
Abstract
Store-operated calcium entry (SOCE) is a ubiquitous Ca2+ entry pathway that is activated in response to depletion of Ca2+ stores within the endoplasmic reticulum (ER) and contributes to the control of various physiological functions in a wide variety of cell types. The transient receptor potential canonical (TRPC) channels (TRPCs 1–7), that are activated by stimuli leading to PIP2 hydrolysis, were first identified as molecular components of SOCE channels. TRPC channels show a miscellany of tissue expression, physiological functions and channel properties. However, none of the TRPC members display currents that resemble I CRAC. Intensive search for the CRAC channel component led to identification of Orai1 and STIM1, now established as being the primary constituents of the CRAC channel. There is now considerable evidence that STIM1 activates both Orai1 and TRPC1 via distinct domains in its C-terminus. Intriguingly, TRPC1 function is not only dependent on STIM1 but also requires Orai1. The critical functional interaction between TRPC1 and Orai1, which determines the activation of TRPC1, has also been identified. In this review, we will discuss current concepts regarding the role of TRPC channels in SOCE, the physiological functions regulated by TRPC-mediated SOCE, and the complex mechanisms underlying the regulation of TRPCs, including the functional interactions with Orai1 and STIM1.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
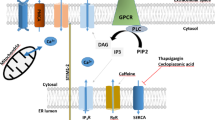
Elevation of cytosolic calcium levels ([Ca2+]i) in response to neurotransmitter stimulation of cells acts as a trigger for activation of many physiological processes, including cell proliferation and differentiation, cell migration, lymphocyte activation, endothelial cell function, as well as protein and fluid secretion from exocrine gland cells. Plasma membrane Ca2+ entry channels contribute to [Ca2+]i elevation and provide critical Ca2+ signals that are utilized for regulation of different cell functions. Store-operated calcium entry (SOCE) is a major ubiquitous Ca2+ entry pathway that contributes to the control of various physiological functions in a wide variety of cell types. SOCE is a unique mechanism in that it is activated in response to depletion of Ca2+ stores within the endoplasmic reticulum (ER). Physiologically, this occurs following stimulation of plasma membrane receptors that lead to phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis and generation of inositol 1,4,5-triphosphate (IP3). IP3 binds to its receptor (IP3R) in the ER membrane and induces Ca2+ release from the ER, resulting in a decrease in ER-[Ca2+] and activation of SOCE. This Ca2+ entry can also be activated by treating cells with sarco-endoplasmic reticulum (SERCA) pump blockers like thapsigargin or cyclopiazonic acid that inhibit Ca2+ uptake into ER, unmasking a passive Ca2+ leak pathway that has not yet been clearly elucidated. Importantly, this leads to loss of ER-Ca2+ and triggers activation of SOCE in the absence of receptor-stimulated signaling. SOCE is inactivated by refilling the Ca2+ stores within the ER, providing further evidence that the activity of channels mediating SOCE are governed by the status of [Ca2+] in the ER [1–3]. The first channel current associated with SOCE, Ca2+-release activated calcium current (I CRAC), was measured in mast cells and T lymphocytes. This highly Ca2+-selective current has a characteristic inward rectification with reversal potential >+40 mV [4–7]. Under the same experimental conditions, currents with varying characteristics and ionic selectivities, ranging from relatively Ca2+-selective to non-selective, have been described in different cell types. These currents have been generally referred to as store-operated calcium current (ISOC) to distinguish them from ICRAC [2, 8].
Search for the molecular components of SOCE, led to the identification of the transient receptor potential canonical (TRPC) channels, part of the superfamily of TRP channels. Mammalian TRPC channels were cloned based on the Drosophila TRP channel, which functions as a light-sensitive Ca2+-permeable channel involved in phototransduction. The Drosophila phototransduction process, a phospholipase C-mediated pathway [9–11], provided further impetus to the search for mammalian TRP channels. The TRPC subfamily consists of seven members (TRPCs 1–7) that are divided into four subsets based on their amino acid (aa) homology: TRPC1, TRPC2, TRPC3/TRPC6/TRPC7 and TRPC4/TRPC5. All TRPC channels display activation in response to receptor-stimulated PIP2 hydrolysis and have six transmembrane domains with a pore-forming domain localized between the fifth and sixth domains. The channels contain N-terminal ankyrin repeats, a highly conserved TRP domain in the C-terminus, several calmodulin (CaM)-binding domains and a putative IP3R binding site [9–11]. TRPC channels show diverse tissue expression, physiological functions and channel properties. Recent reviews have presented a general overview of the molecular components and mechanisms regulating SOCE [1, 12] as well as overviews of the individual TRPC channels: TRPC1 [13], TRPC2 [14], TRPC3 [15], TRPC4 [16], TRPC5 [17], TRPC6 [18], and TRPC7 [19]. We will not be discussing TRPC2, which is a pseudogene in humans [20, 21], in this review. While the discovery of TRPC channels spurred a large number of studies, none of the TRPC family of Ca2+-permeable cation channels generated currents that resembled I CRAC. Thus, identity of the components for this channel, as well as the regulatory proteins in SOCE continued to be a major focus in the field. Intensive search for these finally led to the identification of the CRAC channel component, Orai1, a four-transmembrane domain protein which is assembled as a hexamer to form the pore of the CRAC channel. Two other Orai proteins, Orai2 and Orai3, were also identified and reported to have some similarity with Orai1 and display store-dependent activation. However, since they also contribute to other non-SOCE mechanisms, such as the arachidonic acid-activated channels, further studies are required to fully understand their physiological function. Importantly, the main components involved in sensing ER-[Ca2+] and activating SOCE were also identified. The STIM family of proteins includes two members, STIM1 and STIM2, both of which are Ca2+-sensing proteins that are localized in the ER membrane and sense [Ca2+] within the ER lumen to regulate SOCE. Of the two, STIM1 has been more extensively studied and is now well established as the critical and indispensable regulatory component of SOCE [22]. Furthermore, there is now considerable evidence that STIM1 can activate both Orai1 and TRPC1. The domains of STIM1 involved in gating of these channels are also known. Intriguingly, TRPC1 function is not only dependent on STIM1 but also requires Orai1. The critical functional interaction between TRPC1 and Orai1, which determines the activation of TRPC1, has also been resolved. In the following sections of this review, we will discuss current concepts regarding the role of TRPC channels in SOCE, the physiological functions regulated by TRPC-mediated SOCE, and the complex mechanisms underlying the regulation of TRPCs, including the functional interactions with Orai1 and STIM1.
2 Contribution of TRPC Channels to SOCE
All seven TRPC channels have been implicated as components of SOCE. Furthermore, a variety of physiological functions have been associated with TRPC-mediated SOCE. Recent studies also demonstrate that some human diseases are linked to either loss or gain of function of TRPC channels [23–25]. However, not all the TRPC channels consistently display the hallmarks of SOCE, namely (i) activation by store depletion in response to stimulation with an agonist or treatment with SERCA pump blockers, and (ii) inhibition by Gd3+ (1 μM) and 2-aminophenyl borate (2-APB; ≤10 μM). A large number of the studies assessing the role of TRPCs have used heterologous expression systems where the channels are relatively overexpressed. This does not always result in generation of a functional channel in cells. Some studies have also demonstrated that the mode of regulation of the channels appears to differ depending on the level of their expression. This led to the suggestion that channel overexpression likely results in an unbalanced stoichiometry between TRPCs and the endogenous accessory proteins that regulate and/or modulate their activities. In contrast, more consistent and conclusive data have been provided by studies which assess the function of endogenous TRPCs in SOCE by modulating their expression and/or function in cell lines, primary cell preparations, as well as animal models. So far, the strongest evidence for the contribution of TRPC channels to SOCE has been provided for TRPC1 and TRPC4, whereas the contribution of TRPC3 to SOCE appears to be dependent on cell type and level of expression. TRPCs 5, 6 and 7 have been generally described to be store-independent, with a few exceptions. Note that unlike with Orai1 or STIM1, TRPC channel contribution to SOCE is not seen in all cell types.
