
Abstract
Autophagy is a normal physiological process that plays a pivotal role for cell survival, differentiation, development, and homeostasis. Selective or not, canonical or non-canonical, autophagy processes are considerably more complex than originally thought. Depending on favourable or unfavourable cell environment conditions, the autophagy machinery will promote both cell survival and cell death, thus maintaining a decisive balance between manufacture of cellular components and breakdown of damaged or superfluous organelles and other cellular constituents, for example. Autophagy displays complex, still-debated, interwoven links with several other degradative pathways, such as apoptosis and proteasome-mediated systems. Among its many cellular regulatory functions that have been experimentally proven or that are anticipated, autophagy decisively controls immunity and inflammation, and any impaired autophagy signaling can potentially lead to autoimmune-related diseases. Here we review recent progresses that have been made in deciphering existing links between autophagy and autoimmunity. We further discuss how targeting certain hot spots of autophagy processes with appropriate tools might influence the course of autoimmune diseases by controlling both innate and adaptive immune responses, which are improperly oriented in these settings.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Systemic Lupus Erythematosus
- Autoimmune Disease
- Systemic Lupus Erythematosus Patient
- Primary Biliary Cirrhosis
- Synovial Fibroblast
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Autoimmune diseases are not considered as orphan diseases. In general they are even not regarded as rare since as a whole they affect millions people worldwide. As a result of genetic influence, which is mostly polygenic, or environmental and metabolic factors, there is some disequilibrium regarding their incidence or severity in some parts of the world or in a particular group of people. It remains that in the collective perception, they are viewed as a common group of diseases. It is true that it has been estimated that autoimmune diseases are among the top ten leading causes of death among women in all age groups up to 65 years. In fact, under the term autoimmune diseases, there are more than eighty illnesses caused by autoimmunity. Some of them are rare, either as an entity (e.g. Crohn’s disease/CD; primary biliary cirrhosis, myasthenia gravis, immune thrombocytopenic purpura) or by the form they display in affected patients (neuropsychiatric systemic lupus erythematosus, ocular myasthenia gravis, psoriatic arthritis). Also some individuals may have more than one autoimmune disorder at the same time, which complicates the task of follow-up and treatment, and makes each case unique. There is no known prevention for most autoimmune disorders, and in general there is no specific treatment. Despite the complexity and uniqueness of cellular and molecular pathways that are altered in different autoimmune conditions, investigating these mechanisms is very rewarding for scientists. Such studies can effectively reveal new elements and interacting partners of the immune system as well as unexpected abnormalities linked to autoimmune features. These findings can then inspire researchers to design novel strategies of possible intervention developed to mislead and correct the defective immune system.
2 Autoimmune Diseases
Autoimmunity does not systematically leads to autoimmune diseases. In autoimmunity, the patient’s immune system is activated against the body’s own components and only in certain conditions involving genetic, environmental, and hormonal elements, will the individual develop illness, which is often chronic, debilitating, and life-threatening. A large number of autoimmune diseases are recognized. They are said “organ-specific” when they are restricted to certain organs such as thyroid (e.g. Graves’ disease, autoimmune thyroiditis, Hashimoto’s disease), pancreas (e.g. type 1 diabetes in which insulin-producing beta cells are destroyed) and muscles (myasthenia gravis) or involve a particular tissue in different places (e.g. Goodpasture’s disease, which affects the basement membrane in the lung and kidney). In contrast, they are classified as “systemic” when they implicate a variety of organs and tissues in the whole body. The most emblematic representative of the large family of systemic autoimmune diseases is systemic lupus erythematosus (SLE) in which heart, joints, skin, lungs, blood vessels, liver, kidneys, and nervous system can be affected. In fact, between these two commonly described families, there is no sharp delineation. Thus scleroderma, also known as systemic sclerosis, which is a chronic systemic autoimmune disease characterized by hardening of the skin, also affects blood vessels, muscles, and internal organs in severe forms. A continuing debate and a matter of controversy remain about when a disease should be considered autoimmune. Within the usually reported list of somewhat 80 autoimmune diseases that are currently described [36], very few in fact do respond to the strict Witebsky’s postulates formulated in 1957 and modified 35 years later [126]. The passive transfer of T lymphocytes, which should lead to disease development in the recipient, is generally hardly observed.
According to the American Autoimmune Related Diseases Association, autoimmune diseases affect up to 50 million Americans. The overall cumulative prevalence of all autoimmune diseases is around 5 %, with about 3 % for males and 7 % for females [36]. There is a sexual dimorphism among autoimmune diseases with a well-established disequilibrium toward the female population. This female bias occurs in 59 % of autoimmune diseases, probably in relation with hormonal influence and X-chromosome encoded genes. In general the onset for autoimmune diseases occurs in young people (20–29 year age-group).
Deciphering the molecular and cellular mechanisms leading to immune tolerance breaking and evolution toward autoimmune disease remains a vast area of investigations in the scientific and clinical community. Nowadays, no universal signature could be identified, and clues are largely lacking regarding the reasons of their tropism as well as on the elements triggering their initiation and maintenance. Relatively few is also known regarding the events governing the successive periods of flares and remission occurring in certain autoimmune diseases such as SLE. The multifactorial and polymorphic nature of most autoimmune diseases dramatically complicates their diagnosis and the treatment that can be applied to mitigate the symptoms. Except in very rare cases, the treatments are largely palliative and do not target the cause of illness. Although immense progresses have been made over the last decades leading to patients’ survival rates that have considerably augmented, innovative therapeutic solutions are still awaiting that would combine efficacy, selectivity -and thus less secondary effects- and reliability. Without adapted treatment, the quality-of-life can be relatively poor in autoimmune patients and decreases as the disease evolves (fatigue, pain, fever associated to specific symptoms). Unfortunately, the medications required to minimize symptoms and slow-down inflammatory syndrome (i.e. corticosteroids, immunosuppressive drugs and tumor necrosis factor (TNF)-α blockers used for long-term periods) induce an alteration of the whole immune system leading to intestinal bleeding, kidney failure, increased blood pressure, insomnia, depression, psychosis, osteoporosis, muscle loss, and diabetes, not to mention overwhelming repetitive infection episodes and cancer development. In certain autoimmune diseases such as those affecting the central nervous system, or in anti-phospholipid syndrome that can be associated to SLE, the therapeutic solutions are limited, not specific, and unfortunately sometimes inefficient [9, 35, 40, 44]. Intense research is currently ongoing to develop novel immunomodulatory strategies based on molecular targets that are engaged in deregulated autoimmune processes and can be specifically re-orientated. In this context, a better knowledge of cellular and molecular mechanisms that underline autoimmune responses and most particularly the homeostasis and regulation of autoimmune cells is central. Although the picture is immensely complex, studying the autophagic process, which is involved in the establishment and maintenance of immune tolerance and the proper effectiveness of the immune system, has particular importance in autoimmunity and might reveal decisive hot spots for therapeutic intervention.
3 Autophagy and Its Implication in Autoimmune Diseases
Autophagy is a lysosome-based physiological process, which in basal conditions occurs at low levels to continuously degrade unwanted cytoplasmic constituents and generate substrates for energy production. During oxidative stress, hypoxia or nutritional starvation, its level raises to allow cell survival. Autophagy represents therefore a major hub involved in cellular homeostasis [4, 37, 89, 111, 124]. It also plays a pivotal role in differentiation of many lineages, including adipocytes, erythrocytes and lymphocytes, and tissue remodelling [10, 62, 90, 91, 106, 117]. Under specific environmental conditions, however, autophagy can also mediate cell death and it is mechanistically important to distinguish autophagic cell death, which refers to cell death “by” autophagy from cell death “with” autophagy [58, 83, 128, 134]. Thus, recent studies suggest that autophagy and apoptosis processes are closely nested and share cross-talk between signal transduction elements. It has been shown in particular that certain autophagy-related (ATG) proteins play dual roles in autophagy and apoptosis regulation. This is the case of ATG5 and its binding partner ATG12, BCL-2 interacting myosin/moesin-like coiled-coil protein 1 (BECLIN1/beclin-1), the mammalian ortholog of yeast Atg6/vacuolar protein sorting (Vps)-30 that acts during the formation of autophagosomes by interacting with the class III PI3K pathway, and microtubule-associated-protein light chain 3 (MAP1LC3/LC3) a mammalian ortholog of yeast Atg8, for example [48, 56, 71, 84]. Other forms of cell death are also interconnected with autophagy, such as necrosis, necroptosis (regulated Fas-dependent, caspase-independent non-apoptotic cell death), and pyroptosis (caspase-1-dependent cell death) [128].
Three main types of autophagy have been identified and can be distinguished by both their physiological functions and the mechanisms they use to deliver cytoplasmic cargo to lysosomes (Fig. 1a). They are macroautophagy, microautophagy and chaperone-mediated autophagy or CMA [17, 27, 50, 111]. In fact, many more forms of autophagy have been described. Mention can be made, for example, of aggrephagy (for aggregated proteins), mitophagy (for mitochondria), ribophagy (for ribosomes), pexophagy (for peroxisomes), reticulophagy (for the endoplasmic reticulum, ER), and xenophagy (for pathogens). Thus, we now realize that while originally viewed as a nonselective (random) cytoplasmic degradation system, autophagy actually participates in a highly selective and tightly regulated process of substrate delivery.


