
Abstract
The conversion of somatic cells into pluripotent cells is transforming the way diseases are studied and treated. Owing to their ability to differentiate into any cell type in the body and being patient-specific, induced pluripotent stem cells (iPSCs) hold great promise for disease modeling, drug discovery and regenerative medicine. Since their discovery in 2006, significant efforts have been made to understand the reprogramming process and to generate human iPSCs with potential for clinical use. Additionally, the development of advanced genome-editing platforms to increase homologous recombination efficiency, namely DNA nucleases, is making the generation of gene-corrected patient-specific iPSCs an achievable goal, with potential future therapeutic applications. Here, we review recent developments in the generation, differentiation and genetic manipulation of human iPSCs and discuss their relevance to regenerative medicine and the challenges still remaining for clinical application.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Induced pluripotent stem cells
- Reprogramming
- Retrovirus
- Lentivirus
- Transduction
- Zero-footprint method
- PiggyBac transposase
- Cardiac differentiation
- Targeting vector
- Macula degeneration
3.1 Introduction
The promise of using pluripotent stem cells (PSCs) for regenerative medicine dates back to 1998 when James Thomson (Thomson et al. 1998) first derived human embryonic stem cells (hESCs) from the inner cell mass of developing embryos. Not only do PSCs have the ability to self-renew indefinitely, but in theory they can also differentiate into any cell type in the body, thus providing functional replacement or trophic support to dysfunctional cells and diseased tissues. Ethical concerns surrounding the destruction of human embryos, which occurs in most hESC derivation methods, and safety issues created controversy in developing therapies from hESC lines for many years and stimulated researchers to find alternative approaches to obtain hESC-like cells. In 2006 Kazutoshi Takahashi and Shinya Yamanaka made the seminal discovery that mouse skin fibroblasts can be reprogrammed to an ESC-like state by simple overexpression of master stemness regulators (Takahashi and Yamanaka 2006). They named these cells “induced pluripotent stem cells” (iPSCs). One year later, these same investigators as well as groups headed by James Thomson and George Daley succeeded in converting human fibroblasts into human iPSCs (hiPSCs) (Takahashi et al. 2007; Yu et al. 2007; Park et al. 2008). Reprogramming to pluripotency has now been achieved starting with a variety of somatic cell types (Aasen et al. 2008; Hanna et al. 2008; Utikal et al. 2009; Carette et al. 2010; Miyoshi et al. 2010; Seki et al. 2010; Tsai et al. 2010; Kim et al. 2011a) and generation of patient- and disease-specific hiPSCs is now possible, opening new avenues for exploring disease etiology, developing novel drugs, toxicology screening and cell replacement therapies. Overcoming the ethical difficulties regarding the use of human embryos that are related with hESCs and being genetically matched to the donor, hiPSCs are increasingly used in modern medicine. Latest advances in genome editing of hiPSCs enable researchers to investigate the intricacies of the human genome in a dish and expand the possibilities of combining cell and gene therapies for treating congenital and degenerative disorders.
Here, we review recent developments for the generation of hiPSCs and emphasize those attractive for obtaining translational-grade cells. Furthermore, we summarize the latest advances in their differentiation and the challenges for obtaining functional and safe hiPSC derivatives for therapy, with specific focus on cardiac muscle cells. Finally, we give a brief overview on the latest available genome-editing platforms for generation of gene-corrected patient-specific hiPSCs and discuss their relevance for regenerative medicine purposes.
3.2 Discovery of iPSCs
Pioneering work in cellular reprogramming had demonstrated that somatic cells can be reprogrammed by transferring their nuclear contents into oocytes (Wilmut et al. 1997) or by fusion with ESCs (Tada et al. 2001; Cowan et al. 2005), indicating that unfertilized eggs and ESCs contain factors that can confer pluripotency to somatic cells.
In 2006, Yamanaka and Takahashi hypothesized that factors that play important roles in the maintenance of ESC identity also play pivotal roles in the induction of pluripotency in somatic cells. Using a retroviral system, they forced expression of a selected set of 24 candidate genes in mouse embryonic fibroblasts (MEFs) and were successful in establishing clones that possessed ESC-like morphologies, proliferation rates, expressed ESC markers and had demethylated the promoter of pluripotency genes (Takahashi and Yamanaka 2006). These cells were termed as induced pluripotent stem cells (iPSCs). Removing one factor at a time they further demonstrated that a minimum set of only four factors namely, Klf4, cMyc, Oct4 and Sox2 were necessary for reprogramming MEFs as well as tail-tip fibroblasts from adult mice into iPSCs. Later studies showed that the presence of cMyc is not an absolute reprogramming requirement but its absence significantly reduces the efficiency of the process (Nakagawa et al. 2008).
The successful reprogramming of human somatic cells to hiPSCs was reported within 1 year (Takahashi et al. 2007; Yu et al. 2007; Park et al. 2008). Takahashi and Yamanaka, as well as Daley’s group used KLF4, cMYC, OCT4 and SOX2, the same factors as in the mouse system, to convert human fibroblasts into hiPSCs (Takahashi et al. 2007; Park et al. 2008). Thomson’s group achieved the same results using LIN28, NANOG, OCT4 and SOX2 (Yu et al. 2007).
Since then the field of cellular reprogramming has progressed at an unprecedented pace. An increasing number of studies constantly provide new insights into the molecular mechanism of the reprogramming process (Brambrink et al. 2008; Mikkelsen et al. 2008; Li et al. 2010; Samavarchi-Tehrani et al. 2010; Fussner et al. 2011). Moreover, specific advances have been made to facilitate the transition of this technology into the clinic, including the use of various cell types for reprogramming (Aasen et al. 2008; Hanna et al. 2008; Utikal et al. 2009; Carette et al. 2010; Miyoshi et al. 2010; Seki et al. 2010; Tsai et al. 2010; Kim et al. 2011a), and the replacement of individual factors by other regulators (Zhao et al. 2008; Feng et al. 2009; Heng et al. 2010; Nakagawa et al. 2010; Moon et al. 2011), small molecules (Huangfu et al. 2008; Shi et al. 2008; Ichida et al. 2009; Li et al. 2009; Zhu et al. 2010; Moon et al. 2011; Staerk et al. 2011) or a modified culture condition (Marson et al. 2008). Furthermore, hiPSC lines have been derived from patients affected by various diseases (Moretti et al. 2010; Unternaehrer and Daley 2011; Zhu et al. 2011; Jung et al. 2012; Cherry and Daley 2013; Gramlich et al. 2015) and from species other than mice or humans, including rhesus monkey (Liu et al. 2008), marmoset (Tomioka et al. 2010), rat (Liao et al. 2009; Maherali and Hochedlinger 2009), pig (Esteban et al. 2009; Ezashi et al. 2009; Wu et al. 2009), dog (Shimada et al. 2010; Luo et al. 2011), sheep (Bao et al. 2011), horse (Nagy et al. 2011) and cow (Han et al. 2011).
The following section will focus on developed approaches for the generation of iPSCs from human origin.
3.3 Generation of Human iPSCs: Developments Towards Translational-Grade hiPSCs
Cell Source for Reprogramming
Among the different issues to be considered when reprogramming human somatic cells into hiPSCs an important one is the choice of the starting material. In general, each actively dividing somatic cell type can be used for reprogramming (Haase et al. 2009). Takahashi and Yamanaka used fibroblasts as the starting somatic cell type and to date, owing their easy culture conditions and efficient transduction, dermal fibroblasts are still one of the most commonly used primary cell source. However, the relatively low reprogramming efficiency (0.01–0.5 %) and especially the need of uncomfortable biopsies have stimulated the search for other, “easier accessible” cell sources. Efficient reprogramming has been demonstrated for peripheral blood mononuclear cells (PBMCs) (Loh et al. 2009), exfoliated renal tubular epithelial cells obtained from urine (Zhou et al. 2011), and keratinocytes from plucked hair (Aasen et al. 2008). One advantage of PBMCs is that they can be obtained from routine blood tests or in patient follow- up and can be frozen and stored before reprogramming.