TRPC1 was the first mammalian TRPC channel to be cloned and reported to have a role in SOCE [20, 21, 26, 27]. Among the many studies reported, exogenous expression of TRPC1 did not consistently increase SOCE while knockdown of endogenous TRPC1 significantly decreased SOCE (e.g. in HSG, smooth muscle and endothelial cells, as well as platelets) [26–32]. Further conclusive evidence was provided by studies with mice lacking TRPC1 (TRPC1-/-), which despite having normal viability, development, and behavior [33], showed reduced SOCE in cell preparations from several tissues. Among these, salivary gland and pancreatic acinar cells and aortic endothelial cells from TRPC1−/− mice displayed significant reductions in SOCE as well as attenuation in Ca2+-dependent physiological functions [34–36]. SOCE is fundamentally important for fluid secretion in salivary glands and for protein secretion in the exocrine pancreas. TRPC1−/− mice displayed reduction in salivary gland fluid secretion which was associated with a decrease in SOCE and KCa activity in acinar cells from the mice [36, 37]. Similarly defects in Ca2+-activated Cl− channel activity and protein secretion, as a consequence of reduced SOCE, were reported in pancreatic acinar cells [34]. Notably while there is no change in Orai1 in salivary gland and pancreatic acinar cells from TRPC1−/− mice, the channel does not appear to compensate for the lack of TRPC1 or support cell function on its own. Hence, decreased secretory function in these exocrine glands is primarily due to the loss of TRPC1-mediated SOCE. In endothelial cells, TRPC1 forms a heteromeric channel with TRP vanilloid 4 (TRPV4) to mediate SOCE. This Ca2+ entry was significantly reduced in cells from TRPC1−/− mice which adversely impacted vasorelaxation [35].
The caveolae-residing protein, caveolin-1 (Cav-1), is an important modulator of TRPC1 activity and functions as a plasma membrane scaffold for the channel. In the absence of Cav-1, TRPC1 is mislocalized and is unable to interact with STIM1, which is a requirement for TRPC1 activation [38]. Consistent with this, localization of TRPC1, its interaction with STIM1, as well as SOCE were disrupted in salivary gland acinar cells from Cav-1−/− mice [39]. Together, these findings further establish a role for TRPC1 in mediating SOCE in salivary gland cells. Other physiological functions that are dependent on TRPC1-mediated SOCE are the contractile function of glomerular mesangial cells [40, 41] and osteoclast formation and function [42]. Loss of TRPC1 has also been implicated in aberrant vasorelaxation [43], muscle fatigue and slower regeneration after muscle injury [44, 45], whereas elevated TRPC1 expression has been linked to myopathies such as those observed in patients with Duchenne’s Muscular Dystrophy and mdx mice lacking dystrophin [46–48]. However, it remains to be established whether these effects are due to changes in TRPC1-SOCE.
TRPC3 is reported to contribute to both the store-operated and receptor-activated calcium entry pathways. Loss of endogenous TRPC3 in cell lines and tissue preparations (pancreatic acinar and submandibular gland cells) from TRPC3−/− mice led to significant reductions in SOCE [49, 50]. In contrast, overexpression of TRPC3 increased SOCE in COS, HEK293 and HEK293T cells, as well as DT40 chicken B-lymphocytes. However, when the channel was expressed to very high levels, the regulatory mode was switched from store-operated to receptor-activated. Cells with relatively lower levels of TRPC3 expression displayed Gd3+ (1 μM)-sensitive Ca2+ entry, while those with higher levels of channel expression required higher [Gd3+]. Hence, the mechanism by which TRPC3 is regulated appears to be determined by the level of channel expression in the cells [51–54]. TRPC3-mediated Ca2+ entry can also contribute to pathology and tissue damage. Pancreatic acini from TRPC3−/− mice showed significant protection from acute pancreatitis induced by hyper-activation of SOCE. Similar effects were seen by blocking channel function in TRPC3+/+ mice by treatment with pyrazole 3, a TRPC3 inhibitor [55, 56]. Unlike TRPC3, TRPC6 and TRPC7 channels are largely believed be receptor-activated as both channels are consistently activated by the second messenger, diacylglycerol and its analogs [11, 18, 19, 57].
Both TRPC4 and TRPC5 have been suggested to contribute to SOCE, although there are very few studies reported for either channel. Moreover, TRPC5 can also be directly activated by Ca2+, which makes it difficult to establish conclusively whether TRPC5 is directly regulated by store depletion [58, 59]. Exogenous expression of TRPC4 in HEK293 cells increased SOCE and generated a relatively Ca2+-selective, inwardly rectifying current [60]. Similar results were obtained by overexpression TRPC4 in CHO, RBL cells [61] and Xenopus laevis oocytes [62]. Further evidence for TRPC4-mediated SOCE was provided by studies where TRPC4 expression was suppressed in several cell lines or by knockout of the channel in mice (TRPC4−/− mice). Following siRNA treatment, TRPC4-mediated SOCE was diminished in mouse mesangial cells [63], human adrenal cells [64], both mouse and human endothelial cells [65], human gingival keratinocytes [66], human corneal epithelial cells [67] and human pulmonary artery smooth muscle cells [68]. Additionally, TRPC4 forms a heteromeric channel complex with TRPC1 in human mesangial cells [69] as well as human and mouse endothelial cells [65]. Similar to what has been reported for TRPC1−/−, knockout of TRPC4 did not adversely impact mortality and fertility of the mice. Nonetheless, TRPC4−/− mice show significantly reduced TRPC4-mediated SOCE in aortic [70] and lung endothelial cells [65, 71], resulting in defective regulation of vascular tone and endothelial permeability, respectively.
TRPC channels interact with numerous proteins which can underlie the diversity of calcium channel activity, their regulation, and specificity of downstream signaling events in the cells (recently reviewed in [1]). Not only do TRPC channels have the ability to undergo homomeric interactions to form functional channels, they also interact with other TRPCs to generate functional heteromeric channels. Most of the available data in this regard comes from studies with exogenously expressed channels. TRPC1 interacts with TRPC4 and TRPC5, whereas TRPC3 interacts with TRPC6 and TRPC7 [72–74]. It is presently not clear whether the resulting heteromeric channels have distinct properties and functions as compared to those of the individual channels. Very few studies have elucidated the status of endogenous store-dependent heteromeric TRPC channels and their physiological function. The contribution of endogenous heteromeric channel complexes to SOCE have been reported for TRPC1/TRPC3 in a human parotid gland ductal cell line [75] and rat H19-7 hippocampal cell lines [76]; TRPC1/TRPC5 in vascular smooth muscle [77]; TRPC1/TRPC4 in endothelial cells [65], and TRPC1/TRPC3/TRPC7 in HEK293 cells [50]. Given the overlapping expression of more than one TRPC channel in different cells and tissues, some physiological functions may involve multiple channels. For example, SOCE mediated by both TRPC1 and TRPC4 has been proposed to control endothelial cell permeability [65] and myogenesis [78, 79]. Multiple TRPC channels have been implicated in cardiac hypertrophy [80, 81], but it is not clear whether aberrant TRPC-mediated SOCE underlies this phenomena. A few studies have utilized double TRPC knockout mouse models to determine the role of heteromeric TRPC channels. Knockout of both TRPC1 and TRPC4 in mice severely impaired neuronal burst firing and caused neurodegeneration [82], whereas loss of both TRPC3 and TRPC6 impaired sensitivity to mechanical pressure and hearing [83]. The underlying basis for creating these double knockout mice models was the preponderance of co-expressed TRPC1 and TRPC4 in the brain [82] and TRPC3 and TRPC6 in sensory neurons and cochlear hair cells [83]. Whether the pathophysiological effects observed from double knockouts of endogenous TRPC heteromeric channel complexes can be conclusively linked to impaired regulation of SOCE also remains to be shown.
