
Abstract
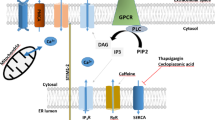
Calcium influx is an essential mechanism for the activation of cellular functions both in excitable and non-excitable cells. In non-excitable cells, activation of phospholipase C by occupation of G protein-coupled receptors leads to the generation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), which, in turn, initiate two Ca2+ entry pathways: Ca2+ release from intracellular Ca2+ stores, signaled by IP3, leads to the activation of store-operated Ca2+ entry (SOCE); on the other hand, DAG activates a distinct second messenger-operated pathway. SOCE is regulated by the filling state of the intracellular calcium stores. The search for the molecular components of SOCE has identified the stromal interaction molecule 1 (STIM1) as the Ca2+ sensor in the endoplasmic reticulum and Orai1 as a store-operated channel (SOC) subunit. Furthermore, a number of reports have revealed that several members of the TRPC family of channels also take part of the SOC macromolecular complex. This introductory chapter summarizes the early pieces of evidence that led to the concept of SOCE and the components of the store-operated signaling pathway.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 1.1 The Concept of Store-Operated Ca2+Entry
A number of physiological agonists activate cellular functions by inducing changes in cytosolic Ca2+ concentration ([Ca2+]c). In general, Ca2+ mobilization consist of the release of Ca2+ from intracellular stores, as well as Ca2+ influx from the extracellular medium through Ca2+-permeable channels. Despite the identification of Ca2+as a second messenger in the excitation-contraction coupling took place in the mid-twentieth century [1], the relevance of extracellular Ca2+in cellular physiology was already highlighted in the publications of Ringer in the Journal of Physiology in the early 1880s [2, 3]. Throughout the 1960s and 1970s, different laboratories determined Ca2+ signals induced by physiological agonists, first using luminescent photoproteins and later on through the use of fluorescent probes, until Ca2+ was recognized as a bona fide second messenger [4].
After Ringer’s findings, the studies concerning the functional role of Ca2+ entry were mainly focused on muscle contraction, especially in the heart and the sartorius muscle, where 45Ca2+ uptake (influx) by the cell was analyzed under different experimental conditions [5, 6]. Among the first analysis of Ca2+ entry in non-electrically excitable cells were performed in HeLa cells in culture, where Borle identified two exchangeable pools, assumed as the extracellular Ca2+ and Ca2+ stored in intracellular compartments, and two unexchangeable Ca2+ pools, corresponding to extracellular and intracellular bound Ca2+ [7]. A year earlier, the same author reported that the extracellular medium was a major source of Ca2+ [8]. Borle defined Ca2+ influx in these cells as a facilitated diffusion process that can be affected by increasing the membrane permeability to this ion and not by stimulating an active or metabolically dependent process [7].
Voltage-dependent Ca2+currents independent of metabolic events were described in the early 70s and a mechanism for Ca2+ influx operated by the occupation of membrane receptors was also soon reported in electrically excitable and non-excitable cells [9–11]. But, it was in 1986, when a mechanism for receptor-operated Ca2+ influx regulated by the filling state of the intracellular Ca2+ stores was proposed [12]. The term capacitative Ca2+entry was coined by James Putney to refer to a process whereby the discharge of Ca2+ stores within a cell secondarily activates Ca2+entry into the cell across the plasma membrane [12], a mechanism analog to the function of a capacitor in an electrical circuit, since this process is characterized by the fact that charged or full intracellular Ca2+ stores prevent Ca2+ current through the plasma membrane, while discharge of the intracellular stores is followed by rapid entry of Ca2+ into the store and, in the continued presence of inositol (1,4,5) trisphosphate (IP3), into the cytosol [12–14].
Analysis of the kinetics of the rises in [Ca2+]c using the intracellular fluorescent probe fura-2 in human platelets revealed that stimulation with the physiological agonist thrombin, which induces active Ca2+ release from the stores via generation of IP3, resulted in Mn2+ entry that starts a few tenths of second after discharge of the intracellular Ca2+stores [15]. This finding was consistent with a mechanism controlled by the agonist-sensitive intracellular Ca2+ stores. Further studies revealed that cell treatment with thapsigargin, a sesquiterpene lactone obtained from Thapsia garganica [16] that inactivates the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) [17] and leads to passive store depletion via Ca2+ leak, resulted in Ca2+ influx across the plasma membrane independently of IP3 generation [18]. Soon after this publication, it was found that Ca2+ influx evoked either by agonist stimulation or by treatment with thapsigargin was mediated via the same mechanism, thus concluding that the mechanism underlying the activation of capacitative Ca2+ entry was the discharge of the intracellular Ca2+ stores per se and not the cellular levels of inositol phosphates [19]. The concept of a Ca2+ entry mechanism regulated by the stores was further confirmed by electrophysiological studies that reported that Ca2+ stores discharge activated a Ca2+ current in mast cells called Ca2+ release-activated Ca2+ current, I CRAC, the first store-operated Ca2+ current identified [20]. In fact, in addition to the physiological pathway to activate capacitative Ca2+ entry via generation of IP3, this mechanism for Ca2+ influx has been reported to be activated by a number of experimental maneuvers leading to a reduction in the amount of free Ca2+ in the intracellular stores. These procedures include treatment with thapsigargin, as well as other SERCA inhibitors, such as cyclopiazonic acid or di-tert-butylhydroquinone [21, 22] (agents that prevent Ca2+ store refilling), loading of Ca2+ stores with the metal Ca2+ chelator N,N,N,N-tetrakis(2-pyridylmethyl)ethylene diamine (which reduces free intraluminal Ca2+ concentration without altering [Ca2+]c [23, 24]), or dialyzing the cytoplasm with the Ca2+ chelators EGTA or BAPTA, which bind Ca2+ leaking from the stores, thus preventing store refilling (see [25]). From that point capacitative Ca2+entry was also known as store-operated Ca2+ entry (SOCE) [26] or store-mediated Ca2+entry [27]. Since SOCE is the most extended term we will use this denomination throughout the chapter.
The manuscript by Kwan and coworkers [19] also raised a key conceptual feature of SOCE. The initial observations had suggested that the transport of extracellular Ca2+ into the cell involved a direct movement of Ca2+ into the intracellular stores, since refilling of the Ca2+ stores were found to occur with no substantial elevation in [Ca2+]c [12, 28]. However, this hypothesis somehow limited the role of Ca2+ entry to the refilling of the intracellular Ca2+ stores. Using the trivalent cation lanthanum, which induces a concentration-dependent inactivation of Ca2+ extrusion through the plasma membrane Ca2+-ATPase, and impairs Ca2+ entry [29, 30], Kwan and coworkers observed that, in the absence of Ca2+ extrusion and entry, Ca2+ stores were refilled by Ca2+ released into the cytoplasm upon agonist stimulation, providing evidence that store refilling did not involve a direct route into the intracellular Ca2+ pools, but rather is the result of a sequential Ca2+ entry into the cytoplasm and subsequent reuptake into the Ca2+stores by active SERCA pumping [19, 31].
SOCE has also been described in a number of excitable cells, including neurons [32], cardiomyocytes [33], smooth muscle cells [34] and both endocrine [35–37] and neuroendocrine cells [38, 39]. In these cells, the functional role of SOCE is not limited to store refilling but it plays an important functional role as revealed by a number of disorders observed in the presence of defective SOCE [40–44].
SOCE activation in electrically excitable cells could trigger Ca2+ entry through voltage-operated channels by causing membrane depolarization [45, 46], which might result directly from the activation of store-operated Ca2+ (SOC) channels, which can be permeable to Ca2+, as well as to other cations, such as Na+, and have reversal potentials near 0 mV. In addition, indirect mechanisms for membrane depolarization associated to rises in [Ca2+]c have been reported, such as a Ca2+-dependent inhibition of voltage-dependent K+ channels [47] reported in pulmonary arterial smooth muscle cells [48]. In contrast to these observations, current evidence supports that the SOCE element STIM1 interacts with the voltage-gated Cav1.2 channels and suppresses its activity [49, 50]. Since functional expression of voltage-operated Ca2+ channels, including Cav1.2, has been reported not only in excitable cells but also in non-electrically excitable cells, such as T and B lymphocytes and mast cells [51–54], these findings further confirm the role of STIM1 as an essential element in the modulation of agonist-induced Ca2+ signals.
