
Abstract
Cell-free protein synthesis based on E. coli cell extracts has been described for the first time more than 50 years ago. To date, cell-free synthesis is widely used for the preparation of toxic proteins, for studies of the translation process and its regulation as well as for the incorporation of artificial or labeled amino acids into a polypeptide chain. Many efforts have been directed towards establishing cell-free expression as a standard method for gene expression, with limited success. In this chapter we will describe the state-of-the-art of cell-free expression, extract preparation methods and recent examples for successful applications of cell-free synthesis of macromolecular complexes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: Motivation and Challenges
The capacity of cell extracts to synthesize proteins has been shown in the 1950s of the last century [1, 2], several years before the identification of ribosomes as protein-synthesizing machines [3]. The cell-free extract was based on the classical S30 fraction obtained by a 30,000× g centrifugation step at 4 °C for 1 h. Initially, endogenous mRNA was used for in vitro translation [4]. Subsequently, Nirenberg and Matthaei developed a protocol to degrade endogenous messenger RNA present in the cell extract and to add exogenous mRNA [5, 6]. The first cell-free protein synthesis (CFPS) from DNA, using a so-called coupled transcription-translation system was developed in the late 1960s by the group of Zubay [7]. They used their coupled transcription-translation system to study the regulation of gene expression by the E. coli lactose operon. Most cell-free extract preparation and in vitro translation protocols are based on this protocol [8, 9].
Significant improvements with respect to protein yields were achieved in the late 1980s, in particular by the group of Spirin, which established the use of phage-specific RNA polymerases, SP6 [10] or T7 RNA polymerases [8]. Using these polymerases a high level of a specific mRNA during the in vitro transcription-translation reaction can be achieved and maintained. Importantly, the Spirin laboratory described the first ‘continuous’ in vitro translation system. It allows for a continuous exchange of small molecules between a ‘feeding compartment’ providing energy and substrates (amino acids) for the translation reaction and a ‘reaction compartment’ from which inhibitory reaction products are removed by dialysis [10, 11]. In a continuous set-up, the in vitro translation reaction can continue for several hours or even days, compared to 40–60 min using the classical reaction set-up. This allows obtaining significantly increased yields: for instance 6 mg chloramphenicol acetyl-transferase protein per milliliter of in vitro translation reaction were synthesized in 21 h [12].
With these advancements, cell-free translation became a very interesting technology for protein production in structural biology. In particular the RIKEN Structural Genomics/Proteomics Initiative (RSGI) in Japan invested into the automation of cell-free protein synthesis and high-throughput screening of protein products with the aim to obtain high yields of isotope-labeled proteins for NMR studies [13–15]. Notably, specific 15N and 13C labeling for any amino acid is trivial as soon as the protein is expressed in vitro. Accordingly, numerous NMR structures have been solved using this approach [16–18]. CFPS also led to several X-ray structures [15, 19].
CFPS allows the rapid and economical screening of a number of different proteins or protein variants (mutants, truncations, etc.) when these are required only in small quantities. Classical sub-cloning of constructs into plasmids is not required since the in vitro transcription/translation reaction can be started from PCR products [20] which significantly improves the screening capacities, a high-throughput set-up and automation.
Cell-free expression remains a powerful approach for the production of toxic and insoluble proteins, for instance membrane proteins. The group of F. Bernhard significantly improved in vitro translation protocols to be able to produce membrane proteins in the presence of detergents or of lipids (for review [21]). Subsequent crystallization attempts, for instance of G-protein coupled receptors, remained mostly unsuccessful. To date, three membrane proteins which were produced by in vitro translation have been crystallized: VDAC, diacylglycerol kinase and EmrE [22–24]. In the case of EmrE, cell-free synthesis was used to generate a seleno-methionine derivative in order to phase already existing crystallographic data.
Cell-free expression has thus several very attractive applications, related to the expression of toxic proteins, rapid production of small quantities of proteins for screening and protein engineering purposes as well as for the incorporation of unnatural amino acids in structural and synthetic biology. In this chapter, we describe the different in vitro translation reaction set-ups used in the field, and we present successful applications applied to study large macromolecular complexes.
2 Basics of E. coli Transcription/Translation Systems
2.1 The Classical S30-Based Cell-Free Expression System
The most common method for cell-free expression is using E. coli S30 extract [7]. This classical cell-free expression system has been only slightly modified since the first description of the protocol by Nirenberg [5]. The S30 extract is composed of a soluble fraction which is obtained after lysis of E. coli cells and centrifugation of the lysate at 30,000× g. Thus, this extract contains all the cytosolic enzymes required for transcription and translation. However, without further treatment, the extract contains endogenous mRNAs which will also be translated, leading to unwanted side products. Nirenberg and Matthaei established a protocol to remove endogenous mRNAs without destabilizing the ribosomal RNAs [6]: After centrifugation, the lysate is treated with high-salt concentrations resulting in the release of mRNAs from the ribosomes. The endogenous mRNAs is then degraded by the RNases present in the cell extract (e.g., by incubation for 1 h at 25 °C). For the cell-free transcription/translation reaction, the S30 extract needs to be supplemented with the 20 amino acids, the E. coli tRNAs, nucleotides (ATP, GTP, CTP and UTP) required as energy sources as well as building blocks for the RNA synthesis, as well as an energy-regenerating system composed of phosphoenol pyruvate and pyruvate kinase, and the T7 RNA polymerase for efficient in vitro transcription [9]. Alternative energy regeneration systems have been reported such as acetyl kinase and acetyl phosphate or creatine phosphate and creatine kinase [12, 25].
The template used in the classical cell-free expression system can be either plasmid DNA, linear DNA (PCR products) or mRNA. Usually, in vitro translated proteins are tagged for subsequent affinity purification directly from the cell-free expression reaction. In addition to the basic components, co-factors or regulatory proteins which are not present or under-represented in the E. coli S30 extract can be added for the production of specific proteins. For instance, Yang and Zubay showed that the araC protein which is required for gene expression of the ara operon was lost during the S30 extract preparation, thus inhibiting the expression of proteins from the ara operon [26]. The addition of chaperones such as Trigger Factor, DnaK, DnaJ, GrpE, GroEL/GroES and protein disulfide isomerase often increases the amount of soluble proteins and helps folding of disulfide-containing proteins (e.g. immunoglobulin domains, see applications) [27]. In conclusion, the composition of the classical cell-free expression system can be optimized and tailored according to the specific requirements of the expressed protein.
