
Abstract
Juvenile Polyposis is a hamartomatous polyposis syndrome with an autosomal dominant inheritance pattern. Patients commonly present with anemia or rectal bleeding in the first two decades of life. These polyps may range from a few to over a 100 in number, and most often are located within the colon or rectum, but may also be found in the stomach and occasionally in the small intestine. Germline mutations in BMPR1A or SMAD4 are responsible for approximately one-half of cases, and the predisposing gene for the other half is unknown. The diagnosis of JP carries with it an increased risk of GI malignancy, particularly of the colon, rectum, and stomach. Screening with colonoscopy and upper endoscopy begins when symptoms develop or at age 15, and colon polyps should be removed endoscopically when possible. Surgical treatment is reserved for those with large numbers of polyps, polyps with dysplasia or cancer, significant anemia, or extensive gastric involvement.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Juvenile polyps are the most common polyps seen in children [1]. It has been estimated that as many as 1 % of the population will have one of these polyps in their lifetime, but in most cases these disappear, and patients do not have ongoing issues related to them, such as bleeding or prolapse. In some individuals, these polyps are multiple, and may continue to form throughout a person’s life. These people have a different situation, where they are born with an autosomal dominant syndrome predisposing them to developing these polyps. It has been estimated that this condition, Juvenile Polyposis (JP), affects approximately 1 in 100,000 individuals [2]. There is an equal incidence between males and females, and an increased incidence in individuals of Northern European descent [3]. JP patients most commonly develop hamartomatous polyps throughout the colon (Fig. 6.1), but may also have polyps in other portions of the GI tract as well, usually within the stomach (Fig. 6.2). Patients with JP have an increased risk of GI cancers, with the highest risk being for colorectal cancers, but there is also increased risk for gastric and pancreatic tumors.

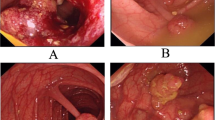
Multiple juvenile polyps in the cecum. Note many diminutive polyps and several larger, red, pedunculated, and multilobular polyps

Gastric polyposis at the GE junction, sparing the more distal stomach in a JP patient. Note the diffuse, frond-like nature, rather than the pedunculated polyps as seen in the colon
The earliest case of JP described in the literature is unclear, but some attribute this to Hertz et al. in 1914. He described a family consisting of four children all having rectal polyps and bleeding, and asymptomatic parents [4]. These polyps were never confirmed to be juvenile histologically, but this was very likely the earliest description of a JP family. In 1939, Diamond reported a 30-month-old child with constipation and hematochezia with a pedunculated, sessile polyp of the rectum, prone to prolapse [5]. This 2.5 cm polyp, although described as an adenoma, had the histologic features we have come to know as a juvenile polyp.
Helwig described the histologic findings of hamartomatous polyps in 1946, including stroma embedded with mucus-filled, glandular structures, and associated inflammatory cell infiltration. There were no dysplastic or adenomatous changes noted within the epithelium [6]. Ravitch described a 10 month old with upper and lower GI juvenile polyps in 1948, who suffered from severe anemia, malnutrition, and prolapsing rectal polyps, and subsequently died from this at an early age [7]. In 1957, Horilleno first introduced the term hamartomatous polyp [8], and shortly thereafter, Morson spelled out the differences between adenomatous, inflammatory, Peutz-Jeghers, and Juvenile polyps [9].
Despite these previous observations and Hertz’s early report, it was not until 1966 that an autosomal dominant inheritance pattern was suggested by Smilow and associates, after studying a three generation family with JP [10]. This was further reinforced by a 1975 report by Stemper and associates, who described a kindred with ten affected individuals with colorectal or gastric polyps. There were also 11 members of the family who developed GI cancer, predominantly of the colon, but also of the stomach, duodenum, and pancreas [11]. The link between hamartomatous polyps and GI cancer was strengthened by the finding of Liu et al. of a focus of signet ring cell carcinoma within a juvenile polyp in a 16-year-old boy [12]. Since then, multiple reports have confirmed this relationship between JP and the development of GI cancer [13–16].
Morphology and Histology
Juvenile polyps are frequently rounded and pedunculated on a stalk. They may range in size from a few mm to up to 5 cm. They can also be sessile, especially when in the stomach. Their surface has a thin mucosa, which may become eroded, leading to bleeding [17, 18]. In those with JP, there may be just a few polyps at a time, or there may be a 100 or more, even within members of the same family. Looking at them microscopically, there is an expansion of the lamina propria with abundant stroma, cystically dilated glands, and infiltration of inflammatory cells (Fig. 6.3). Overlying this markedly expanded lamina propria is a relatively normal layer of epithelium. This epithelium can occasionally become dysplastic, but this is relatively rare. When biopsied, these hamartomatous polyps may be diagnosed as juvenile polyps, hyperplastic polyps, or inflammatory polyps. They differ from hamartomatous polyps in Peutz-Jeghers patients in that the latter contain areas of smooth muscle within the lamina propria.

