
Abstract
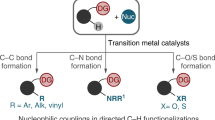
As an alternative to the classical Grignard reaction, transition-metal catalyzed direct addition of aryl C-H bonds to carbonyl groups and their analogues have recently attracted increasing attention due to its atom economy, environmental benefits, abundance of C-H bonds as well as the scientific challenging to achieve mild C-H activations. This chapter briefly summarizes the recent progress in this field, categorized according to different metal catalysts. Applications to synthesize heterocycles and other useful molecules were highlighted as examples. Both the major challenges and the strategies to solve them are discussed in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The Grignard reaction is an important and classical method to construct C‐C bonds by nucleophilic addition of Grignard reagents to carbonyl groups or their derivatives. Despite its high efficiency and broad application in organic synthesis, some limitations still remain. For example, Grignard reagents require strict anhydrous and anaerobic handling, and organic halides, commonly derivatized from C‐H compounds, are employed as the precursors to generate Grignard reagents, which would release stoichiometric halide wastes in hydrolysis step. From the perspective of atom economy, using C‐H bonds rather than C‐X as the precursor of Grignard reagents, bearing an overall 100 % theoretical atom efficacy [1], is a greener methodology to overcome these drawbacks (Scheme 1.1). Over the past decades, transition metal-catalyzed coupling of C‐H bonds to unsaturated alkenes and alkynes was well developed [2–4]; however, the addition of aryl C‐H bonds to polar C=Y (Y = O, N) bonds was relatively rare due to the more covalent nature and weaker nucleophilicity of C‐M (M = transition metal ) bonds derived from C‐H bonds, which make the addition untoward and the addition to C=O bond step reversible, thus the catalytic cycle difficult to proceed [5–8]. In order to solve this dilemma, various methods have been developed. Among these methods, silane capture, C‐H activation /intramolecular annulations (or aromatization), and utilizing highly electron-deficient aldehydes as reaction partners have been the three important strategies.

Comparison of catalytic Grignard-type reaction and classical Grignard reaction
This chapter will give a brief review of catalytic Grignard-type addition of aryl C‐H bonds to C=Y bonds based on different types of transition metals . Due to very different mechanistic pathways, the catalytic addition of aryl C‐H bonds to the C=O bonds of carbon monoxide and carbon dioxide is not included.
1.1 Iridium Catalyzed
In 2002, Murai and co-workers reported the first elegant example of Ir-catalyzed formal direct addition of imidazoles onto aldehydes in the presence of a co-reactant hydrosilane to produce 2-substituted imidazoles (Scheme 1.2) [9]. An air-stable iridium species, tetrairidium dodecacarbonyl [Ir4(CO)12], was applied as the metal catalyst . Using dimethyl acetylenedicarboxylate (DMAD) as a ligand greatly improved the yields. Experimental results indicated that diethylmethylsilane was essential to the reaction because no products could be detected in its absence. The authors speculated that the reaction was initiated by the reduction of aldehydes rather than the activation of the relative acidic C2 position C‐H bonds of imidazoles by the iridium catalyst . The reaction gave higher yields with aliphatic aldehydes than aromatic aldehydes , whereas ketones had the lowest reactivity, which could react with silane to form the side products.

Ir-catalyzed formal direct addition of imidazole onto aldehyde
Using the same iridium catalyst , Ir4(CO)12, but a different ligand, Shi and co-workers successfully coupled pyridines with aryl aldehydes at the C3 position in 2011 (Scheme 1.3) [10]. This reaction fully displayed the power of silane capture methodology in the overall catalytic Grignard-type addition of aryl C‐H bond to C=O bond. Since most intermolecular C‐H functionalizations of pyridines without activating or directing groups generally occur at the C2 position, Shi’s work was the first example to functionalize the C3 C‐H bond. The reaction gave higher yields for electron-deficient aromatic aldehydes than electron-rich aliphatic aldehydes, and aryl halides were compatible despite the tendency to undergo silylation or dehalogenation. Through mechanistic studies, the authors proposed that the reaction started from an active silyl iridium complexes and then underwent a meta selectively pyridyl C‐H bond oxidative addition resulting in the key intermediates. The oxophilic property of silyl group in this key intermediate promoted the insertion of aldehyde C=O bond through the formation of strong Si-O bond. Finally, forming C‐C bond through reductive elimination led to the addition products and an iridium hydride species which reacted with hydrosilane to regenerate the active silyl iridium catalyst .