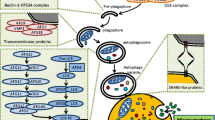
Schematic depiction of autophagic pathways. (a) The three main autophagy axes, macroautophagy, microautophagy and CMA. The process of macroautophagy is initiated with the formation of the so-called isolation membrane. This structure is elongated to engulf cytosolic materials, forming a characteristic double-membrane structure termed autophagosome. The latter next fuses with a lysosome to become an autolysosome, after which the engulfed material is degraded. The molecular pathways regulating autophagy are highly conserved from yeast to higher eukaryotic cells. In CMA, proteins carrying the pentapeptide KFERQ-like signal sequence are recognized by the HSPA8 chaperone, which then associates to LAMP-2A, triggering its oligomerization. This event permits to the targeted protein to be translocated into the lysosome lumen through a process that requires HSPA8. Microautophagy involves the direct sequestration of cellular components by the lysosome through invagination of the lysosomal membranes; (b) Main steps of the macroautophagic process (c) Autophagy as the major sources of peptides for presentation by MHCII molecules to T cells. Abbreviations: CMA chaperone-mediated autophagy, ER endoplasmic reticulum, HLA human leukocytes antigen, HSPA8/HSC70 heat shock cognate protein of 70 KDa, LAMP-2A lysosome-associated membrane protein-2A, MIIC major histocompatibility complex class II compartment, MHCII major histocompatibility complex class II, TCR T cell receptor
Macroautophagy (commonly referred as “autophagy”, which can in some cases create confusion in the literature) remains the major autophagic process through its ability to massively entrap macromolecules and entire organelles. The latter are captured into double-membrane autophagosomes where they are degraded. It therefore represents an alternative mechanism of proteasomal degradation, which rather treats short-lived intracellular proteins, although a cross-talk that is being increasingly understood, has been described to occur between the ubiquitin-proteasome system (UPS) and macroautophagy [19, 52, 57, 69, 124]. The fusion of autophagosomes with lysosomes leads to the formation of autolysosomes in which engulfed cellular constituents -including lipid droplets and protein aggregates- are degraded by lysosomal glycosidases, proteases, lipases and sulfatases (Fig. 1b). Concerning the CMA process, proteins containing a specific peptide motif biochemically related to KFERQ are recognized by the HSPA8/HSC70 chaperone protein prior being internalized and degraded in lysosomes (Fig. 1a). By contrast, in microautophagy, cytosolic components are directly taken up by invaginations of the lysosomal membrane (Fig. 1a).
Autophagic pathways are genetically regulated by proteins belonging to the ATG gene family and are well characterized in yeast and mammals [14, 55, 61, 94, 110, 135]. ATG proteins are evolutionary conserved and each of them has a specific function during autophagy. It is mainly through the discovery that certain ATG genes could be associated to autoimmune syndromes that further studies have been generated to understand the links existing between autophagy and autoimmunity. Genetic analyses effectively reported that some polymorphisms in ATG genes might confer susceptibility to different autoimmune disorders. Thus genome-wide association studies (GWAS) performed in SLE patients identified several single nucleotide polymorphisms (SNPs) located on ATG genes, which have been associated with the disease occurrence [41, 113]. One SNP located in the intergenic region between ATG5 and PRDM1 was found to correlate with a greater expression of ATG5 mRNA [159]. The genetic association between ATG5 and susceptibility to SLE has been confirmed in individual studies, but not found in others [43]. Interestingly, a recent meta-analysis in Asians showed strong association of SNPs on DRAM1 with SLE susceptibility [156]. This gene encodes an activator of macroautophagy in response to p53-mediated stress signals. In patients with CD, a GWA study identified rs2241880, mapping to the ATG16L1 locus, as a susceptibility variant [34]. A statistically significant interaction with respect to CD risk between rs2241880 and the established CARD15/NOD2 (nucleotide-binding oligomerization domain containing 2) susceptibility variants was shown. Interestingly there was no association between rs2241880 and ulcerative colitis, another closely related inflammatory bowel disease. Recent data showed that Atg16L1 mutant mice are resistant to intestinal disease induced by the model bacterial pathogen Citrobacter rodentium [82]. The hyperimmune phenotype and protective effects developed in these mice were lost in Atg16L1/Nod2 double-mutant mice, indicating that the susceptibility from Nod2-deficiency is dominant over the benefit of Atg16L1 deficiency. ATG16L1 is central in the autophagosome formation, being part of the ATG12-ATG5 complex, which is required for the recruitment of MAP1LC3 [94]. Removal of ATG16L1 abrogates the ability of cells to form autophagosomes [130]. More recently it was described that the variant protein that contains a Thr → Ala substitution at position 300 is highly sensitive to cleavage by caspase 3, which is activated during cell stress [105]. Destruction of ATG16L1T300A impaired autophagy and increased release of pro-inflammatory cytokines TNF-α and IL-1β. Several SNPs have been described in association with CD, notably in the so called immunity-related GTPase family M (IRGM) gene [30, 77]. The results indicated that autophagy gene-IRGM polymorphisms confer susceptibility to CD but not ulcerative colitis, especially in Europeans. IRGM is a member of the interferon-inductible GTPase family conferring autophagic defence against intracellular pathogens like M. Tuberculosis. IRGM controls the latter by enhancing mycobacterial phagosome maturation [137].
Altogether these data argue for a strong impact of autophagy elements in several aspects of immunity, including protection to infectious agents and control of inflammatory and autoimmune responses, as well as in tumorigenesis and cancer. Paradoxically, it is only recently that experimental studies based on cellular and molecular investigation shed some light on the involvement of autophagy in immunity. A number of comprehensive review articles have been recently published on this topic with a particular emphasis on the role of autophagy in infection and inflammation [10, 22, 23, 32, 66, 112, 123, 124]. The present review mainly focuses on autophagy in autoimmunity, in relation with possible manipulation of immune system by small molecules and peptides in order to divert deleterious immune responses and at least partly restore impaired tolerance to self.
Innate immune responses importantly influence the adaptive immunity in the induction and regulation of autoimmune diseases. In innate immunity, autophagy works at different levels, notably by controlling activation and release of certain cytokines and chemokines [22, 23, 32, 47, 129]. Autophagy would activate the secretion of TNFα, interleukin (IL)-6, IL-8 and type I interferon (IFN) while it controls the production of IL-1α and β (the latter by regulating inflammasome activation and by targeting pro-IL-1β for degradation), IL-18 and type I IFN. In turn, some secreted cytokines influence autophagy. Thus, T helper type 1 (Th1) and pro-inflammatory cytokines such as IFN-γ (via IRGM), TNFα, IL-1α and β, IL-23, reactive oxygen species (ROS) and engagement of some TLRs (mechanisms that are still poorly understood) induce autophagy. TWEAK (the TNF-like weak inducer of apoptosis, in C2C12 myotubes), IL-2 in CD4+ T cells, IL-6 in peripheral blood mononuclear cells (PBMCs) and TGF-β in hepatocarcinoma cell lines also promote autophagy. Conversely, Th2 and regulatory cytokines such as IL-4, IL-13 and IL-10, via an effect on STAT-3 or −6 pathways and the serine/threonine-protein kinase (AKT) pathway were found to activate mammalian target of rapamycin (mTOR), which inhibits the serine/threonine protein kinase ULK1 and therefore autophagosome formation [33, 47]. Via its effect on cytokine secretion, particularly in antigen-presenting cells (APCs), autophagy represents a pivotal regulator of immune responses [10, 23, 32, 66, 106, 124, 129].
Although not yet recognized to such a level of crucial importance in current text books, autophagy in fact exerts profound effects on different aspects of adaptive immunity. It is a major player in thymic selection of T cells, affecting also T cell homeostasis, repertoire and polarization, survival of B cells, immune tolerance, and antigen presentation.
The discovery that autophagy is a key regulatory element for delivering self-antigens to major histocompatibility complex II (MHCII) molecules has been a critical turning point [21, 116, 158]. At the time of this finding, it was established classically that MHCI molecules presented peptides from intracellular source proteins to T cells while MHCII molecules presented antigenic peptides from exogenous and membrane proteins. The overall picture of T cell activation by MHCII peptide was thus considerably reconsidered and new nexus between immune response and cellular stress, cell metabolism, cell nutrient and cell environment were suggested and analysed further. Incidentally, it is interesting to note that following experiments in which potent macroautophagy inhibitors acting on PI 3-kinase activity, i.e. wortmannin, LY294002 and 3-methyladenine (3-MA) were incubated with macrophage cell line BMC-2 transfected with Eα52-68-eGFP (a peptide fragment issued from transmembrane protein I-Eα) and shown to have no effect, it was concluded that macroautophagy was not a mechanism for cytoplasmic expressed proteins to gain access to the luminal peptide binding site of MHCII molecules [20]. At that time conflicting data were published, which could result from the inherent properties of the antigen that was studied, its half-life and intracellular (vesicular or not) trafficking, and the type of APCs [24, 64, 116]. More recent data have shown that in APCs that are less proteolytically active than other cells such as macrophages, cleavage by lysosomal cysteine proteases – generally known as cathepsins – of particles and proteins that finally reach autolysosomes give rise to protein fragments, which will constitute the major source of peptides for MHCII molecules (Fig. 1c). Lysosomes and autolysosomes have a pH of 4–4.5, which is optimum for cathepsins. Thus, and of importance in the context of autoimmunity, MHCII molecules can bind peptides generated from endogenous antigens that are generated by lysosomal proteolysis. Such endogeneous antigens can be from membranous, cytoplasmic (including vesicle components) or nuclear origin and can have trafficked into the endo-lysosomal network via several forms of autophagy for subsequent processing and presentation by MHCII molecules to promote CD4+ T cells priming [7, 104]. Interestingly, in their pioneer work, Stevanovic, Rammensee and coll. already demonstrated that the induction of autophagy by starvation altered the balance of active proteases in lysosomes [21], which as a matter of consequence, can change the quality of peptides that are loaded onto MHCII molecules. Over the last decade, the role and regulation of specific proteases on the liberation and processing of self-antigens has been studied extensively [148, 150] and it was shown in particular that a distinct set of cathepsins is at work in different APCs, e.g. dendritic cells (DCs) and B cells [8, 81]. There are also multiple mechanisms (including gene up-regulation or down-regulation governed by the environment), that are involved for controlling proteases activity, even in individual endosomes, and strongly affect antigen presentation [21, 148]. Endo-lysosomal proteases are thus key players to generate antigens that in fine will be presented to T cells. Via a stepwise process involving asparagine endopeptidase (AEP) also known as legumain, cystatin C, specific cathepsins and other still unspecified proteases, endo-lysosomal proteases act for processing the invariant (Ii) chain linked to MHCII molecule into class-II associated invariant chain peptide (CLIP), thus generating peptide-receptive MHCII molecules in which the CLIP peptide is exchanged for a high affinity peptide by the enzyme HLA-DM (Fig. 1c) prior its transport to the cell surface of APCs for display to CD4+ T cells [107]. Endo-lysosomal proteases, including AEP, also act to generate epitopes that will be presented by functional MHCII molecules [15, 86, 148]. In the many examples of antigens that have been examined so far, stability was found to be a determining factor that influences antigen presentation. Furthermore because the cleavage via cathepsins can liberate epitopes but also destroy some others, cathepsins regulation is even more strategic for defining the final panel of antigenic peptides that are delivered.