TRPC channels have also been found to associate with other TRP channels, including TRPV6, TRPV4, although in most of these cases it is not clear whether the associating channels form a single channel pore and/or contribute to SOCE. Co-expression of TRPC1 and TRPV4 resulted in formation of a heteromeric channel complex that is activated in response to store depletion in HEK293, vascular smooth muscle and endothelial cells. Moreover, the TRPC1/TRPV4 heteromeric channel exhibited distinct current characteristics when compared to currents mediated by either TRPV4 or TRPC1 alone [35, 84, 85]. TRPC1 was reported to interact with TRPV6 and exert negative regulation of TRPV6 function [86]. A critical heteromeric interaction involving TRPC channels is the TRPC-Orai1 interaction. TRPC1, TRPC3 and TRPC6 functionally interact with Orai1. TRPC1-Orai1 interaction has been confirmed by co-immunoprecipation data as well as TIRFM measurements, where store depletion-dependent clustering of the two channels has been observed in several cell types [87–92]. Further, and more importantly, Orai1 is required for TRPC function, as knockdown of endogenous Orai1 abolished TRPC1 channel activation [88, 92]. A similar requirement for Orai1 has been reported for activation of TRPC3 and TRPC6 in response to ER-Ca2+ store depletion [93–95]. The mechanism underlying the Orai1-dependent regulation of TRPC1 has now been resolved (more details will be presented below). Importantly, TRPC1 and Orai1 have been shown to generate two distinct channels that appear to contribute to specific cellular functions [88, 96]. Interestingly, a recent study reported that a splice variant of TRPC1 interacts with and positively regulates Orai1 channel activity in HEK293 cells. This splice variant, TRPC1ɛ, was first identified in early pre-osteoclasts and together with I-mfa (an inhibitor of MyoD family), has been proposed to function antagonistically to decrease Orai1 channel activity, fine tuning the Ca2+ signaling process that regulates osteoclastogenesis [42]. The multiplicity of interactions between various TRPC channels, as well as between TRPC and other channels or regulatory proteins, lead to the generation of a plethora of signaling complexes that can regulate a wide variety of cellular functions. It is possible that the composition of these heteromeric channels as well as the interacting signaling proteins depends on the type of cell and the particular physiological function to be regulated. There are an increasing number of studies that highlight the importance of spatial and temporal aspects as well as the magnitude of Ca2+ signals as major determining factors in the regulation of cellular responses to different physiological stimuli. It is important that these should be taken into account when the physiological functions of TRPC channels are being assessed.
3 Role of STIM1 and STIM2 in SOCE and TRPC Channel Regulation
STIM1 and STIM2 were discovered in studies using siRNA screening to identify proteins required for SOCE. Both proteins reside within the ER and during resting (unstimulated) conditions, have Ca2+ bound to the luminal N-terminal EF hand domains. Following store depletion, Ca2+ is released from the EF hand which leads to multimerization of the protein and translocation to the peripheral region of the cells where it concentrates in the form of puncta within distinct ER-plasma membrane (ER-PM) junctions. Within this microdomain, the ER membrane and plasma membrane are in close apposition to each other [22, 97, 98]. More importantly, the proximity between the two membranes allows STIM1 in the ER to interact with and gate both TRPC and Orai1 channels. Different aa regions in the cytosolic C-terminus of STIM1 are involved in activating Orai1 and TRPC1. Orai1 activation is mediated by the STIM1 Orai1 Activating Region (SOAR; aa 344–442) [99], whereas TRPC channel gating occurs via the polybasic domain (aa 672–685) [91, 100]. It has been suggested that TRPC1 and TRPC4 are the main TRPC channels that can interact with and be gated by STIM1. However, if other TRPC channels are assembled in a heteromeric channel complex with either TRPC1 or TRPC4, they appear to become store-dependent due to the activation of TRPC1 and TRPC4 by STIM1. For example, although STIM1 does not interact with TRPC3 and TRPC6, STIM1 can activate TRPC1/TRPC3 or TRPC4/TRPC6 channels [101].
Strong evidence for STIM1 in gating TRPC channels comes from studies showing an effect on channel activity of either knockdown or overexpression of STIM1 or STIM1 with mutations that impair STIM1-TRPC channel interactions. TRPC1 was the first TRPC channel shown to be regulated by STIM1. Knockdown of endogenous STIM1 severely reduced endogenous TRPC1-mediated SOCE and Ca2+ currents, whereas co-expression of TRPC1 and STIM1 increased SOCE [87, 88, 91]. Store depletion induced interaction between TRPC1 and STIM1, shown by co-immunoprecipitation experiments as well as FRET and TIRFM measurements. Conversely, store refilling terminated TRPC1 function as well as STIM1-TRPC1 association [91, 100–104]. Thus the TRPC1-STIM1 interaction is dictated by the ER-[Ca2+] status. Conclusive studies by Muallem and co-workers resolved the mechanism by which STIM1 gates TRPC1. Their findings demonstrated that gating of TRPC1 involves electrostatic interactions between the negatively charged aspartate residues in TRPC1 (639DD640) with the positively charged lysines in the STIM1 polybasic domain (684KK685) [100]. Further studies showed that these negatively charged residues are conserved in TRPC3, TRPC4, TRPC5, TRPC6 and TRPC7, suggesting the possibility that STIM1 can also gate these other TRPC channels [100]. Consistent with this study, TRPC4-mediated SOCE in murine and human endothelial cells was suppressed by knockdown of STIM1 or expression of a charge-swap mutant of STIM1 (KK684,685EE). Similarly, disrupting the electrostatic interaction between STIM1 and TRPC4 by mutation of the conserved negatively charged residues on TRPC4 (EE647,648KK) significantly reduced SOCE and STIM1-TRPC4 interactions [65]. Co-immunoprecipitation of STIM1 and TRPC3 was also increased following store depletion in salivary gland duct cells, although this could be due to TRPC1-TRPC3 interaction in these cells [105]. In yet another study, mutations in the conserved negative residues in the C-terminus of TRPC3 (DD697,698KK), TRPC4 (EE648,649KK), TRPC5 (DE651,652KK) and TRPC6 (EE755,756KK) prevented electrostatic interactions with and gating by STIM1. While these mutants did not respond to store depletion induced by cyclopiazonic acid, they could still be activated by muscarinic receptor stimulation in a STIM1-independent manner. However, co-expression of the charge-swap STIM1 mutant (KK684,685EE) restored store responsiveness to these TRPC mutants [106].
Collectively, these data demonstrate that STIM1 has the ability to gate all TRPC channels via similar electrostatic interactions, even though not all channels appear to interact directly with STIM1. Since TRPC channels are widely expressed in tissues and species, it is not yet clear what determines the interaction of any particular TRPC channel with STIM1 and thus their mode of activation. In neuroblastoma cells, STIM1 promoted SOCE mediated via TRPC1 and TRPC6, while inhibiting TRPC6-mediated store-independent Ca2+ entry [107]. TRPC5 was also reported to contribute to SOCE in RBL cells. In these cells co-expression of STIM1 with TRPC5 increased, while knockdown of STIM1 abolished, thapsigargin-induced cation entry [108]. Thus, STIM1 might not only be involved in gating some TRPC channels but also determine their recruitment into a store-dependent mode. However, further studies are required to establish this role of STIM1 on the mode of activation of TRPC channels. Involvement of other factors that might contribute to this switch in the mode of regulation also needs further detailed studies. Since several TRP channels show polymodal regulation of their function, possible activation of TRPC channels by mechanisms other than store depletion appears to be quite feasible.
There is considerable information regarding the exact intramolecular rearrangements and molecular domains involved in activation of STIM1. Further, a large number of structure-function studies have now been reported describing the configuration of STIM1 required for binding and activation of Orai1 (recently reviewed in [109]). However, a similar detailed understanding of STIM1-TRPC channel interaction is currently lacking. An ezrin/radixin/moesin (ERM) domain (aa 251–535) was shown to mediate the binding of STIM1 to TRPC channels [91]. However, since the SOAR domain resides within this ERM region of STIM1 it was suggested that the SOAR domain might also be involved in mediating STIM1 binding to TRPC channels [12, 105]. The coiled-coil (CC) motif within the C-terminus of Orai1 is proposed to interact with STIM1 [110]. TRPC channels also have CC domains in both their N- and C-termini. Further, co-immunoprecipitation studies have revealed strong interactions between exogenously expressed SOAR and endogenous TRPC1, TRPC4 and TRPC5, minimal interactions with endogenous TRPC3 and TRPC6, and no interactions with TRPC7 [101, 105]. Mutation of residues in the N-terminal CC domains severely weakened SOAR interactions with TRPC1, TRPC4 and TRPC5 but enhanced association of TRPC3 and TRPC6 with SOAR. Based on this data, it was proposed that interaction of TRPC1 with TRPC3 induces a structural change which exposes a domain in TRPC3 that promotes its binding to STIM1 [105]. A recent study investigating the stoichiometry of TRPC, STIM1, and CaM assembly in a signaling complex reported TRPC channel activation using recombinantly purified SOAR [111]. This study demonstrated that only TRPC channel complexes containing TRPC1, TRPC4 and TRPC5 could be activated by SOAR. Each TRPC tetrameric complex required two SOAR domains for activation and four CaMs for inactivation. SOAR and CaM appeared to reciprocally regulate TRPC channel activity when co-expressed in HEK293 cells. Following application of tenfold higher amounts of CaM, TRPC1 channel activity was reduced, even though SOAR was still bound to the tetramers at the initial stages of inhibition. SOAR eventually detached from TRPC1, which led to further CaM-dependent decline in channel activity [111]. However, to conclusively establish that SOAR directly affects TRPC channel activity, data need to be provided to exclude SOAR-Orai1 effects in the same cell since SOAR domain will also gate Orai1 and Orai1-mediated Ca2+ entry will lead to TRPC channel activation. It is worth noting that some studies suggest that Orai1 binding to STIM1 might limit availability of STIM1 for TRPC channels. However, these studies have yet to be confirmed at the level of endogenous proteins. Further studies are required to establish whether STIM1 is indeed a limiting factor for channels contributing to SOCE and whether different physiological conditions favor binding of STIM1 to one type of channel vs the other.