The yields (between few micrograms up to several milligrams per milliliter reaction) are dependent on the expressed protein, its mRNA stability, the composition of the cell-free reaction mixture and on the experimental set-up. For cell-free expression, two configurations can be used. The first configuration, which is easier to implement, is the batch method. The bottleneck of this set-up is the yield, which is quite low due to the consumption of energy and amino acids as well as to the accumulation of by-products, which have an inhibitory effect on the in vitro transcription/translation reaction. As a consequence, using the batch method protein is produced mostly during the first 60 min of in vitro translation, thus limiting the yield.
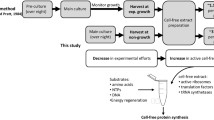
The second configuration was developed to overcome this problem: the continuous exchange cell-free (CECF) system [11] (Fig. 6.1). This system is divided in two compartments that can exchange low molecular weight compounds through a dialysis membrane. The reaction compartment contains all the high molecular weight species required for the reaction such as the cell extract, the enzymes and the nucleic acids as well as the low molecular weight substrates required for the reaction. The feeding compartment contains only the low molecular weight compounds, i.e., the NTPs, substrates of the energy regeneration system and the amino acids. Usually, the feeding compartment is more than ten-times larger than the reaction compartment. Consequently, during the cell-free expression reaction, which is subjected to mixing or shaking, the by-products are dialyzed from the reaction mixture into the feeding compartment. At the same time, the NTPs, energy substrates and amino acids in the reaction mixture are constantly replenished in the reaction compartment. Using this technique, the cell-free expression reaction can be maintained for tens of hours yielding more than 10 mg of protein per milliliter of reaction [28].

Scheme of the continuous exchange cell-free (CECF) system. Two compartments exist separated by a dialysis membrane: the reaction compartment contains the cell extract with the translation machinery, the template (DNA), the RNA polymerase and the low molecular weight substrates required for in vitro transcription and translation. The feeding chamber contains NTPs, substrates of the energy regeneration system and the amino acids in the same reaction buffer as used in the reaction chamber. The feeding chamber is usually more than ten-times larger than the reaction chamber. During protein synthesis, inhibitory side products of the transcription/translation reaction can diffuse into the feeding chamber and thus are diluted. Substrates are consumed during the reaction and are restocked from the feeding compartment
2.2 The PURE Cell-Free Expression System
The PURE (Protein synthesis Using Recombinant Elements) system has been developed with the idea to use only purified components of the transcription and translation machinery for in vitro synthesis [29]. To this end, initiation, elongation and termination factors as well as the 20 aminoacyl-tRNA synthetases, the methyl-tRNA transformylase and the T7 RNA polymerase were expressed as recombinant proteins with a hexahistidine-tag and affinity purified. In total 31 proteins are added to reconstitute the in vitro transcription/translation reaction. The ribosomal subunits were purified from E. coli cells and added to the translation reaction. The resulting PURE system can produce about 100 μg of model proteins per ml reaction in one hour (GFP and DHFR). In addition, the PURE system contains 46 tRNAs, NTPs, creatine phosphate, 10-formyl-5,6,7,8-tetrahydrofolic acid, 20 amino acids, creatine kinase, myokinase, nucleoside-diphosphate kinase and pyrophosphatase. Chaperones, heat shock proteins and other factors can be added to the reaction mixture to keep proteins soluble and assist in protein folding.
The use of histidine-tagged translation components offers the possibility to produce the protein of interest without any tag for affinity purification. The newly synthesized protein can still be easily purified in two steps: ultrafiltration to remove the ribosomes and a Ni-NTA affinity chromatography step to remove the recombinant, his-tagged translation factors. Importantly, RNases and proteases are not present in the PURE system. Thus, mRNA of limited stability can still be a template for translation, and proteins which are rapidly degraded in vivo can be produced in vitro.
The PURE system allows the efficient production of proteins with artificial amino acids. To this end, release factor 1 is omitted from the reaction and a chemically synthesized mis-acylated amino-acyl-tRNA specific for UGA (amber stop codon) is added. This is particularly useful to incorporate fluorescent dyes or specific cross-linkers in the proteins for instance with the aim to analyze protein-protein interactions. Recent improvements of the system aimed at the in vitro synthesis of membrane proteins in the presence of lipids. In summary, the PURE system is highly versatile. It can be modified as specific proteins and other factors can be omitted or added to the reaction according to the needs of the proteins to be produced.
3 Considerations for Cell-Free Protein Synthesis Experiments and Challenges to Produce Protein Complexes
As outlined above, two major approaches exist for cell-free protein expression using the E. coli transcription/translation machinery. The S30 cell extract-based and PURE cell-free systems differ significantly by the degree of purification of the components used. The PURE system has the advantage of being protease- and nuclease-free compared to the S30 cell extract where all cytosolic components are present in the extract. Thus, linear nucleic acids (PCR products and mRNAs) are more stable in the PURE reaction system. Also, proteolytic cleavage of the synthesized protein can be avoided using the PURE system. An additional advantage of the PURE system is the absence of ATP-consuming proteins, which are responsible for the rapid energy depletion in the S30-based system [29]. However, because the PURE system is based on purified components, it is conceivable that some important cofactors or chaperones are missing in this purified system, leading to inefficient folding of the protein. Addition of Trigger Factor, DnaJ, DnaK and GrpE as well as GroEL/GroES may help to improve the yield of soluble, functional protein [30].
The cell extract-based system has the advantage that it is possible to produce the cell extract in large amounts in a standard molecular biology laboratory in a relatively short time (2–3 days for cell extract production and testing). This can be cost-saving, and it allows for upscaling of the in vitro translation reaction. The disadvantage of such cell-extract preparations is that batch-dependent differences in translation activity need to be taken into account. This limits the reproducibility of the method.
The expression of multi-protein complexes is challenging in vitro and in vivo. The correct stoichiometry is difficult to achieve, and the least expressed protein subunit of the complex determines the overall yield of complex. The cell-free systems can be used to express protein complexes: Several DNA templates encoding the protein subunits of the complex can be added simultaneously to the cell-free reaction. In this context, the main advantage of the cell-free expression system compared to the cell-based system is the possibility to precisely adjust the expression of the different subunits of the complex by optimizing the amounts and the ratio of the DNA templates added to the translation reaction. Initial small-scale trials are usually used to optimize the production of the protein subunits in order to achieve stoichiometric expression and homogenous complex formation. In contrast to a cell-based expression in which the different subunits are mostly expressed at the same time, cell-free expression systems allows the sequential addition of DNA templates to the reaction mixture. Moreover, chaperones and additives can be added to the reaction mixture for the efficient integration of the subunits in the complex.