Finally, another important role of endo-lysosomal proteases in antigen-presentation lies to their influence on TLR-receptor signaling. Initially claimed while observing the effect of chloroquine (CQ) on TLR9 signaling [38, 85], it has been demonstrated later that endo-lysosomal proteases also activate endosomal TLRs 3, 7, and 8 [80] and that the mode of action was not the one proposed in the first studies. In fact, whether for TLR9 or for endosomal TLRs, endo-lysosomal proteases would act by converting the receptor from a non-signaling full-length form to a shorter form deleted from an N-terminal region [26, 118]. Although the precise mechanisms that are behind this effect -notably considering the specific proteases that are involved- are still a continuing matter of debates, it remains that such an effect can be strategic as TLR-signaling is central for DC maturation that dictates protease activity and consequently influences the quality of peptides that are presented onto MHCII molecules. These data highlight the importance of TLRs in autophagy processes in conjunction with both innate (see above; [153]) and adaptive immunity.
The importance of autophagy in immunity also came from experiments performed with mice or cells that have been manipulated to under-express Atg genes. Using this strategy, associated to our growing knowledge of genes that appear defective in some individuals, it has been possible to better approach the potential role of some ATG proteins and establish some links with human diseases [12, 45, 79]. Thus, using mice with a B-cell-specific deletion of Atg5, a gene implicated in the elongation of autophagosome membrane, it has been shown that in autophagy-deficient B-cell progenitors the transition from the pro-B to the pre-B cell stage in the bone marrow was defective [87]. Studies of mice in which Atg5 was conditionally deleted in B lymphocytes revealed further that this gene is essential for plasma cells (PC) homeostasis [16]. Class-switch did occur in these mice but antibody responses were strongly decreased after specific immunisation, parasitic infection and mucosal inflammation. These data and others [119] highlight the importance of ATG5 not only in early B cell development but also in late B cell activation and PC differentiation. Conditional deletion of essential autophagy genes Atg5 [139], Atg7 [46, 122], Atg3 [46] also showed that macroautophagy is critical to the survival of peripheral T cells. Some Atg genes are important in infection setting. Thus, using mouse embryonic fibroblasts (MEFs) lacking human ATG16L1 or murine Atg7, Atg9a, or Atg14 [109] showed the importance of ATG16L1, ATG7 and ATG16L1, but not of ATG9A and ATG14, in the IFN-γ-induced recruitment of the immunity-related GTPases to the intracellular pathogen T. gondii. A number of examples in different forms of autophagy processes, including macroautophagy, CMA, and mitophagy have been described in which autophagy genes have been deleted or over-expressed, in some cases in specific tissues. Examples are Pink1/parkin knockout (KO) mice, the Atg16L1 mutant and Atg16L1/Nod2 double-mutant mice described above, Sqstm1/p62/A170 (encoding SQSTM1 multifunctional protein, also known as signaling adaptor/scaffold protein) mutant mice, conditional deletion models invalidating Beclin-1 or Vps34, to quote just a few. Some mutations affecting binding partners of key elements of autophagy pathways were also introduced. Thus, deletion of the gene encoding lysosome-associated membrane protein-2 (LAMP-2A) in T cells was shown recently to cause deficient in vivo responses to immunization or infection with L. monocytogenes [147]. In these mice, CMA in T cells was found to be altered with age. It should be mentioned here that mice invalidated for HSPA8 are not viable, as are Beclin-1 KO mice that die in utero or Atg5 KO mice that die within 24 h after birth due at least in part to deficient amino acid production.
At this stage of our thoughts, it seems important to insist on the fact that if investigations with such mutated mice provide decisive information, it remains that in general much more additional observations are needed to establish direct links between autophagy and certain pathologies, since mutations and polymorphisms of the ATG (or Atg in mice) genes can have many indirect effects as described above. Consistent with these considerations, important caveats have also been warranted regarding the interpretation of data that can be generated using RNA interference-based KO of Atg mRNAs in mammalian cell lines [138].
The close relationships between autophagy and immunity reported above easily explain that any deregulation of autophagy machinery can affect various aspects of immune responses and lead to autoimmunity development [32, 72, 121]. Enhanced autophagy, allowing survival of self-reactive lymphocytes, can promote autoimmunity. Moreover, autophagy, which produces autoantigens through intracellular protein digestion can participate in the initiation or maintenance of autoimmunity. In addition to SNPs and susceptibility genes, a number of studies have highlighted that expression of some genes related to autophagic process is modified during autoimmunity. In rheumatoid arthritis (RA), it has been shown that both ATG7 and BECLIN-1 gene expression is increased in osteoclasts from patients [70]. Atg7 expression was found to be increased by pro-inflammatory cytokine TNF-α, a critical element for the pathogenesis through the regulation of synovial inflammation. Other studies have also demonstrated that in autoimmune demyelination syndrome and in multiple sclerosis (MS), ATG5 gene expression is also significantly elevated compared to healthy controls [2].
Based on genetic evidences, potential links between autophagy and autoimmunity have been suggested for a decade. In general, however, experimental arguments at the cellular and molecular level showing a role of autophagy in the initiation and/or progression of autoimmune diseases are still scarce (Table 1). In SLE patients and two genetically unrelated mouse models of lupus, namely MRL/lpr and (NZBxNZW)F1 (NZB/W) mice, we showed in a seminal report that autophagy is deregulated in T lymphocytes [31]. Autophagic vacuoles were found to be over-represented in T cells indicating that autophagy is hyperactivated. This deregulation was even more obvious when T cells were stimulated by chemical activators of T cell receptor (TCR)-related signalling pathways. The elevated autophagic compartment was not found in all T cells but was restricted to a subset of them. As autophagy is known to be involved in cell survival, these results suggest that autophagy could promote the survival of autoreactive T cells during the disease. A few months after our results came out, independent studies were published describing some deregulated autophagy features in lupus T and B cells. Alessandri et al. [1] showed an increase of the autophagosome-associated MAP1LC3-II isoform in T cells, which mainly occurred in naïve CD4 T cells isolated from SLE patients. These results, which confirm our own data, suggest that there is an intrinsic deregulation of autophagic activity in SLE T cells. The authors proposed another interpretation in concluding that SLE T cells are resistant to macroautophagy induction and could thus become more prone to apoptosis. They came to this conclusion by re-stimulating T cells with rapamycin or with autologous (pro-autophagic) serum. It is possible, however, that SLE T cells are already at the maximum level of autophagosome loading and that re-exposure to their own serum had no further effect on autophagic activity. In any case, these data confirm the pro-autophagic role of SLE serum on normal T cells. Pierdominici and her colleagues also observed that the increase of autophagy was correlated with disease activity scores, important information that could be exploited in future therapeutic strategies [1, 120, 121].
More recent studies have reinforced and extended the pioneered works described above. Thus, for the first time, Clarke et al. [13] showed in NZB/W mice that macroautophagy activation also occurs in B cells, and more particularly in early developmental and transitional stages of B cell development (before disease onset). In patients with lupus, autophagy was also activated compared to healthy individuals, and again this activation occurred mainly in naïve B cells. When autophagy inhibitors such as 3-MA, bafilomycin A1 or CQ were used, plasmablast differentiation and survival hardly occurred. These findings must be related to the overproduction of autoantibodies in the serum of lupus prone mice and patients with lupus. In their study, the authors confirmed that in addition to B cells, autophagy was increased in T cells from lupus patients, and that in both cases, this activation could be correlated to disease activity. Li et al. [67] also described convincing results demonstrating that compared to controls, autophagy was significantly activated in the macrophages collected from an induced mouse model of lupus (BALB/c mice that develop a lupus-like disease after administration in Freund’s adjuvant of homologous activated lymphocyte-derived DNA) and in the PBMCs of patients with lupus. Adoptive transfer of Beclin-1 KO macrophages significantly ameliorates the clinical conditions of recipient mice (decrease of proteinuria levels, reduction of typical renal complex deposition, amelioration of glomerulonephritis) as well as the biological features (decrease of serum anti-dsDNA antibody levels and circulating proinflammatory cytokines IL-6 and TNF-α, as measured by ELISA).