STIM1 shares significant homology with another family member, STIM2. While functional domains such as the EF hand, CC domains, SOAR and polybasic domain are conserved between STIM1 and STIM2 there are several key differences which determine their diverse physiological function and role in SOCE. For example, STIM2 can interact with Orai1 but is a poor activator of the channel compared to STIM1. The difference in the gating efficiency of STIM1 and STIM2 was shown to be due to a single aa difference in their respective SOAR domains; F394 in SOAR1 vs L485 in SOAR2 [112]. The EF hand of STIM2 has a lower affinity for Ca2+ than STIM1. Thus, STIM2 can sense and respond to small changes in ER-[Ca2+]. The triggering threshold level of [Ca2+]ER for STIM2 is >400 μM, while STIM1 responds when ER-[Ca2+] is around 200 μM. Based on this, STIM2 has been suggested to aggregate and translocate to ER-PM junction under conditions when there is minimal depletion ER-Ca2+ [113] One reported function for STIM2 is the regulation of Orai1 in resting cells to maintain [Ca2+]i [114]. Another study suggests that STIM2 gates Orai1 in cells stimulated with low agonist where there is less depletion of ER-Ca2+ stores while STIM1 is involved in gating Orai1 at high agonist concentration when there is greater depletion [115]. A recent study provides a novel role for STIM2 showing that STIM2 associates with STIM1 and promotes the clustering of STIM1 in ER-PM junctions in cells stimulated with low [agonist]. STIM2 co-clusters with Orai1 and promotes STIM1-Orai1 interactions at low levels of stimulation while STIM1 aggregates efficiently, in a STIM2-independent manner, and interacts with Orai1 in cells stimulated with high [agonist] [116]. Knockdown of STIM2 in HEK293 cells or targeted knockout of STIM2 in mouse salivary glands attenuated STIM1-mediated activation of Orai1 and decreased the agonist sensitivity of SOCE activation. This was especially prominent at lower levels of agonist. On the other hand, knockdown of STIM1 completely eliminated SOCE at low and high levels of stimulus. Hence, STIM2 appears to tune the agonist-sensitivity of the STIM1-Orai1 interactions and associated Ca2+ signals [116]. Few studies have investigated the direct contribution of STIM2 to TRPC-mediated SOCE. STIM1 has been proposed to regulate TRPC1 and TRPC3 channel function, whereas STIM2 regulated only TRPC1 function in HEK293 cells [117]. Modulation of the STIM1:STIM2 ratio appears to determine the store responsiveness of TRPC1 channel function in intestinal epithelial cells [118]. Thus, the exact role of STIM2 in TRPC channel function and regulation remains to be determined.
4 Orai1-TRPC Channel Interactions in SOCE
The pore-forming component of CRAC channels, Orai1, is indispensable for SOCE. A naturally occurring mutation of the channel, (R91W), which leads to loss of channel function, has been linked to severe combined immune deficiency (SCID) [119–121]. Orai1 has two closely related family members, Orai2 and Orai3, although there is a paucity of data regarding their contribution to SOCE when compared to Orai1 [122]. While endogenous Orai1 function is supported by endogenous STIM1, exogenously expressed Orai1 does not by itself increase SOCE in cells unless STIM1 is co-expressed with it. The reason for this is not yet clear as cells appear to express STIM1 in excess of Orai1. Subsequent studies identified the pore region of Orai1 by showing that E106Q mutation generates a channel with a non-functional pore, while E106D changes Ca2+ selectivity [22, 110, 123, 124]. An additional interesting observation that has been reported is that STIM1 increases the Ca2+ selectivity of Orai1 [125]. Again the latter study was carried out with overexpressed protein and needs to be more fully examined using the endogenous channel.
Intriguingly, a number of studies demonstrate that Orai1 is also required for TRPC1 function. Knockdown of endogenous Orai1 abolished SOCE, despite the presence of endogenous or exogenously expressed STIM1 and TRPC1. Further, it was reported that Orai1 and STIM1 form a complex with TRPC1 in response to ER-Ca2+ store depletion in HSG cells [103], mouse pulmonary arterial smooth muscle cells [126], human parathyroid cells [127], human liver cells [128] rat kidney fibroblast [129], pancreatic acinar cells and salivary gland acinar cells [34, 39]. Notably assembly of the TRPC1-Orai1 complex requires STIM1 which also gates both channels. Co-localization of the three proteins in ER-PM junctions, as detected by TIRFM, suggests that TRPC1 is also localized in same ER-PM junctions where Orai1-STIM1 complex is assembled. The requirement of Orai1 in TRPC1 function was further revealed by data showing that non-functional Orai1 mutants, either Orai1E106Q or Orai1R91W, abrogated store-dependent activation of TRPC1 [87, 88, 92, 103]. Based on this, it was first proposed that TRPC1 and Orai1 assemble into a heteromeric channel where both proteins contribute to the channel pore. There was also the suggestion that TRPC channel forms the pore while Orai1 serves as a regulator. While this led to an extensive debate regarding the assembly of these putative channels, neither of these proposals was supported by conclusive data. Finally, the mechanism underlying the requirement of Orai1 in TRPC1 function was demonstrated in a study where Ca2+ influx mediated by Orai1 triggers plasma membrane insertion of TRPC1 [88]. The insertion presumably occurs within the same ER-PM junctions where the Orai1-STIM1 complex is assembled, to allow for TRPC1 gating by STIM1. Moreover, recruitment of TRPC1 into these junctions brings TRPC1 in close proximity to Orai1, such that Ca2+ entry via Orai1 can be sensed locally to trigger plasma membrane recruitment of TRPC1. This requirement of Orai1-mediated Ca2+ entry for TRPC1 insertion into the plasma membrane also accounts for the lack of TRPC1 activity when non-functional mutants of Orai1 are expressed. Importantly, Ca2+ entry mediated by TRPC1 and Orai1 are utilized by cells to regulate separate functions. Orai1-mediated SOCE is sufficient for activation of NFAT, whereas Ca2+ entry via both Orai1 and TRPC1 are required for NFκB expression and function, with TRPC1 contribution being predominant [88, 96]. Thus, Orai1 and TRPC1 form two separate STIM1-regulated channel complexes (Fig. 5.1). TRPC1 and STIM1 form a SOC channel that generates I SOC while Orai1 and STIM1 form the highly Ca2+-selective CRAC channel mediating I CRAC. The smaller I CRAC is masked by the larger I SOC current and unmasked when TRPC1 function is suppressed. It should be noted that true TRPC1 currents have not yet been described as most reported measurements of ISOC include currents generated by both TRPC1+STIM1 and Orai1+STIM1 channels [88]. A requirement for Orai1 in other TRPC channels such as TRPC3 and TRPC6 [93], as well as heteromeric TRPC channels TRPC1/TRPC4 [130], have been reported. Whether TRPC channel trafficking is involved in these cases is not yet known. The exact proteins involved in regulating and mediating exocytosis of TRPC1 have not yet been elucidated.