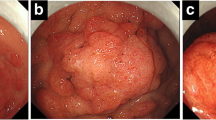
Microscopic view of a polyp showing an expanded lamina propria with cystically dilated glands with inflammatory infiltrate, covered by thin layer of epithelium
Clinical Presentation
The most common presentation is anemia, which is often accompanied by rectal bleeding. Other signs and symptoms may also include abdominal pain, diarrhea, prolapse of a rectal polyp, and intussusception [19, 20]. The most frequent associated anomalies include macrocephaly, mental retardation, Meckel’s diverticulum, malrotation, pulmonary arteriovenous malformations (AVMs), telangiectasias, atrial and ventricular septal defects, pulmonic stenosis, and cryptorchidism [21, 22]. The diagnosis of JP is made based on the clinical criteria initially proposed by Sachatello et al. [23], with the number of polyps later reduced from 10 to 5 by Jass and colleagues [24]. These criteria for JP require
-
1.
At least 5 juvenile polyps within the colorectum; or
-
2.
Juvenile polyps in both the upper and lower GI tract; or
-
3.
Any number of juvenile polyps in a patient with a family history of JP
After one of these conditions is satisfied, JP patients can be further subclassified into Juvenile Polyposis Coli (where patients have only colorectal polyps), generalized Juvenile Polyposis (where patients have polyps in both the upper and lower GI tract), and JP of infancy. These first two usually present with rectal bleeding, prolapse, anemia, diarrhea, or abdominal pain within the first two decades of life [25, 26]. The latter condition is uncommon, but has an earlier and more severe presentation, including protein losing enteropathy, anemia, anasarca, bloody diarrhea, failure to thrive, and often death before age 1 [23, 27, 28].
Other conditions that may present in a similar fashion include the PTEN Hamartoma Tumor syndromes of Bannayan–Riley–Ruvalcaba syndrome (BRRS) and Cowden syndrome (CS). Polyps in patients with these conditions are histologically indistinguishable, but these syndromes can be differentiated by genetic testing and other characteristic phenotypic features. Patients with BRRS may have macrocephaly, developmental delay, prominent corneal nerves, lipid myopathy, lipomas, genital pigmentation, and angiolipomas [29]. Patients with CS have facial trichilemmomas, acral keratoses, papules, breast cancer, fibrocystic disease of the breast, benign or malignant thyroid lesions, mental retardation, lipomas, or fibromas [30].
Genetics
The autosomal dominant inheritance pattern for JP was first revealed in the 1966 publication of three affected members in three generations by Smilow et al. [10]. Approximately 75 % of patients with JP have a family history, and the remainder have de novo mutations leading to JP [31]. Clues to the genes predisposing to JP remained elusive until the late 1990s. In 1997, Jacoby described a patient with features of JP and macrocephaly, who had a deletion of chromosome 10q22 [32]. Olschwang et al. then reported three patients with germline PTEN mutations (which maps to 10q22) thought to have JP [33], but further scrutiny suggested that these patients might actually have had CS rather than JP [34]. Additional studies examining JP patients for germline PTEN mutations were negative, confirming the idea that PTEN was not the JP gene [35, 36].
SMAD4
It was in 1998 that the first hard evidence for the location of a JP gene was established. Howe et al. studied 43 members (including 13 affected) of the family originally reported by Stemper et al. [11], and established genetic linkage with 6 markers from chromosome 18q21, with a maximum lod score of 5.00 with D18S1099 (at θ = 0.001). Critical recombinants placed the JP gene within a 11 cM region between D18S118 and D18S487 [37, 38]. Sequencing of candidate genes from this region revealed that all affected members of this kindred shared a frameshift mutation, a 4 base pair deletion in exon 9 of the SMAD4 gene. In this report, 5 of 9 total JP families tested were found to have germline SMAD4 mutations [39]. This finding was soon confirmed by several other investigators in additional JP families [40–42].
SMAD4 was originally called deleted in pancreatic cancer 4 (DPC4) , because of the fact that it is inactivated in approximately 50 % of pancreatic adenocarcinoma specimens [43]. DPC4 was later renamed SMAD4 when it became clear that this was the common intracellular mediator of the transforming growth factor-beta (TGFβ) superfamily, which signals through SMAD genes [44, 45]. SMAD4 is comprised of 11 coding exons, encoding a 552-amino acid cytoplasmic protein. Within the TGFβ, activin, bone morphogenetic protein (BMP), and inhibin pathways, the function of SMAD4 is to bind to SMAD proteins phosphorylated by the type 1 receptor, after it has been phosphorylated by the type 2 receptor following ligand binding. SMAD4 binds to co-SMADs 1, 5, and 8 in the BMP pathway, and with co-SMADs 2 and 3 in the TGFβ pathway. The complex of SMAD4 and these co-SMADs then migrates to the nucleus, where it recruits a DNA-binding protein, and then binds directly to specific promoters to regulate transcription (Fig. 6.4).