Ir-catalyzed pyridine C3 C–H bond addition onto aryl aldehyde
In 2009, Shibata and co-workers successfully activated phenyl C‐H bonds using a cationic [Ir(cod)2]BARF catalyst with the assistance of directing groups [11]. Different from the silane capture strategy, they took the advantage of dehydration to form stable aromatic rings as the driving force to achieve the goal. A variety of substituted benzofurans and indoles were synthesized in high yields through this protocol intramolecularly (Scheme 1.4).

Ir-catalyzed intramolecular addition of phenyl C–H bond onto ketone
1.2 Rhenium Catalyzed
In 2006, Takai and co-workers reported a groundbreaking result of a rhenium-catalyzed aromatic ketimines C‐H bonds inserting into polar unsaturated C=O bond (Scheme 1.5) [12], representing an example of C‐H activation /intramolecular annulations/aromatization methodology. Coordination of nitrogen atom from imine group to rhenium facilitated ortho C‐H bond activation; and, more importantly, addition of the generated rhenium-carbon bond to aromatic aldehyde C=O bond was promoted by further intramolecular nucleophilic cyclization and aromatization process. Because of the formed aniline which reacted with 1 equiv aldehydes, 2 equiv aldehydes were thus required to complete the reaction. Addition of molecular sieves to remove water greatly enhanced the product yields. Using this rhenium catalyst , naphthalene derivatives were synthesized by further adding dienophiles to trap the cyclization product [13], whereas phthalimidine derivatives were formed in high yields when replacing the aldehyde by isocyanates [14].

Re-catalyzed ketimine C–H bond addition onto aldehyde C=O bond
Recently, Wang and co-workers reported a rhenium-catalyzed C‐H bond activation of azobenzenes through azo functional group chelating assistance to obtain 2H-indazoles (Scheme 1.6) [15]. They characterized the cyclic ReI complexes to confirm a reversible deprotonation process, and further mechanistic studies revealed an irreversible aldehyde insertion process, which was in sharp contrast to Shi’s and Bergman’s work [16, 17]. Base was crucial to the reaction and acetate acted as a catalytic proton shuttle. The required prolonged reaction time at high temperature might be due to the low reactivity of aldehydes.

Re-catalyzed addition of azobenzene C–H bond onto aldehyde C=O bond
1.3 Manganese Catalyzed
Manganese belongs to the fourth row d block transition metal , which is abundant and inexpensive compared to the fifth and sixth ones. However, manganese complexes are scarcely used as catalysts in C‐H bond activation [18]. In 2007, Takai and co-workers reported a manganese-catalyzed phenyl C‐H addition to aldehyde C=O bonds through using imidazole as directing group and co-reactant silanes to provide silyl ethers (Scheme 1.7) [19]. Similarly with the previous work, the success of this system was also attributed to the oxylic capture by silane.

Mn-catalyzed phenyl C–H bond addition onto aldehyde C=O bond
1.4 Rhodium Catalyzed
Rhodium catalysts have been widely employed for C‐H bond activation . At the beginning, nonpolar unsaturated double or triple carbon-carbon bonds were usually selected as reaction partners to form C‐C bonds [20]. Then, moderately polar molecules such as α,β-unsaturated carbonyl compounds were used to synthesize acyclic or cyclic compounds [21, 22]. However, nucleophilic addition of C‐H bonds to polar unsaturated C=O and C=N bonds in aldehydes , ketones, ketimines, and aldimines was difficult to achieve. Such additions were first achieved in 2011 independently by three groups: C=N bond addition by Bergman/Ellman [23] and Shi [24] and C=O bond addition by our group [25].
1.4.1 Addition onto C=N Bond
In 2011, Bergman and Ellman published a protocol, in which through using nitrogen heterocycles as directing groups and rhodiumIII as the catalyst , aryl C‐H bond could be activated inserting into N-Boc-imines to afford α-branched amine products (Scheme 1.8) [23]. They indicated that [Cp*RhCl2]2/AgSbF6 mixture was an efficient catalytic system and silver additive was crucial by removing halides from the rhodium. The reaction is robust and can tolerate various reactive functional groups such as esters, amides, aldehydes , and aryl chlorides. Further exploration of this reaction revealed that the addition was also effective for polar C=N bond of isocyanates to produce benzamides. Subsequently, they found that N-acyl amino directing groups were also effective for such reactions and they were also more readily available and easily removable [26].