Physiological function of Orai1 and TRPC1 in SOCE. Stimulation with agonists generates [Ca2+]i changes that occur locally (i.e. close to the channel pore) and globally (i.e. throughout the cell cytosol). Local SOCE mediated by Orai1 has been shown to activate calcineurin, which subsequently induces NFAT translocation into the nucleus to drive gene expression. Local Orai1-SOCE also promotes insertion of TRPC1 into the plasma membrane. Ca2+ entry via both Orai1 and TRPC1 contribute to increase in global [Ca2+]i, which has been shown to activate NFκB and NFκB-driven gene expression. While the Ca2+-activated ion channels in the plasma membrane are also activated by global [Ca2+]i, it is not clear whether the activating Ca2+ comes from those situated in the vicinity of neighboring Orai1 and TRPC1 channels and/or from the deeper regions of the cell cytosol
One interesting suggestion which has been made is that Orai1 can regulate TRPC channels by determining their recruitment into specialized microdomains in the plasma membrane, such as the lipid raft domains (LRDs). This suggestion is consistent with previous studies showing that SOCE requires intact LRDs [39, 88, 90, 104, 131, 132]. Further, STIM1-TRPC1 interaction also takes place within LRDs as disruption of LRD leads to abrogation of STIM1-TRPC1 interaction and loss of SOCE [104]. Similarly, disruption of LRD in human platelets and HEK293 cells reduced interactions between Orai1, TRPC1, TRPC6 and STIM1 [90, 132, 133]. Orai1 interacts with the cytosolic termini of TRPC1 and TRPC6 to modulate their sensitivity to store depletion and STIM1 [93, 134, 135]. Thus, Lutz Birnbaumer and co-workers suggested the hypothesis that recruitment of Orai1, TRPC and STIM1 into LRD confers store-responsiveness to the channels [135]. At this stage, it remains unclear whether Orai1-STIM1 and TRPC-STIM1 complexes are initially formed outside the lipid rafts and subsequently recruited into these microdomains following store depletion or are maintained within this domain by interactions with Orai1 and STIM1. Presence of PIP2-interacting domains in the C-terminus of STIM1 and STIM2 [132], which are proposed to enable anchoring of the proteins to the plasma membrane within ER-PM junctions, led to several studies examining the role of PIP2 in SOCE. Effects of PIP2 depletion on SOCE were inconsistent with some studies showing no effect on SOCE mediated by Orai1 while others demonstrated decreased function and STIM1 clustering [136–139]. Nevertheless, STIM1 or STIM2 lacking the polybasic tail domain do not form puncta within ER-PM junctional domains. However, when Orai1 is expressed with this mutant of STIM1, it rescues STIM1 clustering and CRAC channel activity. It has been suggested that the Orai1-STIM1ΔK complexes might be localized outside the ER-PM junctions, suggesting that the PIP2 is not required for STIM1-dependent gating of Orai1 [140]. It is unclear whether TRPC-STIM1 interactions can also take place outside the junctional domains. A recent study suggests that dynamic changes in PIP2 levels within ER-PM junctions mediated by proteins such as septin, impact not only assembly of the Orai1-STIM1 complexes but also regulation of CRAC channel activity [141–143]. It has also been recently reported that the ER-PM junctions might contain different PIP2 microdomains. This study showed that Orai1-STIM1 complex assembled in ER-PM junctions is transferred from relatively PIP2-poor to a PIP2-rich microdomain which dictates the Ca2+-dependent regulation of the channel. Interestingly, this recruitment is determined by Cav-1 and septin [143]. Further studies will be required to fully elucidate the dynamic lipid and protein remodeling that occurs with the ER-PM junctions that critically impact Orai1 and TRPC1 interaction with STIM1 and their function.
Trafficking of TRPC channels has been proposed as a major mode of regulation of their function in the plasma membrane. In addition to the trafficking proteins, scaffolding and regulatory proteins also modulate the magnitude and duration of TRPC-mediated SOCE. The main regulatory pathways that modulate surface expression and function of TRPC channels comprise of constitutive and regulated intracellular trafficking mechanisms. The enhancement of Ca2+ influx through TRPC channels can be due to increased exocytosis, retention via interaction with scaffolding proteins, and/or decreased channel endocytosis. As discussed above, TRPC1 function is dependent on LRD [144]. The cholesterol-binding LRD protein Cav-1 is reported to play a pivotal role in plasma membrane localization and activity of TRPC1. TRPC1 interacts with Cav-1 through binding sites located in its N- and C-terminal domains. The N-terminal Cav-1 binding site is involved in scaffolding and localization of TRPC1 in the plasma membrane while the C-terminal domain has been proposed to control channel function and/or inactivation. Knockdown of Cav-1, and mutations in Cav-1 or Cav-1 binding sites in TRPC1 resulted in mislocalization of TRPC1 and impairment of channel activity. Hence, Cav-1 is suggested as an LRD scaffolding protein for TRPC1 that determines plasma membrane localization [38, 39, 131, 145–149]. The current model proposes that in resting cells, constitutive trafficking mechanisms target TRPC1 to cellular regions close to the plasma membrane where the inactive channel interacts with Cav-1 and is retained at that location intracellularly. Following store depletion, STIM1 translocates to ER-PM junctions and activates Orai1. Ca2+ entry mediated by Orai1 is a pivotal step in TRPC1 insertion into the plasma membrane and channel activation [88], as it triggers the insertion of TRPC1 into the plasma membrane. Under these conditions, TRPC1 dissociates from Cav-1, interacts with and is gated by STIM1. Following ER-Ca2+ store refilling, SOCE is inactivated and TRPC1 disassembles from STIM1. LRDs are essential for STIM1 translocation to the ER-PM junctions as deleting the C-terminal lysine-rich region of STIM1, which contains a PIP2-binding sequence, impairs puncta formation in these junctions and also alters partitioning of STIM1 into detergent insoluble fractions from cells. In addition, disruption of lipid rafts by cholesterol depletion also affects the ability of STIM1 to interact with TRPC1 [104]. These data demonstrate the importance of structural integrity for caveolar lipid rafts to act as scaffolding platforms for TRPC1-mediated SOCE. Based on the recent study that showed Cav-1 is required for recruitment of Orai1-STIM1 channel to a PIP2-rich domain [143], it is possible that this event can bring TRPC1 in close proximity to Orai1 such that it can sense local [Ca2+]i elevation due to Orai1-mediated Ca2+ entry. Indeed, Cav-1 might be of utmost importance in Orai1-dependent activation of TRPCs as almost all TRPC channels have once or more Cav-1 binding domains, some of which are fairly well conserved. Nonetheless, Ca2+ sensor proteins, as well as the identity of vesicles and intracellular compartments related to TRPC1 trafficking, remain to be determined.
In addition to Cav-1, another scaffolding protein that regulates TRPC1 function is Homer1. The C-terminus of TRPC1 (aa 644–650) forms a complex with Homer1 and IP3R in resting cells. However, following store depletion, this complex dissociates to enable subsequent TRPC1 interaction with and gating by STIM1 [150, 151]. Further evidence for the contribution of Homer1 came from a study with knockout mice (Homer1−/−) that reported impaired SOCE in skeletal muscle cells [47]. Soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins, such as synaptosome-associated protein (SNAP-25), are involved in membrane fusion within intracellular compartments or between vesicles and plasma membrane. Interaction of SNAP-25 with TRPC1 is vital for channel function as botulinum toxin treatment, which cleaves and inactivates SNAP-25, decreased SOCE in platelets [152]. Several cytoskeletal and microtubule proteins have also been shown to modulate the TRPC1 channel trafficking and activity. The monomeric GTPase protein, RhoA, regulates TRPC1 translocation to the plasma membrane in endothelial cells [29]. Interaction of β-tubulin with TRPC1 determines surface expression of TRPC1 retinal epithelial cells [153]. Disrupting TRPC1 interaction with either RhoA or β-tubulin significantly decreased SOCE. In aggregate, the data show that proper localization of TRPC1 in the plasma membrane, as well as trafficking to the specific domains where SOCE is regulated, are vital for its interaction with Orai1-STIM1 and its activation.