The electrophysiological analysis of store-operated currents has revealed the presence of two types of currents: the Ca2+ release-activated Ca2+ current (I CRAC) and a heterogeneous set of non-CRAC currents, described in different cell types with diverse biophysical properties grouped under the denomination of store-operated currents (I SOC) [25]. The I CRAC current was the first store-operated Ca2+ current identified using combined patch-clamp and fura-2 measurements to monitor membrane currents in mast cells [20]. I CRAC is a non-voltage-operated current, unlike Ca2+ currents through the Cav family of channels, that exhibits large current amplitude at negative potentials and approaches the zero current level at very positive potentials. In addition, the current-voltage relationship for I CRAC reveals a significant inward rectification at negative voltages [20, 25]. The channels conducting I CRAC (CRAC channels) show a single channel conductance <1pS and are highly selective for Ca2+ over monovalent cations. The Ca2+:Na+ permeability ratio has been estimated around 1,000:1. Removal of extracellular Ca2+ in the presence of external Na+ and Mg2+ has been reported to abolish this current [55, 56]. However, CRAC channels lose this selectivity in divalent-free solutions, which allow Na+ to permeate the channels, leading to whole cell currents that are five to eightfold larger than the Ca2+ currents [57, 58].
The I SOC currents are mediated by poorly selective cation channels that exhibit a significantly greater conductance than CRAC channels and show different biophysical properties. I SOC currents have been described in different cell types, including vascular endothelial cells [59, 60], pancreatic acinar cells [61], human A431 carcinoma cells [62, 63], smooth muscle cells [64, 65], submandibular and parotid gland cells [66], liver cells [67], skeletal muscle cells [68], neurons [69] and adrenal chromaffin cells [70]. The Ca2+:Na+ permeability ratio of the channels conducting this currents is very heterogeneous, ranging from 1:0.07 to 50:1.
SOCE has been reported to play a number of important roles in cell physiology, including, among others, Ca2+ store refilling upon agonist stimulation, support for the sustained elevations in [Ca2+]c required for a number of cellular functions and maintenance of the amplitude of Ca2+ oscillations [71, 72]. In addition, a variety of functions have been reported to be regulated by SOCE in a number of cell types, including endothelial cell permeability [73], vascular smooth muscle cell proliferation and contractility [34, 74], platelet function [75, 76], immunological response [77] or exocytosis [70, 78, 79], among many others.
2 1.2 Activation Mechanisms: Initial Studies
Since the discovery of SOCE, the mechanism underlying the regulation of SOC channels in the plasma membrane by distantly located intracellular Ca2+ stores has been intensely investigated and debated. The description that STIM1 is the Ca2+ sensor of the intracellular Ca2+ stores in 2005 [80, 81] was a milestone in the identification of the events that regulate SOC channels following a reduction of the intraluminal Ca2+ concentration and marked an inflection point between the initial models and the current one. The initial studies can be grouped into those supporting the indirect coupling between the stores and SOC channels, those that propose a direct or conformational coupling between SOC channels and elements in the stores, and those reporting the insertion of preformed channels in the plasma membrane [25, 82].
The indirect coupling hypothesis assumes that Ca2+ store depletion results in the generation or activation of diffusible molecules that participate in SOC channel gating. This model includes roles for cGMP [83, 84], a product of cytochrome P450 [85], probably the metabolite 5,6-epoxyeicosatrienoic acid [86], tyrosine kinases [87–89], monomeric GTP-binding proteins [90–92], calmodulin [93] and a still uncharacterized Ca2+-influx factor (CIF) [94, 95].
The conformational coupling hypothesis proposes a physical and constitutive interaction between elements in the membrane of the Ca2+ stores and SOC channels in the plasma membrane. This model was originally proposed by Irvine as a mechanism for the activation of SOCE involving IP3 receptors in the endoplasmic reticulum (ER) and inositol 1,3,4,5-tetrakisphosphate (IP4) receptors in the plasma membrane [96, 97]. Later on, Birnbaumer and colleagues demonstrated that the association of the IP3 receptor with transiently expressed TRPC1, TRPC3, and TRPC6 plays a relevant role in the activation of SOCE [98]. This hypothesis received support from studies providing evidence for a role in the activation of SOCE of the protein junctate, an ER Ca2+-binding protein that induces and/or stabilize the interaction between the ER and the plasma membrane [99]. Junctate has been proposed to play an important role in the association of the IP3 receptors and TRPC3 [99]. Consistent with the conformational coupling are a number of studies reporting that TRPC3 channels present in excised patches can be activated by IP3 [100]. Furthermore, the N-terminus of IP3 receptors, containing the IP3-binding domain, is essential for the activation of plasma membrane TRPC3 channels [101].
In addition to the indirect and conformational coupling hypotheses, a number of studies also supported the activation of SOCE by the translocation and insertion of preformed channels, initially located in intracellular vesicles, into the plasma membrane. This model, that was proposed by Penner and colleagues [90], requires the participation of the synaptosome associated protein SNAP-25 [102]. In hippocampal neurons, HEK 293 cells stably expressing TRPC6 and neuronal and epithelial cells expressing TRPC3 depletion of the intracellular Ca2+ stores results in the expression of TRPC channels in the plasma membrane [103–105].
The conformational coupling model assumed that the IP3 receptor in the stores and SOC channels are permanently associated, so that a decrease in the intraluminal Ca2+ concentration is communicated to the SOC channels via a conformational change in the IP3 receptor. Analyzing the role of the actin cytoskeleton in the activation of SOCE in different cell types, including smooth muscle cell lines, human platelets and other non-excitable cells, such as pancreatic acinar cells and the human hepatocellular carcinoma cell line HepG2, an alternative to the constitutive conformational coupling model called secretion-like coupling or de novo conformational coupling was described. This model assumes a reversible physical coupling between the ER and the plasma membrane where the actin cytoskeleton plays an essential role [106–109]. The de novo coupling between elements in the ER and the plasma membrane was found to be strongly dependent on actin filament reorganization. Thus, the actin network located underneath the plasma membrane acts as a negative clamp that prevents constitutive coupling [106, 109, 110]. According to this, stabilization of the cortical actin network has been reported to impair the activation of SOCE in a variety of cell types, including smooth muscle cell lines, human platelets, corneal endothelial cells, the human prostate adenocarcinoma cell line LNCaP, pancreatic acinar cells, smooth muscle cells or neutrophils [39, 45, 106, 107, 109, 111, 112]; however, in pancreatic β cells, thyroid FRTL-5 cells and Aplisia bag cell neurons, SOCE has been reported to be insensitive to stabilization of the cortical actin cytoskeleton by treatment with jasplakinolide [113–115], and stabilization of the peripheral cytoskeleton and disassembly of actin microfilaments have been shown to fail to alter the rate or extent of activation of I CRAC in the RBL-1 rat basophilic cell line [116]. This discrepancies might be attributed to the different idiosyncrasy of the cells investigated. The role of the cortical cytoskeleton in the modulation of SOCE has been further supported by analysis of the actin polymerization and Ca2+ mobilization on a subsecond time scale. In human platelets stimulated with the physiological agonist thrombin we detected an initial decrease in the actin filament content within 0.1 s after stimulation that reached a minimum 0.9 s after the addition of thrombin. Actin depolymerization, involving the actin-binding protein cofilin, was observed before the initiation of SOCE, which occurred with a latency of 2.1 s after agonist stimulation [117], which is consistent with a role for the actin cortical cytoskeleton in the modulation of SOCE.