[Cp*RhCl2]2/AgSbF6-catalyzed aryl C–H bond addition onto C=N bond
Independently, Shi’s group reported a similar C=N bond addition in 2011 (Scheme 1.9) [24]. In Shi’s work, they preformed catalyst [Cp*Rh(CH3CN)3][SbF6]2 instead of combining [Cp*RhCl2]2 with AgSbF6. Notably, the reaction could be handled under air despite a small decrease of yield.

[Cp*Rh(CH3CN)3][SbF6]2-catalyzed aryl C–H bond addition onto C=N bond
The two groups proposed two slightly different catalytic cycles both involving the same key steps (Scheme 1.10) [16, 17]. Firstly, pyridinyl directed reversible C‐H activation and deprotonation forms the C‐Rh bond. Secondly, coordination of aldimine to cationic Rh intermediates activates the C=N bond insertion to C‐Rh bond to form the fused 7-membered rhodacycle complex intermediate. Both groups isolated and characterized this key intermediate, and further studies confirmed this insertion step was the rate-determining step. Finally, protonation releases the desired product and regenerates the active catalyst . In addition to pyridinyl, amide [27], N,N-dimethylcarbamoyl [28], and azophenyl [29] were also developed as directing groups to facilitate the rhodium-catalyzed addition of aryl C‐H bond to imines .

Mechanistic cycles proposed by Bergman/Ellman and Shi
1.4.2 Addition onto C=O Bond
Through using highly electron-deficient aldehyde as reaction partner, the challenge of the reversible nature of inserting C‐H bond across the C=O bond was overcome. In 2011, our group developed a rhodium-catalyzed Grignard-type arylation through directly adding C‐H bond to C=O bond to produce alcohols in high yields (Scheme 1.11) [25]. A variety of nitrogen-containing heterocycles were effective as directing groups to give the Grignard-type addition products high yields. Ethyl glyoxylate and highly electron-deficient aromatic aldehydes were found to be satisfied substrates, while benzaldehyde failed. Remarkably, the reaction could proceed efficiently in the presence of water under an air atmosphere and could tolerate a variety of functional groups such as ester, halides, nitro, an additional aldehyde, and free hydroxyl. The proposed mechanism involves the following sequences: oxidative addition of the rhodium catalyst to the ortho C‐H bond assisted by the nitrogen chelation to generate the arylrhodium complex, then aldehyde coordination to rhodium to activate both C=O and facilitate nucleophilic addition, and finally, protonation of rhodium alkoxide to release the Grignard-type addition product and regenerate the rhodium catalyst .

Rh-catalyzed directly C–H bond addition onto C=O bond to produce alcohol
A similar rhodium-catalyzed aryl C‐H addition to highly electron-deficient aldehydes C=O bond to produce biaryl methanols was also reported by Shi’s group in 2012 (Scheme 1.12) [30]. N-directing group was essential to this transformation. Intra- and intermolecular isotopic studies showed that the first step of C‐H bond cleavage was reversible. Compared with aldehydes , ketones are less reactive because of their lower electrophilicity and higher steric hindrance. Based on their previous studies, Shi and co-workers successively developed an approach to directly add phenyl C‐H bonds to ketones intermolecularly by using 2.5 mol% [Cp*RhCl2]2 and 20 mol% AgSbF6 catalysts [31].

Rh-catalyzed directly C–H bond addition onto C=O bond to produce biaryl methanol
Inspired by these transformations, our group further reported a tandem rhodium-catalyzed Grignard-type arylation of aldehydes , which was followed by an intramolecular lactonization to synthesize phthalides in 2012 (Scheme 1.13) [32]. Carboxyl group acted as both a directing group to activate ortho aryl C‐H bond and a nucleophile intramolecularly to form the lactones. Although the reaction requires relatively high reaction temperature, long reaction time, and high loading of expensive rhodium catalyst , this cascade cyclization strategy provides a novel alternative to prepare phthalides.