5 Conclusions
The mechanism(s) underlying SOCE involves multiple interactions that allow cells to display dynamic regulatory modes for each physiological stimulus. The multiplicity of channel-protein and protein-protein interactions underscores the variety of signaling complexes that can be generated within a subregion of the cell. Indeed, TRPC channels interact with a wide range of channels and proteins involved in Ca2+ signaling, as well as scaffolding and trafficking processes. Such complexity underlies the physiological functions that have been ascribed to TRPC channels. Many studies have investigated the contributions of STIM1 and Orai1 to TRPC channel function. The functional relevance of STIM2, as well as Orai2 and Orai3, in SOCE remains to be resolved. It is worth noting that many cells and tissues express both STIM proteins and more than one Orai protein. Therefore, depending on the type and intensity of the cell stimulus, TRPC channels may also form dynamic signaling complexes with these STIMs and Orais to generate SOCE. Nonetheless, much remains to be elucidated to expand our current understanding of the exact sequence of molecular events involved in the regulation and function of TRPC channels in response to ER-Ca2+ depletion. As TRPC channels have been implicated in a number of human diseases, understanding the mechanism(s) involved in regulating and modulating channel function will provide potentially important information and lead to novel targets for the development of effective therapeutic interventions.
References
Ong HL, de Souza LB, Cheng KT, Ambudkar IS (2014) Physiological functions and regulation of TRPC channels. Handb Exp Pharmacol 223:1005–1034
Parekh AB, Putney JW Jr (2005) Store-operated calcium channels. Physiol Rev 85(2):757–810
Putney JW Jr (1990) Capacitative calcium entry revisited. Cell Calcium 11(5–6):611–624
Parekh AB, Fleig A, Penner R (1997) The store-operated calcium current I(CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell 89(6):973–980
Penner R, Matthews G, Neher E (1988) Regulation of calcium influx by second messengers in rat mast cells. Nature 334(6182):499–504
Matthews G, Neher E, Penner R (1989) Second messenger-activated calcium influx in rat peritoneal mast cells. J Physiol 418:105–130
Lewis RS, Cahalan MD (1989) Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul 1(1):99–112
Liu X, Groschner K, Ambudkar IS (2004) Distinct Ca2+-permeable cation currents are activated by internal Ca2+-store depletion in RBL-2H3 cells and human salivary gland cells, HSG and HSY. J Membr Biol 200(2):93–104
Minke H, Cook B (2002) TRP channel proteins and signal transduction. Physiol Rev 82:429–472
Montell C (2005) The TRP superfamily of cation channels. Sci STKE 272:re3
Venkatachalam K, Montell C (2007) TRP channels. Annu Rev Biochem 76:387–417
Choi S, Maleth J, Jha A, Lee KP, Kim MS, So I, Ahuja M, Muallem S (2014) The TRPCs-STIM1-Orai interaction. Handb Exp Pharmacol 223:1035–1054
Nesin V, Tsiokas L (2014) Trpc1. Handb Exp Pharmacol 222:15–51
Miller BA (2014) Trpc2. Handb Exp Pharmacol 222:53–65
Lichtenegger M, Groschner K (2014) TRPC3: a multifunctional signaling molecule. Handb Exp Pharmacol 222:67–84
Freichel M, Tsvilovskyy V, Camacho-Londono JE (2014) TRPC4- and TRPC4-containing channels. Handb Exp Pharmacol 222:85–128
Zholos AV (2014) Trpc5. Handb Exp Pharmacol 222:129–156
Dietrich A, Gudermann T (2014) TRPC6: physiological function and pathophysiological relevance. Handb Exp Pharmacol 222:157–188
Zhang X, Trebak M (2014) Transient receptor potential canonical 7: a diacylglycerol-activated non-selective cation channel. Handb Exp Pharmacol 222:189–204
Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten S, Montell C (1995) TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci U S A 92:9652–9656
Zhu X, Chu PB, Peyton M, Birnbaumer L (1995) Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett 373:193–198
Hogan PG, Lewis RS, Rao A (2010) Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol 28:491–533
Nilius B, Owsianik G (2010) Transient receptor potential channelopathies. Pflugers Arch 460(2):437–450
Greka A, Mundel P (2011) Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J Am Soc Nephrol 22(11):1969–1980
Beech DJ (2013) Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ J 77(3):570–579
Liu X, Singh BB, Ambudkar IS (2003) TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5-S6 region. J Biol Chem 278(13):11337–11343
Liu X, Wang W, Singh BB, Lockwich T, Jadlowiec J, O’Connell B, Wellner R, Zhu MX, Ambudkar IS (2000) Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J Biol Chem 275(5):3403–3411
Dietrich A, Chubanov V, Kalwa H, Rost BR, Gudermann T (2006) Cation channels of the transient receptor potential superfamily: their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol Ther 112(3):744–760
Mehta D, Ahmmed GU, Paria BC, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, Minshall RD, Malik AB (2003) RhoA interaction with inositol 1,4,5-triphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. J Biol Chem 278(35):33492–33500
Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB (2006) Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13(8):693–708
Brownlow SL, Harper AG, Harper MT, Sage SO (2004) A role for hTRPC1 and lipid raft domains in store-mediated calcium entry in human platelets. Cell Calcium 35(2):107–113
Rosado JA, Brownlow SL, Sage SO (2002) Endogenously expressed Trp1 is involved in store-mediated Ca2+ entry by conformational coupling in human platelets. J Biol Chem 277(44):42157–42163
Dietrich A, Fahlbusch M, Gudermann T (2014) Classical transient receptor potential 1 (TRPC1): channel or channel regulator? Cells 3(4):939–962
Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, Muallem S (2011) Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12(2):232–245
Ma X, Cheng KT, Wong CO, O’Neil RG, Birnbaumer L, Ambudkar IS, Yao X (2011) Heteromeric TRPV4-C1 channels contribute to store-operated Ca2+ entry in vascular endothelial cells. Cell Calcium 50(6):502–509
Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1−/− mice. Proc Natl Acad Sci U S A 104(44):17542–17547
Sun Y, Birnbaumer L, Singh BB (2015) TRPC1 regulates calcium-activated chloride channels in salivary gland cells. J Cell Physiol. doi:10.1002/jcp.25017
Pani B, Ong HL, Brazer SC, Liu X, Rauser K, Singh BB, Ambudkar IS (2009) Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc Natl Acad Sci U S A 106(47):20087–20092
Pani B, Liu X, Bollimuntha S, Cheng KT, Niesman IR, Zheng C, Achen VR, Patel HH, Ambudkar IS, Singh BB (2013) Impairment of TRPC1-STIM1 channel assembly and AQP5 translocation compromise agonist-stimulated fluid secretion in mice lacking caveolin1. J Cell Sci 126(Pt 2):667–675
Du J, Sours-Brothers S, Coleman R, Ding M, Graham S, Kong DH, Ma R (2007) Canonical transient receptor potential 1 channel is involved in contractile function of glomerular mesangial cells. J Am Soc Nephrol 18(5):1437–1445
Sours S, Du J, Chu S, Ding M, Zhou XJ, Ma R (2006) Expression of canonical transient receptor potential (TRPC) proteins in human glomerular mesangial cells. Am J Physiol Renal Physiol 290(6):F1507–F1515
Ong EC, Nesin V, Long CL, Bai CX, Guz JL, Ivanov IP, Abramowitz J, Birnbaumer L, Humphrey MB, Tsiokas L (2013) A TRPC1-dependent pathway regulates osteoclast formation and function. J Biol Chem 288(31):22219–22231
Kochukov MY, Balasubramanian A, Noel RC, Marrelli SP (2013) Role of TRPC1 and TRPC3 channels in contraction and relaxation of mouse thoracic aorta. J Vasc Res 50(1):11–20
Zanou N, Schakman O, Louis P, Ruegg UT, Dietrich A, Birnbaumer L, Gailly P (2012) Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J Biol Chem 287(18):14524–14534
Zanou N, Shapovalov G, Louis M, Tajeddine N, Gallo C, Van Schoor M, Anguish I, Cao ML, Schakman O, Dietrich A, Lebacq J, Ruegg U, Roulet E, Birnbaumer L, Gailly P (2010) Role of TRPC1 channel in skeletal muscle function. Am J Physiol Cell Physiol 298(1):C149–C162
Sabourin J, Lamiche C, Vandebrouck A, Magaud C, Rivet J, Cognard C, Bourmeyster N, Constantin B (2009) Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem 284(52):36248–36261
Stiber JA, Zhang ZS, Burch J, Eu JP, Zhang S, Truskey GA, Seth M, Yamaguchi N, Meissner G, Shah R, Worley PF, Williams RS, Rosenberg PB (2008) Mice lacking Homer 1 exhibit a skeletal myopathy characterized by abnormal transient receptor potential channel activity. Mol Cell Biol 28(8):2637–2647
Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, Raymond G, Cognard C, Bourmeyster N, Constantin B (2007) Regulation of capacitative calcium entries by {alpha}1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of {alpha}1-syntrophin. FASEB J 20:136–138