It has been acknowledged that reprogrammed iPSCs can retain specific DNA methylation profiles associated with their parental source cell type (Bar-Nur et al. 2011; Kim et al. 2011b; Lister et al. 2011). Variations in these signatures also appear to account for intra-line variability among different clones originating from the same iPSC line (Kim et al. 2011b; Lister et al. 2011). The long-term effect of epigenetic pattern retention, such as methylation profiles from the originating somatic cell type, is not yet fully understood. However, the somatic source cell type is known to affect differentiation efficiency into specific iPSC derivatives and epigenetic memory is a key determinant of iPSC differentiation into lineages that are distinct from the parental one (Ohi et al. 2011; Sanchez-Freire et al. 2014). For example, cardiac progenitor cell-derived iPSC lines have shown an enhanced ability to differentiate into cardiomyocytes compared to fibroblast-derived iPSC lines (Sanchez-Freire et al. 2014). Prolonged propagation of iPSCs through many passages reduces these effects, suggesting that residual epigenetic memory is attenuated in the course of long-term culture (Ohi et al. 2011; Sanchez-Freire et al. 2014). This issue, clearly important for therapeutic applications, will require further study in order to determine to what extent the ultimate transplantable cell type should influence the source of patient-specific cells for reprogramming. As this remains unclear, the choice of the starting tissue material should be based, first, on the most accessible and least invasive, and then, depending on the future use of the hiPSCs, an epigenetically related cell source should be considered if available.
Reprogramming Methodologies
The common aim of all reprogramming methods is the forced expression of the reprogramming factors. As mentioned above, hiPSCs were initially derived from fibroblasts by retrovirus- and lentivirus-mediated transduction of genes encoding transcriptional regulators of stem cells: OCT4, SOX2, LIN28, and NANOG (OSLN) (Yu et al. 2007) or OCT4, SOX2, KLF4, and c-MYC (OSKM) (Takahashi et al. 2007; Park et al. 2008). However, viral delivery of transgenes results in the integration of vector sequences into the genome, which is a source of potential insertion mutagenesis, residual expressions, and reactivation of transgenes during differentiation. Therefore, cells generated by permanent and random integration of exogenous genes have a certain oncogenic potential and are not suitable for therapeutic applications.
Safer non-integrating reprogramming methods have since been developed using minimal footprint systems, such as excisable viruses (Chang et al. 2009; Soldner et al. 2009; Somers et al. 2010), and zero footprint technologies, including adenovirus (Stadtfeld et al. 2008), Sendai virus (Fusaki et al. 2009), plasmids (Okita et al. 2008; Gonzalez et al. 2009; Si-Tayeb et al. 2010), episomal or minicircle vectors (Yu et al. 2009; Jia et al. 2010), piggyBac transposons (Kaji et al. 2009; Woltjen et al. 2009; Yusa et al. 2009), microRNA mimics (Miyoshi et al. 2011), synthetic mRNAs (Warren et al. 2010), and proteins (Zhou et al. 2009) (Fig. 3.1).

Generation of human iPSCs. Different starting cell types are available for the generation of human induced pluripotent stem cells (hiPSCs). Fibroblasts were the first and still the most commonly used cell source. Amongst others, three easily accessible starting cell type are blood cells (T lymphocytes), exfoliated renal tubular epithelial cells obtained from urine, and keratinocytes from plucked hair. The reprogramming can be obtained through the expression of several combinations of pluripotency regulators (OCT4, SOX2, NANOG, LIN28, cMYC AND KLF4) and different methods are available to induce their expression. They can be divided into two major groups: integrating methods, which consist of retrovirus or lentivirus delivery of transgenes that randomly integrate into the genome, and non-integrating methods that enable the generation of hiPSCs without any permanent genetic modification
Minimal footprint approaches mostly use lentiviruses containing loxP sites in the 5′ and 3′ LTR of the viral vectors. The presence of loxP sites provides a substrate to remove most of the transgene sequences by Cre-mediated recombination. However, one loxP site flanked by small portions of the 5′ and 3′ LTRs remains in the iPSC genome following Cre-mediated excision (Soldner et al. 2009; Somers et al. 2010). Thus, the continued presence of exogenous transgene sequences (no matter how minimal) could be a concern if differentiated cells derived from these hiPSCs are to be transplanted into a patient.
Zero-footprint methods include adenoviruses, which are non-integrating viruses that infect both replicating and non-replicating cells. Human iPSCs created via adenovirus showed no signs of transgene integration (Zhou and Freed 2009), which is a favorable result for translational applications. However, adenovirus-based reprogramming has low efficiency. More suitable are Sendai viruses, which are negative sense, single-stranded RNA viruses that produce large amounts of protein without entering the nucleus of the infected cells, thus being completely lost after several cell passages. Generation of translational-grade hiPSCs from multiple somatic cell types, including PBMCs (Seki et al. 2010; Orban et al. 2015), has been successfully and efficiently achieved with this method (Fusaki et al. 2009; Ban et al. 2011; Seki et al. 2012).
Another way to generate zero-footprint hiPSCs is the overexpression of the reprogramming factors by episomal plasmids. Yu et al. (2009) developed an oriP/EBNA (Epstein–Barr nuclear antigen)-based plasmid that allows their expression for a long enough period of time sufficient to initiate the reprogramming process. The plasmid will be lost from proliferating cells if drug selection is removed, therefore leaving no footprint. Further modifications of the episomal plasmid reprogramming method (Chen et al. 2011; Okita et al. 2011) have made this approach also very attractive for generation of iPSCs that could be used in translational studies. Episomal vectors are particularly appealing because they are easy to manipulate, allow a relatively high efficiency of reprogramming, and have been proven to work for many somatic cell types, including blood cells (Chou et al. 2011).
Additional DNA-based zero-footprint systems that have been tested for cellular reprogramming are minicircle vectors and piggyBac transposons. Minicircle vectors are circularized constructs in which the plasmid backbone has been released leaving only the eukaryotic promoter and cDNA(s) that are to be expressed. A minicircle vector was produced with LIN28, NANOG, SOX2, and OCT4 and used to reprogram human adipose stem cells (Jia et al. 2010; Narsinh et al. 2011). However, more validation is required since this method worked at lower efficiency for neonatal fibroblasts and no data of successful reprogramming exist for any other cell types. On the hand, hiPSCs have been generated at a reasonable reprogramming efficiency using a piggyBac transposon (Mali et al. 2010), which is a mobile genetic element that in the presence of the piggyBac transposase can be integrated into chromosomal TTAA sites. Re-expression of the transposase after the transposon has been stably integrated results in its excision with no sequence vestiges at the integrated site. Limitations of this system that hamper any clinical translation are the additional step required for excision of the transposon plus the dearth of information on successful excision in hiPSCs (Mali et al. 2010).
More recently, new zero-footprint tools, which are virus- and DNA-free, have emerged. Direct expression of reprogramming factors as proteins has been used to successfully generate hiPSCs (Kim et al. 2009; Zhou et al. 2009). However, this method is limited by the lengthy timeline, low efficiency, and special technical skills required for the synthesis of bioactive reprogramming proteins. Synthetic modified mRNAs have also been explored (Warren et al. 2010). In mRNA reprogramming, cells are transfected with in vitro-transcribed mRNAs encoding for the reprogramming factors. Several chemical measures are employed to limit activation of the innate immune system by foreign nucleic acids and, due to the very short half-life of mRNAs, daily transfections are required to induce hiPSCs. Also miRNAs have been proven useful for generation of hiPSCs without genome-integrating DNA elements (Miyoshi et al. 2011). It is worth noting that since miRNA-mediated reprogramming are mostly dependent on endogenous pathways and, hence, maintain a smooth epigenetic modification, it shows certain advantages in producing better, safer hiPSCs, but more studies are needed to tackle this issue. Finally, a recent report by Hou et al. (2013) described a gene-free, small molecule–based method for generation of mouse iPSCs, demonstrating that the field of pluripotency induction continues to evolve at a rapid pace.