A few studies have highlighted the role of autophagy in other autoimmune diseases, notably in human RA [49, 70, 151] and in experimental autoimmune encephalomyelitis, a model of MS [6]. Autophagy appears to be activated in osteoclasts from patients with RA and regulates osteoclasts differentiation [70]. This increased autophagic process, also found in RA synovial fibroblast compared to osteoarthritis synovial fibroblast by Kato et al. [49] correlates with a reduced apoptosis level in RA synovial tissues [152]. It was concluded from these observations that the activation of autophagy induced by overproduced TFN-α leads to the reduction of apoptosis in joints and more importantly causes the survival of synovial fibroblasts, which are responsible for the pathology. This again highlights the dual effect of autophagy, which is cytoprotective when it eliminates misfolded or too abundant cellular components, but in excess, can become deleterious and generate negative effects.
4 Targeting Autophagy for Intervention in Autoimmune Diseases
A number of recent findings underlined the pivotal role of macroautophagy in the control of muscle mass, and misregulation of autophagy has been described in myopathies and muscular dystrophies [131]. Information in relation to possible autophagy process dysfunction is scarce, however, regarding patients with fibromyalgia, for example, or with polymyositis [73, 143], a rare disease with an autoimmune component which is characterized by inflammation and degeneration of the muscles. On the other hand, autophagy defects have been observed (or suspected) in several autoimmune settings, including CD, SLE, possibly RA and MS (Table 1), as well as in inflammatory syndromes, notably in pulmonary diseases [88]. It is strongly anticipated that in all these situations, modulation of autophagy, in order to re-establish a proper flux regulation in particular, might rescue alterations and improve the clinical status of treated patients.
As underlined recently [32], some molecules used for years to treat inflammatory and autoimmune diseases have been found much later to target one or another type of autophagy processes. Nowadays, in fact, there are very few specific compounds targeting precise steps of autophagy pathways, and even a single pathway in particular [3], and quite surprisingly, the targets of some autophagy regulators that are widely prescribed to patients are not really known. This is the case, in particular, of CQ and hydroxychloroquine (HCQ) or of dexamethasone, which mode of action (MOA) is still being debated (see below).
A number of comprehensive review articles have recently exhaustively covered various aspects, structural and functional, of families of compounds, activators and inhibitors, which have been generated to modulate autophagy directly or indirectly [5, 11, 29, 32, 45, 125, 127, 148, 149]. Evaluated in rigorously calibrated assays performed both in vitro and in vivo [53, 93], some of these small molecules might prove to be relevant to modulate autoimmune diseases in appropriate settings. In the examples shown in the next section we will limit ourselves to a few pharmacological regulators of autophagy with established or promising clinical efficacy in autoimmune diseases.
Before providing a short description of these selected pharmacological autophagy modulators, several conceptual and practical comments should be made. Firstly, this field of possible intervention is new (or newly rediscovered) and autophagy processes, which are complex and somehow confusing, are not well perceived by decision makers of technology companies and Big Pharmas, of course even less by the general public and informed users. Communication including education towards professionals and patients is certainly much easier when, for example, one describes the activity of a therapeutic antibody specific for a soluble molecule or a surface receptor that is raised in inflammatory and autoimmune conditions. Important efforts of clarification and simplification have thus to be made as it was the cases some decades ago for apoptosis.
Secondly, it is well appreciated for a long time in the field that pharmacological small molecules rarely exert their action on only one single target. This is the case of HCQ and dexamethasone, for example, and many others (see below). These multi-target effects can explain their strong efficacy, but they also complicate the description of the said-molecule and of its safety file.
Thirdly, it is often argued that small molecules (<900–1000 daltons) and short peptides (<20–40 amino acid residues) will be eliminated rapidly from the body and therefore will have a too short period of possible action. This statement regarding pharmacokinetics and pharmacodynamics of molecules may be correct but if it is the case, there are numerous carrier systems or novel devices that increase molecule bioavailability and trafficking leading to improving their efficacy. It should be noted here that conversely, their low molecular mass can be an advantage when the desired objective is to develop a strategy supposed to target the central nervous system, for example [44].
Fourthly, and most importantly, solubility of small molecules and peptides remains a limiting factor, as it is also the case of antibodies and fusion proteins that are designed and produced for therapeutic purposes. This aspect has to be taken into consideration at the very early stage of molecule selection as in general, it cannot be solved easily in the downstream steps of development.
On the other hand, pharmacological small molecules and peptides display a number of advantageous properties that makes them excellent therapeutics, notably for autoimmune diseases. In addition to their synthesis and production that can be highly optimized, and in some cases remarkably simple in comparison to some biologics, and automatable, small molecules and peptides selected as active components of pharmaceutical compositions are characterized by their stability and robustness, easy handling, the relatively low doses that have to be administrated to patients and their cost, which remains reasonable with regard to most biologics. Small molecules and short peptides are not immunogenic per se, another considerable advantage for treating patients with chronic autoimmune diseases [132].
Finally, it must be stressed that, as it is the case for all new therapies that emerge, standardized and universalized animal models of the related human disease have to be developed -if they do not already exist-, a consensus position regarding the most promising modality to be tested has to be established, and formation of a cooperative international network of committed clinical investigators has to be gathered to evaluate these new therapies in a pre-designed rigorous fashion.
5 Existing Pharmacological Regulators of Autophagy
Herein, we briefly describe the characteristics of some chemical molecules that are established pharmacological regulators of autophagy (Fig. 2) and are given to patients with autoimmune diseases. Further details on these and other compounds can be found in recent reviews and articles [5, 11, 29, 32, 125, 127, 140, 146].

Pharmacological regulators of autophagy. A diagram illustrating possible sites of intervention of pharmacological autophagy regulators. From the left to the right: rapamycin and dexamethasone inhibit the kinase activity of mTOR, leading to the upregulation of macroautophagy. Dexamethasone is also known as acting on pre-autophagosomal structure. Trehalose, the target of which still remains debated, is an activator of autophagy through an mTOR-independent pathway. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes. It acts by inhibiting vacuolar H+ ATPase. P140 peptide (▲), the uptake into B lymphocytes by clathrin-mediated endocytosis and homing into lysosomes has been demonstrated after administration to mice, and DSG, both interact with HSPA8 in vitro and alter intralysosomal pH. P140 provokes the accumulation of autophagy markers p62/sequestosome 1 and MAP1LC3-II in MRL/lpr B cells, consistent with a down-regulation of autophagic flux. This peptide affects both CMA and macroautophagy. CQ and HCQ are lysosomotropic agents that prevent endosomal acidification. They accumulate inside endosomes and lysosomes, leading to inhibition of lysosomal enzymes, which requires an acidic pH, defective fusion of endosomes and lysosomes and maturation of autolysosomes. Abbreviations: CMA chaperone-mediated autophagy, CQ chloroquine, DSG 15-deoxyspergualin, HCQ hydroxychloroquine, HSPA8 heat shock protein 8, LAMP-2A lysosome-associated membrane protein-2A, MAP1LC3 microtubule-associated protein light chain 3, mTOR mammalian target of rapamycin
CQ and HCQ
These two small molecules are lipophilic weak bases that easily pass through the lipid cell membrane and preferentially concentrate in acidic cytoplasmic vesicles. As lysosomotropic agent, they raise intralysosomal pH, leading to defective autophagic protein degradation. CQ/HCQ may also affect peptide degradation within lysosomes due to the pH effect on lysosomal cathepsins and therefore the entire process of antigen presentation by MHC molecules in the MIIC compartment leading to activation of autoreactive T cells. HCQ is used for years in the treatment of inflammatory autoimmune diseases, SLE, RA and Sjögren’s syndrome. CQ has been shown to reduce the severity of experimental autoimmune encephalomyelitis, a model for MS, and the mechanism of action that was previously known to involve in part regulatory T cells has been recently established in much more details [144]. CQ and HCQ also operate by interacting directly with TLR ligands [59]. Other characteristics of CQ and derivatives, such as radiosensitising and chemosensitising properties also receive attention in anti-cancer indications [45]. It should be reminded, however, that CQ/HCQ toxicity, in particular in the eye (cornea and macula) and in the occurrence of cardiomyopathies [142], remains a major break. Observed ocular toxicity is related to the total cumulative dose rather than the daily dose; therefore it becomes a serious potential problem in the cases of long-term use. A number of HCQ analogs and mimics have been tentatively designed that keep the molecule activity without secondary effects. Ongoing research should provide such safe molecules in the future.