Kim EY, Alvarez-Baron CP, Dryer SE (2009) Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol 75(3):466–477
Zagranichnaya TK, Wu X, Villereal ML (2005) Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. J Biol Chem 280(33):29559–29569
Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Zhu MX, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L (1999) Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc Natl Acad Sci U S A 96(26):14955–14960
Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S (1998) Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 396(6710):478–482
Vazquez G, Wedel BJ, Trebak M, St John Bird G, Putney JW Jr (2003) Expression level of the canonical transient receptor potential 3 (TRPC3) channel determines its mechanism of activation. J Biol Chem 278(24):21649–21654
Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L (1996) trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 85(5):661–671
Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S (2009) Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137(4):1509–1517
Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, Birnbaumer L, Mori Y, Muallem S (2011) Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology 140(7):2107–2115, 2115 e2101-2104
Ambudkar IS, Ong HL (2007) Organization and function of TRPC channelosomes. Pflugers Arch 455(2):187–200
Okada T, Shimizu S, Wakamori M, Maeda A, Kurosaki T, Takada N, Imoto K, Mori Y (1998) Molecular cloning and functional characterisation of a novel receptor-activated TRP Ca2+ channel from mouse brain. J Biol Chem 273(17):10279–10287
Zeng F, Xu SZ, Jackson PK, McHugh D, Kumar B, Fountain SJ, Beech DJ (2004) Human TRPC5 channel activated by a multiplicity of signals in a single cell. J Physiol 559(Pt 3):739–750
Philipp S, Cavalie A, Freichel M, Wissenbach U, Zimmer S, Trost C, Marquart A, Murakami M, Flockerzi V (1996) A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J 15(22):6166–6171
Warnat J, Philipp S, Zimmer S, Flockerzi V, Cavalie A (1999) P.henotype of a recombinant store-operated channel : highly selective permeation of Ca2+. J Physiol 518(3):631–638
Kinoshita M, Akaike A, Satoh M, Kaneko S (2000) Positive regulation of capacitative Ca2+ entry by intracellular Ca2+ in Xenopus oocytes expressing rat TRP4. Cell Calcium 28(3):151–159
Wang J, Shimoda LA, Sylvester JT (2004) Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 286(4):L848–L858
Philipp S, Trost C, Warnat J, Rautmann J, Himmerkus N, Schroth G, Kretz O, Nastainczyk W, Cavalie A, Hoth M, Flockerzi V (2000) TRP4 (CCE1) protein is part of native calcium release-activated Ca2+-like channels in adrenal cells. J Biol Chem 275(21):23985–23972
Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, Malik AB, Tiruppathi C (2012) The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol 81(4):510–526
Fatherazi S, Presland RB, Belton CM, Goodwin P, Al-Qutub M, Trbic Z, Macdonald G, Schubert MM, Izutsu KT (2007) Evidence that TRPC4 supports the calcium selective ICRAC-like current in human gingival keratinocytes. Pflugers Arch 453(6):879–889
Yang H, Mergler S, Sun X, Wang Z, Lu L, Bonanno JA, Pleyer U, Reinach PS (2005) TRPC4 knockdown suppresses epidermal growth factor-induced store-operated channel activation and growth in human corneal epithelial cells. J Biol Chem 280(37):32230–32237
Zhang S, Remillard CV, Fantozzi I, Yuan JX (2004) ATP-induced mitogenesis is mediated by cyclic AMP response element-binding protein-enhanced TRPC4 expression and activity in human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 287(5):C1192–C1201
Sours-Brothers S, Ding M, Graham S, Ma R (2009) Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med (Maywood) 234(6):673–682
Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weibgerber P, Biel M, Philipp S, Freise D, Droogmans G, Hofmann F, Flockerzi V, Nilius B (2001) Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol 3:121–127
Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB (2002) Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ Res 91(1):70–76
Goel M, Sinkins WG, Schilling WP (2002) Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem 277(50):48303–48310
Hofmann T, Schaefer M, Schultz G, Gudermann T (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A 99(11):7461–7466
Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE (2003) Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem 278(40):39014–39019
Liu X, Bandyopadhyay BC, Singh BB, Groschner K, Ambudkar IS (2005) Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J Biol Chem 280(22):21600–21606
Wu X, Zagranichnaya TK, Gurda GT, Eves EM, Villereal ML (2004) A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J Biol Chem 279(42):43392–43402
Beech DJ (2005) Emerging functions of 10 types of TRP cationic channel in vascular smooth muscle. Clin Exp Pharmacol Physiol 32(8):597–603
Antigny F, Koenig S, Bernheim L, Frieden M (2013) During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca2+ entry. J Cell Sci 126(Pt 11):2525–2533
Louis M, Zanou N, Van Schoor M, Gailly P (2008) TRPC1 regulates skeletal myoblast migration and differentiation. J Cell Sci 121(Pt 23):3951–3959
Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, Birnbaumer L, Rosenberg P (2009) TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res 105(10):1023–1030
Wu X, Eder P, Chang B, Molkentin JD (2010) TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A 107(15):7000–7005
Phelan KD, Mock MM, Kretz O, Shwe UT, Kozhemyakin M, Greenfield LJ, Dietrich A, Birnbaumer L, Freichel M, Flockerzi V, Zheng F (2012) Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol Pharmacol 81(3):384–392
Quick K, Zhao J, Eijkelkamp N, Linley JE, Rugiero F, Cox JJ, Raouf R, Gringhuis M, Sexton JE, Abramowitz J, Taylor R, Forge A, Ashmore J, Kirkwood N, Kros CJ, Richardson GP, Freichel M, Flockerzi V, Birnbaumer L, Wood JN (2012) TRPC3 and TRPC6 are essential for normal mechanotransduction in subsets of sensory neurons and cochlear hair cells. Open Biol 2(5):120068
Ma X, Cao J, Luo J, Nilius B, Huang Y, Ambudkar IS, Yao X (2010) Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-c1 heteromeric channels to the plasma membrane. Arterioscler Thromb Vasc Biol 30(11):2249–2255
Ma X, Nilius B, Wong JW, Huang Y, Yao X (2011) Electrophysiological properties of heteromeric TRPV4-C1 channels. Biochim Biophys Acta 1808(12):2789–2797
Schindl R, Fritsch R, Jardin I, Frischauf I, Kahr H, Muik M, Riedl MC, Groschner K, Romanin C (2012) Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J Biol Chem 287(42):35612–35620
Cheng KT, Liu X, Ong HL, Ambudkar IS (2008) Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem 283(19):12935–12940
Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS (2011) Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol 9(3):e1001025
Galan C, Zbidi H, Bartegi A, Salido GM, Rosado JA (2009) STIM1, Orai1 and hTRPC1 are important for thrombin- and ADP-induced aggregation in human platelets. Arch Biochem Biophys 490(2):137–144
Jardin I, Salido GM, Rosado JA (2008) Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels 2(6):401–403
Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF (2006) STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol 8(9):1003–1010
Kim MS, Zeng W, Yuan JP, Shin DM, Worley PF, Muallem S (2009) Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J Biol Chem 284(15):9733–9741
Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L (2007) Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A 104(11):4682–4687
Jardin I, Gomez LJ, Salido GM, Rosado JA (2009) Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem J 420(2):267–276
Woodard GE, Lopez JJ, Jardin I, Salido GM, Rosado JA (2010) TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J Biol Chem 285(11):8045–8053
Ong HL, Jang SI, Ambudkar IS (2012) Distinct contributions of Orai1 and TRPC1 to agonist-induced [Ca2+]i signals determine specificity of Ca2+-dependent gene expression. PLoS ONE 7(10):e47146
Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15(13):1235–1241
Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169(3):435–445
Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11(3):337–343
Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell 32(3):439–448
Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9(6):636–645
Lopez JJ, Salido GM, Pariente JA, Rosado JA (2006) Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem 281(38):28254–28264
Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS (2007) Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem 282(12):9105–9116
Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS, Singh BB (2008) Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of Store-operated Ca2+ Entry (SOCE). J Biol Chem 283(25):17333–17340
Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S, Yuan JP (2014) Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J Biol Chem 289(10):6372–6382
Lee KP, Yuan JP, So I, Worley PF, Muallem S (2010) STIM1-dependent and STIM1-independent function of transient receptor potential canonical (TRPC) channels tunes their store-operated mode. J Biol Chem 285(49):38666–38673
Albarran L, Dionisio N, Lopez E, Salido GM, Redondo PC, Rosado JA (2014) STIM1 regulates TRPC6 heteromultimerization and subcellular location. Biochem J 463(3):373–381
Ma HT, Peng Z, Hiragun T, Iwaki S, Gilfillan AM, Beaven MA (2008) Canonical transient receptor potential 5 channel in conjunction with Orai1 and STIM1 allows Sr2+ entry, optimal influx of Ca2+, and degranulation in a rat mast cell line. J Immunol 180(4):2233–2239
Shim AH, Tirado-Lee L, Prakriya M (2015) Structural and functional mechanisms of CRAC channel regulation. J Mol Biol 427(1):77–93
Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C (2008) Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem 283(12):8014–8022
Asanov A, Sampieri A, Moreno C, Pacheco J, Salgado A, Sherry R, Vaca L (2015) Combined single channel and single molecule detection identifies subunit composition of STIM1-activated transient receptor potential canonical (TRPC) channels. Cell Calcium 57(1):1–13
Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, Gill DL (2014) Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat Commun 5:3183–3193
Collins SR, Meyer T (2011) Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol 21(4):202–211
Brandman O, Liou J, Park WS, Meyer T (2007) STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131(7):1327–1339
Kar P, Bakowski D, Di Capite J, Nelson C, Parekh AB (2012) Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc Natl Acad Sci U S A 109(18):6969–6974
Ong HL, de Souza LB, Zheng C, Cheng KT, Liu X, Goldsmith CM, Feske S, Ambudkar IS (2015) STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci Signal 8(359):ra3
Shalygin A, Skopin A, Kalinina V, Zimina O, Glushankova L, Mozhayeva GN, Kaznacheyeva E (2014) STIM1 and STIM2 proteins differently regulate endogenous store-operated channels in HEK293 cells. J Biol Chem 290(8): 4717–4727
Rao JN, Rathor N, Zhuang R, Zou T, Liu L, Xiao L, Turner DJ, Wang JY (2012) Polyamines regulate intestinal epithelial restitution through TRPC1-mediated Ca2+ signaling by differentially modulating STIM1 and STIM2. Am J Physiol Cell Physiol 303(3):C308–C317
Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R (2006) CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol 16(20):2073–2079
Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD (2006) Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci U S A 103(24):9357–9362
Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441(7090):179–185
Hoth M, Niemeyer BA (2013) The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr Top Membr 71:237–271
Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A (2007) Biochemical and functional characterization of Orai proteins. J Biol Chem 282(22):16232–16243
Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443(7108):230–233
McNally BA, Somasundaram A, Yamashita M, Prakriya M (2012) Gated regulation of CRAC channel ion selectivity by STIM1. Nature 482(7384):241–245
Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, Shen XM, Hume JR (2009) TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J Physiol 587(Pt 11):2429–2442
Lu M, Branstrom R, Berglund E, Hoog A, Bjorklund P, Westin G, Larsson C, Farnebo LO, Forsberg L (2010) Expression and association of TRPC subtypes with Orai1 and STIM1 in human parathyroid. J Mol Endocrinol 44(5):285–294
Zhang ZY, Pan LJ, Zhang ZM (2010) Functional interactions among STIM1, Orai1 and TRPC1 on the activation of SOCs in HL-7702 cells. Amino Acids 39(1):195–204
Almirza WH, Peters PH, van Zoelen EJ, Theuvenet AP (2012) Role of Trpc channels, Stim1 and Orai1 in PGF2α-induced calcium signaling in NRK fibroblasts. Cell Calcium 51(1):12–21
Cioffi DL, Wu S, Chen H, Alexeyev M, St Croix CM, Pitt BR, Uhlig S, Stevens T (2012) Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ Res 110(11):1435–1444
Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS (2000) Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem 275(16):11934–11942
Dionisio N, Galan C, Jardin I, Salido GM, Rosado JA (2011) Lipid rafts are essential for the regulation of SOCE by plasma membrane resident STIM1 in human platelets. Biochim Biophys Acta 1813(3):431–437
Galan C, Woodard GE, Dionisio N, Salido GM, Rosado JA (2010) Lipid rafts modulate the activation but not the maintenance of store-operated Ca2+ entry. Biochim Biophys Acta 1803(9):1083–1093
Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L (2008) Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A 105(8):2895–2900
Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L (2009) A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci U S A 106(9):3202–3206
Chvanov M, Walsh CM, Haynes LP, Voronina SG, Lur G, Gerasimenko OV, Barraclough R, Rudland PS, Petersen OH, Burgoyne RD, Tepikin AV (2008) ATP depletion induces translocation of STIM1 to puncta and formation of STIM1-ORAI1 clusters: translocation and re-translocation of STIM1 does not require ATP. Pflugers Arch: Eur J Physiol 457(2):505–517
Korzeniowski MK, Popovic MA, Szentpetery Z, Varnai P, Stojilkovic SS, Balla T (2009) Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J Biol Chem 284(31):21027–21035
Walsh CM, Chvanov M, Haynes LP, Petersen OH, Tepikin AV, Burgoyne RD (2010) Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem J 425(1):159–168
Calloway N, Owens T, Corwith K, Rodgers W, Holowka D, Baird B (2011) Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P2 between distinct membrane pools. J Cell Sci 124(Pt 15):2602–2610
Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136(5):876–890
Sharma S, Quintana A, Findlay GM, Mettlen M, Baust B, Jain M, Nilsson R, Rao A, Hogan PG (2013) An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 499(7457):238–242
Wollman R, Meyer T (2012) Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat Cell Biol 14(12):1261–1269
Maleth J, Choi S, Muallem S, Ahuja M (2014) Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat Commun 5:5843
Ong HL, Ambudkar IS (2011) The dynamic complexity of the TRPC1 channelosome. Channels 5(5):424–431
Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS (2003) Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J Biol Chem 278(29):27208–27215
Kwiatek AM, Minshall RD, Cool DR, Skidgel RA, Malik AB, Tiruppathi C (2006) Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor potential channel-1 in endothelial cells. Mol Pharmacol 70(4):1174–1183
Weihuang Y, Chang SJ, Harn HI, Huang HT, Lin HH, Shen MR, Tang MJ, Chiu WT (2015) Mechanosensitive store-operated calcium entry regulates the formation of cell polarity. J Cell Physiol. doi:10.1002/jcp.24936
Rathor N, Chung HK, Wang SR, Wang JY, Turner DJ, Rao JN (2014) Caveolin-1 enhances rapid mucosal restitution by activating TRPC1-mediated Ca2+ signaling. Physiol Rep 2(11):e12193
Sundivakkam PC, Kwiatek AM, Sharma TT, Minshall RD, Malik AB, Tiruppathi C (2009) Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am J Physiol Cell Physiol 296(3):C403–C413
Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF (2003) Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 114(6):777–789
Yuan JP, Lee KP, Hong JH, Muallem S (2012) The closing and opening of TRPC channels by Homer1 and STIM1. Acta Physiol 204:237–247
Redondo PC, Harper AG, Salido GM, Pariente JA, Sage SO, Rosado JA (2004) A role for SNAP-25 but not VAMPs in store-mediated Ca2+ entry in human platelets. J Physiol 558(Pt 1):99–109
Bollimuntha S, Cornatzer E, Singh BB (2005) Plasma membrane localization and function of TRPC1 is dependent on its interaction with beta-tubulin in retinal epithelium cells. Vis Neurosci 22(2):163–170
Acknowledgements
Work in ISA’s laboratory is supported by the Intramural Research Program of the NIH, NIDCR.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ong, H.L., de Souza, L.B., Ambudkar, I.S. (2016). Role of TRPC Channels in Store-Operated Calcium Entry. In: Rosado, J. (eds) Calcium Entry Pathways in Non-excitable Cells. Advances in Experimental Medicine and Biology, vol 898. Springer, Cham. https://doi.org/10.1007/978-3-319-26974-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-26974-0_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26972-6
Online ISBN: 978-3-319-26974-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)