Diagram of the signaling pathway of TGFβ depicting its interactions with BMPR1A and SMAD 4 to facilitate changes in nuclear transcription. BMPR1A works via SMAD 1, 5, and 8 while TGFβ works through SMAD 2 and 3. These join a common pathway with SMAD4 which creates an oligomer that translocates into the nucleus to regulate transcription [31]
In a 2009 paper by Calva et al., 77 of 357 JP probands (21.6 %) were found to have germline SMAD4 mutations by sequencing. Of these mutations, 78 % were within exons 8–11 (encoding for the mad homology 2 domain), 17 % within exons 3–7, and 5 % in exons 1 and 2 (Fig. 6.5) [46]. A smaller percentage of JP patients have been found to have deletions of SMAD4, which have been identified using the mixed ligation-dependent probe amplification assay (MLPA) [47, 48]. Combining the largest studies using this technique, 7 of 128 (7 %) of JP probands were found to have SMAD4 deletions [31].

Distribution of mutations in SMAD4. The rectangle above represents the coding exons of the gene, with nucleotides at each exon boundary shown underneath. The rectangle below represents the protein, with corresponding nucleotide numbers listed above and below domain boundaries. Mutations listed above the exons are those described in the present study, and those below the exons are from the literature. MH1, mad homology 1; MH2, mad homology 2 (Figure originally from Clinical Genetics) [46]
BMPR1A
After it became apparent that only 20 % of JP families had germline changes in SMAD4, it seemed likely that there must be other JP genes. In 2001, Howe et al. found a suggestion of a second JP locus in four unrelated JP families, with a lod score of 2.33 at θ = 0.10 with the marker D10S573. This was in the vicinity of the PTEN gene on chromosome 10q22-23, but these families had already been sequenced for PTEN and found to not have mutations. Another gene involved in the TGFβ superfamily was found to map to this general region, and they identified 2 polymorphic simple tandem repeat markers 49 and 76 kb upstream from this gene. When these markers were tested in these four families, the maximum lod scores were 4.17 and 4.74 at θ = 0, proving linkage at this locus. Sequencing in these families revealed that all affected members had germline mutations of BMPR1A, which were frameshift in two families and nonsense mutations in the two others [49]. This finding was confirmed in other JP families shortly thereafter [50, 51]. BMPR1A encodes for the type I receptor in the BMP pathway, and is comprised of 11 coding exons. This receptor is a 532-amino acid transmembrane protein that associates with and is phosphorylated by the type II receptor (BMPR2) after it binds to extracellular BMP ligands. BMPR1A then phosphorylates co-SMADs 1, 5, and 8, which form oligomers that bind to intracellular SMAD4. This complex then migrates to the nucleus, recruits DNA-binding proteins, then binds directly to promoters to activate transcription (Fig. 6.4) [31, 37].
Calva et al. found that 62 of 336 JP probands (18.5 %) had germline BMPR1A mutations by sequencing. Of these mutations, 52 % were within the intracellular protein kinase domain (exons 7–11), and 31 % in the Mad homology I domain (exons 2–4). No mutations have been described within the transmembrane region of the receptor, and the mutations seen in BMPR1A are more uniformly spread out and more likely to be unique than seen with SMAD4 mutations in JP patients (Fig. 6.6) [46]. A small percentage of JP patients have been found to have larger deletions of BMPR1A by MLPA, affecting 8 of 128 probands tested (6.3 %) [31, 47, 48]. Another small group of JP patients have been found with mutations in the promoter of BMPR1A, including one family with ten affected individuals all sharing a 12,433 bp deletion lying 119 kb upstream from the coding region, deleting a non-coding exon and a promoter. This paper reported five unrelated probands with deletion or missense mutations of the BMPR1A promoter which led to reduced luciferase activity in in vitro promoter constructs [52]. One study recreating JP patient BMPR1A missense mutations in a cell line demonstrated that the protein was held up intracellularly and did not efficiently translocate to the cell membrane, suggesting one potential mechanism through which BMP signaling may be reduced [53].

Distribution of mutations in BMPR1A. The rectangle above represents the coding exons of the gene, with nucleotides at each exon boundary shown underneath. The rectangle below represents the protein, with corresponding nucleotide numbers listed above and below domain boundaries. Mutations listed above the exons are those described in the present study, and those below the exons are from the literature (Figure originally from Clinical Genetics) [46]
Other JP Genes
Sweet et al. reported that 2 of 14 JP patients that did not have either SMAD4 or BMPR1A germline mutations had changes in the endoglin (ENG) gene , one of the genes responsible for hemorrhagic hereditary telangiectasia (HHT); neither of these two probands had signs of HHT [54]. Howe et al., by sequencing ENG, found substitutions in ENG in 6 of 31 JP probands (without SMAD4 or BMPR1A mutations), but these substitutions were also found in control patients, and this study concluded that it was not clear whether ENG was a predisposing gene for JP [55]. No confirmatory studies have been published since these reports to confirm that ENG really is a gene predisposing to JP.