Bafilomycin A
This compound isolated from Streptomyces sp. is a member of the plecomacrolide sub-class of macrolide antibiotics. Early studies showed that at a 100 nM-concentration and short incubation time (1 h), in a rat hepatoma H-4-II-E cell line, it specifically acts by inhibiting the vacuolar H+ ATPase (V-ATPase) that is essential for acidifying lumen lysosomes and blocks the fusion of autophagosomes with lysosomes [155]. Used at the same or higher concentration and other settings, in the same cell line or other types of cell lines, effects targeted other key steps of the autophagy axes have been observed as summarized and analyzed by Klionsky et al. [54]. This in-depth analysis of published data led these authors to propose that at early time-points, bafilomycin could mainly interfere with the autophagic flux by slowing the degradation of MAP1LC3-II within existing autolysosomes, while at later time-points, its effect on acidification of lysosomes and possibly also of endosomes and amphisomes could impair the fusion of autophagosomes with both late endosomes and lysosomes as shown [42]. Altogether this sequence of events highlights again the fact that the pleiotropic effects of certain molecules, as a function of concentration, treatment time, or environment, have to be taken into account when mechanistic studies are performed, notably with the objective to elaborate therapeutic strategies.
P140 peptide/Lupuzor
This 21-mer linear peptide encompassing the sequence 131–151 of the spliceosomal U1-70 K protein and containing a phosphoserine residue at position 140, was found to be safe and significantly ameliorated lupus patients’ clinical status when administrated subcutaneously in the presence of mannitol as excipient [102, 103, 160]. All appropriate preclinical studies were done in the widely used MRL/lpr model, a mouse that develops a strong and rapid lupus disease. The capacity of P140 to ameliorate biological and clinical parameters in these mice, and to enhance their survival, was demonstrated in a robust manner [99, 133]. After P140 treatment, an accumulation of autophagy markers SQSTM1 and MAP1LC3 was observed in MRL/lpr B cells, consistent with a down-regulation of autophagic flux [114]. CMA was also recently found to be a target of P140 and it was demonstrated that P140 peptide inhibitory effect on CMA is likely tied to its ability to interact with HSPA8 [115] and to alter the composition of HSPA8 heterocomplexes [78]. Expression of both HSPA8 and the limiting CMA component LAMP-2A, which is increased in MRL/lpr B cells, is down-regulated after treating mice with P140 peptide. It was shown further that P140, but not the non-phosphorylated peptide that is not protective against disease development in mice [99], uses the clathrin-dependent endo-lysosomal pathway to enter into MRL/lpr B lymphocytes and accumulates in the lysosomal lumen where it may directly hamper lysosomal HSPA8 chaperoning functions, and also destabilize LAMP-2A in lysosomes as a result of its effect on HSP90. This dual effect may interfere with the endogenous (auto)antigen processing and loading to MHCII molecules and as a consequence, lead to the lower activation of autoreactive T cells that was previously shown experimentally [100, 101].
Interestingly, earlier work also indicated that ex vivo, P140 does not induce proliferation of human peripheral T cells (in contrast to the non-phosphorylated form that does) but generates secretion of high levels of regulatory cytokine IL-10 in cell cultures [98]. This observation and others generated in our own studies might indicate that beside its effect on autophagy processes, P140 might also act as a so-called ‘peptide altered ligand’ of the TCR. Our first studies showed that the nominal peptide 131–151 contains an epitope that is effectively recognized by CD4+ T cells from MRL/lpr and NZBxW mice [96, 97]. The phosphate moiety introduced at position 140 in the P140 peptide might have no effect on MHC presentation (as experimentally demonstrated) but induce qualitatively different activation of T cells with changes in cytokine production and T cell responsiveness [98, 99]. Altogether, these considerations point out again the multi-target functions of efficient immunomodulator molecules. Thus, in the case of P140 peptide, both specific CD4+ T cell clones recognizing the sequence 131–151 of U1-70 K protein and T cell clones with a broader specificity for various self-components generated in autolysosomes and lysosomes and loaded onto MHC class II molecules in the MIIC compartment (Fig. 1c), could be simultaneously involved in the mechanism of peptide action.
15-Deoxyspergualin (DSG)
This compound (1-amino-19-guanidino-11-hydroxy-4, 9, 12-triazanona-decane-10, 1–3-dione) is a synthetic analogue of spergualin, a natural product of the bacterium Bacillus laterosporus. A long list of more stable analogs have been designed, synthesized and evaluated over years. 15-DSG is a potent immunosuppressant, which showed immunosuppressive activity both in vitro and in vivo, affecting B lymphocytes, T lymphocytes and macrophage/monocyte functions. It was shown to bind to the EEVD domain of HSPA8, a site that is apparently different from the one(s) recognized by P140 peptide [140], with an affinity of approximately 4 μM, and increase its ATPase activity of 20–40 %. It also binds to HSP90. 15-DSG blocks the NF-κB pathway and antigen presentation, causing alteration in the activation of immune cells, notably monocytes, DCs and T cells. It also inhibits AKT activation and phosphatidylcholine synthesis [51]. DSG was also shown to suppress the progression of polyclonal B cell activation and lupus nephropathy in lupus-prone MRL/lpr mice. In patients, in an first short clinical trial, two of three patients treated with DSG showed infectious episodes and the trial was interrupted [74]. Later, another phase-I/II study including a total of 21 patients was engaged [75]. After the first DSG injection, one patient was excluded from the study due to renal failure. Five patients dropped out due to adverse events or serious adverse events including fever, leukopenia, oral candidiasis, herpes zoster or pneumonia. Eleven of 20 patients achieved partial (4) or complete responses (7), 8 were judged as treatment failures and 1 patient was not assessable. In the 12 patients who completed all nine cycles, proteinuria was statistically decreased and the Selena-SLEDAI SLE responder score was decreased from 17.6 to 11.7. These data led the authors to conclude that although the number of patients still remained small, the improvement of their clinical status, particularly their proteinuria, was encouraging, supporting further investigations with large cohorts. At this stage, however, and although some promising data were also obtained in patients with anti-neutrophil cytoplasmic autoantibodies-associated vasculitis and cancer conditions, careful studies designed to better characterize toxicity and side-effects generated by DSG will be determining [63].
Dexamethasone
This potent immunosuppressive drug is widely used to treat many different inflammatory and autoimmune conditions such as inflammatory bowel diseases (ulcerative colitis and CD), RA, SLE, chronic skin conditions (e.g. dermatitis herpetiformis, pemphigus, severe psoriasis and seborrheic dermatitis). It is also given in severe allergic conditions and certain types of cancer. Its MOA is multiple, complex and still a matter of some controversies. It was found in particular that dexamethasone induces the expression of a gene encoding the stress response protein Dig2/RTP801/REDD1 [95], and the elevation of Dig2/RTP801/REDD1, a negative regulator of mTOR signaling pathway, contributes to the induction of macroautophagy. It should be mentioned herein that depending on the dose and the type of cells, the effect of dexamethasone on Dig2/RTP801/REDD1 is not equivalent (less dependence at high dexamethasone dose, for example). Other dexamethasone effects were described. Thus, dexamethasone was shown to increase expression of several autophagy genes, including ATG5, MAP1LC3, BECLIN1n and SQSTM1, and to trigger 5’ AMP-activated protein kinase-dependent mitochondrial fragmentation associated with increased levels of dynamin-1-like protein, a GTPase that regulates mitochondrial fission [145]. Thus, certain steps of the mitophagy axis would be targeted by dexamethasone as well. The anti-inflammatory actions of dexamethasone are also thought to involve phospholipase A2 inhibitory proteins, lipocortins, which control the biosynthesis of potent mediators of inflammation such as prostaglandins and leukotrienes.
Rapamycine/sirolimus
This macrolide antibiotic is a safe and well-tolerated drug clinically used for rejection prophylaxis in renal transplantation. It is also used as immunosuppressant and anti-fungal agent. It forms a complex with the immunophilin FKBP-12 and inhibits the kinase activity of mTOR complex 1 (mTORC1), leading thus to autophagy induction (mTORC2 is largely resistant to rapamycin). It regulates mitochondrila transmembrane potential and calcium influx. Its potent effect on the development of nephritis in NZB/W mice was shown [76]. Twelve-week-old female NZB/W mice were treated by oral gavage for 20 weeks with rapamycin (3 mg/kg body weight). Rapamycin treatment markedly reduced proteinuria, improved renal function, decreased serum anti-dsDNA antibody levels and diminished splenomegaly. Rapamycin-treated mice had near normal renal histology, with marked reduction in glomerular immune deposition and the infiltration by T cells, B cells and macrophages. These data were reinforced by recent mechanistic findings published independently [141]. In humans, rapamycin treatment showed some benefit in the treatment of nine SLE patients with refractory disease [28]. In a recent prospective open-label study based on 59 patients and 54 matched healthy subjects (for a total of 274 visits), rapamycin was shown to mainly block IL-4 production and necrosis of double negative (DN) T cells in patients with SLE. In addition, rapamycin was found to enhance FoxP3 expression in CD25+/CD4+ T cells and expand CD25+CD19+ B cells, suggesting that mTOR activation can trigger IL-4 production and necrosis of DN T cells in active SLE [60]. Further investigation in large cohorts of patients with lupus and also in patients with other immune-mediated disorders, including type 1 diabetes and RA are awaited for consolidating these data. If we only take into account the role exerted by rapamycin on the autophagy flux (Fig. 2), and considering that basal autophagy seems to be activated in different subsets of lymphocytes in murine and human lupus (Table 1), rapamycin administration should not be beneficial in lupus. It might even make the illness more severe. This leads us to conclude that rapamycin probably modulates another pathway and not autophagy as main target.