Rh-catalyzed Grignard-type arylation of aldehyde and intramolecular lactonization
Through choosing proper directing groups , Bergman, Ellman, and co-workers successfully extended the substrate scope to electron-rich aromatic aldehydes and unactivated aliphatic aldehydes (Scheme 1.14) [33]. Imidate directing groups were found to not only activate the ortho C‐H bonds of benzimidates but also capture the alcohol intermediate. In addition, such directing groups were also easier to remove than pyridinyl group. They also found that azo groups can also accomplish the C‐H bond addition to aldehydes; by using 5 mol% [Cp*RhCl2]2, 20 mol% AgSbF6 catalysts , and MgSO4 as the additive in THF for 20 h at 110 °C, 2H-indazole derivatives could be prepared [34]. Similarly, azo group not only played the role of directing group but also served as an intramolecular capture group for aromatization to form stable products. In 2014, Zhu and co-workers reported another example of this strategy. By using N-nitroso as the directing group, through a cascade [2+2] cycloaddition/fragmentation reaction, the indazoles can be synthesized in modest to high yields [35].

Rh-catalyzed imidate directed Grignard-type arylation of aldehyde to form phthalide
1.5 Cobalt Catalyzed
Although cationic Cp*RhIII-catalyzed processes are powerful, the high price of rhodium limits their applications. Therefore, inexpensive and robust alternative catalysts are highly desirable. High-valent cobalt catalyst, being homologous with rhodium but a more abundant first-row transition metal , might possess the ability to promote similar C‐H bond functionalization. Several groups investigated and achieved fruitful results.
In 2012, Yoshikai and co-workers reported a cobalt-N-heterocyclic carbene (NHC)-catalyzed arylation of aldimines through C‐H bond functionalization (Scheme 1.15) [36]. The reaction, however, required the combination with a specific Grignard reagent tBuCH2MgBr; unlike the Rh-catalyzed reactions reported by Bergman/Ellman and Shi, aldimines with N-Boc or N-Ts did not react under such conditions.

Cobalt/NHC-catalyzed aryl C–H bond addition to aldimine C=N bond
Using cationic high-valent Cp*CoIII complex, two groups, Kanai and Ellman, successfully developed Grignard-type addition of aryl C‐H bonds to C=O bonds or C=N bonds (Scheme 1.16). In 2013, Kanai and co-workers established a new cationic high-valent Cp*CoIII complex to promote 2-phenylpyridine [37] and indole [38] C‐H bond addition to sulfonyl imines . In 2015, Ellman and co-workers reported a novel air-stable cationic cobalt(III)-catalyzed C‐H bond addition to aldehydes followed by in situ cyclization and aromatization to produce N-aryl-2H-indazoles [39]. Compared with rhodium catalyst , unfortunately, the cobalt catalyst system gave lower regioselectivities and inferior yields. A catalytic amount of acetic acid (10 mol%) was required as an additive and the reaction could be handled on benchtop in large scale. Using this cationic cobalt complex, Ellman also expanded isocyanates as the reaction partners to produce amides [40]. In addition to N-aryl-1H-pyrazole directing group , other nitrogen heterocycles such as 2-pyridinyl and 2-pyrimidinyl could also achieve good results.