The confusion surrounding whether PTEN is a JP gene was discussed earlier, and patients with juvenile polyps and germline PTEN mutations are likely to have CS or BRRS rather than JP. However, some patients with JP have been described that have contiguous deletions of both PTEN and BMPR1A, which lie within 1.1 Mb of one another on chromosome 10q22-23. Delnatte et al. described four patients with deletions of both these genes, all of who had presentation at an early age, upper and lower GI juvenile polyps, and macrocephaly, suggestive of JP of infancy [56]. Salviati et al. described a JP patient with deletion of both genes, presenting at age 3 with mild dysmorphic features, but not macrocephaly [57]. Menko et al. described four additional cases, in which all patients had macrocephaly and dysmorphic features [58]. The effect of contiguous deletion of these two genes is not entirely clear, but appears to result in a more severe phenotype, combining some features of BRRS with those of JP, and sometimes with JP of infancy [59].
Genotype–Phenotype Correlations
Patients with mutations of SMAD4 are more likely to have gastric polyposis than patients with BMPR1A mutations [48, 51, 60, 61]. In 2007, Aretz et al. found that 72 % of patients who had upper endoscopy results and a SMAD4 mutation were found to have gastric polyps while only 8 % of patients with BMPR1A mutations that had upper endoscopy were found to have polyps. On average, gastric polyps were found much later in life with a median age of 41 years at the time of discovery [48]. Sayed et al. showed that JP patients with germline SMAD4 mutations had a higher rate of positive family history of UGI polyps than those with BMPR1A mutations (86 % vs. 10 %, p < 0.01) [60]. Juvenile polyps from patients with SMAD4 germline mutations generally have a more proliferative epithelium and decreased stroma when compared to polyps from patients with BMPR1A germline mutations [62]. Handra-Luca et al. found that patients with germline SMAD4 mutations had more low-grade adenomas than those with BMPR1A mutations, and that only patients with SMAD4 mutations had high-grade dysplasia or carcinomas within their polyps [61].
Combined JP and HHT
In the early 1980s, Cox et al. and Conte et al. described individuals with JP that also had pulmonary AVMs, telangiectasias, and digital clubbing [63, 64]. In 1999, Inoue described a teenage girl who presented with nosebleeds beginning at age 6 and rectal bleeding at age 14. Work-up revealed 30 colonic juvenile polyps, and features of HHT (telangiectasias of the skin, a dilated hepatic artery, and pulmonary AVMs) [65].
Gallione et al. studied 14 individuals from 7 families with combined JP and HHT, none of whom had mutations of the two known HHT genes, ENG or the activin receptor type I (ACVR1). All of the patients were found to have germline SMAD4 mutations, three of which were de novo [66]. Gallione et al. later tested 30 unselected patients with HHT who did not have ENG or ACVR1 mutations and found that 3 had SMAD4 mutations and endoscopic evidence of JP [67]. The frequency of HHT in JP patients with known SMAD4 mutations has been reported to be between 15 % and 22 % [48, 66]. The majority of the SMAD4 mutations resulting in this combined syndrome are within the MH2 domain of the gene involving exons 8–11 [66]. Mutations at other sites within the gene have been described but are less common [68]. The phenotypic presentation of combined JP/HHT due to SMAD4 mutations is variable, but includes multiple juvenile polyps, mucocutaneous telangiectasias, pulmonary AVMs, hepatic AVMs, cerebral AVMs, GI AVMs, and epistaxis [37]. O’Malley et al. analyzed 21 individuals with JP/HHT and found epistaxis in 71 %, pulmonary AVMs in 81 %, visceral AVMs in 86 %, and telangiectasias in 57 % of patients [69]. Wain et al. found that 76 % of JP patients with SMAD4 mutations had some feature of HHT [70]. Based upon the results of this study, combined JP and HHT appears to be much more common than originally thought, and screening for HHT should be strongly considered in any JP patient found with germline SMAD4 mutation.
The Malignant Potential of Juvenile Polyps
When JP was first described, most people felt that since these polyps were hamartomatous, that they had no malignant potential. This continued despite several examples of patients having both JP and GI cancers, and even the publication of the large Iowa kindred in 1975 with 11 individuals with GI cancer was careful not to stress the connection between juvenile polyps and GI cancer [11]. Further histologic investigation of 1032 polyps from 80 JP patients by Jass et al. revealed that 840 were typical juvenile polyps, 169 were multilobulated or villous polyps, 21 were adenomas, and 2 hyperplastic polyps. A total of 9 % of the juvenile polyps harbored dysplastic changes while 47 % of the villous polyps had dysplasia. They estimated the risk of developing colorectal cancer to be greater than 50 % for patients with JP, with a mean age of onset of colorectal cancer at 34 years of age [24]. Giardiello and colleagues found that only 4 % of patients with 1–2 juvenile polyps and no family history of JP had adenomas or adenocarcinoma, versus 29 % of those with >3 polyps or a family history of JP. The mean age at diagnosis of neoplasia was 37 years old for the JP patients within this study [71].
