems that decreased FOXP3 expression causes defects in the function of Treg cells, and their differentiation into effector cells results in an augmented immune response that produces a loss of tolerance and probable development of the autoimmune disease.
10.9 Toll-Like Receptors (TLR) and Regulatory T Cells
Innate and acquired immune response are induced and regulated by the TLR. MyD88, a protein associated with TLR-mediated signal transduction pathway in dendritic cells, is involved in the suppression of Treg cell activity, resulting in an augmented immune response. TLR signaling is required for the maturation of dendritic cells, and mature dendritic cells are potent inhibitors of Treg cell function. However, mature dendritic cells induce Treg cells expansion in association with TLR, IL-1, and IL-6. Small doses of IL-2 are required to maintain the suppressive function of Treg cells, which is inhibited by high doses of IL-2. The mechanism of IL-2-mediated loss of Treg cell activity is not known and is not mediated via FoxP-3 gene. Furthermore, IL-6 and the strength of TCR signal help overcome the suppressive effects of Treg cells on effector cells. The suppression of Treg cells by TLR is attributed to TLR-2, which can recognize bacterial lipoproteins, and the removal of the TLR-2 influence results in the reestablishment of the suppressive abilities of Treg cells. TLR-2 is expressed on CD4+ and CD8+ T cells and can activate TCR-primed T cells as well as memory T cells. Treg cells and effector T cells are distinctly regulated by TLR-2-dependent signal transduction. Although T-cell function is not affected by TLR-2 signaling alone, the proliferation of TCR-primed Treg cells is strongly augmented by the agonists of TLR-2 which makes the Treg cells temporarily inactive. During infection bacterial lipoproteins also increase the proliferation and IL-2 production from the TCR-triggered effector cells. This IL-2 increases the proliferation of both the effector and Treg cells; however, Treg cells are not able to suppress the effector cell function, which is a unique mechanism by which TLR regulates the function of Treg cells. After the bacterial infection is under control and the pathogens have been eliminated, Treg cells regain their suppressive function and IL-6 plays a role in this process. Consequently, this avoids the development of autoimmune disease that may result from the unregulated activity of effector T cells.
10.10 CTLA-4 and Regulatory T Cells
CTLA-4 is constitutively expressed on Treg cells; its deficiency is associated with fatal autoimmune proliferative disease, and inhibition of CTLA-4 by specific antibodies results in the development of autoimmune disease. Its polymorphism has a role in the development of autoimmune diseases including diabetes, Addison’s disease, and thyroid disease. Treg cells are involved in the development of disease resulting from antagonisms of CTLA-4, which may be the result of depletion of Treg cells by antibodies to CTLA-4. The activation of CD4+CD25− T cells as a result of blockade of CTLA-4 receptors may block the suppressive effects of Treg cells on CD4+CD25− T cells.
10.11 T-bet, GATA-3, and Regulatory T Cells
The function of the expression of T-bet and GATA-3 on regulatory T cells is of interest. Expression of T-bet and GATA-3 is regulated by the environment of cytokines. The deletion of genes for either T-bet or GATA-3 does not affect Treg function. However, when both are deleted, there is development of severe autoimmune-like disease in animal models. There is reduced expression of FOXP3 and increased expression of RORϒt, which correlates with the loss of Treg function. In the steady state, Treg cells transiently upregulate one of the two transcription factors for maintaining T-cell homeostasis.
10.12 Regulatory T Cells and Disease States
10.12.1 Allergic Disease
The role of Treg cells in the prevention of allergic disease and asthma is of considerable interest as the prevalence of these diseases continues to rise. On the basis of the ability of Treg cells to prevent sensitization to allergens, they could be potentially used for the treatment of the allergic disease, and the prevention and regulation of TH2-mediated responses may be possible. Mouse CD4+CD25+ T cells, after pre-activation with differentiated TH2 cells, inhibit TH2 cytokine production and suppress TH2 cell differentiation from naïve CD4+ T cells without the requirement of cytokines. CD25+ cells also inhibit IgE production in transgenic mice with monoclonal populations of T and B cells, and Tr1 cells inhibit TH2 sensitization and IgE production provided that their adoptive transfer is before sensitization.
In humans, undesired TH2 responses to environmental allergens are prevented by Treg cells, and the allergic disease may result from inadequate suppression of unwanted TH2 responses both by naturally occurring Treg cells and Tr1 cells. An overall defect in regulatory ability of Treg cells is not present in atopic individuals but a diminished suppressive ability of CD4+CD25+ T cells is observed in atopic individuals when compared with their nonatopic counterparts. Treg cells from both the asthmatic and non-asthmatic individuals have the same ability to suppress anti-CD3−- and anti-CD28−-stimulated cytokine production. Nonetheless, T-cell activation by allergens is suppressed by Treg cells, and atopic individuals may be deficient in these regulatory mechanisms. Nonatopic individuals possess higher numbers of allergen-specific IL-10-producing CD4+ cells as compared to the atopic individuals. In addition, allergen-activated TH2 cells can be inhibited by IL-10-producing T cells, and this can be reversed by either anti-IL-10 or TGF-β.