It is worth mentioning that, based on a recent systematic evaluation of the three so far most widely used techniques for generating integration-free hiPSCs (Sendai viruses, episomal plasmids, and synthetic modified mRNAs) (Schlaeger et al. 2015), significant differences exist in aneuploidy rates, reprogramming efficiency, reliability and workload, but all methods generate high-quality hiPSCs. Thus the choice of the reprogramming method should depend on each laboratory’s particular requirements.
Genomic stability is critical for clinical applications of hiPSCs. There are evidences that hiPSCs may harbor epigenetic and transcriptional abnormalities (Kim et al. 2010; Polo et al. 2010; Stadtfeld et al. 2010; Bar-Nur et al. 2011; Kim et al. 2011b) as well as genomic aberrations that are either pre-existing or generated during reprogramming (Mayshar et al. 2010; Gore et al. 2011; Laurent et al. 2011; Lister et al. 2011; Pera 2011), raising significant concerns about their safety for potential clinical applications. However, most of the hiPSCs described in these studies have genomic abnormalities generated from integrating reprogramming methods. A recent comparative work demonstrated, using high resolution HD genotyping, that hiPSC lines obtained by non-integrating approaches have lower incidences of genomic aberrations (Kang et al. 2015). The use of high-resolution methods to monitor genomic aberrations in hiPSCs intended for clinical applications will be necessary. Moreover, the focus of current technology development efforts should be the identification of novel pathways that can be manipulated to augment the efficiency and completeness of reprogramming (Jiang et al. 2013), possibly leading to improved methodologies for safe clinical translation.
3.4 iPSC Differentiation and Challenges for Translational-Grade Derivatives: Cardiomyocytes as an Example
Owing their potential of differentiating into virtually all cell types found in the human body (neurons, cardiac muscle cells, hepatocytes, chondrocytes, retinal pigment epithelial cells, and many others), hiPSCs serve as an unlimited source of human cells for both biomedical research and regenerative medicine purposes. There are a number of ways in which the fate of hiPSCs can be directed towards specific cell lineages and the methodologies are being continuously optimized to improve differentiation efficiency and scalability and enable clinical applications. Depending on the tissue of interest, various differentiation approaches have been explored, ranging from two-dimensional (2D) monolayer cultures with specific growth factors/cytokines and signaling inhibitors, co-culture with supporting cells, up to three-dimensional (3D) differentiation systems. In some cases, when organ development and differentiation pathways are well characterized, it is also feasible to isolate precursor cells at intermediate stages and then direct them further to terminal differentiation (Cao et al. 2013; Reinhardt et al. 2013a). More recently, through the development of 3D culture systems, structures exhibiting multiple cell types that self-organize to form an organ-like tissue, termed “organoids ”, have been generated from hPSCs (Lancaster and Knoblich 2014; Huch and Koo 2015). To date, derivation methods specific for obtaining intestinal (Spence et al. 2011), kidney (Humphreys 2014), brain (Lancaster et al. 2013), and retinal (Nakano et al. 2012) organoids, as well as liver organoid-like tissues called liver buds (Takebe et al. 2013) have been established, making the therapeutic promise of organoids an area of greatest potential for personalized regenerative medicine.
It is important to keep in mind that the differentiation process of PSCs is considered to mimic developmental processes. Therefore, most of the differentiated cells from hPSCs tend to be a reflection of the early stage of development (i.e., embryonic or infant stage). Such immature cells significantly differ from adult cells. Establishment of mature phenotypes is an important challenge for obtaining functional cells for cell therapy. Likewise, the purity and cell number are critical issues for any translational application. Below, we discuss these aspects in details for the differentiation of hiPSCs towards the cardiac lineage.
Cardiac Differentiation of hiPSCs
Historically, the most common method by which cardiomyocytes have been derived from PSCs has involved the formation of three-dimensional aggregates, so-called embryoid bodies (EBs) (Mummery et al. 2003) (Fig. 3.2). Spontaneous EB differentiation relies on a combination of physical and chemical cues to modulate cell signaling pathways and directs PSCs toward various cell types, with 5–70 % of EBs contain beating cardiomyocytes (Kawamura et al. 2012). High variability between experiments, low cardiomyocyte yield (often, 1 %) and immature cardiomyocyte phenotype (Laflamme et al. 2007; Kawamura et al. 2012) have stimulated researchers to explore alternative methods (Moretti et al. 2013). Coculture systems with END-2 stromal cells (Mummery et al. 2003), cardiac fibroblasts (Ou et al. 2011) or human umbilical-vein endothelial cells (Stevens et al. 2009) have been tested in order to mimic microenvironmental factors that are potentially important for cardiac differentiation (Fig. 3.2). In the last decade, knowledge from in vivo developmental studies (Garry and Olson 2006; Evans et al. 2010; Noseda et al. 2011) has guided the establishment of novel 2D and 3D cardiomyocyte differentiation approaches that rely on specific temporal and dose dependent modulation of key pathways involved in cardiogenesis, such as activin/nodal/transforming growth factor-β, Wnt, and bone morphogenetic protein (Kehat et al. 2001; Mummery et al. 2007; Paige et al. 2010; Burridge et al. 2012; Zhang et al. 2012) (Fig. 3.2). Most recently, specific small molecules have been employed to replace growth factors as modulators of these signaling pathways (Lian et al. 2012; Burridge et al. 2014). Using distinct growth factors and small molecules to specifically direct hPSCs towards the cardiac lineages has allowed to achieve more efficient cardiomyocyte differentiation, with yields as high as 85–95 %, and, thanks to the fully defined culture conditions, more reproducible results (Laflamme et al. 2007; Lian et al. 2012, 2013; Cao et al. 2013; Burridge et al. 2014). Although different hiPSC lines can respond differently to developmental signals because of the intrinsic differences in their genetic background, directed differentiation protocols have been successfully applied to various hiPSCs derived from distinct sources of somatic cells and reprogramming methods (Passier et al. 2005; Paige et al. 2010; Lian et al. 2012; Xu et al. 2012; Okano et al. 2013).

Differentiation of human iPSCs into the cardiac lineage. Methods for differentiation of human iPSCs into cardiomyocytes are based on a combination of physical and chemical cues able to induce temporal and dose dependent modulation of specific signaling pathways with pivotal roles during cardiovascular development (Activin, WNT, transforming growth factor β (TGF-β) and bone morphogenic protein (BMP)). The current differentiation protocols can be divided into three main categories: three-dimensional (3D) systems, which mainly include embryoid bodies and the more recently developed 3D systems (Microtissues, engineered heart muscle and cardiac microchambers); two-dimensional (2D) systems, which include several monolayer directed differentiation protocols; and co-culture systems with cells able to promote cardiogenesis (e.g. END-2 stromal cells, cardiac fibroblasts or human umbilical-vein endothelial cells (HUVEC))
Yet despite the advances in differentiation efficiency , major challenges still remain for safe clinical translation of hiPSC-derived cardiomyocytes. One important issue is their purity and risk of teratomas arising from residual undifferentiated hiPSCs. Several non-genetic methods has been reported to improve cardiomyocyte purity after directed hiPSC differentiation, including cell-surface markers (Mummery et al. 2003; Graichen et al. 2008), mitochondria-specific dyes (Kawamura et al. 2012), fluorescent probes (Laflamme et al. 2007), and g lucose deprivation (Burridge et al. 2014).