Further evidence that carcinoma may develop from juvenile polyps has been provided by several case reports. In 1978, Liu et al. described a 16 years old with an adenocarcinoma arising from within a juvenile polyp [12]. In 1979, Goodman et al. described a case of a 23-year-old woman with multiple upper and lower GI juvenile polyps who underwent proctocolectomy and antrectomy. Several different kinds of polyps were seen, including small hyperplastic polyps, typical juvenile polyps, juvenile polyps with adenomatous epithelium, and adenomas. There was a rectal adenocarcinoma among the polyps, and the authors suggested that there was likely a progression from hyperplastic to adenomatous change in JP that eventually leads to adenocarcinoma [13]. In support of this, Jarvinen and Fransilla reported 2 JP patients with severe dysplastic change in juvenile polyps [14] and Ramaswamy et al. described a 19 years old with generalized JP and dysplastic changes with carcinoma in situ [15]. Jones et al. described a case of an intramucosal carcinoma arising within a typical juvenile polyp in a patient without JP, suggesting that cancer can arise in these lesions, albeit very rarely [72]. Longo et al. reported a case of a patient with generalized JP and osteoarthropathy who had a subtotal colectomy at age 6, and then a proctectomy and Swenson pull-through at age 12 (leaving 2 cm of rectum). At age 17, he developed a large tubulovillous adenoma in the rectal remnant which was removed, then 2 years later underwent completion proctectomy for what proved to be a poorly differentiated adenocarcinoma arising within a juvenile polyp [73].
Coburn studied 218 JP patients and found that the mean age of diagnosis was 18.5 years for JP and 35.5 years for GI cancer. A total of 36 patients (17 %) developed GI cancer, 34 of the colorectum, 1 gastric, and 1 duodenal [74]. Howe et al. found that within the Iowa JP kindred, that 16/29 affected members (55 %) developed GI cancers, including 11 (38 %) with colorectal cancer, and 6 (21 %) with upper GI cancers (4 gastric, 1 pancreatic, 1 duodenal). The median age at presentation or diagnosis of JP was 32.7 years (range 6.0–68.2 years), and the median age of colorectal cancer was 42.0 years (the youngest was 17.4 years old) and 57.6 years for upper GI cancers (the youngest was 20.5 years old) [75]. Brosens et al. studied 84 patients from 44 families and found 8 with colorectal cancer (mean age of 43.9 years) and calculated the lifetime colorectal cancer risk to be 38 % (and a 34-fold increased relative risk compared to the normal population). They had no cases of upper GI cancer in their cohort [76].
The process by which juvenile polyps undergo transformation to malignant polyps has not been thoroughly established, although the development of adenomatous elements and later adenocarcinoma as suggested by Goodman et al. seems most plausible [13]. The specific mechanisms of how germline mutations in SMAD4 or BMPR1A lead to polyps and cancer continue to be a matter of speculation. One theory, the landscaper hypothesis, postulates that stromal changes lead to cancer in the overlying epithelium. This is based upon the observation that the majority of histologic changes within juvenile polyps are found within the stroma, which might then create an abnormal landscape, and aberrant signaling (presumably in the BMP or TGFβ pathway) in this layer leads to neoplastic change within the adjacent epithelium [77]. Another possibility is a tumor suppressor model, where germline mutation of one copy followed by somatic loss of the other within epithelial cells leads to cancer [78, 79]. Although neither of these models has been definitively proven, both should be considered useful paradigms of how polyps may transform into cancers.
Management
Patients presenting with symptoms of JP, including rectal bleeding, changes in stool, abdominal pain, and intussusception should be worked up with a thorough history and physical. If colonoscopic evaluation yields findings consistent with a possible diagnosis of JP (see diagnostic criteria) then follow-up should be performed as described below. In addition, individuals who are first-degree relatives of those with JP should also be worked up.
Individuals from a JP family with a known mutation in either SMAD4 or BMPR1A should undergo genetic testing within the first 5 years of life [31]. If the results of the genetic test are negative, then the patient does not need enhanced surveillance and may follow the same recommendations for colorectal cancer screening as the normal population. Patients who are found to have a mutation should follow the same screening regimen recommended for all JP patients. This includes colonoscopy and upper endoscopy beginning either at age 15 or earlier if symptoms develop (such as anemia, bleeding, and abdominal pain). If an individual is found to have polyps, the gastroenterologist or surgeon should attempt to remove them colonoscopically. If polyps are found, then repeat screening in 1 year is recommended, unless the polyps cannot be cleared. If no polyps are found, then colonoscopy and upper endoscopy can be extended out to every 3 years. If no mutation has been found within a family with JP, but a patient is at risk by virtue of having an affected first-degree relative, then this person should undergo the same surveillance as recommended for someone with a known mutation above (Fig. 6.7) [80].

Recommended screening algorithm for patients at risk for JP. This screening guideline is recommended for anyone who meets the criteria for JP or who otherwise has a family history of JP with unknown polyp status. CBC complete blood count (Figure originally from Surgery) [79]
When polyps are found, the management has been evolving. The early recommendations from Sachatello et al. were for polypectomy or fulguration with resection of the affected bowel outside the rectum [28]. Grosfield et al. were more aggressive and recommended subtotal colectomy with ileorectal anastomosis (IRA) for patients with anemia from chronic rectal bleeding, hypoproteinemia resulting in failure to thrive, and recurrent intussusceptions [81]. These indications were expanded to all JP patients by Jarvinen et al. in 1993 when they recommended prophylactic colectomy with IRA for patients with JP in their early 20s, in order to reduce the risk of colorectal cancer [82]. Oncel et al. compared their results of performing subtotal colectomy with IRA with total proctocolectomy and ileoanal pouch (IPAA) in JP patients. The functional results were better with IRA, and although 4 of 7 IRA patients eventually underwent completion proctectomy, the authors concluded that both procedures were reasonable options [83]. Howe et al. supported the use of subtotal colectomy with IRA in severe cases (100 or more polyps, significant anemia, dysplasia), but recommended aggressive colonoscopic polypectomy as initial treatment for colorectal polyps in JP patients. In those having resection and IRA, screening would include flexible sigmoidoscopy and upper endoscopy every 3 years [75]. Patients who have colonoscopic polypectomy should be screened yearly until polyp free, and then every 3 years thereafter.