The mechanisms that alter the intricate balance between regulatory and suppressive responses after allergen exposure have not been elucidated, although a number of possibilities exist that may lead to atopic state. According to the hygiene hypothesis, microorganisms alter antigen-presenting cells, which may result in the production of Treg cells when exposed to allergens. As a consequence, it is feasible that regulation by Treg cells is restricted by LPS-induced activation of Toll-like receptors, specifically TLR4. IL-10-producing Treg cells are also involved in the suppression of allergic responses as a result of prior exposure to allergens and mycobacterial antigens. In young children, exposure to cat antigens protect them from later development of allergies to cats due to a dominant IL-10 response resulting in modified TH2 responses where it seems that both Treg and Tr1 cells are responsible in developing tolerance.
Corticosteroids remain the main hallmark for the treatment of allergic disease/asthma despite their adverse side effects. While their administration via inhalers and nebulizers has alleviated some concerns because of their less detrimental side effects, the development of drugs that will induce Treg cells will provide an attractive alternative. Corticosteroids, in addition to inhibiting TH2 cytokines, stimulate Tr1 cells and increase the effector function of Treg cells by increasing IL-10 production. Inhaled corticosteroids also increase Foxp3 expression in asthmatics. Glucocorticoid-resistant asthma patients have impaired Tr1 cells, and they do not show improvement after treatment with corticosteroids. These observations provide a rationale for the development of a new class of drugs that may selectively increase the effector function of naturally occurring as well as IL-10-induced Treg cells and enhance FoxP-3 expression. The concept is further strengthened by the observation that immunotherapy for allergic disease results in induction of IL-10 production from Treg cells, and these patients have increased numbers of IL-10-producing Treg cells after they have received injections for allergen extracts.
Lastly, under naturally induced circumstances of tolerance such as beekeepers receiving multiple bee stings or children who are no longer allergic to cow’s milk, there is an increase in IL-10-producing Treg cells. The evidence is convincing that Treg and Tr1 cells regulate responses to allergens in nonatopic individuals and their function may be impaired in atopy, specifically after prolonged antigen exposure. This suggests the need for the development of either new corticosteroid-like drugs, which only target these mechanisms or alternate novel immunotherapeutic regimens. In Fig. 10.3 is shown the association of Treg cells with various disease states.

Treg cells and disease states. This figure depicts the association of Treg cells with some disease states. The induction and proliferation of naïve T cells and their differentiation to effector T cells is inhibited by natural Treg cells (FOXP3 +). This includes the production of TH1, TH2, and TH17 cells. These subsets of T cells play a role in normal biological function as well as in a number of disease states (Reproduced with permission. Source: Sakaguchi S, Powerie F (2007) Emerging challenges in regulatory T cell function and biology. Science 317: 627–629. American Association for the Advancement of Science)
10.12.2 Autoimmune Diseases
Human autoimmune diseases are a number of complex genetic disorders strongly associated with the MHC complex on the chromosomes. Defects in the function of CD4+CD25+ Treg cells are associated with various autoimmune diseases including multiple sclerosis, type 1 diabetes, and myasthenia gravis. The patients with multiple sclerosis have a significant decrease in the effector function of Treg cells, despite no differences in their frequency as compared to the normal controls. The patients with rheumatoid arthritis and juvenile idiopathic arthritis also have altered CD4+CD25+ Treg cells. Adult patients with rheumatoid arthritis exhibit high numbers of Treg cells in synovial fluid as opposed to the peripheral blood. The patients with juvenile arthritis disease have similar increases of Treg cells in the synovial fluid. These cells also contain CD27 marker with higher expression of Foxp3. Treg cells function differentially in various autoimmune diseases; for example, in human autoimmune polyglandular syndromes and multiple sclerosis, the function of Treg cells is decreased, whereas Treg cells isolated from patients with autoimmune arthritic diseases exhibit augmented effector function.
10.12.3 Inflammatory Diseases
The expression of Id2 and Id3 in Treg cell is responsible for the inhibition of the development of fatal inflammatory disease. It has been reported that TCR-induced signaling at first inhibited Id3, causing the stimulation of follicular regulatory T-cell-dependent transcription signature. But continued lower levels of Id2 and Id3 infringed with follicular regulatory T-cell development. The maintenance and localization of the Treg cell population was inhibited by the suppression of id2 and id3 expression. This suggests that Id2 and Id3 regulate follicular regulatory T-cell checkpoints and modulate the survival and homing of Treg cells.