Another fundamental concern regarding iPSC cardiac differentiation is the varying degree of heterogeneity achieved in the generated cardiomyocyte population. Current hiPSC differentiation strategies yield a heterogeneous mixture of atrial-like and ventricular-like lineages, as well as pacemaker-like lineages such as atrioventricular node-like, sinoatrial node-like, and Purkinje fiber-like cells (Burridge et al. 2012). A deeper understanding of directed lineage differentiation, followed by its modulation, would facilitate subtype-specific cardiac differentiation. In this respect, recent reports suggest that hiPSCs could be directed either to atrial- or to ventricular-like cardiomyocytes by modulating the retinoic acid (Cao et al. 2013; Lian et al. 2013; Devalla et al. 2015) and Wnt signaling pathways (Kim et al. 2013). Additionally, direct manipulations at the epigenetic level or by achieving mRNA-based delivery of lineage-specific factors have also been tested (Ong et al. 2015).
The most immediate need for potential translational applications of hiPSC -derived cardiomyocytes, however, is to achieve defined culture conditions and standardized protocols that address the issue of cellular maturation. These cells begin contracting in the first 2 weeks of differentiation (Burridge et al. 2014), but have a relative immature phenotype more similar to fetal than to adult cardiomyocytes (Robertson et al. 2013). For instance, at the structural level, hPSC-derived cardiomyocytes have a smaller length-to-width aspect ratio (3:1 compared to 15:1), are mononuclear, have fewer mitochondria, and have poor sarcomere organization (Lundy et al. 2013). Also their global gene expression profile is closer to embryonic than adult cardiomyocytes (Gupta et al. 2010). Finally, from the functional point of view, they show underdeveloped Ca2+ handling, low Ca2+ buffering capacity in the sarcoplasmic reticulum, slow beat rates (∼40 BPM), immature action potential characteristics, abnormal levels of ionic currents, and negative force–frequency relationships (Lundy et al. 2013). Attempts to bypass this limitation have demonstrated that long-term culture enhances the appearance of more mature sarcomeric structural organization and change in global gene expression profile (Otsuji et al. 2010; Lundy et al. 2013). In addition, external cues such as electrical stimulation and mechanical cyclic stretching have been reported to aid in obtaining functionally mature hiPSC -derived cardiomyocytes (Lieu et al. 2013; Hirt et al. 2014a). Improvements in maturation were also achieved via genetic overexpression of distinct factors (Fu et al. 2011; Bett et al. 2013; Lieu et al. 2013) and novel 3D culture methods (Nunes et al. 2013; Rao et al. 2013). Moreover, 3D differentiation systems have also been scaled up to generate three-dimensional microtissues (3D-MTs) (Emmert et al. 2013; Thavandiran et al. 2013), engineered heart muscle (EHM) (Kensah et al. 2013; Hirt et al. 2014a), and more recently cardiac microchambers (Ma et al. 2015). Since low retention rate of transplanted single-cell suspensions remains a major issue for clinical translation, the concept of scaffold-free cellular self-assembly into 3D-MTs or EHMs prior to transplantation may be also beneficial to enhance cellular engraftment and survival. These approaches are currently subjects of intense research (Hirt et al. 2014b).
3.5 Genetic Engineering of iPSCs and Personalized Medicine
With the advent of hPSCs, it has become clear that efficient and precise genome editing is crucial for realizing their full potential in research and therapy.
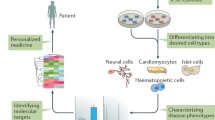
In particular for hiPSCs, genetic correction of the disease-associated mutation(s) in patient-specific lines serves several purposes (Fig. 3.3). First, it will generate isogenic cells that share a common genotype with the exception of the disease-causing mutation, thereby eliminating confounding effects from genetic heterogeneity. These disease-corrected hiPSCs are the perfect control for any comparative analyses of disease phenotype and allow generation of accurate, reliable, and less expensive in vitro human models for understanding diseases and studying genotype/phenotype relationships. Second, genomic modification to directly correct disease-specific point mutations in vitro is also valuable for exploring drug development and performing toxicology tests in patient-specific cells. A large majority of identified candidate drugs fail to reach the market because of safety concerns (about one third of pharmaceuticals are withdrawn due to cardiotoxicity (Guo et al. 2011) and efficacy issues). Human iPSC-derived cardiomyocytes are currently being utilized as a system to evaluate novel and existing medications and to test patient-specific drug responses (Liang et al. 2013; Sinnecker et al. 2013; Wang et al. 2014). Finally, genome editing may accelerate the future clinical application of integration-free cell-based gene therapy, including the autologous transplantation of patient-specific, genome-corrected hiPSC-derived target cells. Of note, genetic correction directly in hiPSCs is however not always achievable, because some genetic diseases imply a reprogramming barrier (e.g. Fanconi anemia (Raya et al. 2009)). In those cases, the cells of origin could be corrected before generating patient-specific hiPSCs.

Genetic engineering of hiPSCs and applications in personalized medicine. Patient-specific iPSCs and isogenic control iPSC lines generated through genome editing approaches can be differentiated toward a specific cell type of interest. Patient-specific and corrected iPSC derivatives can then be used for disease modeling studies, tissue engineering approaches, and high-throughput drug/toxicity screenings, thus facilitating personalized therapy and ultimately autologous cell transplantation for regenerative purposes
3.5.1 Genetic Manipulation of hiPSCs and Gene Correction Approaches
Owing to the fragile nature of hPSCs when dissociated into single cells and their low transfection frequency, gene targeting in hPSCs present a bigger challenge than in the mouse counterparts. An important contribution to improving the handling of hPSCs was made by Yoshiki Sasai’s team whit the discovery of a selective inhibitor of Rho-associated kinase (ROCKi) Y-27632 (Watanabe et al. 2007). The inhibitor significantly suppressed the apoptosis of hPSCs when dissociated, enabling cells to be electroporated and subcloned more easily.
At present, various strategies have been tested and proven for genetic manipulation of patient-specific hiPSCs (Hotta and Yamanaka 2015). Below, we focus exclusively on the approaches that have been used for site-specific genome modification via homologous recombination (HR) and emphasize their advantages and limitations.
Targeting Vector Approach
Initial triumphs in gene targeting of hPSCs were achieved by using a targeting vector , which employs long (5–10 kb) and short (1–4 kb) homology arms on both sides (Zwaka and Thomson 2003). Owing to the low frequency of targeting events in general, the classical targeting vector also contained a drug-selection cassette, such as a neomycin resistance gene derived from a ubiquitous PGK gene promoter, for positive selection. Flanking of the selection cassette by two loxP sequences allowed, after successful targeting, its excision by a Cre recombinase. Using this targeting strategy, only few disease-causing mutations have been corrected in patient-specific hiPSCs (Yusa et al. 2011; Bellin et al. 2013), merely due to the inherently low HR efficiencies. In fact, the propensity of a genomic region to undergo HR is dependent on the local chromatin structure and the generation of a double strand break (DSB) at the specific target site (Carroll 2011b), as well as on transit through the S–G2 phase of the cell cycle (Delacote and Lopez 2008). Thus, the nonhomologous end-joining pathway (NHEJ), which is several orders of magnitude more efficient than HR, is responsible for random integration of targeting vectors. Improved HR efficiency in hPSCs has been achieved by using viral vector-mediated targeting approaches (adeno and adeno-associated viruses) (Mitsui et al. 2009; Khan et al. 2010), which have the advantage of high transduction efficiency, and bacterial artificial chromosome-based strategy (Song et al. 2010), which allows increasing the length of homology between targeting vectors and endogenous loci.
Engineered DNA Nucleases
Another strategy to enhance the efficiency of gene targeting is to introduce site specific DSBs to target loci. DSBs are highly recombinogenic and can stimulate HR in hPSCs by three orders of magnitude (Zou et al. 2009). Cells are obligated to repair the introduced DSBs either by NHEJ, generating small deletions or insertions, or by the homology-directed repair pathway when a homologous donor template is provided. The endonucleases employed must then recognize DNA sequences that occur uniquely at target loci and have minimal off-target activity. Several endonucleases have been engineered to meet these requirements. Currently, the most used for site-specific gene targeting in hPSCs are: zinc finger nucleases (ZFNs), transcription activator-like (TAL) effector nucleases (TALENs), and more recently the clustered regulatory inter-spaced short palindromic repeats (CRISPRs)/Cas9 nucleases (Li et al. 2014). Importantly, owing to the high HR efficiency of genome editing achieved by such nucleases, the HR donor template can be supplied as a single-strand oligonucleotide (ssODN) as short as 80–150 bases, thus facilitating gene correction applications in hiPSCs (Soldner et al. 2011; Ding et al. 2013a, b).