The treatment of gastric polyps is more technically difficult due to their sessile nature and the fact that polyps are more diffuse and without well-defined stalks. If significant anemia develops, or polyps develop dysplastic changes, then subtotal or total gastrectomy is recommended. As described earlier, patients with SMAD4 mutations are at increased risk and may need more frequent screening than patients with BMPR1A or unknown mutations.
Summary
Our understanding of JP has come a long way over the past 2 decades. It has been recognized that despite the fact that patients have hamartomatous polyps, they are at significant risk for colorectal and upper GI malignancies. Two predisposing genes have been clearly identified which cause JP, and there are likely others since these only explain roughly one-half of cases. Understanding the genetics of JP has helped clarify and allowed separation from other hamartomatous polyposis syndromes, and suggested potential mechanisms by which cancers may develop in these patients. Screening algorithms have been suggested for JP based upon the age of onset of cancers and symptoms, and taking into account the results of genetic testing.
References
Adolph VR, Bernabe K. Polyps in children. Clin Colon Rectal Surg. 2008;21(4):280–5.
Burt RW, Bishop DT, Lynch HT, Rozen P, Winawer SJ. Risk and surveillance of individuals with heritable factors for colorectal cancer. WHO Collaborating Centre for the Prevention of Colorectal Cancer. Bull World Health Organ. 1990;68(5):655–65.
Jass J. Pathology of polyposis syndromes with special reference to juvenile polyposis. Hereditary Colorectal Cancer. 1990;345–250.
Hertz AF. Four cases of rectal polypus occurring in one family. Proc R Soc Med. 1914;7(Surg Sect):255–6.
Diamond M. Adenoma of the rectum in children: report of a case in a thirty month old girl. Am J Dis Child. 1939;57:360–7.
Helwig EB. Adenomas of the large intestine in children. Am J Dis Child. 1946;72:289–95.
Ravitch MM. Polypoid adenomatosis of the entire gastrointestinal tract. Ann Surg. 1948;128(2):283–98.
Horrilleno EG, Eckert C, Ackerman LV. Polyps of the rectum and colon in children. Cancer. 1957;10(6):1210–20.
Morson BC. Some prominent personalities in the history of St. Mark’s Hospital. Dis Colon Rectum. 1962;5:173–83.
Smilow PC, Pryor Jr CA, Swinton NW. Juvenile polyposis coli. A report of three patients in three generations of one family. Dis Colon Rectum. 1966;9(4):248–54.
Stemper TJ, Kent TH, Summers RW. Juvenile polyposis and gastrointestinal carcinoma. A study of a kindred. Ann Intern Med. 1975;83(5):639–46.
Liu TH, Chen MC, Tseng HC, Chou L, Lu C. Malignant change of juvenile polyp of colon: a case report. Chin Med J (Engl). 1978;4(6):434–9.
Goodman ZD, Yardley JH, Milligan FD. Pathogenesis of colonic polyps in multiple juvenile polyposis: report of a case associated with gastric polyps and carcinoma of the rectum. Cancer. 1979;43(5):1906–13.
Jarvinen H, Franssila KO. Familial juvenile polyposis coli; increased risk of colorectal cancer. Gut. 1984;25(7):792–800.
Ramaswamy G, Elhosseiny AA, Tchertkoff V. Juvenile polyposis of the colon with atypical adenomatous changes and carcinoma in situ. Report of a case and review of the literature. Dis Colon Rectum. 1984;27(6):393–8.
Rozen P, Baratz M. Familial juvenile colonic polyposis with associated colon cancer. Cancer. 1982;49(7):1500–3.
Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–62. quiz 63.
Brosens LA, Langeveld D, van Hattem WA, Giardiello FM, Offerhaus GJ. Juvenile polyposis syndrome. World J Gastroenterol. 2011;17(44):4839–44.
Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100(2):476–90.
Chow E, Macrae F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol. 2005;20(11):1634–40.
Brosens LA, van Hattem WA, Jansen M, de Leng WW, Giardiello FM, Offerhaus GJ. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7(1):29–46.
Desai DC, Murday V, Phillips RK, Neale KF, Milla P, Hodgson SV. A survey of phenotypic features in juvenile polyposis. J Med Genet. 1998;35(6):476–81.
Sachatello CR. Polypoid diseases of the gastrointestinal tract. J Ky Med Assoc. 1972;70(7):540–4.