The ability of Treg cells to suppress the function of various effector T cells has not been clearly established. A PTEN-mTORC2 axis is responsible for the maintenance of Treg cells and modulates the regulation of effector cells by Treg cells. The phosphatase PTEN is involved in the stability of Treg cells and suppression of the activity of TH1 cells and follicular helper T cells. When PTEN is removed from the Treg cells, there is increased follicular helper T-cell response, resulting in spontaneous inflammatory disease. This is resolved by inhibition (deletion) of IFNϒ, suggesting that the control of TH1 cells and follicular helper T cells is coordinated. PTEN is responsible for maintaining the metabolism and stability of Treg cells. Furthermore, the deficiency of PTEN results in the upregulation of the metabolic checkpoint kinase complex mTORC2 and Akt (serine–threonine kinase), and its inhibition will renew the effector function of PTEN-deficient Treg cells.
10.12.4 Infections
Infections present a challenge to the immune system which requires measured pro-inflammatory anti-infectious agent response without being detrimental to self. This intricate balance is subject to control by Treg cells and a role of Treg cells in chronic viral and bacterial infections has been suggested. An increase in peripheral Treg cells is observed in patients with hepatitis B and C infections. Furthermore, Treg cells prevent antiviral response, since T-cell responses to HCV, HBV, and HIV antigens and cytomegalovirus are induced after removal of Treg cells from peripheral blood of patients with viral infection (Table 10.4). Treg cells have a protective effect in HIV infection, where a decrease in Treg cells results in immune hyperactivity in HIV-affected patients. A strong HIV-specific Treg cell activity is associated with lower levels of virus in plasma and higher CD4+/CD8+ ratios in HIV-infected patients. Consequently, intact Treg cell activity is desirable in patients infected with HIV.
Following HIV infection, Treg cells control the levels of the activation of the immune response to avoid immune system exhaustion as well as tissue damage due to a robust immune response. However, this causes the dysfunction of immune response specifically due to inhibition of the generation of HIV-specific effector cells. Furthermore, the role of Treg cells changes during the different stages of HIV infection, as HIV infection causes alterations in the frequencies of Treg cells in the peripheral blood. As the disease progresses, there is a decrease in the frequency of Treg cells in the peripheral blood despite elevated CD25bright expression on CD4+ T cells, which may be attributed to their expression of CD4 and other chemokine receptors that are targets for HIV.
Chronic infection such as HIV results in immunosuppression as a result of induction of CD4+ Treg cells. Increased risk of Kaposi’s sarcoma, non-Hodgkin’s lymphoma, and liver cancer is associated with long-term infection such as HIV. Immunosuppression in HIV-infected individuals occurs before the development of AIDS, which proceeds before the depletion of CD4+ T cells, and the induction of Treg cells may play a role in this process. The mechanisms are IL-10 independent and include the involvement of TGF-β secreted via signaling through cell–cell interaction involving CTLA-4.
CD8+ T-cell response plays an important role in the viral replication of hepatitis B virus (HBV) resulting in liver damage. These CD8+ T cells are virus specific and a defect is found in these cells in the patients with chronic HBV infection as opposed to the recovered individuals. This may be due to a high antigen dose deletion and a lack of help from CD4+ T cells or may be a result of Treg cells. For example, in herpes simplex virus-infected mice, the clonal expansion and effector function of virus-specific CD8+ T cells is augmented by Treg cells, suggesting that Treg cells can be modulated in the periphery after viral infection. Another example is the regulation of hepatitis C virus (HCV)-specific T cells by Treg cells in patients with HCV infection. The circulating CD4+CD25+ Treg cells in patients with HBV infection suppress activation of HBV-specific CD8+ T cells. This may also serve as a feedback to avoid excessive pathogenic responses in chronic HBV infection and also helps to avoid complete clearance of the viral antigen in patients who have resolved HBV infection.
10.12.5 Cancer
Immune dysfunction and poor tumor-specific immune responses are observed in cancer patients with enhanced Treg cell activity. Furthermore, many different types of tumors possess high frequency of Treg cells that inhibit a variety of immune functions including T-cell proliferation, cytokine production, and cytotoxic activity. Regulatory T cells play an important role in the escape of tumor cells. The expression of CD69 is induced on T lymphocytes and natural killer cells. CD69 is rapidly and transiently expressed on activated but not on resting lymphocytes and functions as signal-transmitting receptors in lymphocytes. It may produce an activating function to cause pro-inflammatory responses but also produces regulatory function. The number of CD69+CD4+ CD25− T cells increases dramatically during the progression of the tumor. As opposed to other regulatory T-cell subsets, they express high numbers of CD122 but do not express FOXP3, and they secrete IL-2, IL-10, IFN-ϒ, IL-10, and TGF-β1. CD69+CD4+ CD25− T cells inhibit proliferation of T cells via cell–cell interaction. The antibodies directed against TGF-β1 neutralize their suppressor function. These cells express high levels of membrane-bound TGF-β1, which plays a role in the inhibition of T-cell proliferation. In addition, ERK activation is involved in the maintenance of high expression of CD69+CD4+ CD25− T cells via engagement of CD69. The subset is expanded in tumor-bearing hosts and may play a role in an inadequate response to tumors.