E. coli membrane proteins are mostly dependent on the presence of the conserved Sec translocation machinery for their proper integration into the membrane bilayer [31]. Traditionally, microsomal membranes from dog pancreas treated with high salt and partial trypsin digestion were added to the in vitro translation reaction to achieve co-translational protein translocation [32]. E. coli inverted membrane vesicles and proteoliposomes reconstituted from components of the translocation machinery also have been successfully used for protein translocation and secretion [33, 34]. However, the specific requirements of membrane proteins for efficient translocation and folding are still poorly understood. Cell-free systems are commonly used to study the process of co-translational membrane protein insertion and folding which is rather inefficient [33]. The presence of membrane protein chaperones and additional translocation factors may be crucial. For instance, the subunit c of the F1F0-ATPase has been shown to be dependent on the function of YidC, which is an insertase, integrating small membrane proteins into the membrane of E. coli [35]. In Sect. 6.4.2, we describe the application of cell-free expression systems for membrane protein synthesis and their integration into a lipid bilayer or detergent micelles.
4 Applications
4.1 Ribosome-Nascent Chain Complexes
One important application of cell-free synthesis relates to the preparation of ribosome-nascent chain complexes (RNCs) for structural and functional studies as well as for synthetic biology applications including protein engineering, selection and evolution. To this end, mRNA-ribosome-nascent polypeptide complexes are produced which are stalled in a specific translational state. For this application, cell extract is required containing high concentrations of active ribosomes. The aim is not high yields of newly synthesized protein, but every ribosome is supposed to translate a mRNA template once and then get stalled before translation termination. For RNC production, the in vitro transcription and translation reaction are often uncoupled [36]. In a first step, the mRNA template is generated by in vitro transcription, using T7 RNA polymerase for instance. The mRNA is subsequently purified by LiCl precipitation followed by ethanol precipitation and added to the in vitro translation reaction. The purified mRNA is then added to the in vitro translation reaction. The translation reaction can be stopped by addition of high concentrations of magnesium, chloramphenicol or other antibiotics. Alternatively, stalling motifs like SecM or TnaC, or mRNA templates without a stop codon are used to arrest translation. To stabilize the RNCs in the in vitro translation reaction mix, it is recommended to add oligonucleotides that inhibit the transfer-messenger-RNA complex of E. coli which recognizes ribosomes stalled during protein translation [37]. Subsequently, RNCs can be purified by traditional sucrose gradient centrifugation, or affinity purification via the nascent polypeptide which contains a specific purification tag or an epitope recognized by an antibody [36].
Homogenous RNCs stalled in a specific translational state and complexes with translation factors or factors in co-translational events are mostly studied by single-particle electron cryo-microscopy (cryo-EM). Thanks to recent advances in single-particle cryo-EM it is now possible to reach near-atomic resolution. For instance, a translating ribosome stalled with a TnaC-motif acting L-tryptophan sensor was recently reported at 3.8 Å resolution [38]. Stalling motifs like TnaC and SecM are short peptide sequences that interact with the ribosomal tunnel during translation and induce a conformation in the peptidyl transferase center of the large ribosomal subunits that inhibits further elongation of the nascent polypeptide chain. The ribosome is thus trapped in a specific conformation and displays a nascent polypeptide of defined length. The stalling peptide is hidden in the ribosomal tunnel while the N-terminal part of the nascent chain can exit from the ribosomal tunnel (Fig. 6.2). At the exit of the ribosomal tunnel the nascent chain can fold into a functional protein or bind diverse protein factors involved in co-translational folding, targeting and translocation.

In vitro preparation of ribosome-nascent chain complexes. The DNA template used encodes a promoter (T7 if T7 RNA polymerase is used for in vitro transcription), a Shine-Dalgarno sequence (ribosome binding site), an N-terminal triple TAG (Strep3-tag) followed by the sequence encoding the gene of interest. At the 3′ end the gene encoding the protein of interest is fused in frame to a sequence encoding the translation arrest motif of SecM. During in vitro translation, the protein synthesis is not terminated at a stop codon. It is stalled due to the presence of the SecM arrest motif. This results in stable ternary complexes consisting of mRNA, ribosome and nascent polypeptide. The RNCs can be purified via sucrose gradient centrifugation and via the N-terminal tag of the nascent polypeptide by affinity chromatography. Finally, RNCs and binding factors are reconstituted and analysed, for instance by single particle cryo-EM
Such factors can be directly added to the cell-free expression system. This was the case with the trigger factor (TF), for instance, which is the first chaperone interacting with the newly synthesized polypeptide exiting the ribosome tunnel [39]. Structure determination of the RNC-TF complex suggested that the co-translational folding of the nascent chain was favored by a protected environment formed by TF and the ribosome (Fig. 6.3). Using a similar approach, several ribosomal complexes have been solved by cryo-EM providing important insights into the molecular mechanism of co-translational targeting and translocation [31]. For these studies a DNA sequence encoding the N-terminal part which includes the signal-anchor sequence of the E. coli membrane protein FtsQ was used to produce RNCs. Subsequently, ribosomal complexes were reconstituted for cryo-EM studies by adding purified signal recognition particle (SRP) [40] or SRP-SRP receptor complexes [41] to the RNCs (Fig. 6.3).

Cryo-EM reconstructions of E. coli ribosomal complexes in co-translational folding, targeting and translocation. Homogeneous RNC preparations are used to reconstitute complexes with ribosome binding partners. These complexes allowed visualizing how trigger factor binds to the large ribosomal subunit (50S) and arches over the exit of the ribosomal tunnel (a). Together, the ribosome and trigger factor provide a protected folding space for the ribosome [39]. (b) The signal recognition particle (SRP) binds next to the exit of ribosomal tunnel and adopts an elongated conformation stabilized by interactions with 50S [40]. (c) SRP receptor binding leads to formation of an early complex which adopts a V-shape [42]. (d) After successful handover of the translating ribosome, the SecYEG complex binds tightly to the exit of the ribosomal tunnel. The translocation channel is aligned with the ribosomal tunnel such that an almost continuous channel from the PTC into the periplasm is formed for the nascent chain [44]. The scheme also visualizes the increasing resolution that can be achieved by single particle cryo-EM due to significant improvements in the microscope, detectors and image processing
Structural insights into the mechanism of signal sequence surveillance during protein targeting were obtained by using a DNA sequence for the E. coli autotransporter EspP for RNC generation [42]. EspP is not targeted by SRP to the membrane, but its signal sequence can be bound by SRP. The cryo-EM structures of the RNC-SRP-SRP receptor complexes with the EspP nascent chain revealed how RNCs can be rejected from the SRP targeting pathway [42] (Fig. 6.3). These RNCs were crucial to elucidate the conformational states of SRP and its receptor during co-translational targeting by Fluorescence Resonance Energy Transfer (FRET) [43]. These studies revealed that the targeting reaction is tightly controlled in space and time by the ribosome, the translocation machinery and through GTP hydrolysis.