Cationic Cp*CoIII-catalyzed Grignard-type addition reaction
2 Conclusions and Outlook
Catalytic Grignard-type addition of aryl C‐H bonds to polar C=O or C=N bonds mediated by transition metals is a very attractive approach to synthesize alcohols, amines, and their derivatives by avoiding generating stoichiometric amount of metal and halide wastes associated with the classical Grignard-type reaction . In spite of these preliminary success, there are still many challenges to be solved: (1) most reactions require directing groups to assist the coordination of aryl C‐H with transition metal and some of the directing groups such as pyridinyl are not easy to remove; (2) some reactions require super-stoichiometric amounts of substrates in order to completely consume another substrate, which decreases the atom economy ; and (3) the use of precious and toxic late transition metal catalysts , typically with high loadings, hampers their applications. Thus, expanding the substrate scope, without using directing groups, and developing inexpensive catalysts with lower catalyst loading will be the main research directions of this field.
References
Li C-J, Trost BM (2008) Green chemistry for chemical synthesis. Proc Natl Acad Sci 105:13197
Satoh T, Miura M (2010) Oxidative coupling of aromatic substrates with alkynes and alkenes under rhodium catalysis. Chem Eur J 16:11212
Song G, Wang F, Li X (2012) C–C, C–O and C–N bond formation via rhodium(III)-catalyzed oxidative C–H activation. Chem Soc Rev 41:3651
Patureau FW, Wencel-Delord J, Glorius F (2012) Cp*Rh-catalyzed C–H activations: versatile dehydrogenative cross-couplings of Csp2 C–H positions with olefins, alkynes, and arenes. Aldrichim Acta 45:31
Yang L, Huang H (2015) Transition-metal-catalyzed direct addition of unactivated C–H bonds to polar unsaturated bonds. Chem Rev 115:3468
Zhang X-S, Chen K, Shi Z-J (2014) Transition metal-catalyzed direct nucleophilic addition of C–H bonds to carbon–heteroatom double bonds. Chem Sci 5:2146
Colby DA, Tsai AS, Bergman RG, Ellman JA (2012) Rhodium catalyzed chelation-assisted C– H bond functionalization reactions. Acc Chem Res 45:814
Yan G, Wu X, Yang M (2013) Transition-metal-catalyzed additions of C–H bonds to C–X (X = N, O) multiple bonds via C–H bond activation. Org Biomol Chem 11:5558
Fukumoto Y, Sawada K, Hagihara M, Chatani N, Murai S (2002) [Ir4(CO)12]-catalyzed coupling reaction of imidazoles with aldehydes in the presence of a hydrosilane to give 2-substituted imidazoles. Angew Chem Int Ed 41:2779
Li B-J, Shi Z-J (2011) Ir-catalyzed highly selective addition of pyridyl C–H bonds to aldehydes promoted by triethylsilane. Chem Sci 2:488
Tsuchikama K, Hashimoto Y-k, Endo K, Shibata T (2009) Iridium-catalyzed selective synthesis of 4-substituted benzofurans and indoles via directed cyclodehydration. Adv Synth Catal 351:2850
Kuninobu Y, Nishina Y, Nakagawa C, Takai K (2006) Rhenium-catalyzed insertion of aldehyde into a C−H bond: synthesis of isobenzofuran derivatives. J Am Chem Soc 128:12376
Kuninobu Y, Nishina Y, Takai K (2007) Rhenium-catalyzed synthesis of naphthalene derivatives via insertion of aldehydes into a C–H bond. Tetrahedron 63:8463
Kuninobu Y, Tokunaga Y, Kawata A, Takai K (2006) Insertion of polar and nonpolar unsaturated molecules into carbon−rhenium bonds generated by C−H bond activation: synthesis of phthalimidine and indene derivatives. J Am Chem Soc 128:202
Geng X, Wang C (2015) Rhenium-Catalyzed [4 + 1] Annulation of Azobenzenes and Aldehydes via Isolable Cyclic Rhenium(I) Complexes. Org Lett 17:2434
Li Y, Zhang X-S, Li H, Wang W-H, Chen K, Li B-J, Shi Z-J (2012) Mechanistic understanding of Rh-catalyzed N-sulfonylaldimine insertion into aryl C–H bonds. Chem Sci 3:1634
Tauchert ME, Incarvito CD, Rheingold AL, Bergman RG, Ellman JA (2012) Mechanism of the rhodium(III)-catalyzed arylation of imines via C–H bond functionalization: inhibition by substrate. J Am Chem Soc 134:1482
Wang C (2013) Manganese-mediated C–C bond formation via C–H activation: from stoichiometry to catalysis. Synlett 24:1606
Kuninobu Y, Nishina Y, Takeuchi T, Takai K (2007) Manganese-catalyzed insertion of aldehydes into a C-H bond. Angew Chem Int Ed 46:6518