The role of CD69+CD4+ CD25− has been evaluated in leukemia relapse after allogeneic hematopoietic transplantation. The number of CD69+CD4+ CD25− T cells dramatically increases in patients in the relapsed group and the patients with positive minimal residual disease (MRD+) as compared to healthy controls. The therapeutic intervention results in the decrease of this subset. It seems that there is a correlation between CD69+CD4+ CD25− T cells and leukemia relapse after allogeneic hematopoietic transplantation. This suggests the need for adoptive T-cell immunotherapy.
Treg cells are present in high numbers in tumor tissues of lung, breast, pancreatic, gastrointestinal, and liver cancers and malignant melanoma. A poor prognosis for the breast, gastric, and ovarian cancers has been reported, if in the tumor-infiltrating lymphocytes there is a smaller ratio of CD8+T cells to FOXP3+ Treg cells and increased numbers of CD4+ Treg cells. Based on these observations, FOXP3+ Treg cells inhibit cytolytic T cells (CD8+) that kill tumors. There is an improved prognosis for Hodgkin’s lymphoma and head or neck and colon cancer, if there is an enhanced infiltration of FOXP3+ Treg cells. These differing observations may be attributed to different makeups of FOXP3+ Treg cell subsets in various tumors. This concept is supported by the reports that eTreg cells are present in larger numbers as opposed to naïve Treg cells and FOXP3+ non-Treg cells in melanoma tumor-infiltrating lymphocytes, whereas in colon cancers there are high numbers of non-reg T cells and eTreg cells in infiltrating FOXP3+T cells. A role of pro-inflammatory cytokines has been suggested for a better outcome in colon cancer, where there is an increased infiltration of FOXP3+ non-Treg cells, despite possessing increased numbers of FOXP3+ tumor-infiltrating lymphocytes. In cancer patients a variety of tumor molecules, such as Survivin and NY-ESO-1, are recognized by Treg cells, resulting in the inhibition of tumor-specific effector T-cell response.
FOXP3+CD4+ Treg cells express chemokine receptors. CCR4 attracts chemokine ligand 22 (CCL22) secreted by the tumor cells and/or tumor-infiltrating macrophages. Treg infiltration is also mediated by other combinations of chemokine ligands and chemokine receptors, including CCR10-CCL28 and CXCR and CXCL9, CXCL10, and CXCL11. It is not known whether regular T cells can become suppressive FOXP3+ Treg cells in the microenvironment of human tumors. After reaching the tumor, FOXP3+CD4+ Treg cells go through induction and proliferation following recognizing tumor antigens or self-antigens, found due to tumor lysis. The natural FOXP3+CD4+ Treg cells, perhaps, are better in detecting tumor-associated self-antigens than tumor-reactive effector or memory CD4+ T cells. This is attributed to the fact that TCR repertoires are more self-reactive as compared to regular T cells. Furthermore FOXP3+CD4+ Treg cells exhibit enhanced expression of T-cell accessory molecules such as adhesion molecules (LFA-1). It has been suggested that cancer vaccines may activate tumor-specific Treg cells because of immunosuppressive tumor microenvironment and not tumor-specific effector T cells. Data obtained from clinical studies in melanoma and ovarian cancer patients have suggested that both healthy individuals, patients without cancer, and cancer patients possess T cells, which are potentially tumor reactive, but their induction and proliferation are inhibited by natural Treg cells. In these trials depletion of Treg cells resulted in the expansion of a tumor antigen-specific effector T cells from naïve T-cell precursors. These effector T cells harbored potent antitumor function.
CD-25, IL-2 receptor α chain and its high-affinity receptors, chemokine receptors, and other molecules predominantly expressed on Treg cells could be targeted for their depletion to treat cancer. The effector helper and cytolytic T cells express CD25, IL-2 secretion is required for their propagation, and inhibition of CD25 will result in the suppression of both the effector and Treg cells. Blocking CCR4 has been more promising. CCR4+ eTreg cells are in high numbers in the tumor-infiltrating FOXP3+ T cells in melanoma than those in the peripheral blood. As a result clinical trials are underway to assess the ability of anti-CCR4 monoclonal antibody to treat cancer because it induces and enhances antitumor responses. Another molecule of interest is glucocorticoid-induced TNF-receptor family-related protein (GITR), which is a costimulatory molecule with minimal expression on resting helper and cytolytic T cells. GITR is constitutively expressed with high density on FOXP3+CD4+ Treg cells. The use of agonistic anti-GITR monoclonal antibody or GITR ligand can antagonize the inhibitory function of FOXP3+CD4+ Treg cells. This also results in a loss of susceptibility of effector T cells to Treg cells. OX 40, another molecule, is transiently expressed on activated T cells but constitutively expressed on FOXP3+CD4+ Treg cells. The agonistic anti-OX40 monoclonal antibodies have shown antitumor effects by inhibiting FOXP3+CD4+ Treg cells and inducing effector T-cell function.