ZFNs are hybrid nucleases that rely on a series of linked zinc finger motifs to recognize specific DNA sequences and the DNA-cleavage domain FokI restriction enzyme to sever DNA. Because FokI nuclease activity depends on dimerization, the ZFN system works as pairs of two monomers of ZFN in reverse orientation that can be designed to bind to a genomic sequence 18–36 nucleotides in length (Porteus and Carroll 2005; Carroll 2011a). Successful ZNF-mediated gene correction in hiPSCs was achieved for several disease-causing mutations (Sebastiano et al. 2011; Reinhardt et al. 2013b). However, due to the complexity of the required engineering steps, ZFNs have been largely supplanted by TALENs and more recently by the CRISPRs/Cas9 nuclease system.
TALENs have a similar structure to ZFNs, but the DNA-binding domain comes from TAL effector proteins and is a tandem array of amino acid repeats. Each of these units is able to bind to one of the four possible nucleotides. TALENs also cleave as dimers (Li et al. 2011) and display not only the unique advantage of easy modular assembly but also enhanced specificity as well as reduced off-target action compared to ZFNs (Li et al. 2011; Pattanayak et al. 2014). As demonstrated in a recent study, TALENs have greatly simplified genome editing in hiPSCs for generating disease models (Ding et al. 2013a). However, despite the initial enthusiasm, TALEN technology has several limitations for future clinical applications of gene-edited hiPSCs. TALEN target-site selection is restricted by the requirement of a preceding T base (Boch et al. 2009). Although this should not prohibit successful design of TALENs in most cases, it may be an issue when modifying a specific mutation for future cell-based gene therapy. The reported sensitivity of TALENs to 5-methylcytosine could be a more serious drawback of the TALEN technology because of the prevalence of this DNA modification in the genome, though this problem may be overcome by engineering 5-methylcytosine- insensitive TALEN DNA-binding domains (Valton et al. 2012).
CRISPRs/Cas9 are RNA-guided engineered nucleases that have been developed from microbial adaptive immune systems named as clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems (Ishino et al. 1987; Jansen et al. 2002). The system utilizes a protein component Cas9 (CRISPR-associated 9) and two small RNAs, crRNA (CRISPR RNA) and tracrRNA (trans crRNA), to mediate target sequence-specific cleavage of double-stranded DNA. To simplify expression in mammalian cells, crRNA and tracrRNA have been fused into one sgRNA (single guide RNA) by a tetranucleotide loop to generate a DSB at a target site. Remarkably, the RNA component of the CRISPR system determines the target sequence based on the Watson-Crick base pairing. Therefore, the design and construction of a target-specific sgRNA is versatile and straightforward and, because of the small size of the sgRNA (20 nucleotides), it is also possible to deliver multiple sgRNAs at the same time to achieve multiplex targeting (Cong et al. 2013; Mali et al. 2013). This makes CRISPRs/Cas9 system as the most accessible means to facilitate and optimize genetic engineering so far (Hsu et al. 2014) and since 2013, several groups have already demonstrated its usefulness for genome editing in hiPSCs (Ding et al. 2013b; Mali et al. 2013; Flynn et al. 2015; Song et al. 2015a). Despite its versatility, also the CRISPR/CAS9 system has several restraints. First, the targetable sites of Cas9 are constrained by the requirement of a GN20GG sequence motif (Jinek et al. 2012), which may cause a problem when targeting certain loci. Second, up to six mismatches between crRNA and target DNA are tolerated by Cas9, which may result in off-target cleavage (Jinek et al. 2012). Indeed, a recent study showed that CRISPR/CAS9 nucleases induce mutations at off-target sites with up to five mismatches (Fu et al. 2013). More importantly, frequencies of off-target mutations are equal to or higher than those of on-target mutations (Fu et al. 2013). Cas9 mutants with a more stringent requirement of crRNA-target DNA complementation may be engineered. For instance, Cas9 has been converted into a nickase, which reduces mutagenesis at off-target sites (Cong et al. 2013).
Thanks to the rapid development of engineered DNA nucleases, genome editing in hiPSCs has evolved from being a daunting task a few years ago to a routine procedure in most laboratories. However, the use of genome editing in the clinic requires very high levels of inspection to ensure safety. A systemic examination of off-target mutagenesis by whole-genome (Kiskinis et al. 2014; Smith et al. 2014; Suzuki et al. 2014; Veres et al. 2014; Yang et al. 2014) or exome sequencing (Yusa et al. 2011; Li et al. 2015) needs to be performed before any clinical translation of genome editing technologies and patient-specific, genome-corrected hiPSC derivatives will be possible.
3.6 Clinical Applications of hiPSCs in Regenerative Medicine: Where Do We Stand?
Regenerative medicine aims to replace and/or regenerate damaged cells, organs, or tissues in order to restore normal function. Cell therapy is an important regenerative medicine approach. The inherent pluripotency of hiPSCs, along with their genetic identity to specific patients, raises the possibility of autologous transplantation to treat patients suffering from a myriad of disorders characterized by loss of a key cellular function, such as cardiomyocytes in myocardial infarct, dopaminergic neurons in Parkinson’s disease, beta cells in type 1 diabetes, or hematopoietic stem cells in aplastic anemias. In the case of monogenic diseases, in which all the cells from the body initially carry the disease-causing mutation in their genomic DNA, a gene correction approach can be considered to generate disease-free autologous cells, as discussed in the previous paragraph.
However, compared to other cell-based therapies, the Investigational New Drug (IND) review process for hPSCs involves a higher level of scrutiny owing to their potential to form tumors and ectopic tissue. The lack of data on the potential untoward effects of hPSC-derived therapies in humans means that parameters surrounding efficacy, biodistribution, persistence, toxicity, presence of residual pluripotent cell contaminants and tumorigenicity potential all need to be thoroughly addressed before these therapies receive IND approval (Bailey 2012). Preclinical animal models need to be carefully designed to address these issues in a manner that satisfies the regulatory agencies (Frey-Vasconcells et al. 2012). Furthermore, the starting hPSC line itself needs to undergo extensive characterization for assurances of safety, such as analyses of genetic stability, virus and pathogen testing, derivation methods in the spirit of good manufacturing practices (GMPs), maintenance of the line under GMP conditions, and donor screening and eligibility (Carpenter et al. 2009). For hiPSCs, the reprogramming strategy is an additional consideration and those methods that do not involve integration of transgenes into the genome are definitely safer. Another major consideration for hPSC-based therapies is the route of administration. For the time being, therapies that are injected locally or contained within a device that limits their migration may have an easier time achieving IND status than those that are systemically injected, as these approaches help to limit the area in which potential adverse effects may occur. That being said, since 2010 several clinical trails using hPSC-based therapy have been initiated in various countries, as overviewed below.
3.6.1 Clinical Trials Involving hiPSCs
The current wave of clinical trials testing hPSC-based therapy predominantly focuses on hESC-derived cells (Fig. 3.4), including retinal pigment epithelium (for macula degeneration and related diseases) (Schwartz et al. 2012, 2015; Song et al. 2015b), pancreatic endoderm derivatives (for type 1 diabetes) (Schulz et al. 2012; Pagliuca et al. 2014), oligodendrocytes (for spinal cord injury) as well as cardiac progenitors (for severe heart failure) (Menasche et al. 2015). Yet, only one trial using autologous patient-specific hiPSC derivatives exists, which aims at curing the wet form of age-related macular degeneration using retinal pigment epithelium cells transplanted as sheets (Kamao et al. 2014) (Fig. 3.4). It started in September 2014 with the treatment of the first patient at the Riken Institute in Japan (Reardon and Cyranoski 2014), but was recently put on hold because hiPSCs from a second patient were found to carry genetic mutations.