Recently a randomized trial was conducted to investigate the efficacy and safety of rapamycin treatment in adults with chronic immune thrombocytopenia (ITP), an acquired autoimmune disease characterized by an autoantibody-mediated destruction and impaired platelet production [68]. Two groups of 40 patients were examined, the control one that received cyclosporine A plus prednisone and the experimental one that received rapamycin plus prednisone. The overall response was similar in both groups. However, sustained response was more pronounced in the experimental group than in the control group. Both groups showed similar incidence of adverse events (7 % vs. 11 %). The experimental group experienced a significant rise in CD4+CD25+CD127low regulatory T cells level, and there was a strong correlation between the levels of regulatory T cells and TGF-β after the treatment. From these data it was concluded that rapamycin plus low dose prednisone could provide a new promising option for therapy of ITP.
6 Future Prospects and Concluding Remarks
The list of components described briefly above is far to be exhaustive. Excellent recent review articles gave much more structural and functional details on many other molecules (small molecules and peptides), some of them that are already administrated as therapeutics and some others that are under evaluation in autoimmune patients or included in preclinical studies in pertinent animal models [25, 39, 136]. The information we summarized herein underlines that most, if not all of the molecules, exhibit complex pleiotropic properties, and can notably influence different autophagy pathways (e.g. mTOR-dependent and -independent) as well as other quality-control mechanisms affecting the cell live/death balance. Several widely used molecules can exert dual (sometimes opposite) effects on upstream and downstream molecular events of the autophagy axes. It should be kept in mind also that the large majority of these molecules have been initially evaluated in cell culture conditions (some are issued from cellular screens) and it has been seen that their MOA largely depends on the selected cell type (immortalized cell lines, primary cells; cancer cells or non-cancer cells), concentration, and time of exposure. As underlined recently [32], these considerations are fundamental to analyze the conclusions that can be raised with most caution.
Nowadays, a number of pharmaceuticals approved in the European Union and USA, and in regular clinical use for alternative indications, inhibit autophagy and may therefore be novel treatments for autoimmune diseases. Chemical drugs acting on autophagy and/or other pivotal cellular pathways are also often evaluated in association to reinforce their efficacy while lowering dosage to minimize deleterious side effects. Based on our increasing understanding of the physiological autophagy mechanisms and of their dysfunctions in pathological settings [18, 65, 92, 120, 147, 157], we dare believe that molecules that very specifically target key elements of the autophagy process will emerge and, with a minimum of side effects, will efficiently modulate debilitating autoimmune diseases that today affect more than 3 % of the general population worldwide.
Abbreviations
- 3-MA:
-
3-methyladenine
- AEP:
-
Asparagine endopeptidase
- APC:
-
Antigen-presenting cell
- ATG:
-
Autophagy-related protein
- BECLIN1/beclin-1:
-
BCL-2 interacting myosin/moesin-like coiled-coil protein 1
- CD:
-
Crohn’s disease
- CLIP:
-
Class-II associated invariant chain peptide
- CMA:
-
Chaperone-mediated autophagy
- CQ/HCQ:
-
Chloroquine/hydroxychloroquine
- DC:
-
Dendritic cell
- DN:
-
Double negative
- DRAM1:
-
DNA damage-regulated autophagy modulator1
- ds:
-
Double-stranded
- DSG:
-
Deoxyspergualin
- HSP:
-
Heat shock protein
- HSPA8/HSC70:
-
Heat shock cognate protein of 70 KDa
- IFN:
-
Interferon
- IL:
-
Interleukin
- IRGM:
-
Immunity-related GTPase M
- ITP:
-
Immune thrombocytopenia
- KO:
-
Knockout
- LAMP-2A:
-
Lysosome-associated membrane protein-2A
- LNC:
-
Lymph node cells
- LPS:
-
Lipopolysaccharide
- MAP1LC3/LC3:
-
Microtubule-associated protein light chain 3
- MHCI/II:
-
Major histocompatibility complex class I or MHC class II
- MIIC:
-
Major histocompatibility complex class II compartment
- MOA:
-
Mode of action
- MRL:
-
Murphy Roths large
- MS:
-
Multiple sclerosis
- mTOR:
-
Mammalian target of rapamycin
- NZB/W:
-
(NZBxNZW) F1
- PBMCs:
-
Peripheral blood mononuclear cells
- PC:
-
Plasma cell
- RA:
-
Rheumatoid arthritis
- ROS:
-
Reactive oxygen species
- SLE:
-
Systemic lupus erythematosus
- SQSTM1/p62:
-
Sequestosome 1
- TCR:
-
T cell receptor
- Th1/Th2:
-
T helper type 1 and type 2
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- UPS:
-
Ubiquitin-proteasome system.
References
Alessandri C, Barbati C, Vacirca D et al (2012) T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J 26:4722–4732. doi:10.1096/fj.12-206060
Alirezaei M, Fox HS, Flynn CT et al (2009) Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 5:152–158. doi:10.4161/auto.5.2.7348
Anguiano J, Garner TP, Mahalingam M et al (2013) Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 9:374–382. doi:10.1038/nchembio.1230
Awan MUF, Deng Y (2014) Role of autophagy and its significance in cellular homeostasis. Appl Microbiol Biotechnol 98:5319–5328. doi:10.1007/s00253-014-5721-8
Baek K-H, Park J, Shin I (2012) Autophagy-regulating small molecules and their therapeutic applications. Chem Soc Rev 41:3245–3263. doi:10.1039/c2cs15328a
Bhattacharya A, Parillon X, Zeng S et al (2014) Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J Biol Chem 289:26525–26532. doi:10.1074/jbc.M114.575860
Blum JS, Wearsch PA, Cresswell P (2013) Pathways of antigen processing. Annu Rev Immunol 31:443–473. doi:10.1146/annurev-immunol-032712-095910
Burster T, Beck A, Tolosa E et al (2004) Cathepsin G, and not the asparagine-specific endoprotease, controls the processing of myelin basic protein in lysosomes from human B lymphocytes. J Immunol 172:5495–5503. doi:10.4049/jimmunol.172.9.5495
Carrithers MD (2014) Update on disease-modifying treatments for multiple sclerosis. Clin Ther. doi:10.1016/j.clinthera.2014.08.006
Cenci S (2014) Autophagy, a new determinant of plasma cell differentiation and antibody responses. Mol Immunol 62:289–295. doi:10.1016/j.molimm.2014.02.008
Cheong H, Lu C, Lindsten T, Thompson CB (2012) Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol 30:671–678. doi:10.1038/nbt.2285
Choi AMK, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:1845–1846. doi:10.1056/NEJMc1303158
Clarke AJ, Ellinghaus U, Cortini A et al (2014) Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-204343
Codogno P, Mehrpour M, Proikas-Cezanne T (2012) Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol 13:7–12. doi:10.1038/nrm3249
Colbert JD, Matthews SP, Miller G, Watts C (2009) Diverse regulatory roles for lysosomal proteases in the immune response. Eur J Immunol 39:2955–2965. doi:10.1002/eji.200939650
Conway KL, Kuballa P, Khor B et al (2013) ATG5 regulates plasma cell differentiation. Autophagy 9:528–537. doi:10.4161/auto.23484
Cuervo AM (2004) Autophagy: many paths to the same end. Mol Cell Biochem 263:55–72. doi:10.1023/B:MCBI.0000041848.57020.57
Cuervo AM, Macian F (2014) Autophagy and the immune function in aging. Curr Opin Immunol 29:97–104. doi:10.1016/j.coi.2014.05.006
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24:92–104. doi:10.1038/cr.2013.153
Dani A, Chaudhry A, Mukherjee P et al (2004) The pathway for MHCII-mediated presentation of endogenous proteins involves peptide transport to the endo-lysosomal compartment. J Cell Sci 117:4219–4230. doi:10.1242/jcs.01288
Dengjel J, Schoor O, Fischer R et al (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A 102:7922–7927. doi:10.1073/pnas.0501190102
Deretic V (2012) Autophagy: an emerging immunological paradigm. J Immunol 189:15–20. doi:10.4049/jimmunol.1102108
Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737. doi:10.1038/nri3532
Dörfel D, Appel S, Grünebach F et al (2005) Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood 105:3199–3205. doi:10.1182/blood-2004-09-3556
Dowdle WE, Nyfeler B, Nagel J et al (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16:1069–1079. doi:10.1038/ncb3053
Ewald SE, Lee BL, Lau L et al (2008) The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456:658–662. doi:10.1038/nature07405
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24:24–41. doi:10.1038/cr.2013.168
Fernandez D, Bonilla E, Mirza N et al (2006) Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum 54:2983–2988. doi:10.1002/art.22085
Fleming A, Noda T, Yoshimori T, Rubinsztein DC (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 7:9–17. doi:10.1038/nchembio.500
Glas J, Seiderer J, Bues S et al (2013) IRGM variants and susceptibility to inflammatory bowel disease in the German population. PLoS One 8:e54338. doi:10.1371/journal.pone.0054338
Gros F, Arnold J, Page N et al (2012) Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 8:1113–1123. doi:10.4161/auto.20275
Gros F, Muller S (2014) Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br J Pharmacol 171:4337–4359. doi:10.1111/bph.12792
Gutierrez MG, Master SS, Singh SB et al (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766. doi:10.1016/j.cell.2004.11.038
Hampe J, Franke A, Rosenstiel P et al (2007) A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39:207–211. doi:10.1038/ng1954
Hanly JG (2014) Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol 10:338–347. doi:10.1038/nrrheum.2014.15