RNCs displaying a signal-anchor sequence have also been successfully used to reconstitute complexes with the E. coli protein-conducting channel SecYEG and to solve the structure by cryo-EM [44] (Fig. 6.3). Similarly, RNCs that translate the subunit c of the ATP synthase allowed the reconstitution of complexes with the E. coli YidC translocase for cryo-EM [45] and biochemical characterization of the complex using cross-linking agents [35]. In summary, homogenous RNCs are a prerequisite for structural studies. To date, cell-free translation followed by RNC purification and reconstitution of complexes is the method of choice for cryo-EM studies of ribosomal complexes in translation initiation, elongation, termination, recycling and many other ribosomal complexes.
Notably, RNCs are also successfully used to study co-translational folding, targeting and translocation [46]. The dynamic folding of the nascent chain can be studied by FRET and NMR [47, 48]. For NMR, two advantages of the cell-free translation system can be exploited: The specific isotope-labeling of the nascent polypeptide during cell-free synthesis while the ribosome is not labeled, as well as the arrest of the translation reaction to produce nascent chains of different lengths. Moreover, the impact of Trigger Factor and other chaperones on the folding of the nascent chain can be studied with NMR [49].
4.1.1 In Vitro Selection and Evolution Using Ribosome-Nascent Chain Complexes
RNCs provide a link between genotype (mRNA) and phenotype (protein) and thus can be used for in vitro peptide and protein selection experiments. Display techniques such as ribosome display [20] and mRNA-protein fusions [50] allow selecting for antibody single-chain Fv fragments (scFvs) and other proteins that interact with a molecule of interest. The starting library can encode up to 3 × 1011 different proteins which corresponds to a significantly larger library size compared to typical library sizes used for phage display selections (~107–108). Thus, the sequence space explored by in vitro selection is much larger compared to selection methods that involve a transformation or transfection step into a host cell. In ribosome display (Fig. 6.4), a DNA library is first transcribed using the T7 RNA polymerase and then translated in vitro. The mRNA sequences encoding the protein library do not contain a stop codon, but possess a long linker sequence which encodes for a C-terminal spacer peptide that spans the ribosomal exit tunnel. Therefore, the protein part is displayed outside of the ribosomal tunnel and can fold. The RNCs are then mixed with the protein of interest containing an affinity tag and the ribosomal complexes binding to it are therefore co-purified during the subsequent affinity purification. High Mg2+ concentration and low temperature allows preserving the ribosomal complexes such that the mRNA remains bound. After affinity purification, EDTA addition leads to disassembly of the RNCs and the release of the mRNAs. These mRNAs for selected binders and their sequences can be recovered and amplified by RT-PCR. The T7 promoter sequence is reintroduced during the PCR amplification step (Fig. 6.4).

In vitro selection and evolution of protein by ribosome display. A DNA library is transcribed and then translated in vitro. The mRNA sequences lack a stop codon and encode a linker sequence for a C-terminal peptide that spans the ribosomal exit tunnel. Therefore, the proteins encoded by the library can fold. Subsequently, RNCs are mixed with the immobilized target protein of interest. The RNCs interacting with the target protein are co-purified, while the others are washed away during the subsequent affinity purification. EDTA addition leads to dissociation of the RNCs and release of their mRNAs which can be recovered and amplified by RT-PCR. The T7 promoter sequence is reintroduced during the PCR amplification step. The resulting PCR products are subjected to further ribosome display cycles in order to enrich the best binders
The PCR product can then be used for further ribosome display cycles in order to enrich the best binders. Due to PCR errors the protein sequences can evolve in vitro during the selection experiment, and finally proteins with significantly improved affinity are selected which were not encoded by the original library pool [51]. Using PCR mutagenesis protocols, this can of course be exploited for in vitro evolution of proteins towards higher affinity, stability or in case of enzymes improved/altered substrate specificity. For these experiments, it is very important that at each step the diversity of the library is maintained: ideally, each member of the library is present in the experiment in several copies.
The concept of mRNA-protein fusions [50] is very similar to ribosome display. The major difference between the two methods is that a DNA spacer with a 3′ puromycin is fused to the mRNA encoding the protein library. During the in vitro translation reaction, the puromycin can enter the ribosome peptidyl transferase center, and subsequently the nascent polypeptide is transferred to puromycin. Thus, a covalent link is generated between the encoding mRNA and the protein allowing for harsher screening conditions compared to ribosome display where the intactness of the RNCs is crucial.
The ribosome display approach has been successfully used for the generation of high-affinity and highly specific scFvs [51, 52]. More recently, target proteins of bioactive small molecules (drugs) were selected by ribosome display from a library encoding full-length human proteins [53]. Ribosome display was very successfully applied to screen for Designed Ankyrin Repeat Proteins (DARPins), which are designed based on small, concave-shaped, α-helical protein domains typically involved in protein-protein interactions in vivo. The generation of DARPin libraries allows the selection of specific binders to virtually any protein of interest with up to low picomolar affinity. The stability of the core scaffold of DARPins leads to high-level expression and robust folding in ribosome display experiments. Indeed, issues exist with displaying scFvs because of their low folding efficiency. This is partially due to the disulfide bond that needs to be formed in the two immunoglobulin (Ig) domains. Cell-free transcription-translation is routinely performed under reducing conditions, while Ig domains require an oxidative environment for their folding. In ribosome display, this has been addressed by omitting reducing agents (DTT, Dithiothreitol) from the translation reaction and the addition of protein disulfide isomerase (PDI) for improved folding.
Antibody discovery and engineering is of high pharmaceutical interest. Accordingly, many groups developed cell-free expression-based tools to generate antibodies as diagnostics and drugs. For instance the use of the PURE system has several advantages [54] because of its low nuclease and protease activities as well as the absence of the tmRNA complex which increases the stability of the RNCs and allows screening of even larger libraries. The composition of the PURE reaction can be adjusted, release factors are omitted from the reaction, PDI and oxidized glutathione are added leading to proper folding of antibody fragments. A different construct design now also allows to screen libraries of Fabs (Fragment antigen-binding) which are usually more stable than scFvs [54].