The induction of Fcϒ receptors (FcϒRs) enhances the effector function of lymphocytes and phagocytes that include antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis, which in concert cause the removal of tumor cells. The mechanism behind these events involves the antibodies directed against OX40 (CD134). This molecule, OX40 (CD134), is a crucial costimulatory TNF receptor, which is transiently present on activated helper T cells (CD4+). The antibodies to OX40 (CD134) remove intratumoral Treg cells in an activating FcϒR-dependent manner. These observations in a murine model are similar to other observations where Treg cells suppress TH1 effector responses in tumors. The success of checkpoint blockade inhibitors can be assessed by the presentation of TH1 immune polarity with a reduction in the ratio of intratumoral Tregs to effector T cells, mostly cytolytic T cells. It is intriguing that the monoclonal antibodies envisioned to act through inhibiting checkpoints or activating costimulatory molecules may simply be producing their pharmacologic responses through antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis of Tregs, in the tumor microenvironment. Antibodies directed against OX40 and other costimulatory molecules on T lymphocytes inhibit the immunosuppressive effects of Tregs and as a consequence augment the effector function of T cells against tumors. For these effects there seems to be a requirement of FcR-activating receptors and depletion of intratumoral Treg cells. However, immune response is not produced against many types of cancers and the role of Treg cells in these forms of cancers has not been established.
Tr1 cells also participate in a poor anticancer response. The infiltrating lymphocytes in Hodgkin’s lymphoma contain both Treg and Tr1 cells, which suppress a variety of immune functions.
10.12.6 Transplantation
Treg cells play an important role in suppressing alloantigen-specific immune response following an organ graft. The maintenance of tolerance by various protocols in allograft transplantation is mediated via the induction of Treg cells in otherwise alloresponsive T cells. The responding T cells have a suppressive effect if naïve T cells are repetitively stimulated with immature allogeneic dendritic cells. The maximum T-cell response after allograft is dependent on the maturation stage of dendritic cells in the grafted tissue. Immature dendritic cells that result in suboptimal T-cell responses and limited costimulatory signals and cytokine production are ideal for the induction of Treg cells in responding T cells. In addition to the developmental stage, the particular subset of dendritic cells is also important in inducing Treg cells. Treg cells that are naturally occurring play an important role in the generation of induced Treg cells, resulting in a Tr1 suppressor phenotype from the graft-killing effector T cells. Furthermore, TGF-β can convert non-regulatory T cells to CD4+CD25+ suppressor cells. The tolerant grafts contain TGF-β and induce Treg cells in the graft, and there is also a presence of CD8+ Treg cells in allografts. Patients undergoing allogeneic bone marrow transplantation who do not exhibit graft-versus-host disease have T cells that produce IL-10 and IFN-γ but low levels of IL-2.
10.13 Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a rapidly progressing neuro-inflammatory disorder that destroys motor neurons resulting in death. Most of the patients diagnosed with the disease survive only 2–3 years while 10 % of the patients live about 5–10 years. The disease exhibits infiltrating lymphocytes which seem to be related to the enhanced chemokine CCL2 ligand levels and the morphological activation of microglia. A role of Treg cells has emerged as protectors of motor neuron as a result of their anti-inflammatory effects. In rapidly progressing patients, the numbers of Treg cells and the expression of FoxP3 are reduced. Furthermore, there is inverse correlation between the disease progression and the number and expression of Treg cells and FoxP3, respectively. There is reduction in the mRNA levels of FoxP3, IL-4, TGF-β, and GATA3 in rapidly progressing patients with an inverse correlation. The patients who live longer have a higher number of regulatory T cells. It has been suggested that administration of low levels of IL-2 results in the production of Treg cells and a phase I/II randomized placebo-controlled clinical trial is planned to look into this further. In animal models of ALS, there is a change from the protective effects of Treg cells to injury-causing responses of effector T cells. The Treg cells switch the microglial responses from cytolytic effects to neuroprotection.
10.14 Future Direction
Recent understanding of Treg cells provides a number of unique opportunities for their use in therapeutic intervention. The expectation is that the therapeutic application of Treg cells will result in reestablishing tolerance, the breakdown of which has resulted in the development of the disease. The treatment of individuals with antigen-specific Treg cells will be an interesting approach. Treg cells will have several advantages including a long half-life, ability to condition other cell types, induction of non-regulatory cells to secrete IL-10, and suppressive effects on the costimulatory activity of antigen-presenting cells. However, so far the therapeutic use of Treg cells has been limited due to their low frequency in the circulating blood, and this will require industrial cultures for their propagation for clinical therapy. It has not yet been possible to use industrially produced cells therapeutically, since their culture conditions stimulate TH2 responses.