Jass JR, Williams CB, Bussey HJ, Morson BC. Juvenile polyposis—a precancerous condition. Histopathology. 1988;13(6):619–30.
Roth SI, Helwig EB. Juvenile polyps of the colon and rectum. Cancer. 1963;16:468–79.
Reed K, Vose PC. Diffuse juvenile polyposis of the colon: a premalignant condition? Dis Colon Rectum. 1981;24(3):205–10.
Soper RT, Kent TH. Fatal juvenile polyposis in infancy. Surgery. 1971;69(5):692–8.
Sachatello CR, Hahn IS, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: report of a case and review of the literature of a recently recognized syndrome. Surgery. 1974;75(1):107–14.
Gorlin RJ, Cohen Jr MM, Condon LM, Burke BA. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44(3):307–14.
Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70(4):829–44.
Calva D, Howe J. Juvenile polyposis. In Riegert-Johnson DL, Boardman LA, Hefferon T, Roberts M, editors. Cancer syndromes. Bethesda MD: National Center for Biotechnology Information, USA; 2009.
Jacoby RF, Schlack S, Cole CE, Skarbek M, Harris C, Meisner LF. A juvenile polyposis tumor suppressor locus at 10q22 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 1997;112(4):1398–403.
Olschwang S, Serova-Sinilnikova OM, Lenoir GM, Thomas G. PTEN germ-line mutations in juvenile polyposis coli. Nat Genet. 1998;18(1):12–4.
Eng C, Peacocke M. PTEN and inherited hamartoma-cancer syndromes. Nat Genet. 1998;19(3):223.
Marsh DJ, Roth S, Lunetta KL, Hemminki A, Dahia PL, Sistonen P, et al. Exclusion of PTEN and 10q22-24 as the susceptibility locus for juvenile polyposis syndrome. Cancer Res. 1997;57(22):5017–21.
Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997;57(13):2578–80.
Larsen Haidle J, Howe JR. Juvenile polyposis syndrome. In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle, WA: University of Washington, Seattle; 1993.
Howe JR, Ringold JC, Summers RW, Mitros FA, Nishimura DY, Stone EM. A gene for familial juvenile polyposis maps to chromosome 18q21.1. Am J Hum Genet. 1998;62(5):1129–36.
Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280(5366):1086–8.
Houlston RS, Tomlinson IP. Modifier genes in humans: strategies for identification. Eur J Hum Genet. 1998;6(1):80–8.
Roth S, Sistonen P, Salovaara R, Hemminki A, Loukola A, Johansson M, et al. SMAD genes in juvenile polyposis. Genes Chromosomes Cancer. 1999;26(1):54–61.
Friedl W, Kruse R, Uhlhaas S, Stolte M, Schartmann B, Keller KM, et al. Frequent 4-bp deletion in exon 9 of the SMAD4/MADH4 gene in familial juvenile polyposis patients. Genes Chromosomes Cancer. 1999;25(4):403–6.
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–3.
Massaous J, Hata A. TGF-beta signalling through the Smad pathway. Trends Cell Biol. 1997;7(5):187–92.
Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465–71.
Calva-Cerqueira D, Chinnathambi S, Pechman B, Bair J, Larsen-Haidle J, Howe JR. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet. 2009;75(1):79–85.
van Hattem WA, Brosens LA, de Leng WW, Morsink FH, Lens S, Carvalho R, et al. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut. 2008;57(5):623–7.
Aretz S, Stienen D, Uhlhaas S, Stolte M, Entius MM, Loff S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44(11):702–9.
Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28(2):184–7.
Zhou XP, Woodford-Richens K, Lehtonen R, Kurose K, Aldred M, Hampel H, et al. Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet. 2001;69(4):704–11.
Friedl W, Uhlhaas S, Schulmann K, Stolte M, Loff S, Back W, et al. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet. 2002;111(1):108–11.
Calva-Cerqueira D, Dahdaleh FS, Woodfield G, Chinnathambi S, Nagy PL, Larsen-Haidle J, et al. Discovery of the BMPR1A promoter and germline mutations that cause juvenile polyposis. Hum Mol Genet. 2010;19(23):4654–62.
Howe JR, Dahdaleh FS, Carr JC, Wang D, Sherman SK, Howe JR. BMPR1A mutations in juvenile polyposis affect cellular localization. J Surg Res. 2013;184(2):739–45.
Sweet K, Willis J, Zhou XP, Gallione C, Sawada T, Alhopuro P, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005;294(19):2465–73.
Howe JR, Haidle JL, Lal G, Bair J, Song C, Pechman B, et al. ENG mutations in MADH4/BMPR1A mutation negative patients with juvenile polyposis. Clin Genet. 2007;71(1):91–2.
Delnatte C, Sanlaville D, Mougenot JF, Vermeesch JR, Houdayer C, Blois MC, et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet. 2006;78(6):1066–74.
Salviati L, Patricelli M, Guariso G, Sturniolo GC, Alaggio R, Bernardi F, et al. Deletion of PTEN and BMPR1A on chromosome 10q23 is not always associated with juvenile polyposis of infancy. Am J Hum Genet. 2006;79(3):593–6. author reply 6-7.