Hayter SM, Cook MC (2012) Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev 11:754–765. doi:10.1016/j.autrev.2012.02.001
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. doi:10.1146/annurev-genet-102808-114910
Hong Z, Jiang Z, Liangxi W et al (2004) Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int Immunopharmacol 4:223–234. doi:10.1016/j.intimp.2003.12.006
Huss M, Wieczorek H (2009) Inhibitors of V-ATPases: old and new players. J Exp Biol 212:341–346. doi:10.1242/jeb.024067
Inglese M, Petracca M (2014) Therapeutic strategies in multiple sclerosis: a focus on neuroprotection and repair and relevance to schizophrenia. Schizophr Res. doi:10.1016/j.schres.2014.04.040
International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN, Harley JB, Alarcón-Riquelme ME et al (2008) Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40:204–210. doi:10.1038/ng.81
Jahreiss L, Menzies FM, Rubinsztein DC (2008) The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic Cph Den 9:574–587. doi:10.1111/j.1600-0854.2008.00701.x
Järvinen TM, Hellquist A, Zucchelli M et al (2012) Replication of GWAS-identified systemic lupus erythematosus susceptibility genes affirms B-cell receptor pathway signalling and strengthens the role of IRF5 in disease susceptibility in a Northern European population. Rheumatol Oxf Engl 51:87–92. doi:10.1093/rheumatology/ker263
Jeltsch-David H, Muller S (2014) Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol 10:579–596. doi:10.1038/nrneurol.2014.148
Jiang P, Mizushima N (2014) Autophagy and human diseases. Cell Res 24:69–79. doi:10.1038/cr.2013.161
Jia W, He Y-W (2011) Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol 186:5313–5322. doi:10.4049/jimmunol.1002404
Jones SA, Mills KHG, Harris J (2013) Autophagy and inflammatory diseases. Immunol Cell Biol 91:250–258. doi:10.1038/icb.2012.82
Kang R, Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18:571–580. doi:10.1038/cdd.2010.191
Kato M, Ospelt C, Gay RE et al (2014) Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol 66:40–48. doi:10.1002/art.38190
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22:407–417. doi:10.1016/j.tcb.2012.05.006
Kawada M, Masuda T, Ishizuka M, Takeuchi T (2002) 15-Deoxyspergualin inhibits Akt kinase activation and phosphatidylcholine synthesis. J Biol Chem 277:27765–27771. doi:10.1074/jbc.M200318200
Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34:259–269. doi:10.1016/j.molcel.2009.04.026
Klionsky DJ, Abdalla FC, Abeliovich H et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544. doi:10.4161/auto.19496
Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC (2008) Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4:849–850. doi:10.4161/auto.6845
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. doi:10.1126/science.290.5497.1717
Konishi A, Arakawa S, Yue Z, Shimizu S (2012) Involvement of Beclin 1 in engulfment of apoptotic cells. J Biol Chem 287:13919–13929. doi:10.1074/jbc.M112.348375
Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584:1393–1398. doi:10.1016/j.febslet.2009.12.047
Kroemer G, Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9:1004–1010. doi:10.1038/nrm2529
Kuznik A, Bencina M, Svajger U et al (2011) Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 186:4794–4804. doi:10.4049/jimmunol.1000702
Lai Z-W, Borsuk R, Shadakshari A et al (2013) Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol 191:2236–2246. doi:10.4049/jimmunol.1301005
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14:759–774. doi:10.1038/nrm3696
Lee E, Koo Y, Ng A et al (2014) Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy 10:572–587. doi:10.4161/auto.27649
Lee YH, Lee HS, Choi SJ et al (2011) Efficacy and safety of tacrolimus therapy for lupus nephritis: a systematic review of clinical trials. Lupus 20:636–640. doi:10.1177/0961203310389486
Leung CS, Haigh TA, Mackay LK et al (2010) Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U S A 107:2165–2170. doi:10.1073/pnas.0909448107
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42. doi:10.1016/j.cell.2007.12.018
Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469:323–335. doi:10.1038/nature09782
Li B, Yue Y, Dong C et al (2014) Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin Exp Rheumatol 32:705–714
Li J, Wang Z, Dai L et al (2013) Effects of rapamycin combined with low dose prednisone in patients with chronic immune thrombocytopenia. Clin Dev Immunol 2013:548085. doi:10.1155/2013/548085
Lilienbaum A (2013) Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol 4:1–26
Lin N-Y, Beyer C, Giessl A et al (2013) Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann Rheum Dis 72:761–768. doi:10.1136/annrheumdis-2012-201671
Li W, Zou W, Yang Y et al (2012) Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J Cell Biol 197:27–35. doi:10.1083/jcb.201111053
Lleo A, Invernizzi P, Selmi C et al (2007) Autophagy: highlighting a novel player in the autoimmunity scenario. J Autoimmun 29:61–68. doi:10.1016/j.jaut.2007.06.003
Lloyd TE (2010) Novel therapeutic approaches for inclusion body myositis. Curr Opin Rheumatol 22:658–664. doi:10.1097/BOR.0b013e32833f0f4a
Lorenz H-M, Grunke M, Wendler J et al (2005) Safety of 15-deoxyspergualin in the treatment of glomerulonephritis associated with active systemic lupus erythematosus. Ann Rheum Dis 64:1517–1519. doi:10.1136/ard.2005.035329
Lorenz H-M, Schmitt WH, Tesar V et al (2011) Treatment of active lupus nephritis with the novel immunosuppressant 15-deoxyspergualin: an open-label dose escalation study. Arthritis Res Ther 13:R36. doi:10.1186/ar3268
Lui SL, Yung S, Tsang R et al (2008) Rapamycin prevents the development of nephritis in lupus-prone NZB/W F1 mice. Lupus 17:305–313. doi:10.1177/0961203307088289
Lu XC, Tao Y, Wu C et al (2013) Association between variants of the autophagy related gene – IRGM and susceptibility to Crohn’s disease and ulcerative colitis: a meta-analysis. PLoS One 8:e80602. doi:10.1371/journal.pone.0080602
Macri C, Wang F, Tasset I et al (2015) Modulation of deregulated chaperone-mediated autophagy by a phopsphopeptide. Autophagy 11:472–486. http://dx.doi.org/10.1080/15548627.2015.1017179
Majai G, Kiss E, Tarr T et al (2014) Decreased apopto-phagocytic gene expression in the macrophages of systemic lupus erythematosus patients. Lupus 23:133–145. doi:10.1177/0961203313511557
Manoury B (2013) Proteases: essential actors in processing antigens and intracellular toll-like receptors. Front Immunol. doi:10.3389/fimmu.2013.00299
Manoury B, Mazzeo D, Fugger L et al (2002) Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol 3:169–174. doi:10.1038/ni754
Marchiando AM, Ramanan D, Ding Y et al (2013) A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe. doi:10.1016/j.chom.2013.07.013
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15:81–94. doi:10.1038/nrm3735
Marquez RT, Xu L (2012) Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res 2:214–221
Matsumoto F, Saitoh S-I, Fukui R et al (2008) Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun 367:693–699. doi:10.1016/j.bbrc.2007.12.130
Matthews SP, Werber I, Deussing J et al (2010) Distinct protease requirements for antigen presentation in vitro and in vivo. J Immunol 184:2423–2431. doi:10.4049/jimmunol.0901486
Miller BC, Zhao Z, Stephenson LM et al (2008) The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 4:309–314
Mizumura K, Cloonan SM, Haspel JA, Choi AMK (2012) The emerging importance of autophagy in pulmonary diseases. Chest 142:1289–1299. doi:10.1378/chest.12-0809
Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873. doi:10.1101/gad.1599207
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741. doi:10.1016/j.cell.2011.10.026
Mizushima N, Levine B (2010) Autophagy in mammalian development and differentiation. Nat Cell Biol 12:823–830. doi:10.1038/ncb0910-823
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi:10.1038/nature06639
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326. doi:10.1016/j.cell.2010.01.028
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132. doi:10.1146/annurev-cellbio-092910-154005
Molitoris JK, McColl KS, Swerdlow S et al (2011) Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem 286:30181–30189. doi:10.1074/jbc.M111.245423
Monneaux F, Briand JP, Muller S (2000) B and T cell immune response to small nuclear ribonucleoprotein particles in lupus mice: autoreactive CD4(+) T cells recognize a T cell epitope located within the RNP80 motif of the 70 K protein. Eur J Immunol 30:2191–2200. doi:10.1002/1521-4141(2000)30:8<2191::AID-IMMU2191>3.0.CO;2-R
Monneaux F, Dumortier H, Steiner G et al (2001) Murine models of systemic lupus erythematosus: B and T cell responses to spliceosomal ribonucleoproteins in MRL/Fas(lpr) and (NZB x NZW)F(1) lupus mice. Int Immunol 13:1155–1163. doi:10.1093/intimm/13.9.1155
Monneaux F, Hoebeke J, Sordet C et al (2005) Selective modulation of CD4+ T cells from lupus patients by a promiscuous, protective peptide analog. J Immunol 175:5839–5847. doi:10.4049/jimmunol.175.9.5839
Monneaux F, Lozano JM, Patarroyo ME et al (2003) T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70 K snRNP protein administered in MRL/lpr mice. Eur J Immunol 33:287–296. doi:10.1002/immu.200310002
Monneaux F, Parietti V, Briand J-P, Muller S (2004) Intramolecular T cell spreading in unprimed MRL/lpr mice: importance of the U1-70 k protein sequence 131–151. Arthritis Rheum 50:3232–3238. doi:10.1002/art.20510