Development of culture techniques that have allowed the ability of continuous culture of CD4+CD25+ cells and the generation of these Treg cells from CD25− cells after FOXP3 gene is transduced by using a retroviral vector have permitted the production of these cells. A number of protocols have been published utilizing different stimuli for the culture of Treg cells on a large scale and their efficacy in in vivo models has been established. The use of CD86hi dendritic cells as antigen-presenting cells as compared with whole splenocytes is another approach in expanding ovalbumin-specific Treg cells. Furthermore, mature dendritic cells are a better stimulator than immature dendritic cells, and IL-2 or IL-15 secretion from dendritic cells is not required for their successful function. Since these cells remain dormant, unless stimulated, the choice of an expansion agent has been problematic. In various combinations four expansion agents, IL-2, anti-CD3, anti-CD3 plus anti-CD28, and rapamycin, are generally used. These expansion agents cannot be used therapeutically because of their toxicity. But they produce mixed population of CD4+ T cells, which cannot be used therapeutically due to the risk of the release of pro-inflammatory cytokines. Another approach has been to use TNFR2 agonist to produce phenotypically homogeneous population of Treg cells. Nonetheless, obtaining a homogeneous population of Treg cells in large enough numbers to be used as a therapeutic agent for treating a disease remains a challenge.
Bibliography
Allez M, Mayer L (2006) Regulatory T cells, peace keepers in the gut. Inflamm Bowel Dis 10:666–676
Baecher-Allan C, Hafler DA (2004) Suppressor T cells in human disease. J Exp Med 200:273–276
Baecher-Allan C, Hafler DA (2006) Human regulatory T cells and their role in autoimmune disease. Immunol Rev 212:203–216
Barnett Z, Larussa SH, Hogam M, Wei S et al (2004) Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res 64:8451–8455
Battaglia M, Gregori S, Bacchetta R, Roncarolo MG (2006) Tr1 cells: from discovery to their clinical application. Semin Immunol 18:120–127
Beers DR, Henkel JS, Zhao W, Wang J (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134:1293–1314
Beissert S, Schwarz A, Schwarz T (2006) Regulatory T cells. J Invest Dermatol 126:15–24
Belkaid Y, Rouse BJ (2005) Natural regulatory T cells in infectious disease. Nat Immunol 6:353–360
Beyer M, Schultze JL (2007) CD4+CD25+high Foxp3+ regulatory T cells in peripheral blood are primarily of effector memory phenotype. J Clin Oncol 25:2628–2630
Bluestone JA (2004) Therapeutic vaccination using CD4+CD25+ antigen-specific regulatory T cells. Proc Natl Acad Sci U S A 101:14622–14626
Bluestone JA, Abbas AK (2003) Natural versus adaptive regulatory T cells. Nat Rev Immunol 3:253–257
Brimnes J, Allez M, Dotan I, Mayer L (2005) Defects in CD8+ regulatory T cells in the lamina propria of patients with inflammatory bowel disease. J Immunol 174:5814–5822
Buckner JH, Ziegler SF (2004) Regulating the immune system: the induction f regulatory T cells in periphery. Arthritis Res Ther 6:215–222
Cao D, Vollenhoven RV, Klareskog L, Trollmo C, Malstrom V (2004) CD25+CD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther 6:R335–R346
Chatenoud L, Salomon B, Bluestone JA (2001) Suppressor T cells – they’re back and critical for regulation of autoimmunity! Immunol Rev 182:149–163
Chen Z, Benoist C, Mathis D (2005) How defects in central tolerance impinge on a deficiency in regulatory T cells. Proc Natl Acad Sci U S A 102:14735–14740
Cohen IR, Quintana FJ, Mimran A (2004) Tregs in T cell vaccination: exploring the regulation of regulation. J Clin Invest 114:1227–1232
Dejaco C, Duftner C, Grubeck-Loebenstein B, Schrimer M (2006) Imbalance of regulatory T cells in human autoimmune diseases. Immunology 117:289–300
Dieckmann D, Bruett CH, Ploettner H, Lutz MB, Schuler G (2002) Human CD4+CD25+ regulatory, contact-dependent T cells induce interleukin 10 producing, contact-independent type 1-regulatory T cells. J Exp Med 196:247–253
Fang YU, Sharma S, Edwards J, Feigenbaum L et al (2015) Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat Immunol 16:197–206
Fehervari Z, Sakaguchi S (2004) CD4+ Tregs and immune control. J Clin Invest 114:1209–1217
Ferretti G, Felici A, Pino MS, Cognetti F (2006) Does CTLA4 influence the suppressive effects of CD25+CD4+ regulatory T cells? J Clin Oncol 24:5469–5470
Fields RC, Bharat A, Mohanakumar T (2006) The role of regulatory T-cells in transplantation tolerance. ASHI Quarterly (first quarter):10–15