After the identification of STIM1 as the ER Ca2+ sensor [80, 118] and Orai1 as the prototypical CRAC/SOC channel [77, 119–122] the initial hypotheses had to be necessarily reconsidered and/or reinterpreted. Therefore, as described in a number of cells for SOCE, is the interaction between STIM1 and Orai1 modulated by the cortical actin cytoskeleton? There is a growing body of evidence supporting a role for the cytoskeleton in the interaction between STIM1 and Orai1. In HEK-293 cells and platelets, we have demonstrated that stabilization of the cortical actin cytoskeleton impairs de novo association between the ER Ca2+ sensor STIM1 and the SOC channel Orai1 [123, 124], without altering the coupling between plasma membrane-resident STIM1 and Orai1 [125]. These findings might explain why, in the presence of extracellular Ca2+, treatment with jasplakinolide prevented Ca2+ entry but not the influx of other cations, such as Mn2+ or Na+ [126] that might permeate through non-selective cation channels containing TRP subunits. More recently, it has been reported in HeLa cells that calcineurin impairs cytoskeleton remodeling and STIM1/Orai1 puncta-like formation in a KSR-2-dependent manner [127]. Furthermore, in activated T cells, the Rac1 effector protein WAVE2 has been reported to modulate SOCE and STIM1/Orai1 interaction by regulation of actin filament reorganization [128]. Therefore, the participation of the cortical actin cytoskeleton in the modulation of the de novo interaction between STIM1 and Orai1 and, subsequently SOCE, might still be a potential model.
3 1.3 STIM1, Orai and TRPCs
As we discussed in the previous section, STIM1, originally named GOK, was presented in 2005 as an essential protein involved in the activation of SOCE [80, 81]. The initial studies on STIM1 were published in the mid-1990s, where STIM1 was described as a type-1 transmembrane glycoprotein of 685 amino acids involved in cell-cell interactions in hematopoietic cells [129] and as a cell growth suppressor that plays a pivotal role in the establishment and progression of rhabdomyosarcomas and rhabdoid tumors [130, 131]. Since then, several studies were focused on the study of the structure and function of STIM1 and the determination of the signaling pathways involving STIM1. In 2000, immunofluorescence and cell surface biotinylation analysis revealed that STIM1 is a 90-kDa integral transmembrane phosphoprotein ubiquitously expressed both in the plasma membrane and intracellular membranes of a variety human primary cells, including neonatal foreskin fibroblasts and MG63 osteoblast-like cells, and established tumor cell lines, such as the human leukemic K562, HL60 and U937 cell lines [132]. Later on, the same group demonstrated that the luminal N-terminal region of STIM1 includes an ER signal peptide, EF-hand Ca2+-binding motif, and a single sterile alpha motif (SAM) involved in protein-protein interaction, whereas the cytosolic region consists of two coiled-coil domains, a proline/serine-rich region, and a lysine-rich region [132–134]. These studies also showed that STIM1 is subjected to different post-translational modifications, including phosphorylation on serine residues [133], and N-linked glycosylation in two sites within in the SAM domain [134].
The role of STIM1 in the regulation of SOCE was initially discovered in 2005 by Roos and coworkers [80]. In this study, RNA interference (RNAi)-based screen of more than 170 proteins with known signaling motifs, was designed to identify genes that alter SOCE and the signal pathways controlling them in Drosophila S2 cells. With this approach, the stim gene and its product, STIM1, were identified as essential regulators of SOCE and CRAC channel activity since RNAi-mediated knockdown of the stim gene significantly blocked thapsigargin-induced Ca2+ influx in S2 cells. This new promising role of STIM1 was also confirmed in human cells. Suppression of STIM1 expression also significantly reduced thapsigargin-induced SOCE in Jurkat T cells expressing a short RNA hairpin loop (shRNA) targeting human STIM1. Moreover, Ca2+ influx was also diminished when STIM1 expression was down-regulated in HEK-293 and SH-SY5Y human cells transfected with STIM1 siRNA [80].
Since STIM1 overexpression in HEK-293 cells was not associated with an increased Ca2+ influx and detectable SOCE current, it was proposed that STIM1 could not function as SOC channel itself, and the intraluminal location of the EF-hand motif suggested that STIM1 might act as an ER Ca2+ sensor [80]. The report by Roos and coworkers was followed shortly thereafter by two studies from Liou and coworkers [81] and Zhang and coworkers [118]. Cell transfection with a mutated EF-hand motif of STIM1 or Drosophila Stim induced a constitutive Ca2+ influx mediated by CRAC channels and independent of the filling state of the Ca2+ stores, and treatment with thapsigargin failed to further promote SOCE in HeLa [81], Jurkat and S2 cells [118]. Using different techniques, from immunofluorescence and electron microscopy to surface biotinylation, both studies also demonstrated that, once Ca2+ stores are depleted, STIM1 migrates from the ER to the proximity of the plasma membrane. Liou and coworkers first reported that upon store depletion STIM1 redistributes into punctae structures, ER regions enriched in STIM1 oligomers and located in close proximity to the plasma membrane [81]. Later on, it was demonstrated that SAM is an essential STIM1 domain for oligomerization and punctae formation, while the coiled-coil domains are involved in further stabilization of the STIM1 oligomers and also in functional protein-protein interactions of STIM1 with Ca2+ channels located in the plasma membrane [135].
The identification of STIM1 as the intraluminal Ca2+ sensor provided an explanation for the communication between the intracellular Ca2+ stores and the CRAC/SOC channels located in the plasma membrane, whose identity still remained elusive. In 2006, Orai1 was identified as the pore subunit of the CRAC channels [77, 121, 136–138]. Initially Feske and coworkers [77] and soon thereafter Vig et al. [136] presented Orai1, also named as CRAC modulator 1 (CRACM1), as an essential component regulator of the CRAC channel. Both studies identified Drosophila gene olf186-F as an essential gene in the activation of thapsigargin-induced SOCE by using a genome-wide RNAi screen in Drosophila S2 cells to identify the genes encoding the CRAC channel or other proteins involved in its regulation [77, 136]. The Drosophila olf186-F gene has three human homologues that encode the proteins named, for the first time, as Orai1, Orai2 and Orai3 [77]. The term “orai” is referred to the three keepers of heaven’s gate in Greek mythology. The gene encoding human Orai1 was mapped on chromosome region 12q24 and was shown to be mutated in the hereditary severe combined immune deficiency (SCID) syndrome patients. Collected T cells from these patients are characterized by an impaired SOCE and CRAC channel activity. This mutation consists in a C→T transition at position 271 of the coding sequence of the human orai1 gene, leading to replacement of a highly conserved arginine residue by tryptophan at position 91 of the protein (R91W). Expression of an exogenous wild-type Orai1 in SCID T-cells restored store depletion-evoked Ca2+ influx, suggesting that this single point mutation R91W in Orai1 sequence was responsible of the defective I CRAC. However, expression of wild-type Orai1 in SCID T-cells did not promote Ca2+ influx per se in resting conditions, suggesting that Orai1 is not a Ca2+ channel constitutively activated. Furthermore, electrophysiological and pharmacological properties of the restored Ca2+ influx current were fully consistent with those observed in I CRAC in normal T cells [77]. According to these results, the selective small interfering RNA (SiRNA)-mediated knockdown of Orai1 reduced both Ca2+ influx and I CRAC in response to thapsigargin in human HEK-293 and Jurkat T cells [136]. In addition to the demonstration that Orai1 plays a pivotal role in CRAC current generation, both studies also predicted the topology Orai1 as a membrane protein with four transmembrane domains and cytosolic N- and C- termini, which shows no structural homology to other known ion-channels [77, 136].
Soon after the identification of Orai1 as a the pore forming subunit of the CRAC channel published studies demonstrated the earliest pieces of evidence for the functional interaction between STIM1 and Orai1 in the activation of CRAC currents were reported. Co-expression of STIM1 and Orai1 resulted in a significantly enhanced CRAC current in HEK-293, Jurkat T cells [119, 120, 122] and Drosophila S2 cells [139]. The generated current showed Ca2+ selectivity, development and inactivation kinetics, pharmacological profiles and biophysical properties indistinguishable from native CRAC current [120, 139].
According to the idea that conserved acidic residues are essential for ion permeation through known Ca2+ channels, three simultaneous studies analyzed highly conserved glutamate residues across species and the most interesting results come from the residues E178 and E262 in Drosophila and their corresponding human homologues E106 and E190. Mutation of E106 to alanine or glutamine greatly impaired channel function. However, the conservative mutations of E106 to an aspartate (E106D) resulted in a channel with reduced selectivity for Ca2+, thus indicating that this residue functions as part of the selectivity filter, and confirms that Orai1 is a pore forming subunit of the CRAC channel [121, 137, 138]. While mutation of E190 to aspartate or alanine had no significant effect on channel function, the substitution by glutamine (E190Q) resulted in diminished Ca2+ selectivity [121, 138]. These studies also demonstrated, for the first time, the STIM1-Orai1 protein interaction upon Ca2+ store depletion to initiate CRAC channel activation, by using co-immunoprecipitation in co-transfected HEK-293 cells [138] and Drosophila S2 cells [137]. The STIM1/Orai1 complex was reported to be located in the punctae region on the plasma membrane after CRAC current activation by fluorescence microscopy [140, 141].