4.2 Cell-Free Membrane Protein Expression
Membrane proteins represent about one third of the proteome of a cell. However, their study is often hampered by the lack of a suitable expression system. High-level overexpression of membrane proteins is frequently toxic for the cell. Moreover, the copy number of proteins is limited by the translocation and folding machinery as well as the space which is offered by the membrane bilayer of the host. Cell-free expression of membrane proteins allows overcoming several of these difficulties as it can be adapted to the expression of hydrophobic proteins.
Different possibilities exist to express membrane proteins in a cell-free expression system. First, it is possible to refold the precipitate which is formed during the cell-free expression of a membrane protein. This is achieved by solubilization of the aggregated proteins with detergent for a few hours under gentle agitation (precipitation-forming cell-free, P-CF) (Fig. 6.5). Not all detergents are suitable for the refolding step but dodecylymaltoside (DDM), dodecylphosphocholine and lyso-phosphoglycerol derivatives (LMPG, LPPG) have been shown to successfully solubilize precipitates [55]. This approach has been successfully applied to the production of EmrE, a multidrug transporter [55, 56], and to the human histamine-1 receptor [57]. Second, the addition of detergent directly to the cell-free reaction keeps the nascent membrane proteins in solution (detergent-based cell-free, D-CF) (Fig. 6.5). Like in the P-CF approach, not all the detergents can be used in the detergent-based cell-free expression system. Detergents with a high critical micellar concentration (CMC) such as CHAPS have a tendency to destabilize the translation machinery. In contrast, mild detergents like DDM and digitonin are efficient for D-CF expression of EmrE [58]. Other surfactants which are traditionally not used for membrane solubilization because of their low efficiency to solubilize lipid bilayers have been shown to be particularly useful to stabilize membrane protein during the D-CF: MscL, the mechanosensitive channel, is efficiently expressed as a soluble protein in the presence of amphipols [59]. Compared to the cellular expression and the P-CF, the D-CF expression offers several advantages: (i) it avoids the formation of aggregates; (ii) it avoids the membrane integration step which is limited by the targeting and translocation efficiency, thus improving the production of the protein; (iii) the detergent-solubilized membrane protein can be used immediately. A third approach is based on the addition of lipids to the reaction mixture (lipid-based cell-free). Here, the classical cell-free reaction is supplemented with a preformed lipid bilayer (Fig. 6.5). This membrane-like environment can be either liposomes, bicelles or nanodiscs. While the membrane protein is synthesized at the ribosomes, the transmembrane segments are thought to spontaneously insert into the lipid bilayer offered by those lipidic environments. The main advantage of this technique is that the membrane protein will be produced in a “native-like” environment which is necessary to obtain a functional protein. Not only single membrane proteins can be prepared following these protocols. In fact, several membrane protein complexes have been generated using these methods. For instance, the F1F0-ATP synthase complex has been produced using the three techniques, P-CF, D-CF and L-CF [60]. Importantly, the complexes produced by the three cell-free expression protocols were similar to the in vivo complex in terms of enzymatic activity and structural properties. Using the L-CF approach, the SecYEG complex was produced in vitro [61]. Preformed liposomes were added to the reaction mixture and during translation, the SecYEG complex spontaneously inserted into the liposomes bilayer. The SecYEG translocon produced in this way was functionally active in the translocation of other membrane proteins.

Cell-free synthesis of membrane proteins. Three strategies are used to produce membrane proteins in vitro: in a conventional cell-free translation reaction the membrane protein precipitates (left). Subsequently, the aggregated protein is solubilized with detergent, in the presence of which it can fold into its correct structure. Several mild detergents can be added directly to the translation reaction without interfering with translation (middle), thus preventing the aggregation of the hydrophobic membrane proteins. In the presence of membranes, some membrane proteins can spontaneously insert into the lipid bilayer (right). The correct folding of the in vitro produced membrane proteins needs to be verified in functional assays
Taken together, CFPS has been proven to be a very useful approach to overcome common problems faced with the traditional cellular expression system of membrane proteins.
4.3 Synthetic Biology
Synthetic biology is a rapidly expanding field which is currently actively researched. The idea to engineer biology in order to develop new biotechnological tools is indeed very attractive. Cell-free synthesis can be used to reproduce cellular pathways ex-vivo. On the one hand, the PURE system can allow deciphering the components required to realize a specific biological process. On the other hand, the classical cell-free extract can be the basis for the comprehensive synthesis and assembly of cellular macromolecules towards the development of a synthetic cell.
4.3.1 Bottom-Up Approach
Using the PURE system, it was possible to reconstitute bacterial transcription initiation from five different plasmids [62]. The α, β, β′ and ω subunits of the E. coli RNA polymerase as well as a σ factor (σ32 or σ70) were co-expressed by the PURE machinery using T7 promoter. In this study, the expression and correct assembly of the RNA polymerase and the σ-factor-dependent transcription initiation was confirmed by production of luciferase from a linear DNA template under the control of an E. coli promoter [62]. It was found that the ω subunit is dispensable for transcription initiation. It is now possible to assess the activity of point mutants of the different subunits of the E. coli RNA polymerase. This work could not be performed in bacteria since the expressed variants are likely toxic to the cell. The work also paves the way to study the assembly and the function of other bacterial RNA polymerases for which we have little knowledge.
More recently, the co-expression of 13 genes building up a replication machinery was reported [63]. Step-by-step the authors produced a functional Pol III HE, which is composed of nine different proteins and forms an assembly of 17 subunits. Together with the primase DnaG, it was possible to replicate the G4 phage ssDNA. Using this remarkable system, it was demonstrated that all genes but dnaQ, a proofreading exonuclease, are required for replication activity. The initiation machinery consisting of DnaA possessing the initiator activity, DnaB helicase and DnaC, the helicase loader, was also produced in the PURE system [63]. It was demonstrated that these three proteins are essential and sufficient for initiation of replication. The authors were also able to reconstitute replication activity using a mixture of proteins/complexes produced in different tubes. It was possible to detect ssDNA replication using 13 genes (Pol III HE genes and dnaA, dnaB, dnaC and dnaG) when the PURE synthesis reaction was performed in a single tube. Moreover, the dsDNA produced by the neo-synthesized replication machinery possesses a biological activity as shown in a phage-plaque forming assay. Finally, a synthetic gene circuit using GFP as reporter showed the possibility to produce the complete and functional replication machinery producing a dsDNA containing GFP under the control of the T7 promoter, the only polymerase present in the PURE system. The final production of GFP confirmed the in vitro central dogma in a single tube [63].