Franzece O, Kennedy PTF, Gehring AJ, Gotto GM et al (2005) Modulation of the CD8+ T cell response by CD4+CD25+ regulatory T cells in patients with Hepatitis B virus infection. J Virol 79:3322–3328
Gershon RK (1975) A disquisition on suppressor T cells. Transplant Rev 26:170–185
Gershon RK, Kondo K (1970) Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology 18:723–737
Godfrey DI, Kronenberg M (2004) Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest 114:1379–1388
Gould HJ, Sutton BJ et al (2003) The biology of IgE and the basis of the allergic disease. Ann Rev Immunol 21:579–628
Green DR, Flood PM, Gershon RK (1983) Immuno-regulatory T cell pathways. Ann Rev Immunol 1:439–463
Groux H (2003) Type 1 T-regulatory cells: their role in the control of immune response. Transplantation 75:85–125
Han Y, Guo Q, Zhang M, Chen Z et al (2009) CD69+ CD4+ CD25− T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane bound TGF-β1. J Immunol 182:111–120
Hawrylowicz CM, O’Garra A (2005) Potential role of interleukin-10 secreting regulatory T cells in allergy and asthma. Nat Rev Immunol 5:271–283
Henkel JS, Beers DR, Wen S, Rivera AL et al (2012) Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 2:64–79
Hong J, Zang YC, Nie H, Zhong J (2006) CD4+ regulatory T cell responses induced by T cell vaccination in patients with multiple sclerosis. Proc Natl Acad Sci U S A 103:5024–5029
Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057–1061
Ito T, Wang Y-H, Duramad O, Hanabuchi S et al (2006) OX40 ligand shuts down IL-10 producing regulatory T cells. Proc Natl Acad Sci U S A 103:13138–13143
Iwashiro M, Messer RJ, Peterson KE, Stronnes IM et al (2001) Immunosuppression of CD4+ regulatory T cells induced by chronic retroviral infection. Proc Natl Acad Sci U S A 98:9226–9230
Janeway C Jr (1988) Do suppressor T cells exist? A reply. SC J Immunol 27:621–623
Jerne NK (1974) Towards a network theory of the immune system. Ann Immunol 125:373–389
Joosten SA, Meigjaarden KE, Savage N, Boer TD et al (2007) Identification of a humanCD8+ regulatory T cells subset that mediates suppression through the chemokine CC chemokine ligand 4. Proc Natl Acad Sci U S A 104:8029–8034
Karlsson MR, Rugtveit J, Brandtzaeg P (2004) Allergen-responsive CD4+CD25+ regulatory T cells in children who have outgrown cow’s milk allergy. J Exp Med 12:1679–1688
Kim JM, Rudensky A (2006) The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol Rev 212:86–98
Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK (2005) Sequential development of interleukin 2-depdent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med 202:1375–1386
Kretschmer K, Apostolou I, Jaechel E, Khazaie K et al (2006) Making regulatory T cells with defined antigen specificity: role in autoimmunity and cancer. Immunol Rev 212:163–169
Latour RP, Dujardin HC, Mishellany F, Defranoux OB et al (2006) Ontogeny, function, and peripheral homeostasis of regulatory T cells in the absence of interleukin-7. Blood 108:2300–2306
Liu H, Komai-Koma M, Xu D, Liew FY (2005) Toll-like receptor2 signaling modulates the function of CD4+CD25+ regulatory T cells. Proc Natl Acad Sci U S A 103:7048–7053
Lohr J, Knowchel B, Abbas AK (2006) Regulatory T cells in periphery. Immunol Rev 212:149–162
Lu L, Wermeck MBF, Cantor H (2006) The immunoregulatory effects of Qa-1. Immunol Rev 212:51–59
Lu SY, Hunag XJ, Liu KY, Xu LP (2012) High frequency of CD4+ CD25–CD69+ T cells is correlated with a low risk of acute graft-versus-host-disease in allotransplants. Clin Transpl 26:E158–E167
Masaki M, Miyazaki K, Chen S, Itoi M et al (2014) Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol 15:767–776
Masteller EL, Tang Q, Bluestone JA (2006) Antigen-specific regulatory T cells-ex vivo expansion and therapeutic potential. Semin Immunol 18:103–110
Nishikawa H, Sakaguchi S (2014) Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 27:1–7
Nishizuka Y, Sakakura T (1969) Thymus and reproduction: sex linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166:753–755
O’Garra A, Vieira P (2004) Regulatory T cells and mechanisms of immune system control. Nat Med 10:801–805
O’Garra A, Vieira P, Goldfeld A (2004) IL-10 producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest 114:1372–1378
Okubo Y, Mera T, Wang L, Faustman D (2013) Homogenous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep 3:3153. doi:10.1038/srep03153
Picca CC, Larkin J III, Boesteanu A, Lerman MA, Rankin AL, Caton AJ (2006) Role of TCR specificity in CD4+CD25+ regulatory T cell selection. Immunol Rev 212:74–85