Clinical trials with hESC and hiPSC cell derivatives. Current clinical trials involving pluripotent stem cell derivatives target four organ systems: the eye, the heart, the pancreas, and the nervous system. Abbreviation: AMD, age-related macular degeneration; hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell; MMD, myopic macular degeneration; RPE, retinal pigment epithelium
Considering that hESCs took almost 12 years from the first establishment to the first transplantation into a spinal cord injury patient in October 2010, the transition of patient-specific hiPSCs from bench to bedside was relatively quick and, as time goes on, the number of hiPSC-based clinical trials will probably increase.
3.7 Concluding Remarks
Though hiPSC technology is not even a decade old, it has significantly revolutionized the world of stem cells, disease modeling, drug testing and regenerative medicine. The advent of improved reprogramming methods that do not involve integration of transgenes into the genome and the rapid development of large-scale culture systems and efficient differentiation protocols as well as of advanced genome-editing technologies has begun to overcome the shortcomings of using hiPSCs in regenerative medicine.
The year 2014 marked the arrival of patient-specific hiPSCs onto the clinical stage, and this is just the beginning. Further efforts are needed to tap the full potential of hiPSC-mediated cell therapy to benefit human health. In addition, incorporating newly emerging genome-editing technologies might trigger a new era of gene therapy using hiPSCs.
References
Aasen T, Raya A, Barrero MJ et al (2008) Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26:1276–1284
Bailey AM (2012) Balancing tissue and tumor formation in regenerative medicine. Sci Transl Med 4:147fs128
Ban H, Nishishita N, Fusaki N et al (2011) Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci U S A 108:14234–14239
Bao L, He L, Chen J et al (2011) Reprogramming of ovine adult fibroblasts to pluripotency via drug-inducible expression of defined factors. Cell Res 21:600–608
Bar-Nur O, Russ HA, Efrat S et al (2011) Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 9:17–23
Bellin M, Casini S, Davis RP et al (2013) Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J 32:3161–3175
Bett GC, Kaplan AD, Lis A et al (2013) Electronic “expression” of the inward rectifier in cardiocytes derived from human-induced pluripotent stem cells. Heart Rhythm 10:1903–1910
Boch J, Scholze H, Schornack S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Brambrink T, Foreman R, Welstead GG et al (2008) Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2:151–159
Burridge PW, Keller G, Gold JD et al (2012) Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell 10:16–28
Burridge PW, Matsa E, Shukla P et al (2014) Chemically defined generation of human cardiomyocytes. Nat Methods 11:855–860
Cao N, Liang H, Huang J et al (2013) Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res 23:1119–1132
Carette JE, Pruszak J, Varadarajan M et al (2010) Generation of iPSCs from cultured human malignant cells. Blood 115:4039–4042
Carpenter MK, Frey-Vasconcells J, Rao MS (2009) Developing safe therapies from human pluripotent stem cells. Nat Biotechnol 27:606–613
Carroll D (2011a) Genome engineering with zinc-finger nucleases. Genetics 188:773–782
Carroll D (2011b) Zinc-finger nucleases: a panoramic view. Curr Gene Ther 11:2–10
Chang CW, Lai YS, Pawlik KM et al (2009) Polycistronic lentiviral vector for “hit and run” reprogramming of adult skin fibroblasts to induced pluripotent stem cells. Stem Cells 27:1042–1049
Chen G, Gulbranson DR, Hou Z et al (2011) Chemically defined conditions for human iPSC derivation and culture. Nat Methods 8:424–429
Cherry AB, Daley GQ (2013) Reprogrammed cells for disease modeling and regenerative medicine. Annu Rev Med 64:277–290
Chou BK, Mali P, Huang X et al (2011) Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res 21:518–529
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Cowan CA, Atienza J, Melton DA et al (2005) Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 309:1369–1373
Delacote F, Lopez BS (2008) Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle 7:33–38
Devalla HD, Schwach V, Ford JW et al (2015) Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol Med 7:394–410
Ding Q, Lee YK, Schaefer EA et al (2013a) A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 12:238–251
Ding Q, Regan SN, Xia Y et al (2013b) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12:393–394
Emmert MY, Wolint P, Wickboldt N et al (2013) Human stem cell-based three-dimensional microtissues for advanced cardiac cell therapies. Biomaterials 34:6339–6354
Esteban MA, Xu J, Yang J et al (2009) Generation of induced pluripotent stem cell lines from Tibetan miniature pig. J Biol Chem 284:17634–17640
Evans SM, Yelon D, Conlon FL et al (2010) Myocardial lineage development. Circ Res 107:1428–1444
Ezashi T, Telugu BP, Alexenko AP et al (2009) Derivation of induced pluripotent stem cells from pig somatic cells. Proc Natl Acad Sci U S A 106:10993–10998
Feng B, Jiang J, Kraus P et al (2009) Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nat Cell Biol 11:197–203
Flynn R, Grundmann A, Renz P et al (2015) CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol 43(10):838–848.e3
Frey-Vasconcells J, Whittlesey KJ, Baum E et al (2012) Translation of stem cell research: points to consider in designing preclinical animal studies. Stem Cells Transl Med 1:353–358
Fu JD, Rushing SN, Lieu DK et al (2011) Distinct roles of microRNA-1 and -499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PLoS One 6:e27417
Fu Y, Foden JA, Khayter C et al (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:822–826
Fusaki N, Ban H, Nishiyama A et al (2009) Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 85:348–362
Fussner E, Djuric U, Strauss M et al (2011) Constitutive heterochromatin reorganization during somatic cell reprogramming. EMBO J 30:1778–1789
Garry DJ, Olson EN (2006) A common progenitor at the heart of development. Cell 127:1101–1104
Gonzalez F, Barragan Monasterio M, Tiscornia G et al (2009) Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc Natl Acad Sci U S A 106:8918–8922
Gore A, Li Z, Fung HL et al (2011) Somatic coding mutations in human induced pluripotent stem cells. Nature 471:63–67
Graichen R, Xu X, Braam SR et al (2008) Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation 76:357–370
Gramlich M, Pane LS, Zhou Q et al (2015) Antisense-mediated exon skipping: a therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol Med 7:562–576
Guo L, Abrams RM, Babiarz JE et al (2011) Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci 123:281–289
Gupta MK, Illich DJ, Gaarz A et al (2010) Global transcriptional profiles of beating clusters derived from human induced pluripotent stem cells and embryonic stem cells are highly similar. BMC Dev Biol 10:98
Haase A, Olmer R, Schwanke K et al (2009) Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 5:434–441
Han X, Han J, Ding F et al (2011) Generation of induced pluripotent stem cells from bovine embryonic fibroblast cells. Cell Res 21:1509–1512
Hanna J, Markoulaki S, Schorderet P et al (2008) Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133:250–264
Heng JC, Feng B, Han J et al (2010) The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 6:167–174
Hirt MN, Boeddinghaus J, Mitchell A et al (2014a) Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol 74:151–161
Hirt MN, Hansen A, Eschenhagen T (2014b) Cardiac tissue engineering: state of the art. Circ Res 114:354–367
Hou P, Li Y, Zhang X et al (2013) Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341:651–654
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Huangfu D, Maehr R, Guo W et al (2008) Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol 26:795–797
Huch M, Koo BK (2015) Modeling mouse and human development using organoid cultures. Development 142:3113–3125
Humphreys BD (2014) Kidney structures differentiated from stem cells. Nat Cell Biol 16:19–21
Ichida JK, Blanchard J, Lam K et al (2009) A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 5:491–503
Ishino Y, Shinagawa H, Makino K et al (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jansen R, Embden JD, Gaastra W et al (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575
Jia F, Wilson KD, Sun N et al (2010) A nonviral minicircle vector for deriving human iPS cells. Nat Methods 7:197–199
Jiang J, Lv W, Ye X et al (2013) Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of iPS cells as demonstrated by tetraploid complementation. Cell Res 23:92–106