4.3.2 Cell-Like Systems
A completely different strategy has been pursued for the development of a cell-free expression toolbox for synthetic biology [64]. A very simple approach based on bead-beater cell breaking was developed to prepare a reproducible, highly active S30 extract. High expression levels of eGFP were obtained under the control of the sigma factor 70, and therefore endogenous RNA polymerase was used for transcription [64]. The aim is to set up a close to native E. coli system to test synthetic gene circuits and to develop an artificial cell. This system enabled the assembly of the bacterial actin MreB on membranes after cell-free transcription/translation inside large liposomes [65]. Furthermore, it was shown that the presence of MreC is required to obtain filamentous structures (Fig. 6.6a, b). An organized cytoskeleton-like structure could thus be obtained inside liposome vesicles by using cell-free expression system producing MreB and MreC.

Successful examples of cell-free synthetic biology. (a) Scheme of cell-free co-expression of YFP-tagged MreB and MreC inside a liposome. (b) Expression of the YFP-MreB fusion protein together with MreC results in the formation of filamentous structures (left panel), rhodamine-BSA stains the lumen of the lipid vesicle (middle panel). The merged red and green image highlights the localization of the YFP-MreB filament on the surface of the liposome. The scale bar corresponds to 10 μm. (c) General scheme of the coupled in vitro transcription-translation reaction allowing the production of assembled and infectious phage particles from the complete 40 kbp genome. (d) Transmission electron microscope micrograph of PHIX174 phage particles produced by the cell-free system. Inset: close-up view of an in vitro synthesized phage (Panels a and b are adapted with permission from Ref. [65]. Panels c and d are adapted with permission from Ref. [67])
Large vesicles of more than 10 μm encapsulating the extract were formed using dispersion of small droplets in an oil phase as a first step [66]. Expression of α-hemolysin lasting for more than 4 days was achieved in this system by exchange of small, up to 3 kDa molecules across the membrane bilayer leading to a continuous supply of substrates for the transcription and translation reactions. This system is therefore the first step towards a bioreactor encapsulated inside a lipid vesicle and able to express proteins for more than 4 days. A step forward was achieved by the expression of the whole T7 bacteriophage genome, containing about 60 genes encoded by 40 kbp DNA. The complete proteome was synthesized using an E. coli cell-free transcription-translation system. Billions of T7 bacteriophages, assembled spontaneously into well-shaped particles (Fig. 6.6c, d), are produced per milliliter of batch reaction. Importantly, these in vitro assembled phages are as infectious as in vivo synthesized ones [67].
This approach opens up the possibility to directly and rapidly assess genetic circuits and the effects of promoter strength or different substrate concentrations, to help understanding bacterial cell metabolism. Very recently, two-dimensional DNA compartments in silicon were generated [68]. In these compartments protein expression cycles can be auto-regulated using interconnected compartments containing different sets of DNA. This approach aims to study biological networks and communication between cells.
4.3.3 Expansion of the Genetic Code
A clear advantage of the cell-free expression is the possibility to efficiently and specifically synthesize proteins with non-natural amino acids. It is possible to replace a certain amino acid by a non-natural analogue provided that the corresponding amino acyl t-RNA synthase (aaRS) recognizes the unnatural amino acid. This can be easily achieved for seleno-methionine which is used in crystallography to solve the phase problem [69] and to structurally similar analogues of proline, tyrosine, phenylalanine, leucine and valine (reviewed in [70]). To further expand the repertoire of amino acids, stop codon suppressor-tRNAs were employed that recognize the amber stop codon were chemically acylated with artificial amino acids [71]. The advantage of this approach is that the incorporation of the artificial amino acid is site specific. Similarly, pairs of specific tRNAs—recognizing the amber stop codon or even a 4-base codon—and engineered aaRSs were evolved to incorporate the artificial amino acid at a specific site of the protein. Several tRNA/aaRS pairs are required to incorporate two or more unnatural amino acids in one protein for protein folding studies using FRET (e.g., [72]). This represents a very powerful approach for investigation of protein structure, function and dynamics. To improve the efficiency of stop codon suppression, release factor RF1 can be omitted from the cell-free translation reaction. For improved 4-base codon tRNA recognition, ‘orthogonal’ ribosomes have been engineered [73]. Similarly, an engineered elongation factor EF-Tu exhibiting improved affinity for incorporation of phosphoserine was reported.
The application possibilities of such unnatural proteins are manifold: ranging from protein folding and protein-protein interaction studies using amino acids with fluorescent dyes or photo-activatable crosslinkers, to production of protein conjugates with small molecules or synthetic polymers for protein therapeutics. Of particular interest are antibody–drug conjugates and polyethylene glycol-growth factor conjugates with improved bio-kinetics [74].
5 Limitations
In the case of large scale expression for structural studies, the main limitation of E. coli cell free extract resides in the cost of the chemicals that have to be added to the system. Furthermore, the use of bacterial extracts leads to the production of proteins without any post-translational modifications, which are sometimes crucial for proper folding and function of eukaryotic proteins. E. coli cell-free expression is therefore most successful for expression of bacterial proteins. Eukaryotic cell-free expressions are often rather inefficient, resulting in low protein yields—this is most likely due to the lack of translation factors in the cell extracts. Moreover, eukaryotic cell-free expression systems are more labor-intensive, for instance requiring capped and polyadenylated mRNA for in vitro efficient translation.
For expression of protein complexes, cell-free expression is limited to bacterial or phage protein complex expression, notably because of the limited protein size that can be expressed in E. coli per se (proteins larger than 100 kDa are difficult to produce in E. coli). This also applies when the transcription-translation machinery is “purified”. In general, the ribosomal machinery tends to be less efficient as soon as it is extracted from the cell and even more in the case of the PURE system in which it has been shown that the ribosomes are ten-times slower than the ones in the cell, incorporating only two amino acids per second [62]. Furthermore, the different enzymes including the ribosomes become less active over time outside of the cell. Due to this limited efficiency, cell-free protein expression did not become a general method for protein production.
6 Outlook
As highlighted in this review, cell-free expression is particularly suited for specific structural biology applications, in vitro protein screening, selection and evolution as well as for synthetic biology. One main advantage is the possibility of specific protein labeling, for instance, in NMR and the possibility to incorporate unnatural amino acids at specific sites of the protein. Here, we provide several examples that apply cell-free expression to produce large assemblies including phages. In these cases, the cell-free systems are used for production of small quantities for analytical purposes and functional studies, rather than large scale protein production.