Qizhi T, Bluestone JA (2006) Regulatory T cell physiology and application to treat autoimmunity. Immunol Rev 212:217–237
Reibke R, Garbi N, Hammerling GH et al (2006) CD8+ regulatory T cells generated by neonatal recognition of peripheral self antigen. Proc Natl Acad Sci U S A 103:15142–15147
Roncarlo MG, Gregori S, Battaglia M, Bacchetta R et al (2006) Interleukin-10 secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 212:28–50
Rouse BT, Sarangi PP, Suvas S (2006) Regulatory T cells in virus infections. Immunol Rev 212:272–286
Sakaguchi S (2004) Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune response. Ann Rev Immunol 22:531–562
Sakaguchi S (2005) Naturally arising Foxp3-expressing Cd25+CD4+ regulatory T cells in immunological tolerance to self and nonself. Nat Immunol 6:345–352
Sakaguchi S (2006) Regulatory T cells: Meden Agan. Immunol Rev 212:1–5
Sakaguchi S, Ono M, Setoguchi R, Yagi H et al (2006) Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev 212:8–27
Sansom DM, Walker LS (2006) The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T cell biology. Immunol Rev 212:131–148
Schultze JL (2006) Regulatory T cells: timing is everything. Blood 107:857
Shevach EM (2000) Regulatory T cells in autoimmunity. Ann Rev Immunol 18:423–449
Shevach EM (2002) CD4+CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol 2:389–400
Shevach EM, DiPaolo RA, Anderson J, Zhao DM (2006) The lifestyle of naturally occurring CD4+CD25+Foxp3+ regulatory T cells. Immunol Rev 212:60–73
Shevack EM (2004) Fatal attraction: tumors beckon regulatory T cells. Nat Med 10:900–901
Shrestha S, Yang K, Guy C, Vogel P et al (2015) Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol 16:178–187
Smyth MJ, Ngiow SF, Teng MWL (2014) Using FcR to suppress cancer: targeting regulatory T cells in tumor immunotherapy. Immunol Cell Biol 92:473–474
Snoeck V, Peters I, Cox E (2006) The IgA system. Vet Res 37:455–467
Sutmuller RPM, den Brok HMGM, Kramer M, Bennink EJ et al (2006) Toll-like receptors control expansion and function of regulatory T cells. J Clin Invest 116:485–494
Taams LS, Palmer DB, Akbar AN, Robinson DS et al (2006) Regulatory T cells in human disease and their potential for therapeutic manipulation. Immunology 118:1–9
Tarbell KV, Yamazaki S, Steinman RM (2006) The interaction of dendritic cells with antigen specific, regulatory T cells that suppress autoimmunity. Semin Immunol 18:93–102
Thomas D, Zaccone P, Cooke A (2005) The role of regulatory T cell defects in Type I diabetes and the potential of these cells for therapy. Rev Diabet Stud 2:9–18
Ulges A, Klein M, Reuter S, Gerlitzki B (2015) Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat Immunol 16:267–275
Umetsu DT, DeKruyff RH (2006) The regulation of allergy and asthma. Immunol Rev 212:238–255
Vigouroux S, Yvon E, Biagi E, Brenner MK (2004) Antigen-induced regulatory T cells. Blood 104:26–33
Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK (2003) Antigen-dependent proliferation of CD4+CD25+ regulatory T cells in vivo. J Exp Med 198:249–258
Walsh PT, Taylor DK, Turka LA (2004) Tregs and transplantation tolerance. J Clin Invest 114:1398–1403
Walsh PT, Buckler JL, Zhang J, Gelman AE et al (2006) PTEN inhibits IL-2 receptor mediated expansion of CD4+CD25+ Tregs. J Clin Invest 116:2521–2531
Wan YY, Flavell RA (2006) The roles for cytokines in the generation and maintenance of regulatory T cells. Immunol Rev 212:114–130
Wau Y, Flavell RA (2007) Regulatory T cell functions are subverted and converted owing to attenuated FoxP-3 expression. Nature 445:766–770
You S, Thieblemont N, Alayankian MA, Bach J, Chatenoud L (2006) Transforming growth factor-β and T-cell-mediated immunoregulation in the control of autoimmune diabetes. Immunol Rev 212:185–202
Zhao X-S, Wang X-H, Zhao X-Y, Chang Y-J et al (2014) Non –traditional CD4+ CD25–CD69+ regulatory T cells are correlated to leukemia relapse after allogeneic hematopoietic stem cell transplantation. J Transl Med 12:187
Zorn E, Nelson EA, Mohseni M, Porchery F et al (2006) IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expression of these cells in vivo. Blood 108:1571–1579
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Khan, M.M. (2016). Regulatory T Cells and Disease State. In: Immunopharmacology. Springer, Cham. https://doi.org/10.1007/978-3-319-30273-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-30273-7_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30272-0
Online ISBN: 978-3-319-30273-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)