The identification of Orai1 as the pore forming region of the CRAC channel raised the question concerning the architecture of the channel, with reports proposing tetrameric and even higher-order multimeric structures [142–144]. Hou and coworkers [145] reported in 2012 the crystal structure of Drosophila Orai channel, which shares 70 % homology with human Orai1. The crystal structure indicates that the pore of Drosophila Orai is formed by six subunits, each containing four transmembrane regions (M1 to M4). The transmembrane helices are arranged in three concentric circles around the pore, where the inner ring is formed of six M1 helices, the middle ring consists of M2 and M3 helices and the outer ring is formed of M4 helices located at the periphery of the channel. The side chains of M1 line the pore of Orai, which is composed of four different sections: a selectivity filter formed by a ring of glutamates at the extracellular end of the channel, followed by a hydrophobic section, a basic region spanning near the intracellular side of the channel, which might contribute to the stabilization of the closed state of the channel, and a wider section that extends into the cytosol. The pore region of Orai shows biochemical differences with the known K+ channels, which emphasizes the distinct mechanisms of ion selectivity and permeation between Orai and K+ channels.
In addition to the small families of STIM and Orai, another somewhat larger family of ion channels are also linked to SOCE. These are a subfamily of the larger family of TRP channels, called TRPC for classical or canonical TRPs, because it is comprised of those proteins most highly related to the Drosophila TRP channel [146].
The origin of the Drosophila TRP goes back to 1969, when Cosens and coworkers identified a spontaneously formed mutant on the basis of a behavioral phenotype [147], but it was not until 1975 that Minke described and named it as “Transient Receptor Potential” (TRP) because of the transient depolarization of the photoreceptors due to Na+ and Ca2+ entry [148]. The identity of the mutated protein was reported in 1989 by Montell and coworkers who cloned, sequenced and presented a molecular characterization of the Drosophila trp gen and highlighted its similarity with the Ca2+ channels [149]. Later studies characterized TRP and its homologue TRPL as Ca2+ permeable channels activated downstream of phospholipase C [150].
After the initial identification of the Drosophila TRPs, all the attempts were focused on finding its counterpart in mammals. In 1995, regarding its high homology to the TRP protein sequence, an expressed sequence from a human fetal brain cDNA library was reported by two groups as the first homologue in human (TRPC1) [151, 152]. Since the identification of TRPC1, several mammalian homologues have been described and are classified into six subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystin), TRPML (mucolipin). In addition, a less related TRPN has been reported in worms and fish and TRPY in fungi [153].
The DNA and corresponding amino acid sequence of the previously called TRPC1, the expression pattern as well as another general features of the novel protein were described by Wes and coworkers and Zhu and coworkers, who characterized it as a polypeptide of 793 amino acids expressed at the highest levels in the fetal brain and in the adult heart, brain, testis and ovaries [151, 152]. The primary structure was supposed to present six transmembrane domains with a pore region between the fifth (S5) and sixth (S6) transmembrane segments and both C and N termini located intracellularly [154]. This structure is shared by all the members of the TRP family. Furthermore, Wes also described that the most highly conserved domain among all TRP family members contains three ankyrin repeats which are thought to play key roles in protein-protein interactions [151, 155]. Later on the whole structure of the protein was reported, with the N-terminus containing three to four ankyrin repeats, a predicted coiled-coil motif and a caveolin binding region. By the way, the citoplasmic C-terminus includes an EWKFAR TRP box, a highly conserved proline rich domain, a coiled-coil motif and a CaM/IP3 receptor binding (CIRB) domain [98, 156]. The presence of coiled-coil domains, commonly involved in subunit oligomerization suggest that they are probably involved in homo-and heteromerization of TRPCs or in linking TRPCs to other proteins also containing coiled-coil domains [157, 158].
Despite the key role of TRPs in receptor-operated Ca2+ entry (ROCE) and in SOCE has been reported in different cell types, the latter has been a matter of intense debate, and the role of TRPCs in SOCE has not been demonstrated in all the cellular models investigated. Among the first pieces of evidence for a role of TRPC1 in SOCE comes from a study made in Chinese hamster ovary cells (CHO), where expression of TRPC1A, a splice variant of TRPC1, resulted in a linear nonselective cation current with similar permeabilities for Na+, Ca2+, and Cs+, activated by intracellular infusion of either IP3 or thapsigargin to deplete intracellular Ca2+ stores [159]. At the same time, Zhu and coworkers demonstrated that expression in COS cells of full-length cDNA encoding human TRPC1 increased SOCE while expression of antisense sequences suppressed it [160]. Further evidence supporting the role of TRPC1 in SOCE comes from studies based on overexpression of TRPC1 proteins or knockdown of the endogenous TRPC1 channels in several cell types, including human cells. In human submandibular gland cells and vascular endothelial cells, the overexpression of TRPC1 increased SOCE while the silencing of the endogenous protein by using antisense oligonucleotides reduced thapsigargin-evoked influx [161, 162]. In general, the key role of TRPC1 in SOCE has been supported by several research groups by means of different experimental maneuvers in many cell types, such as DT40-B-lymphocytes, endothelial cells, rat cardiac myocytes, the human megakaryoblastic cell line MEG01, the C2C12 mouse myoblastic cell line, rat kidney fibroblasts or human platelets [162–166]. Other members of the TRPC family have also been reported to be activated by store depletion or to be involved in SOCE both in excitable and non-excitable cells. For instance, the transient expression of the full length cDNA of mouse TRPC2, the homologue of the human trp2 pseudogene, was reported to evoke a Ca2+ entry that was shown to be readily activated not only after agonist stimulation but also by store depletion in the absence of an agonist [167]. In addition, in 1996 Zhu and coworkers observed that the heterologous expression of the human TRPC3 in COS cells enhanced SOCE [160] and similar results were obtained by Boulay and coworkers by expressing the murine TRPC6 in this cells [168]. Over the years, some studies have highlighted that there is also a growing body of evidence supporting a role of TRPC6 in the conduction of SOCE in different cells [169, 170].
Nevertheless, several studies have failed to observe store-operated behavior of exogenously expressed TRPC channels in different cell types, thus demonstrating that TRPCs did not account for SOCE in all cellular models investigated [171]. Moreover, the mode of expression of TRPCs might determine its involvement in SOCE or ROCE in the same cell type, as it was reported by Lievremont and coworkers in 2004, who described that TRPC7 activation mode was different in cells transiently or stably expressing this channel [172].
With the identification of STIM1 and Orai1 as the key molecular players of CRAC currents, most studies focused on the possible interaction between these proteins and TRPC channels. In 2006, after Spassova and coworkers confirmed the crucial functional role of STIM1 over the regulation of SOC channels by using electrophysiological analysis [173], two independent studies demonstrated for the first time that STIM1 is associated with TRPC1, in the plasma membrane upon Ca2+ store depletion, and that the STIM1-TRPC1 interaction promotes channel activation and SOCE [174, 175]. In HEK-293 cells expressing exogenous STIM1, Ca2+ store depletion increased the association between STIM1 and TRPC1, while the expression of the EF-hand mutant STIM1 yield a constitutive STIM1-TRPC1 complex formation and subsequent TRPC1 activation and Ca2+ influx [174]. According to this, electrotransjection of human platelets with anti-STIM1 antibody, directed toward the EF-hand Ca2+-binding motif, prevented the migration of STIM1 toward the plasma membrane, the interaction between endogenously expressed STIM1 and TRPC1 and reduced SOCE [175]. Functional interaction between STIM1 and TRPC1 has also been reported, among others cellular types, in intestinal epithelial cells [176], salivary and pancreatic acinar cells [79, 177], pulmonary artery cells [178] or messanglial cells [179].