Cell-free translation is routinely used to study the translation process itself. Recent advances in single molecule techniques may even allow following co-translational processes such as protein folding during active protein synthesis, rather than using stalled RNCs.
For structural biology, cell-free production of complexes comes to the fore when ribosomal complexes are studied. To date, cell-free extracts from eukaryotic species such as yeast, wheat germ, insect cells, rabbit reticulocytes and HeLa cells are constantly improved for protein production. A reconstituted system has been reported for the study of the mechanisms of mammalian protein synthesis [75]. With these cell-free systems, specific eukaryotic RNC complexes can be generated in vitro and structurally and functionally characterized to understand the complex function of the eukaryotic translation machinery.
References
Borsook H (1950) Protein turnover and incorporation of labeled amino acids into tissue proteins in vivo and in vitro. Physiol Rev 30(2):206–219
Winnick T (1950) Studies on the mechanism of protein synthesis in embryonic and tumor tissues. I. Evidence relating to the incorporation of labeled amino acids into protein structure in homogenates. Arch Biochem 27(1):65–74
Palade GE (1955) A small particulate component of the cytoplasm. J Biophys Biochem Cytol 1(1):59–68
Tissieres A, Schlessinger D, Gros F (1960) Amino acid incorporation into proteins by Escherichia coli ribosomes. Proc Natl Acad Sci U S A 46(11):1450–1463
Nirenberg MW (1963) Cell-free protein synthesis directed by messenger RNA. Methods Enzymol 6:17–23
Nirenberg MW, Matthaei JH (1961) The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc Natl Acad Sci U S A 47:1588–1602
Zubay G (1973) In vitro synthesis of protein in microbial systems. Annu Rev Genet 7:267–287
Pratt JM (1984) Coupled transcription–translation in prokaryotic cellfree systems. In: Hemes BD, Higgins SJ (eds) Current protocols. IRL Press, Oxford, pp 179–209
Lesley SA (1995) Preparation and use of E. coli S-30 extracts. Methods Mol Biol 37:265–278
Spirin AS (1991) Cell-free protein synthesis bioreactor. Front Bioprocess II:31–43
Spirin AS, Baranov VI, Ryabova LA, Ovodov SY, Alakhov YB (1988) A continuous cell-free translation system capable of producing polypeptides in high yield. Science 242(4882):1162–1164
Kigawa T et al (1999) Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett 442(1):15–19
Kigawa TIM, Aoki M, Matsuda T, Yabuki T, Seki E, Harada T, Watanabe S, Yokoyama S (2008) The use of the Escherichia coli cell-free protein synthesis for structural biology and structural proteomics. In: Spirin AS, Swartz JR (eds) Cell-free protein synthesis: methods and protocols. Wiley, New York, pp 99–109
Vinarov DA et al (2004) Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nat Methods 1(2):149–153
Yokoyama S et al (2000) Structural genomics projects in Japan. Nat Struct Biol 7(Suppl):943–945
He F et al (2009) Structural and functional characterization of the NHR1 domain of the Drosophila neuralized E3 ligase in the notch signaling pathway. J Mol Biol 393(2):478–495
Koglin A et al (2006) Combination of cell-free expression and NMR spectroscopy as a new approach for structural investigation of membrane proteins. Magn Reson Chem 44 Spec No:S17–S23
Maslennikov I et al (2010) Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc Natl Acad Sci U S A 107(24):10902–10907
Yokoyama S (2003) Protein expression systems for structural genomics and proteomics. Curr Opin Chem Biol 7(1):39–43
Hanes J, Plückthun A (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A 94(10):4937–4942
Klammt C et al (2007) Cell-free production of G protein-coupled receptors for functional and structural studies. J Struct Biol 158(3):482–493
Chen YJ et al (2007) X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A 104(48):18999–19004
Deniaud A, Liguori L, Blesneac I, Lenormand JL, Pebay-Peyroula E (2010) Crystallization of the membrane protein hVDAC1 produced in cell-free system. Biochim Biophys Acta 1798(8):1540–1546
Boland C et al (2014) Cell-free expression and in meso crystallisation of an integral membrane kinase for structure determination. Cell Mol Life Sci 71(24):4895–4910
Kim TW et al (2006) Simple procedures for the construction of a robust and cost-effective cell-free protein synthesis system. J Biotechnol 126(4):554–561
Yang HL, Zubay G (1973) Synthesis of the arabinose operon regulator protein in a cell-free system. Mol Gen Genet 122(2):131–136
Ryabova LA, Desplancq D, Spirin AS, Plückthun A (1997) Functional antibody production using cell-free translation: effects of protein disulfide isomerase and chaperones. Nat Biotechnol 15(1):79–84
Iskakova MB, Szaflarski W, Dreyfus M, Remme J, Nierhaus KH (2006) Troubleshooting coupled in vitro transcription-translation system derived from Escherichia coli cells: synthesis of high-yield fully active proteins. Nucleic Acids Res 34(19), e135
Shimizu Y et al (2001) Cell-free translation reconstituted with purified components. Nat Biotechnol 19(8):751–755
Fedorov AN, Baldwin TO (1998) Protein folding and assembly in a cell-free expression system. Methods Enzymol 290:1–17
Von Loeffelholz O, Botte M, Schaffitzel C (2011) Escherichia coli cotranslational targeting and translocation. ELS. doi:10.1002/9780470015902.a0023170
Walter P, Blobel G (1983) Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol 96:84–93
Müller M, Blobel G (1984) In vitro translocation of bacterial proteins across the plasma membrane of Escherichia coli. Proc Natl Acad Sci U S A 81(23):7421–7425
Schulze RJ et al (2014) Membrane protein insertion and proton-motive-force-dependent secretion through the bacterial holo-translocon SecYEG-SecDF-YajC-YidC. Proc Natl Acad Sci U S A 111(13):4844–4849
Yi L, Celebi N, Chen M, Dalbey RE (2004) Sec/SRP requirements and energetics of membrane insertion of subunits a, b, and c of the Escherichia coli F1F0 ATP synthase. J Biol Chem 279(38):39260–39267
Schaffitzel C, Ban N (2007) Generation of ribosome nascent chain complexes for structural and functional studies. J Struct Biol 158(3):463–471
Giudice E, Mace K, Gillet R (2014) Trans-translation exposed: understanding the structures and functions of tmRNA-SmpB. Front Microbiol 5:113