Menko FH, Kneepkens CM, de Leeuw N, Peeters EA, Van Maldergem L, Kamsteeg EJ, et al. Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin Genet. 2008;74(2):145–54.
Dahdaleh FS, Carr JC, Calva D, Howe JR. Juvenile polyposis and other intestinal polyposis syndromes with microdeletions of chromosome 10q22-23. Clin Genet. 2012;81(2):110–6.
Sayed MG, Ahmed AF, Ringold JR, Anderson ME, Bair JL, Mitros FA, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol. 2002;9(9):901–6.
Handra-Luca A, Condroyer C, de Moncuit C, Tepper M, Flejou JF, Thomas G, et al. Vessels’ morphology in SMAD4 and BMPR1A-related juvenile polyposis. Am J Med Genet A. 2005;138A(2):113–7.
van Hattem WA, Langeveld D, de Leng WW, Morsink FH, van Diest PJ, Iacobuzio-Donahue CA, et al. Histologic variations in juvenile polyp phenotype correlate with genetic defect underlying juvenile polyposis. Am J Surg Pathol. 2011;35(4):530–6.
Conte WJ, Rotter RJ, Schwartz AG, Congelton JE. Hereditary generalized juvenile polyposis, arteriovenous malformations and colonic carcinoma. Clin Res. 1982;30(93A).
Cox KL, Frates Jr RC, Wong A, Gandhi G. Hereditary generalized juvenile polyposis associated with pulmonary arteriovenous malformation. Gastroenterology. 1980;78(6):1566–70.
Inoue S, Matsumoto T, Iida M, Hoshika K, Shimizu M, Hisamoto N, et al. Juvenile polyposis occurring in hereditary hemorrhagic telangiectasia. Am J Med Sci. 1999;317(1):59–62.
Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363(9412):852–9.
Gallione CJ, Richards JA, Letteboer TG, Rushlow D, Prigoda NL, Leedom TP, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43(10):793–7.
Gallione C, Aylsworth AS, Beis J, Berk T, Bernhardt B, Clark RD, et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet A. 2010;152A(2):333–9.
O’Malley M, LaGuardia L, Kalady MF, Parambil J, Heald B, Eng C, et al. The prevalence of hereditary hemorrhagic telangiectasia in juvenile polyposis syndrome. Dis Colon Rectum. 2012;55(8):886–92.
Wain KE, Ellingson MS, McDonald J, Gammon A, Roberts M, Pichurin P, et al. Appreciating the broad clinical features of SMAD4 mutation carriers: a multicenter chart review. Genet Med. 2014;16(8):588–93.
Giardiello FM, Hamilton SR, Kern SE, Offerhaus GJ, Green PA, Celano P, et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Arch Dis Child. 1991;66(8):971–5.
Jones MA, Hebert JC, Trainer TD. Juvenile polyp with intramucosal carcinoma. Arch Pathol Lab Med. 1987;111(2):200–1.
Longo WE, Touloukian RJ, West AB, Ballantyne GH. Malignant potential of juvenile polyposis coli. Report of a case and review of the literature. Dis Colon Rectum. 1990;33(11):980–4.
Coburn MC, Pricolo VE, DeLuca FG, Bland KI. Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol. 1995;2(5):386–91.
Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5(8):751–6.
Brosens LA, van Hattem A, Hylind LM, Iacobuzio-Donahue C, Romans KE, Axilbund J, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56(7):965–7.
Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280(5366):1036–7.
Langeveld D, van Hattem WA, de Leng WW, Morsink FH, Ten Kate FJ, Giardiello FM, et al. SMAD4 immunohistochemistry reflects genetic status in juvenile polyposis syndrome. Clin Cancer Res. 2010;16(16):4126–34.
Woodford-Richens K, Williamson J, Bevan S, Young J, Leggett B, Frayling I, et al. Allelic loss at SMAD4 in polyps from juvenile polyposis patients and use of fluorescence in situ hybridization to demonstrate clonal origin of the epithelium. Cancer Res. 2000;60(9):2477–82.
Howe JR, Ringold JC, Hughes JH, Summers RW. Direct genetic testing for Smad4 mutations in patients at risk for juvenile polyposis. Surgery. 1999;126(2):162–70.
Grosfeld JL, West KW. Generalized juvenile polyposis coli. Clinical management based on long-term observations. Arch Surg. 1986;121(5):530–4.
Jarvinen H. Juvenile gastrointestinal polyposis. Probl Gen Surg. 1993;10:749–57.
Oncel M, Church JM, Remzi FH, Fazio VW. Colonic surgery in patients with juvenile polyposis syndrome: a case series. Dis Colon Rectum. 2005;48(1):49–55; discussion -6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Keck, K., Howe, J.R. (2016). Juvenile Polyposis Syndrome. In: Boardman, L. (eds) Intestinal Polyposis Syndromes. Springer, Cham. https://doi.org/10.1007/978-3-319-28103-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-28103-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28101-8
Online ISBN: 978-3-319-28103-2
eBook Packages: MedicineMedicine (R0)