Ritleng V, Sirlin C, Pfeffer M (2002) Ru-, Rh-, and Pd-catalyzed C−C bond formation involving C−H activation and addition on unsaturated substrates: reactions and mechanistic aspects. Chem Rev 102:1731
Lim S-G, Ahn J-A, Jun C-H (2004) Ortho alkylation of aromatic ketimine with functionalized alkene by Rh(I) catalyst. Org Lett 6:4687
Ueura K, Satoh T, Miura M (2007) An efficient waste-free oxidative coupling via regioselective C−H bond cleavage: Rh/Cu-catalyzed reaction of benzoic acids with alkynes and acrylates under air. Org Lett 9:1407
Tsai AS, Tauchert ME, Bergman RG, Ellman JA (2011) Rhodium(III)-catalyzed arylation of Boc-Imines via C−H bond functionalization. J Am Chem Soc 133:1248
Li Y, Li B-J, Wang W-H, Huang W-P, Zhang X-S, Chen K, Shi Z-J (2011) Rhodium-catalyzed direct addition of aryl C–H bonds to N-Sulfonyl aldimines. Angew Chem Int Ed 50:2115
Yang L, Correia CA, Li C-J (2011) Grignard-type arylation of aldehydes via a rhodium-catalyzed C–H activation under mild conditions. Adv Synth Catal 353:1269
Hesp KD, Bergman RG, Ellman JA (2011) Expedient synthesis of N-acyl anthranilamides and β-enamine amides by the Rh(III)-catalyzed amidation of aryl and vinyl C–H bonds with isocyanates. J Am Chem Soc 133:11430
Hesp KD, Bergman RG, Ellman JA (2012) Rhodium-catalyzed synthesis of branched amines by direct addition of benzamides to imines. Org Lett 14:2304
Zhou B, Yang Y, Lin S, Li Y (2013) Rhodium-catalyzed direct addition of indoles to N-sulfonylaldimines. Adv Synth Catal 355:360
Wangweerawong A, Bergman RG, Ellman JA (2014) Asymmetric Synthesis of α-Branched Amines via Rh(III)-Catalyzed C–H Bond Functionalization. J Am Chem Soc 136:8520
Li Y, Zhang X-S, Chen K, He K-H, Pan F, Li B-J, Shi Z-J (2012) N-directing group assisted rhodium-catalyzed aryl C–H addition to aryl aldehydes. Org Lett 14:636
Zhang X-S, Zhu Q-L, Luo F-X, Chen G, Wang X, Shi Z-J (2013) Aromatic C–H addition to ketones: the effect of directing groups. Eur J Org Chem 2013:6530
Shi X, Li C-J (2012) A novel rhodium-catalyzed cascade cyclization: direct synthesis of 3-substituted phthalides from aldehydes and aromatic acids. Adv Synth Catal 354:2933
Lian Y, Bergman RG, Ellman JA (2012) Rhodium(III)-catalyzed synthesis of phthalides by cascade addition and cyclizationof benzimidates with aldehydes. Chem Sci 3:3088
Lian Y, Bergman RG, Lavis LD, Ellman JA (2013) Rhodium(III)-catalyzed indazole synthesis by C–H bond functionalization and cyclative capture. J Am Chem Soc 135:7122
Chen J, Chen P, Song C, Zhu J (2014) Rhodium(III)-catalyzed N-nitroso-directed C–H addition to ethyl 2-oxoacetate for cycloaddition/fragmentation synthesis of indazoles. Chem Eur J 20:14245
Gao K, Yoshikai N (2012) Cobalt-catalyzed arylation of aldimines via directed C–H bond functionalization: addition of 2-arylpyridines and self-coupling of aromatic aldimines. Chem Commun 48:4305
Yoshino T, Ikemoto H, Matsunaga S, Kanai M (2013) A cationic high-valent Cp*CoIII complex for the catalytic generation of nucleophilic organometallic species: directed C–H bond activation. Angew Chem Int Ed 52:2207
Yoshino T, Ikemoto H, Matsunaga S, Kanai M (2013) Cp*CoIII-catalyzed C2-selective addition of indoles to imines. Chem Eur J 19:9142
Hummel JR, Ellman JA (2015) Cobalt(III)-catalyzed synthesis of indazoles and furans by C–H bond functionalization/addition/cyclization cascades. J Am Chem Soc 137:490
Hummel JR, Ellman JA (2015) Cobalt(III)-catalyzed C–H bond amidation with isocyanates. Org Lett 17:2400
Acknowledgments
We are indebted to our colleagues, whose name are given in the list of references. We also thank the Canada Research Chair (Tier I) foundation; the CFI, NSERC, and FQRNT; and the CSC (China Scholarship Council) for a postdoctoral scholarship to Feng Wang.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Wang, F., Liu, W., Li, CJ. (2016). Catalytic Grignard-Type Addition of Aryl C‐H Bonds to C=O and C=N Bonds. In: Tundo, P., He, LN., Lokteva, E., Mota, C. (eds) Chemistry Beyond Chlorine. Springer, Cham. https://doi.org/10.1007/978-3-319-30073-3_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-30073-3_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-30071-9
Online ISBN: 978-3-319-30073-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)