STIM1 also interacts directly with other members of TRPC family involved in SOCE, such as TRPC4 and TRPC5 in HEK293 cells [180] and TRPC6 in human platelets [24], regulating their activation. STIM1 also indirectly regulates TRPC3 and TRPC6 by mediating the heteromultimerization of TRPC3 with TRPC1 and TRPC4 with TRPC6 [180]. Direct STIM1/TRPC interaction has been reported to require interaction of the STIM1 SOAR region with the TRPC C-terminal coiled-coil domains, as well as electrostatic interactions involving the lysine-rich domain of STIM1 [181, 182]. This association is supported by the actin cytoskeleton [175] and plasma membrane lipid rafts domains [183–185].
In addition to the STIM1-TRPC coupling previously described, there is a body of evidence supporting the formation of ternary complexes among the three main elements involved in SOCE, STIM, Orai and TRPCs. In HEK293 cells, SOCs were reported to be built by TRPC pore-forming subunits and Orai regulatory subunits that transduce the Ca2+ store depletion signal from STIM1 to TRPCs [186]. Supporting this hypothesis, a dynamic assembly among STIM1, Orai1 and TRPC1 has been proposed to be essential for SOCE in human salivary gland cells [187] and human platelets [123]. Interestingly, the association of these proteins has been reported to occur in specific plasma membrane microdomains known as lipid rafts which provide the adequate environment for the formation of the store-operated Ca2+ influx complex (SOCIC) or receptor-operated Ca2+ entry complexes [184, 185]. The following chapters will highlight recent advances in the Ca2+ entry mechanisms, involving store-operated and receptor-operated signaling pathways, in non-excitable cells.
References
Ebashi S (1961) Calcium binding activity of vesicular relaxing factor. J Chir (Paris) 50:236–244
Ringer S (1883) A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol 4(1):29–42.3
Ringer S, Buxton DW (1885) Concerning the action of small quantities of calcium, sodium, and potassium salts upon the vitality and function of contractile tissue and the cuticular cells of fishes. J Physiol 6(4–5):154–161
Hofer AM, Lefkimmiatis K (2007) Extracellular calcium and cAMP: second messengers as “third messengers”? Physiology (Bethesda) 22:320–327
Bianchi CP, Shanes AM (1959) Calcium influx in skeletal muscle at rest, during activity, and during potassium contracture. J Gen Physiol 42(4):803–815
Winegrad S, Shanes AM (1962) Calcium flux and contractility in guinea pig atria. J Gen Physiol 45:371–394
Borle AB (1969) Kinetic analyses of calcium movements in HeLa cell cultures. I. Calcium influx. J Gen Physiol 53(1):43–56
Borle AB (1968) Calcium metabolism in HeLa cells and the effects of parathyroid hormone. J Cell Biol 36(3):567–582
Shigenobu K, Sperelakis N (1972) Calcium current channels induced by catecholamines in chick embryonic hearts whose fast sodium channels are blocked by tetrodotoxin or elevated potassium. Circ Res 31(6):932–952
Hogestatt ED (1984) Characterization of two different calcium entry pathways in small mesenteric arteries from rat. Acta Physiol Scand 122(4):483–495
Hallam TJ, Rink TJ (1985) Agonists stimulate divalent cation channels in the plasma membrane of human platelets. FEBS Lett 186(2):175–179
Putney JW Jr (1986) A model for receptor-regulated calcium entry. Cell Calcium 7(1):1–12
Holda JR, Klishin A, Sedova M, Huser J, Blatter LA (1998) Capacitative calcium entry. News Physiol Sci 13:157–163
Takemura H, Putney JW Jr (1989) Capacitative calcium entry in parotid acinar cells. Biochem J 258(2):409–412
Sage SO, Merritt JE, Hallam TJ, Rink TJ (1989) Receptor-mediated calcium entry in fura-2-loaded human platelets stimulated with ADP and thrombin. Dual-wavelengths studies with Mn2+. Biochem J 258(3):923–926
Thastrup O, Foder B, Scharff O (1987) The calcium mobilizing tumor promoting agent, thapsigargin elevates the platelet cytoplasmic free calcium concentration to a higher steady state level. A possible mechanism of action for the tumor promotion. Biochem Biophys Res Commun 142(3):654–660
Lytton J, Westlin M, Hanley MR (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem 266(26):17067–17071
Takemura H, Hughes AR, Thastrup O, Putney JW Jr (1989) Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J Biol Chem 264(21):12266–12271
Kwan CY, Takemura H, Obie JF, Thastrup O, Putney JW Jr (1990) Effects of MeCh, thapsigargin, and La3+ on plasmalemmal and intracellular Ca2+ transport in lacrimal acinar cells. Am J Physiol 258(6 Pt 1):C1006–C1015
Hoth M, Penner R (1992) Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355(6358):353–356
Okon EB, Golbabaie A, Breemen C (2008) Paracrine effects of endothelial cells in a diabetic mouse model: capacitative calcium entry stimulated thromboxane release. Horm Metab Res 40(9):645–650
Rosado JA, Lopez JJ, Harper AG, Harper MT, Redondo PC, Pariente JA, Sage SO, Salido GM (2004) Two pathways for store-mediated calcium entry differentially dependent on the actin cytoskeleton in human platelets. J Biol Chem 279(28):29231–29235
Hofer AM, Fasolato C, Pozzan T (1998) Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. J Cell Biol 140(2):325–334
Jardin I, Gomez LJ, Salido GM, Rosado JA (2009) Dynamic interaction of hTRPC6 with the Orai1/STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem J 420:267–276
Parekh AB, Putney JW Jr (2005) Store-operated calcium channels. Physiol Rev 85(2):757–810
Montero M, Garcia-Sancho J, Alvarez J (1993) Transient inhibition by chemotactic peptide of a store-operated Ca2+ entry pathway in human neutrophils. J Biol Chem 268(18):13055–13061
Davis W, Sage SO, Allen JM (1994) Cytosolic calcium elevation in response to Fc receptor cross-linking in undifferentiated and differentiated U937 cells. Cell Calcium 16(1):29–36
Merritt JE, Rink TJ (1987) Regulation of cytosolic free calcium in fura-2-loaded rat parotid acinar cells. J Biol Chem 262(36):17362–17369
van Breemen C, De Weer P (1970) Lanthanum inhibition of 45Ca efflux from the squid giant axon. Nature 226(5247):760–761
Lansman JB (1990) Blockade of current through single calcium channels by trivalent lanthanide cations. Effect of ionic radius on the rates of ion entry and exit. J Gen Physiol 95(4):679–696
Rosado JA (2006) Discovering the mechanism of capacitative calcium entry. Am J Physiol Cell Physiol 291(6):C1104–C1106
Emptage NJ, Reid CA, Fine A (2001) Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29(1):197–208
Hunton DL, Lucchesi PA, Pang Y, Cheng X, Dell’Italia LJ, Marchase RB (2002) Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem 277(16):14266–14273
Rodriguez-Moyano M, Diaz I, Dionisio N, Zhang X, Avila-Medina J, Calderon-Sanchez E, Trebak M, Rosado JA, Ordonez A, Smani T (2013) Urotensin-II promotes vascular smooth muscle cell proliferation through store operated calcium entry and EGFR transactivation. Cardiovasc Res 100:297–306
Liu YJ, Gylfe E (1997) Store-operated Ca2+ entry in insulin-releasing pancreatic beta-cells. Cell Calcium 22(4):277–286
Tornquist K, Malm AM, Pasternack M, Kronqvist R, Bjorklund S, Tuominen R, Slotte JP (1999) Tumor necrosis factor-alpha, sphingomyelinase, and ceramide inhibit store-operated calcium entry in thyroid FRTL-5 cells. J Biol Chem 274(14):9370–9377
Zerbes M, Bunn SJ, Powis DA (1998) Histamine causes Ca2+ entry via both a store-operated and a store-independent pathway in bovine adrenal chromaffin cells. Cell Calcium 23(6):379–386
Kim JH, Shin SY, Nam JH, Hong EK, Chung YS, Jeong JY, Kang J, Uhm DY, Kim SJ (2003) Adrenergic regulation of the intracellular [Ca2+] and voltage-operated Ca2+ channel currents in the rat prostate neuroendocrine cells. Prostate 57(2):99–110
Vanoverberghe K, Lehen’kyi V, Thebault S, Raphael M, Vanden Abeele F, Slomianny C, Mariot P, Prevarskaya N (2012) Cytoskeleton reorganization as an alternative mechanism of store-operated calcium entry control in neuroendocrine-differentiated cells. PLoS ONE 7(9):e45615
Feske S (2009) ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev 231(1):189–209
McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, Kutok J, Hivroz C, Ledeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A, Feske S (2009) ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol 124(6):1311–1318, e1317
Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, Fischer A, Feske S (2009) STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 360(19):1971–1980
Ariano P, Zamburlin P, D’Alessandro R, Meldolesi J, Lovisolo D (2010) Differential repression by the transcription factor REST/NRSF of the various Ca2+ signalling mechanisms in pheochromocytoma PC12 cells. Cell Calcium 47(4):360–368
Hao B, Lu Y, Wang Q, Guo W, Cheung KH, Yue J (2014) Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1-mediated Ca2+ entry. Stem Cell Res 12(2):452–466
Morales S, Camello PJ, Rosado JA, Mawe GM, Pozo MJ (2005) Disruption of the filamentous actin cytoskeleton is necessary for the activation of capacitative calcium entry in naive smooth muscle cells. Cell Signal 17(5):635–645
Chakraborty S, Berwick ZC, Bartlett PJ, Kumar S, Thomas AP, Sturek M, Tune JD, Obukhov AG (2011) Bromoenol lactone inhibits voltage-gated Ca2+ and transient receptor potential canonical channels. J Pharmacol Exp Ther 339(2):329–340
Post JM, Gelband CH, Hume JR (1995) [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 77(1):131–139
Lu W, Wang J, Peng G, Shimoda LA, Sylvester JT (2009) Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 297(1):L17–L25
Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL (2010) The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330(6000):105–109
Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136(5):876–890
Suzuki Y, Yoshimaru T, Inoue T, Ra C (2009) Ca v 1.2 L-type Ca2+ channel protects mast cells against activation-induced cell death by preventing mitochondrial integrity disruption. Mol Immunol 46(11–12):2370–2380
Yoshimaru T, Suzuki Y, Inoue T, Ra C (2009) L-type Ca2+ channels in mast cells: activation by membrane depolarization and distinct roles in regulating mediator release from store-operated Ca2+ channels. Mol Immunol 46(7):1267–1277
Suzuki Y, Inoue T, Ra C (2010) L-type Ca2+ channels: a new player in the regulation of Ca2+ signaling, cell activation and cell survival in immune cells. Mol Immunol 47(4):640–648
Matza D, Flavell RA (2009) Roles of Ca(v) channels and AHNAK1 in T cells: the beauty and the beast. Immunol Rev 231(1):257–264
Fierro L, Lund PE, Parekh AB (2000) Comparison of the activation of the Ca2+ release-activated Ca2+ current ICRAC to InsP3 in Jurkat T-lymphocytes, pulmonary artery endothelia and RBL-1 cells. Pflugers Arch 440(4):580–587
Zweifach A, Lewis RS (1993) Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A 90(13):6295–6299
Bakowski D, Parekh AB (2002) Monovalent cation permeability and Ca(2+) block of the store-operated Ca(2+) current I(CRAC) in rat basophilic leukemia cells. Pflugers Arch 443(5–6):892–902
Prakriya M, Lewis RS (2002) Separation and characterization of currents through store-operated CRAC channels and Mg2+ -inhibited cation (MIC) channels. J Gen Physiol 119(5):487–507
Vaca L, Kunze DL (1994) Depletion of intracellular Ca2+ stores activates a Ca(2+)-selective channel in vascular endothelium. Am J Physiol 267(4 Pt 1):C920–C925
Fasolato C, Nilius B (1998) Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embryonic kidney cell lines. Pflugers Arch 436(1):69–74
Krause E, Pfeiffer F, Schmid A, Schulz I (1996) Depletion of intracellular calcium stores activates a calcium conducting nonselective cation current in mouse pancreatic acinar cells. J Biol Chem 271(51):32523–32528
Luckhoff A, Clapham DE (1994) Calcium channels activated by depletion of internal calcium stores in A431 cells. Biophys J 67(1):177–182
Kiselyov KI, Semyonova SB, Mamin AG, Mozhayeva GN (1999) Miniature Ca2+ channels in excised plasma-membrane patches: activation by IP3. Pflugers Arch 437(2):305–314
Trepakova ES, Gericke M, Hirakawa Y, Weisbrod RM, Cohen RA, Bolotina VM (2001) Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J Biol Chem 276(11):7782–7790
Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX (2001) Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280(2):H746–H755
Liu X, Groschner K, Ambudkar IS (2004) Distinct Ca(2+)-permeable cation currents are activated by internal Ca(2+)-store depletion in RBL-2H3 cells and human salivary gland cells, HSG and HSY. J Membr Biol 200(2):93–104
To MS, Aromataris EC, Castro J, Roberts ML, Barritt GJ, Rychkov GY (2010) Mitochondrial uncoupler FCCP activates proton conductance but does not block store-operated Ca(2+) current in liver cells. Arch Biochem Biophys 495(2):152–158
Hopf FW, Reddy P, Hong J, Steinhardt RA (1996) A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. J Biol Chem 271(37):22358–22367
Putney JW Jr (2003) Capacitative calcium entry in the nervous system. Cell Calcium 34(4–5):339–344
Fomina AF, Nowycky MC (1999) A current activated on depletion of intracellular Ca2+ stores can regulate exocytosis in adrenal chromaffin cells. J Neurosci 19(10):3711–3722
Bird GS, Putney JW Jr (2005) Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J Physiol 562(Pt 3):697–706
Wedel B, Boyles RR, Putney JW Jr, Bird GS (2007) Role of the store-operated calcium entry proteins Stim1 and Orai1 in muscarinic cholinergic receptor-stimulated calcium oscillations in human embryonic kidney cells. J Physiol 579(Pt 3):679–689
Wu S, Cioffi EA, Alvarez D, Sayner SL, Chen H, Cioffi DL, King J, Creighton JR, Townsley M, Goodman SR, Stevens T (2005) Essential role of a Ca2+ -selective, store-operated current (ISOC) in endothelial cell permeability: determinants of the vascular leak site. Circ Res 96(8):856–863
Dominguez-Rodriguez A, Diaz I, Rodriguez-Moyano M, Calderon-Sanchez E, Rosado JA, Ordonez A, Smani T (2012) Urotensin-II signaling mechanism in rat coronary artery: role of STIM1 and Orai1-dependent store operated calcium influx in vasoconstriction. Arterioscler Thromb Vasc Biol 32(5):1325–1332
Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, Bosl M, Stoll G, Nieswandt B (2009) Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 113(9):2056–2063
Galan C, Zbidi H, Bartegi A, Salido GM, Rosado JA (2009) STIM1, Orai1 and hTRPC1 are important for thrombin- and ADP-induced aggregation in human platelets. Arch Biochem Biophys 490(2):137–144
Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441(7090):179–185
Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc Natl Acad Sci U S A 104(44):17542–17547
Pani B, Liu X, Bollimuntha S, Cheng KT, Niesman IR, Zheng C, Achen VR, Patel HH, Ambudkar IS, Singh BB (2013) Impairment of TRPC1-STIM1 channel assembly and AQP5 translocation compromise agonist-stimulated fluid secretion in mice lacking caveolin1. J Cell Sci 126(Pt 2):667–675
Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169(3):435–445
Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15(13):1235–1241
Salido GM, Sage SO, Rosado JA (2009) Biochemical and functional properties of the store-operated Ca2+ channels. Cell Signal 21(4):457–461
Pandol SJ, Schoeffield-Payne MS (1990) Cyclic GMP mediates the agonist-stimulated increase in plasma membrane calcium entry in the pancreatic acinar cell. J Biol Chem 265(22):12846–12853
Rosado JA, Porras T, Conde M, Sage SO (2001) Cyclic nucleotides modulate store-mediated calcium entry through the activation of protein-tyrosine phosphatases and altered actin polymerization in human platelets. J Biol Chem 276(19):15666–15675
Alvarez J, Montero M, Garcia-Sancho J (1992) Cytochrome P450 may regulate plasma membrane Ca2+ permeability according to the filling state of the intracellular Ca2+ stores. FASEB J 6(2):786–792
Ben Amor N, Redondo PC, Bartegi A, Pariente JA, Salido GM, Rosado JA (2006) A role for 5,6-epoxyeicosatrienoic acid in calcium entry by de novo conformational coupling in human platelets. J Physiol 570(Pt 2):309–323
Sargeant P, Farndale RW, Sage SO (1993) The tyrosine kinase inhibitors methyl 2,5-dihydroxycinnamate and genistein reduce thrombin-evoked tyrosine phosphorylation and Ca2+ entry in human platelets. FEBS Lett 315(3):242–246
Sargeant P, Farndale RW, Sage SO (1993) ADP- and thapsigargin-evoked Ca2+ entry and protein-tyrosine phosphorylation are inhibited by the tyrosine kinase inhibitors genistein and methyl-2,5-dihydroxycinnamate in fura-2-loaded human platelets. J Biol Chem 268(24):18151–18156
Rosado JA, Graves D, Sage SO (2000) Tyrosine kinases activate store-mediated Ca2+ entry in human platelets through the reorganization of the actin cytoskeleton. Biochem J 351(Pt 2):429–437