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jung CB, Moretti A, Mederos Y, Schnitzler M et al (2012) Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med 4:180–191
Kaji K, Norrby K, Paca A et al (2009) Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458:771–775
Kamao H, Mandai M, Okamoto S et al (2014) Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep 2:205–218
Kang X, Yu Q, Huang Y et al (2015) Effects of integrating and non-integrating reprogramming methods on copy number variation and genomic stability of human induced pluripotent stem cells. PLoS One 10:e0131128
Kawamura M, Miyagawa S, Miki K et al (2012) Feasibility, safety, and therapeutic efficacy of human induced pluripotent stem cell-derived cardiomyocyte sheets in a porcine ischemic cardiomyopathy model. Circulation 126:S29–S37
Kehat I, Kenyagin-Karsenti D, Snir M et al (2001) Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest 108:407–414
Kensah G, Roa Lara A, Dahlmann J et al (2013) Murine and human pluripotent stem cell-derived cardiac bodies form contractile myocardial tissue in vitro. Eur Heart J 34:1134–1146
Khan IF, Hirata RK, Wang PR et al (2010) Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther 18:1192–1199
Kim D, Kim CH, Moon JI et al (2009) Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4:472–476
Kim K, Doi A, Wen B et al (2010) Epigenetic memory in induced pluripotent stem cells. Nature 467:285–290
Kim J, Lengner CJ, Kirak O et al (2011a) Reprogramming of postnatal neurons into induced pluripotent stem cells by defined factors. Stem Cells 29:992–1000
Kim K, Zhao R, Doi A et al (2011b) Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol 29:1117–1119
Kim HT, Lee KI, Kim DW et al (2013) An ECM-based culture system for the generation and maintenance of xeno-free human iPS cells. Biomaterials 34:1041–1050
Kiskinis E, Sandoe J, Williams LA et al (2014) Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14:781–795
Laflamme MA, Chen KY, Naumova AV et al (2007) Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25:1015–1024
Lancaster MA, Knoblich JA (2014) Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345:1247125
Lancaster MA, Renner M, Martin CA et al (2013) Cerebral organoids model human brain development and microcephaly. Nature 501:373–379
Laurent LC, Ulitsky I, Slavin I et al (2011) Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8:106–118
Li W, Wei W, Zhu S et al (2009) Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell 4:16–19
Li R, Liang J, Ni S et al (2010) A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7:51–63
Li T, Huang S, Zhao X et al (2011) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res 39:6315–6325
Li M, Suzuki K, Kim NY et al (2014) A cut above the rest: targeted genome editing technologies in human pluripotent stem cells. J Biol Chem 289:4594–4599
Li HL, Fujimoto N, Sasakawa N et al (2015) Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep 4:143–154
Lian X, Hsiao C, Wilson G et al (2012) Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 109:E1848–E1857
Lian X, Zhang J, Azarin SM et al (2013) Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc 8:162–175
Liang P, Lan F, Lee AS et al (2013) Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 127:1677–1691
Liao J, Cui C, Chen S et al (2009) Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell 4:11–15
Lieu DK, Fu JD, Chiamvimonvat N et al (2013) Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol 6:191–201
Lister R, Pelizzola M, Kida YS et al (2011) Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471:68–73
Liu H, Zhu F, Yong J et al (2008) Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell 3:587–590
Loh YH, Agarwal S, Park IH et al (2009) Generation of induced pluripotent stem cells from human blood. Blood 113:5476–5479
Lundy SD, Zhu WZ, Regnier M et al (2013) Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev 22:1991–2002
Luo J, Suhr ST, Chang EA et al (2011) Generation of leukemia inhibitory factor and basic fibroblast growth factor-dependent induced pluripotent stem cells from canine adult somatic cells. Stem Cells Dev 20:1669–1678
Ma Z, Wang J, Loskill P et al (2015) Self-organizing human cardiac microchambers mediated by geometric confinement. Nat Commun 6:7413
Maherali N, Hochedlinger K (2009) Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr Biol 19:1718–1723
Mali P, Chou BK, Yen J et al (2010) Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells 28:713–720
Mali P, Yang L, Esvelt KM et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Marson A, Foreman R, Chevalier B et al (2008) Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell 3:132–135
Mayshar Y, Ben-David U, Lavon N et al (2010) Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 7:521–531
Menasche P, Vanneaux V, Fabreguettes JR et al (2015) Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: a translational experience. Eur Heart J 36:743–750
Mikkelsen TS, Hanna J, Zhang X et al (2008) Dissecting direct reprogramming through integrative genomic analysis. Nature 454:49–55
Mitsui K, Suzuki K, Aizawa E et al (2009) Gene targeting in human pluripotent stem cells with adeno-associated virus vectors. Biochem Biophys Res Commun 388:711–717
Miyoshi N, Ishii H, Nagai K et al (2010) Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci U S A 107:40–45
Miyoshi N, Ishii H, Nagano H et al (2011) Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 8:633–638
Moon JH, Heo JS, Kim JS et al (2011) Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res 21:1305–1315
Moretti A, Bellin M, Welling A et al (2010) Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 363:1397–1409
Moretti A, Laugwitz KL, Dorn T et al (2013) Pluripotent stem cell models of human heart disease. Cold Spring Harb Perspect Med 3(11):a014027
Mummery C, Ward-Van Oostwaard D, Doevendans P et al (2003) Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation 107:2733–2740
Mummery C, Van Der Heyden MA, De Boer TP et al (2007) Cardiomyocytes from human and mouse embryonic stem cells. Methods Mol Med 140:249–272
Nagy K, Sung HK, Zhang P et al (2011) Induced pluripotent stem cell lines derived from equine fibroblasts. Stem Cell Rev 7:693–702
Nakagawa M, Koyanagi M, Tanabe K et al (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26:101–106
Nakagawa M, Takizawa N, Narita M et al (2010) Promotion of direct reprogramming by transformation-deficient Myc. Proc Natl Acad Sci U S A 107:14152–14157
Nakano T, Ando S, Takata N et al (2012) Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10:771–785
Narsinh KH, Jia F, Robbins RC et al (2011) Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat Protoc 6:78–88
Noseda M, Peterkin T, Simoes FC et al (2011) Cardiopoietic factors: extracellular signals for cardiac lineage commitment. Circ Res 108:129–152
Nunes SS, Miklas JW, Liu J et al (2013) Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 10:781–787
Ohi Y, Qin H, Hong C et al (2011) Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol 13:541–549
Okano H, Nakamura M, Yoshida K et al (2013) Steps toward safe cell therapy using induced pluripotent stem cells. Circ Res 112:523–533
Okita K, Nakagawa M, Hyenjong H et al (2008) Generation of mouse induced pluripotent stem cells without viral vectors. Science 322:949–953
Okita K, Matsumura Y, Sato Y et al (2011) A more efficient method to generate integration-free human iPS cells. Nat Methods 8:409–412
Ong SG, Lee WH, Kodo K et al (2015) MicroRNA-mediated regulation of differentiation and trans-differentiation in stem cells. Adv Drug Deliv Rev 88:3–15
Orban M, Goedel A, Haas J et al (2015) Functional comparison of induced pluripotent stem cell- and blood-derived GPIIbIIIa deficient platelets. PLoS One 10:e0115978
Otsuji TG, Minami I, Kurose Y et al (2010) Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: qualitative effects on electrophysiological responses to drugs. Stem Cell Res 4:201–213
Ou DB, He Y, Chen R et al (2011) Three-dimensional co-culture facilitates the differentiation of embryonic stem cells into mature cardiomyocytes. J Cell Biochem 112:3555–3562
Pagliuca FW, Millman JR, Gurtler M et al (2014) Generation of functional human pancreatic beta cells in vitro. Cell 159:428–439
Paige SL, Osugi T, Afanasiev OK et al (2010) Endogenous Wnt/beta-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS One 5:e11134
Park IH, Zhao R, West JA et al (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451:141–146
Passier R, Oostwaard DW, Snapper J et al (2005) Increased cardiomyocyte differentiation from human embryonic stem cells in serum-free cultures. Stem Cells 23:772–780
Pattanayak V, Guilinger JP, Liu DR (2014) Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Methods Enzymol 546:47–78