Monneaux F, Parietti V, Briand J-P, Muller S (2007) Importance of spliceosomal RNP1 motif for intermolecular T-B cell spreading and tolerance restoration in lupus. Arthritis Res Ther 9:R111. doi:10.1186/ar2317
Muller S, Monneaux F, Schall N et al (2008) Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial. Arthritis Rheum 58:3873–3883. doi:10.1002/art.24027
Muller S, Wallace D (2014) The importance of implementing proper selection of excipients in lupus clinical trials. Lupus. doi:10.1177/0961203314525249
Münz C (2012) Antigen processing for MHC class II presentation via autophagy. Front Antigen Present Cell Biol 3:9. doi:10.3389/fimmu.2012.00009
Murthy A, Li Y, Peng I et al (2014) A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506:456–462. doi:10.1038/nature13044
Nedjic J, Aichinger M, Emmerich J et al (2008) Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455:396–400. doi:10.1038/nature07208
Neefjes J, Jongsma MLM, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11:823–836. doi:10.1038/nri3084
Nogalska A, D’Agostino C, Terracciano C et al (2010) Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol 177:1377–1387. doi:10.2353/ajpath.2010.100050
Ohshima J, Lee Y, Sasai M et al (2014) Role of mouse and human autophagy proteins in IFN-γ-induced cell-autonomous responses against Toxoplasma gondii. J Immunol 192:3328–3335. doi:10.4049/jimmunol.1302822
Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24:9–23. doi:10.1038/cr.2013.169
Okamoto K (2014) Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol 205:435–445. doi:10.1083/jcb.201402054
Oliva L, Cenci S (2014) Autophagy in plasma cell pathophysiology. Front Immunol 5:103. doi:10.3389/fimmu.2014.00103
Orozco G, Eyre S, Hinks A et al (2011) Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 70:463–468. doi:10.1136/ard.2010.137174
Page N, Gros F, Schall N et al (2011) HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann Rheum Dis 70:837–843. doi:10.1136/ard.2010.139832
Page N, Schall N, Strub J-M et al (2009) The spliceosomal phosphopeptide P140 controls the lupus disease by interacting with the HSC70 protein and via a mechanism mediated by γδ T cells. PLoS One 4:e5273. doi:10.1371/journal.pone.0005273
Paludan C, Schmid D, Landthaler M et al (2005) Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307:593–596. doi:10.1126/science.1104904
Pampliega O, Orhon I, Patel B et al (2013) Functional interaction between autophagy and ciliogenesis. Nature 502:194–200, doi:10.1038/nature12639
Park B, Brinkmann MM, Spooner E et al (2008) Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol 9:1407–1414. doi:10.1038/ni.1669
Pengo N, Scolari M, Oliva L et al (2013) Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 14:298–305. doi:10.1038/ni.2524
Pierdominici M, Barbati C, Vomero M et al (2014) Autophagy as a pathogenic mechanism and drug target in lymphoproliferative disorders. FASEB J 28:524–535. doi:10.1096/fj.13-235655
Pierdominici M, Vomero M, Barbati C et al (2012) Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J 26:1400–1412. doi:10.1096/fj.11-194175
Pua HH, Guo J, Komatsu M, He Y-W (2009) Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 182:4046–4055. doi:10.4049/jimmunol.0801143
Puleston DJ, Simon AK (2014) Autophagy in the immune system. Immunology 141:1–8. doi:10.1111/imm.12165
Ravikumar B, Sarkar S, Davies JE et al (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435. doi:10.1152/physrev.00030.2009
Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC (2010) Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem 285:11061–11067. doi:10.1074/jbc.R109.072181
Rose NR, Bona C (1993) Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today 14:426–430. doi:10.1016/0167-5699(93)90244-F
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730. doi:10.1038/nrd3802
Ryter SW, Mizumura K, Choi AMK (2014) The impact of autophagy on cell death modalities. Int J Cell Biol 2014:502676. doi:10.1155/2014/502676
Saitoh T, Akira S (2010) Regulation of innate immune responses by autophagy-related proteins. J Cell Biol 189:925–935. doi:10.1083/jcb.201002021
Saitoh T, Fujita N, Jang MH et al (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456:264–268. doi:10.1038/nature07383
Sandri M, Coletto L, Grumati P, Bonaldo P (2013) Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci 126:5325–5333. doi:10.1242/jcs.114041
Schall N, Muller S (2015) Resetting the autoreactive immune system with a therapeutic peptide in lupus. Lupus 24:412–418. doi: 10.1177/0961203314556138
Schall N, Page N, Macri C et al (2012) Peptide-based approaches to treat lupus and other autoimmune diseases. J Autoimmun 39:143–153. doi:10.1016/j.jaut.2012.05.016
Shen S, Kepp O, Kroemer G (2012) The end of autophagic cell death? Autophagy 8:1–3. doi:10.4161/auto.8.1.16618
Shibutani ST, Yoshimori T (2014) A current perspective of autophagosome biogenesis. Cell Res 24:58–68. doi:10.1038/cr.2013.159
Shoji-Kawata S, Sumpter R, Leveno M et al (2013) Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494:201–206. doi:10.1038/nature11866
Singh SB, Davis AS, Taylor GA, Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313:1438–1441. doi:10.1126/science.1129577
Staskiewicz L, Thorburn J, Morgan MJ, Thorburn A (2013) Inhibiting autophagy by shRNA knockdown: cautions and recommendations. Autophagy 9:1449–1450. doi:10.4161/auto.24895
Stephenson LM, Miller BC, Ng A et al (2009) Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 5:625–635. doi:10.4161/auto.5.5.8133
Stricher F, Macri C, Ruff M, Muller S (2013) HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy 9:1937–1954. doi:10.4161/auto.26448
Stylianou K, Petrakis I, Mavroeidi V et al (2011) The PI3K/Akt/mTOR pathway is activated in murine lupus nephritis and downregulated by rapamycin. Nephrol Dial Transplant 26:498–508. doi:10.1093/ndt/gfq496
Sumpter MD, Tatro LS, Stoecker WV, Rader RK (2012) Evidence for risk of cardiomyopathy with hydroxychloroquine. Lupus 21:1594–1596. doi:10.1177/0961203312462757
Temiz P, Weihl CC, Pestronk A (2009) Inflammatory myopathies with mitochondrial pathology and protein aggregates. J Neurol Sci 278:25–29. doi:10.1016/j.jns.2008.11.010
Thomé R, Issayama LK, DiGangi R et al (2014) Dendritic cells treated with chloroquine modulate experimental autoimmune encephalomyelitis. Immunol Cell Biol 92:124–132. doi:10.1038/icb.2013.73
Troncoso R, Paredes F, Parra V et al (2014) Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle 13:2281–2295. doi:10.4161/cc.29272
Tsvetkov AS, Miller J, Arrasate M et al (2010) A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A 107:16982–16987. doi:10.1073/pnas.1004498107
Valdor R, Mocholi E, Botbol Y et al (2014) Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 15:1046–1054. doi:10.1038/ni.3003
Van Kasteren SI, Overkleeft HS (2014) Endo-lysosomal proteases in antigen presentation. Curr Opin Chem Biol 23C:8–15. doi:10.1016/j.cbpa.2014.08.011
Vidal RL, Matus S, Bargsted L, Hetz C (2014) Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci. doi:10.1016/j.tips.2014.09.002
Villadangos JA, Bryant RA, Deussing J et al (1999) Proteases involved in MHC class II antigen presentation. Immunol Rev 172:109–120. doi:10.1111/j.1600-065X.1999.tb01360.x
Xu K, Xu P, Yao J-F et al (2013) Reduced apoptosis correlates with enhanced autophagy in synovial tissues of rheumatoid arthritis. Inflamm Res 62:229–237. doi:10.1007/s00011-012-0572-1
Xu X, Kobayashi S, Chen K et al (2013) Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 288:18077–18092. doi:10.1074/jbc.M113.474650
Xu Y, Jagannath C, Liu X-D et al (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27:135–144. doi:10.1016/j.immuni.2007.05.022
Yamahara K, Yasuda M, Kume S et al (2013) The role of autophagy in the pathogenesis of diabetic nephropathy. J Diabetes Res 2013:193757. doi:10.1155/2013/193757
Yamamoto A, Tagawa Y, Yoshimori T et al (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23:33–42
Yang W, Tang H, Zhang Y et al (2013) Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet 92:41–51. doi:10.1016/j.ajhg.2012.11.018
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12:814–822. doi:10.1038/ncb0910-814
Zhou D, Li P, Lin Y et al (2005) Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 22:571–581. doi:10.1016/j.immuni.2005.03.009
Zhou X, Lu X, Lv J et al (2011) Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis 70:1330–1337. doi:10.1136/ard.2010.140111
Zimmer R, Scherbarth HR, Rillo OL et al (2013) Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202460
Acknowledgments
The authors would like to thank Jean-Paul Briand, Frédéric Gros and Jean-Louis Pasquali for useful discussion and a critical reading of the manuscript. Research in the SM’s laboratory is financially supported by the French Centre National de la Recherche Scientifique and the Laboratory of Excellence Medalis (ANR-10-LABX-0034), Initiative of Excellence (IdEx), Strasbourg University. MW was supported by a Region Alsace and ImmuPharma-France doctoral fellowship
Conflicts of Interests
SM is a consultant of ImmuPharma. MW declares no conflicts of interests
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Wilhelm, M., Muller, S. (2016). Target Autophagy as a Novel Therapeutic Strategy in Autoimmune Diseases. In: Maiuri, M., De Stefano, D. (eds) Autophagy Networks in Inflammation. Progress in Inflammation Research. Springer, Cham. https://doi.org/10.1007/978-3-319-30079-5_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-30079-5_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30077-1
Online ISBN: 978-3-319-30079-5
eBook Packages: MedicineMedicine (R0)