Fasolato C, Hoth M, Penner R (1993) A GTP-dependent step in the activation mechanism of capacitative calcium influx. J Biol Chem 268(28):20737–20740
Bird GS, Putney JW Jr (1993) Inhibition of thapsigargin-induced calcium entry by microinjected guanine nucleotide analogues. Evidence for the involvement of a small G-protein in capacitative calcium entry. J Biol Chem 268(29):21486–21488
Rosado JA, Sage SO (2000) Farnesylcysteine analogues inhibit store-regulated Ca2+ entry in human platelets: evidence for involvement of small GTP-binding proteins and actin cytoskeleton. Biochem J 347(Pt 1):183–192
Vaca L (1996) Calmodulin inhibits calcium influx current in vascular endothelium. FEBS Lett 390(3):289–293
Randriamampita C, Tsien RY (1993) Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature 364(6440):809–814
Csutora P, Zarayskiy V, Peter K, Monje F, Smani T, Zakharov SI, Litvinov D, Bolotina VM (2006) Activation mechanism for CRAC current and store-operated Ca2+ entry: calcium influx factor and Ca2+ -independent phospholipase A2beta-mediated pathway. J Biol Chem 281(46):34926–34935
Irvine RF, Moor RM (1987) Inositol(1,3,4,5)tetrakisphosphate-induced activation of sea urchin eggs requires the presence of inositol trisphosphate. Biochem Biophys Res Commun 146(1):284–290
Irvine RF (1990) ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates – a possible mechanism. FEBS Lett 263(1):5–9
Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Zhu MX, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L (1999) Modulation of Ca(2+) entry by polypeptides of the inositol 1,4, 5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca(2+) entry. Proc Natl Acad Sci U S A 96(26):14955–14960
Treves S, Franzini-Armstrong C, Moccagatta L, Arnoult C, Grasso C, Schrum A, Ducreux S, Zhu MX, Mikoshiba K, Girard T, Smida-Rezgui S, Ronjat M, Zorzato F (2004) Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J Cell Biol 166(4):537–548
Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S (1998) Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 396(6710):478–482
Kiselyov K, Mignery GA, Zhu MX, Muallem S (1999) The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Mol Cell 4(3):423–429
Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY (1999) Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell 98(4):475–485
Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE (2004) Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 6(8):709–720
Cayouette S, Lussier MP, Mathieu EL, Bousquet SM, Boulay G (2004) Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem 279(8):7241–7246
Singh BB, Lockwich TP, Bandyopadhyay BC, Liu X, Bollimuntha S, Brazer SC, Combs C, Das S, Leenders AG, Sheng ZH, Knepper MA, Ambudkar SV, Ambudkar IS (2004) VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol Cell 15(4):635–646
Patterson RL, van Rossum DB, Gill DL (1999) Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell 98(4):487–499
Rosado JA, Jenner S, Sage SO (2000) A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Evidence for conformational coupling. J Biol Chem 275(11):7527–7533
Rosado JA, Rosenzweig I, Harding S, Sage SO (2001) Tumor necrosis factor-alpha inhibits store-mediated Ca2+ entry in the human hepatocellular carcinoma cell line HepG2. Am J Physiol Cell Physiol 280(6):C1636–C1644
Redondo PC, Lajas AI, Salido GM, Gonzalez A, Rosado JA, Pariente JA (2003) Evidence for secretion-like coupling involving pp60src in the activation and maintenance of store-mediated Ca2+ entry in mouse pancreatic acinar cells. Biochem J 370(Pt 1):255–263
Rosado JA, Sage SO (2000) The actin cytoskeleton in store-mediated calcium entry. J Physiol 526(Pt 2):221–229
Xie Q, Zhang Y, Zhai C, Bonanno JA (2002) Calcium influx factor from cytochrome P-450 metabolism and secretion-like coupling mechanisms for capacitative calcium entry in corneal endothelial cells. J Biol Chem 277(19):16559–16566
Wang JP, Hsu MF, Ko HH, Lin CN (2004) Stimulation of cellular free Ca2+ elevation and inhibition of store-operated Ca2+ entry by kazinol B in neutrophils. Naunyn Schmiedebergs Arch Pharmacol 370(6):500–509
Gratschev D, Blom T, Bjorklund S, Tornquist K (2004) Phosphatase inhibition reveals a calcium entry pathway dependent on protein kinase A in thyroid FRTL-5 cells: comparison with store-operated calcium entry. J Biol Chem 279(48):49816–49824
Kachoei BA, Knox RJ, Uthuza D, Levy S, Kaczmarek LK, Magoski NS (2006) A store-operated Ca(2+) influx pathway in the bag cell neurons of Aplysia. J Neurophysiol 96(5):2688–2698
Henquin JC, Mourad NI, Nenquin M (2012) Disruption and stabilization of beta-cell actin microfilaments differently influence insulin secretion triggered by intracellular Ca2+ mobilization or store-operated Ca2+ entry. FEBS Lett 586(1):89–95
Bakowski D, Glitsch MD, Parekh AB (2001) An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current I(CRAC) in RBL-1 cells. J Physiol 532(Pt 1):55–71
Redondo PC, Harper MT, Rosado JA, Sage SO (2006) A role for cofilin in the activation of store-operated calcium entry by de novo conformational coupling in human platelets. Blood 107(3):973–979
Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437(7060):902–905
Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW Jr (2006) Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem 281(34):24979–24990
Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol 8(7):771–773
Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443(7108):230–233
Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem 281(30):20661–20665
Jardin I, Lopez JJ, Salido GM, Rosado JA (2008) Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J Biol Chem 283(37):25296–25304
Galan C, Dionisio N, Smani T, Salido GM, Rosado JA (2011) The cytoskeleton plays a modulatory role in the association between STIM1 and the Ca2+ channel subunits Orai1 and TRPC1. Biochem Pharmacol 82(4):400–410
Jardin I, Lopez JJ, Redondo PC, Salido GM, Rosado JA (2009) Store-operated Ca2+ entry is sensitive to the extracellular Ca2+ concentration through plasma membrane STIM1. Biochim Biophys Acta 1793(10):1614–1622
Harper AG, Sage SO (2007) A key role for reverse Na+/Ca2+ exchange influenced by the actin cytoskeleton in store-operated Ca2+ entry in human platelets: evidence against the de novo conformational coupling hypothesis. Cell Calcium 42(6):606–617
Giurisato E, Gamberucci A, Ulivieri C, Marruganti S, Rossi E, Giacomello E, Randazzo D, Sorrentino V (2014) The KSR2-calcineurin complex regulates STIM1-ORAI1 dynamics and store-operated calcium entry (SOCE). Mol Biol Cell 25(11):1769–1781
Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, Burkhardt JK, Freedman BD, Billadeau DD (2006) The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol 16(1):24–34
Oritani K, Kincade PW (1996) Identification of stromal cell products that interact with pre-B cells. J Cell Biol 134(3):771–782
Parker NJ, Begley CG, Smith PJ, Fox RM (1996) Molecular cloning of a novel human gene (D11S4896E) at chromosomal region 11p15.5. Genomics 37(2):253–256
Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M (1997) GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res 57(20):4493–4497
Manji SS, Parker NJ, Williams RT, van Stekelenburg L, Pearson RB, Dziadek M, Smith PJ (2000) STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta 1481(1):147–155
Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA (2001) Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J 357(Pt 3):673–685
Williams RT, Senior PV, Van Stekelenburg L, Layton JE, Smith PJ, Dziadek MA (2002) Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta 1596(1):131–137
Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M, Kurosaki T (2006) Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci U S A 103(45):16704–16709
Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312(5777):1220–1223
Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443(7108):226–229
Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R (2006) CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol 16(20):2073–2079
Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD (2006) Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A 103(24):9357–9362
Luik RM, Wu MM, Buchanan J, Lewis RS (2006) The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol 174(6):815–825
Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C (2008) Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem 283(12):8014–8022
Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD (2008) The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456:116–120
Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, Chen Y, Hille B, Xu T, Chen L (2008) Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci U S A 105(36):13668–13673
Mignen O, Thompson JL, Shuttleworth TJ (2008) Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol 586(2):419–425
Hou X, Pedi L, Diver MM, Long SB (2012) Crystal structure of the calcium release-activated calcium channel Orai. Science 338(6112):1308–1313
Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, Clapham DE, Harteneck C, Heller S, Julius D, Kojima I, Mori Y, Penner R, Prawitt D, Scharenberg AM, Schultz G, Shimizu N, Zhu MX (2002) A unified nomenclature for the superfamily of TRP cation channels. Mol Cell 9(2):229–231
Cosens DJ, Manning A (1969) Abnormal electroretinogram from a Drosophila mutant. Nature 224(5216):285–287
Minke B, Wu C, Pak WL (1975) Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature 258(5530):84–87
Montell C, Rubin GM (1989) Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2(4):1313–1323
Hardie RC, Minke B (1993) Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: implications for phosphoinositide-mediated Ca2+ mobilization. Trends Neurosci 16(9):371–376
Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C (1995) TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci U S A 92(21):9652–9656
Zhu X, Chu PB, Peyton M, Birnbaumer L (1995) Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett 373(3):193–198
Montell C (2001) Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci STKE 2001 (90):re1
Vannier B, Zhu X, Brown D, Birnbaumer L (1998) The membrane topology of human transient receptor potential 3 as inferred from glycosylation-scanning mutagenesis and epitope immunocytochemistry. J Biol Chem 273(15):8675–8679
Bennett V (1992) Ankyrins. Adaptors between diverse plasma membrane proteins and the cytoplasm. J Biol Chem 267(13):8703–8706
Montell C, Birnbaumer L, Flockerzi V (2002) The TRP channels, a remarkably functional family. Cell 108(5):595–598
Burkhard P, Stetefeld J, Strelkov SV (2001) Coiled coils: a highly versatile protein folding motif. Trends Cell Biol 11(2):82–88
Engelke M, Friedrich O, Budde P, Schafer C, Niemann U, Zitt C, Jungling E, Rocks O, Luckhoff A, Frey J (2002) Structural domains required for channel function of the mouse transient receptor potential protein homologue TRP1beta. FEBS Lett 523(1–3):193–199
Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Luckhoff A, Schultz G (1996) Cloning and functional expression of a human Ca2+ -permeable cation channel activated by calcium store depletion. Neuron 16(6):1189–1196
Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L (1996) trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 85(5):661–671
Liu X, Wang W, Singh BB, Lockwich T, Jadlowiec J, O’Connell B, Wellner R, Zhu MX, Ambudkar IS (2000) Trp1, a candidate protein for the store-operated Ca(2+) influx mechanism in salivary gland cells. J Biol Chem 275(5):3403–3411
Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, Stevens T (2001) Contribution of endogenously expressed Trp1 to a Ca2+ -selective, store-operated Ca2+ entry pathway. FASEB J 15(10):1727–1738
Mori Y, Wakamori M, Miyakawa T, Hermosura M, Hara Y, Nishida M, Hirose K, Mizushima A, Kurosaki M, Mori E, Gotoh K, Okada T, Fleig A, Penner R, Iino M, Kurosaki T (2002) Transient receptor potential 1 regulates capacitative Ca(2+) entry and Ca(2+) release from endoplasmic reticulum in B lymphocytes. J Exp Med 195(6):673–681
Almirza WH, Peters PH, van Zoelen EJ, Theuvenet AP (2012) Role of Trpc channels, Stim1 and Orai1 in PGF(2alpha)-induced calcium signaling in NRK fibroblasts. Cell Calcium 51(1):12–21
Lopez E, Berna-Erro A, Salido GM, Rosado JA, Redondo PC (2013) FKBP52 is involved in the regulation of SOCE channels in the human platelets and MEG 01 cells. Biochim Biophys Acta 1833(3):652–662
Rosado JA, Brownlow SL, Sage SO (2002) Endogenously expressed Trp1 is involved in store-mediated Ca2+ entry by conformational coupling in human platelets. J Biol Chem 277(44):42157–42163
Vannier B, Peyton M, Boulay G, Brown D, Qin N, Jiang M, Zhu X, Birnbaumer L (1999) Mouse trp2, the homologue of the human trpc2 pseudogene, encodes mTrp2, a store depletion-activated capacitative Ca2+ entry channel. Proc Natl Acad Sci U S A 96(5):2060–2064
Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L (1997) Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem 272(47):29672–29680
Brechard S, Melchior C, Plancon S, Schenten V, Tschirhart EJ (2008) Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell Calcium 44(5):492–506
Jardin I, Redondo PC, Salido GM, Rosado JA (2008) Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim Biophys Acta 1783(1):84–97
DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, Putney JW Jr (2009) TRPC channels function independently of STIM1 and Orai1. J Physiol 587(Pt 10):2275–2298
Lievremont JP, Bird GS, Putney JW Jr (2004) Canonical transient receptor potential TRPC7 can function as both a receptor- and store-operated channel in HEK-293 cells. Am J Physiol Cell Physiol 287(6):C1709–C1716
Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL (2006) STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci U S A 103(11):4040–4045
Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF (2006) STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol 8(9):1003–1010
Lopez JJ, Salido GM, Pariente JA, Rosado JA (2006) Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem 281(38):28254–28264
Rao JN, Rathor N, Zou T, Liu L, Xiao L, Yu TX, Cui YH, Wang JY (2010) STIM1 translocation to the plasma membrane enhances intestinal epithelial restitution by inducing TRPC1-mediated Ca2+ signaling after wounding. Am J Physiol Cell Physiol 299(3):C579–C588
Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, Muallem S (2011) Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12(2):232–245
Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, Shen XM, Hume JR (2009) TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J Physiol 587(Pt 11):2429–2442
Sours-Brothers S, Ding M, Graham S, Ma R (2009) Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med (Maywood) 234(6):673–682
Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9(6):636–645
Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell 32(3):439–448
Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S, Yuan JP (2014) Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J Biol Chem 289(10):6372–6382
Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS, Singh BB (2008) Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE). J Biol Chem 283(25):17333–17340
Jardin I, Salido GM, Rosado JA (2008) Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels (Austin) 2(6):401–403
Sampieri A, Zepeda A, Saldaña C, Salgado A, Vaca L (2008) STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: moving TRPC1 in and out of lipid rafts. Cell Calcium 44(5):479–491
Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L (2008) Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A 105(8):2895–2900
Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS (2007) Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem 282(12):9105–9116
Acknowledgments
This work was supported by MINECO (Grant BFU2013-45564-C2-1-P) and Gobierno de Extremadura-FEDER (GR15029). JJL and LA are supported by Juan de la Cierva Program (JCI-2012-12934) and MINECO fellowship BES-2011-043356, respectively.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Albarran, L., Lopez, J.J., Salido, G.M., Rosado, J.A. (2016). Historical Overview of Store-Operated Ca2+ Entry. In: Rosado, J. (eds) Calcium Entry Pathways in Non-excitable Cells. Advances in Experimental Medicine and Biology, vol 898. Springer, Cham. https://doi.org/10.1007/978-3-319-26974-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-26974-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26972-6
Online ISBN: 978-3-319-26974-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)