Pera MF (2011) Stem cells: the dark side of induced pluripotency. Nature 471:46–47
Polo JM, Liu S, Figueroa ME et al (2010) Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 28:848–855
Porteus MH, Carroll D (2005) Gene targeting using zinc finger nucleases. Nat Biotechnol 23:967–973
Rao C, Prodromakis T, Kolker L et al (2013) The effect of microgrooved culture substrates on calcium cycling of cardiac myocytes derived from human induced pluripotent stem cells. Biomaterials 34:2399–2411
Raya A, Rodriguez-Piza I, Guenechea G et al (2009) Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 460:53–59
Reardon S, Cyranoski D (2014) Japan stem-cell trial stirs envy. Nature 513:287–288
Reinhardt P, Glatza M, Hemmer K et al (2013a) Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 8:e59252
Reinhardt P, Schmid B, Burbulla LF et al (2013b) Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12:354–367
Robertson C, Tran DD, George SC (2013) Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells 31:829–837
Samavarchi-Tehrani P, Golipour A, David L et al (2010) Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7:64–77
Sanchez-Freire V, Lee AS, Hu S et al (2014) Effect of human donor cell source on differentiation and function of cardiac induced pluripotent stem cells. J Am Coll Cardiol 64:436–448
Schlaeger TM, Daheron L, Brickler TR et al (2015) A comparison of non-integrating reprogramming methods. Nat Biotechnol 33:58–63
Schulz TC, Young HY, Agulnick AD et al (2012) A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One 7:e37004
Schwartz SD, Hubschman JP, Heilwell G et al (2012) Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379:713–720
Schwartz SD, Regillo CD, Lam BL et al (2015) Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 385:509–516
Sebastiano V, Maeder ML, Angstman JF et al (2011) In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29:1717–1726
Seki T, Yuasa S, Oda M et al (2010) Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 7:11–14
Seki T, Yuasa S, Fukuda K (2012) Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat Protoc 7:718–728
Shi Y, Do JT, Desponts C et al (2008) A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell 2:525–528
Shimada H, Nakada A, Hashimoto Y et al (2010) Generation of canine induced pluripotent stem cells by retroviral transduction and chemical inhibitors. Mol Reprod Dev 77:2
Sinnecker D, Goedel A, Laugwitz KL et al (2013) Induced pluripotent stem cell-derived cardiomyocytes: a versatile tool for arrhythmia research. Circ Res 112:961–968
Si-Tayeb K, Noto FK, Nagaoka M et al (2010) Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51:297–305
Smith C, Gore A, Yan W et al (2014) Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell 15:12–13
Soldner F, Hockemeyer D, Beard C et al (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136:964–977
Soldner F, Laganiere J, Cheng AW et al (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146:318–331
Somers A, Jean JC, Sommer CA et al (2010) Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 28:1728–1740
Song H, Chung SK, Xu Y (2010) Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell 6:80–89
Song B, Fan Y, He W et al (2015a) Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev 24:1053–1065
Song WK, Park KM, Kim HJ et al (2015b) Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: preliminary results in Asian patients. Stem Cell Rep 4:860–872
Spence JR, Mayhew CN, Rankin SA et al (2011) Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470:105–109
Stadtfeld M, Nagaya M, Utikal J et al (2008) Induced pluripotent stem cells generated without viral integration. Science 322:945–949
Stadtfeld M, Apostolou E, Akutsu H et al (2010) Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465:175–181
Staerk J, Lyssiotis CA, Medeiro LA et al (2011) Pan-Src family kinase inhibitors replace Sox2 during the direct reprogramming of somatic cells. Angew Chem Int Ed Engl 50:5734–5736
Stevens KR, Kreutziger KL, Dupras SK et al (2009) Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proc Natl Acad Sci U S A 106:16568–16573
Suzuki K, Yu C, Qu J et al (2014) Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 15:31–36
Tada M, Takahama Y, Abe K et al (2001) Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol 11:1553–1558
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Takahashi K, Tanabe K, Ohnuki M et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Takebe T, Sekine K, Enomura M et al (2013) Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499:481–484
Thavandiran N, Dubois N, Mikryukov A et al (2013) Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proc Natl Acad Sci U S A 110:E4698–E4707
Thomson JA, Itskovitz-Eldor J, Shapiro SS et al (1998) Embryonic stem cell lines derived from human blastocysts. Science 282:1145–1147
Tomioka I, Maeda T, Shimada H et al (2010) Generating induced pluripotent stem cells from common marmoset (Callithrix jacchus) fetal liver cells using defined factors, including Lin28. Genes Cells 15:959–969
Tsai SY, Clavel C, Kim S et al (2010) Oct4 and klf4 reprogram dermal papilla cells into induced pluripotent stem cells. Stem Cells 28:221–228
Unternaehrer JJ, Daley GQ (2011) Induced pluripotent stem cells for modelling human diseases. Philos Trans R Soc Lond B Biol Sci 366:2274–2285
Utikal J, Maherali N, Kulalert W et al (2009) Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci 122:3502–3510
Valton J, Dupuy A, Daboussi F et al (2012) Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J Biol Chem 287:38427–38432
Veres A, Gosis BS, Ding Q et al (2014) Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 15:27–30
Wang Y, Liang P, Lan F et al (2014) Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol 64:451–459
Warren L, Manos PD, Ahfeldt T et al (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7:618–630
Watanabe K, Ueno M, Kamiya D et al (2007) A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25:681–686
Wilmut I, Schnieke AE, Mcwhir J et al (1997) Viable offspring derived from fetal and adult mammalian cells. Nature 385:810–813
Woltjen K, Michael IP, Mohseni P et al (2009) piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458:766–770
Wu Z, Chen J, Ren J et al (2009) Generation of pig induced pluripotent stem cells with a drug-inducible system. J Mol Cell Biol 1:46–54
Xu H, Yi BA, Wu H et al (2012) Highly efficient derivation of ventricular cardiomyocytes from induced pluripotent stem cells with a distinct epigenetic signature. Cell Res 22:142–154
Yang L, Grishin D, Wang G et al (2014) Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat Commun 5:5507
Yu J, Vodyanik MA, Smuga-Otto K et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920
Yu J, Hu K, Smuga-Otto K et al (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science 324:797–801
Yusa K, Rad R, Takeda J et al (2009) Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods 6:363–369
Yusa K, Rashid ST, Strick-Marchand H et al (2011) Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature 478:391–394
Zhang J, Klos M, Wilson GF et al (2012) Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res 111:1125–1136
Zhao Y, Yin X, Qin H et al (2008) Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 3:475–479
Zhou W, Freed CR (2009) Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27:2667–2674
Zhou H, Wu S, Joo JY et al (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4:381–384
Zhou T, Benda C, Duzinger S et al (2011) Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol 22:1221–1228
Zhu S, Li W, Zhou H et al (2010) Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 7:651–655
Zhu H, Lensch MW, Cahan P et al (2011) Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet 12:266–275
Zou J, Maeder ML, Mali P et al (2009) Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 5:97–110
Zwaka TP, Thomson JA (2003) Homologous recombination in human embryonic stem cells. Nat Biotechnol 21:319–321
Acknowledgments
AM would like to acknowledge and thank the German Research Foundation and the German Ministry for Education and Research for their ongoing support of research in the laboratory. AM also acknowledge the Munich Heart Alliance, a member of the German Center for Cardiovascular Research.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Pane, L.S., My, I., Moretti, A. (2016). Induced Pluripotent Stem Cells in Regenerative Medicine. In: Steinhoff, G. (eds) Regenerative Medicine - from Protocol to Patient. Springer, Cham. https://doi.org/10.1007/978-3-319-27610-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-27610-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27608-3
Online ISBN: 978-3-319-27610-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)