Bischoff L, Berninghausen O, Beckmann R (2014) Molecular basis for the ribosome functioning as an L-tryptophan sensor. Cell Rep 9(2):469–475
Merz F et al (2008) Molecular mechanism and structure of Trigger Factor bound to the translating ribosome. EMBO J 27(11):1622–1632
Schaffitzel C et al (2006) Structure of the E. coli signal recognition particle bound to a translating ribosome. Nature 444(7118):503–506
Estrozi LF, Boehringer D, Shan SO, Ban N, Schaffitzel C (2011) Cryo-EM structure of the E. coli translating ribosome in complex with SRP and its receptor. Nat Struct Mol Biol 18(1):88–90
von Loeffelholz O et al (2013) Structural basis of signal sequence surveillance and selection by the SRP-FtsY complex. Nat Struct Mol Biol 20(5):604–610
Zhang X, Schaffitzel C, Ban N, Shan SO (2009) Multiple conformational switches in a GTPase complex control co-translational protein targeting. Proc Natl Acad Sci U S A 106(6):1754–1759
Frauenfeld J et al (2011) Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat Struct Mol Biol 18(5):614–621
Kohler R et al (2009) YidC and Oxa1 form dimeric insertion pores on the translating ribosome. Mol Cell 34(3):344–353
Clark PL, Ugrinov KG (2009) Measuring cotranslational folding of nascent polypeptide chains on ribosomes. Methods Enzymol 466:567–590
Hsu ST et al (2007) Structure and dynamics of a ribosome-bound nascent chain by NMR spectroscopy. Proc Natl Acad Sci U S A 104(42):16516–16521
Hsu ST, Cabrita LD, Fucini P, Christodoulou J, Dobson CM (2009) Probing side-chain dynamics of a ribosome-bound nascent chain using methyl NMR spectroscopy. J Am Chem Soc 131(24):8366–8367
O’Brien EP, Christodoulou J, Vendruscolo M, Dobson CM (2012) Trigger factor slows co-translational folding through kinetic trapping while sterically protecting the nascent chain from aberrant cytosolic interactions. J Am Chem Soc 134(26):10920–10932
Roberts RW, Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A 94(23):12297–12302
Hanes J, Schaffitzel C, Knappik A, Pluckthun A (2000) Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat Biotechnol 18(12):1287–1292
Schaffitzel C et al (2001) In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc Natl Acad Sci U S A 98(15):8572–8577
Wada A, Hara S, Osada H (2014) Ribosome display and photo-cross-linking techniques for in vitro identification of target proteins of bioactive small molecules. Anal Chem 86(14):6768–6773
Kanamori T, Fujino Y, Ueda T (2014) PURE ribosome display and its application in antibody technology. Biochim Biophys Acta 1844(11):1925–1932
Klammt C et al (2004) High level cell-free expression and specific labeling of integral membrane proteins. Eur J Biochem 271(3):568–580
Elbaz Y, Steiner-Mordoch S, Danieli T, Schuldiner S (2004) In vitro synthesis of fully functional EmrE, a multidrug transporter, and study of its oligomeric state. Proc Natl Acad Sci U S A 101(6):1519–1524
Sansuk K et al (2008) GPCR proteomics: mass spectrometric and functional analysis of histamine H1 receptor after baculovirus-driven and in vitro cell free expression. J Proteome Res 7(2):621–629
Klammt C et al (2005) Evaluation of detergents for the soluble expression of alpha-helical and beta-barrel-type integral membrane proteins by a preparative scale individual cell-free expression system. FEBS J 272(23):6024–6038
Park KH et al (2007) Fluorinated and hemifluorinated surfactants as alternatives to detergents for membrane protein cell-free synthesis. Biochem J 403(1):183–187
Matthies D et al (2011) Cell-free expression and assembly of ATP synthase. J Mol Biol 413(3):593–603
Matsubayashi H, Kuruma Y, Ueda T (2014) In vitro synthesis of the E. coli Sec translocon from DNA. Angew Chem Int Ed Engl 53(29):7535–7538
Asahara H, Chong S (2010) In vitro genetic reconstruction of bacterial transcription initiation by coupled synthesis and detection of RNA polymerase holoenzyme. Nucleic Acids Res 38(13), e141
Fujiwara K, Katayama T, Nomura SM (2013) Cooperative working of bacterial chromosome replication proteins generated by a reconstituted protein expression system. Nucleic Acids Res 41(14):7176–7183
Shin J, Noireaux V (2010) Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. J Biol Eng 4:8
Maeda YT et al (2012) Assembly of MreB filaments on liposome membranes: a synthetic biology approach. ACS Synth Biol 1(2):53–59
Noireaux V, Libchaber A (2004) A vesicle bioreactor as a step toward an artificial cell assembly. Proc Natl Acad Sci U S A 101(51):17669–17674
Shin J, Jardine P, Noireaux V (2012) Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth Biol 1(9):408–413
Karzbrun E, Tayar AM, Noireaux V, Bar-Ziv RH (2014) Synthetic biology. Programmable on-chip DNA compartments as artificial cells. Science 345(6198):829–832
Watanabe M et al (2010) Cell-free protein synthesis for structure determination by X-ray crystallography. Methods Mol Biol 607:149–160
Hendrickson TL, de Crecy-Lagard V, Schimmel P (2004) Incorporation of nonnatural amino acids into proteins. Annu Rev Biochem 73:147–176
Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244(4901):182–188
Wang K et al (2014) Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat Chem 6(5):393–403
Chin JW (2014) Expanding and reprogramming the genetic code of cells and animals. Annu Rev Biochem 83:379–408
Kim CH, Axup JY, Schultz PG (2013) Protein conjugation with genetically encoded unnatural amino acids. Curr Opin Chem Biol 17(3):412–419
Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV (2006) In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell 125(6):1125–1136
Acknowledgements
C.S. acknowledges funding by the European Research Council Starting grant (ComplexNMD, 281331); and by a Sinergia grant from the Swiss National Science Foundation (CRSII3_136254/1).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Botte, M., Deniaud, A., Schaffitzel, C. (2016). Cell-Free Synthesis of Macromolecular Complexes. In: Vega, M. (eds) Advanced Technologies for Protein Complex Production and Characterization. Advances in Experimental Medicine and Biology, vol 896. Springer, Cham. https://doi.org/10.1007/978-3-319-27216-0_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-27216-0_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27214-6
Online ISBN: 978-3-319-27216-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)