
Abstract
Breast cancer is the most common cancer in women worldwide. Although various subgroups are defined according to the expression of hormones and ErbB family receptors, it is well known that this disease is more heterogeneous than its classification system suggests. As new effective therapeutic choices are developed and used clinically, resistance to these new agents is also being observed. The most promising new anti-HER therapies are T-DM1 and pertuzumab, which has been evaluated in trastuzumab-resistant patients and also in a first-line setting with trastuzumab. The dual blockage of HER seems to be a favorable approach for these patients; however, the downstream signaling steps can be activated to overcome the tyrosine kinase inhibition. Because tumor cells can adapt themselves by using alternative pathways to maintain proliferation, providing a sufficient treatment approach also requires the consideration of possible escape mechanisms in tumor cells. By inhibiting tyrosine kinases combined with another agent that affects downstream factors of the PI3K/AKT/mTOR pathway, drug resistance in breast cancer can be overcome or delayed. In this chapter, we discuss the new tyrosine kinase inhibitors that inhibit more than only HER-2 and discuss some ongoing clinical trials in this area. In so doing, we hope to provide information for overcoming tyrosine kinase drug resistance and to identify the ideal settings for these treatment choices according to recent data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
A tyrosine kinase is an enzyme that phosphorylates tyrosine residues in proteins to regulate signaling within a cell. In normal conditions, tyrosine kinase activity is regulated by strict mechanisms; however, this tight control is lost in cancer cells, which results in uncontrolled cell proliferation, differentiation, apoptosis, and invasion [1]. Because tyrosine kinases are the driving step in cell signaling, tyrosine kinase inhibitors (TKIs) can serve as a therapeutic option for breast cancer patients. In this chapter, we will review the monoclonal antibodies targeting tyrosine kinase receptors and the small molecule TKIs that serve as anticancer agents, and we will also discuss the newer therapeutic options developed to overcome drug resistance in breast cancer.
Introduction
Receptor tyrosine kinases have three different major domains—the extracellular domains (domains I–IV), the transmembrane domain, and the juxtamembrane domain. Upon ligand binding, two receptor tyrosine kinases homo- or heterodimerize and tyrosine residues are phosphorylated to activate the downstream signaling cascade. Nearly 90 tyrosine kinases have been identified in humans including receptor tyrosine kinases and cellular tyrosine kinases [2].
Among these, the epidermal growth factor receptor family (EGFR, ErbB) and VEGF are the primary targets studied in breast cancer. Four members of ErbB receptor family have been identified: (1) EGFR (ErbB1), (2) HER-2 (ErbB2), (3) HER-3 (ErbB3), and (4) HER-4 (ErbB4).
Within the ErbB family, HER-2 is the preferred dimerization partner because its kinase catalytic activity is most potent, and it does not require a ligand for dimerization. HER-2 overexpression is observed in 15–30 % of breast cancers and is associated with poorer prognosis. Trastuzumab, a humanized monoclonal anti-HER-2 antibody, binds to the extracellular domain of HER-2 (subdomain IV) and leads to a conformational change. Previous studies have confirmed that via different mechanisms, including antibody-dependent cell-mediated cytotoxicity, downstream pathway inhibition, and dimerization prevention, trastuzumab exerts antitumor activity. Although the agent achieves its efficacy by various mechanisms, the majority of HER-2-positive tumors develop resistance to treatment. Increased MUC-4 expression, alternative downstream PI3K–AKT pathway activation, PTEN loss, truncated p95 expression, and p27 downregulation are possible reasons for trastuzumab resistance.
The majority of targeted therapy studies have attempted to overcome these resistance mechanisms by targeting various steps of the tyrosine kinase activation cascade or by using a combination of new anti-HER-2 therapies targeting the HER-2 signaling network at multiple points.
Anti-HER-2 Therapies
Lapatinib
Lapatinib is an orally active dual inhibitor of EGFR and HER-2. Preclinical studies demonstrated that lapatinib could inhibit trastuzumab-resistant HER-2(+) breast cancer by binding to truncated p95 [3, 4]. In the metastatic first-line setting, a lapatinib/chemotherapy combination is approved for use following disease progression in patients previously treated with trastuzumab. In this setting, paclitaxel–lapatinib combination therapy significantly improved event-free survival (EFS), the time to progression (TTP), and the clinical benefit rate (CBR) without any overall survival (OS) advantage compared with paclitaxel–placebo in a phase III study [5]. In subsequent settings, lapatinib/capecitabine and lapatinib/trastuzumab are possible therapeutic options [6, 7]. The combination of lapatinib and trastuzumab, by blocking HER-2 through different mechanisms, appears to be a good choice. EGF104900 study data demonstrated that lapatinib plus trastuzumab improved PFS, and the clinical benefit was comparable to lapatinib monotherapy [7, 8]. However, it is not clear whether the sequential or combined use of these agents will establish better results. An ongoing phase III study, NCT00968968, evaluating the efficacy of lapatinib plus trastuzumab versus trastuzumab alone to enable continuous HER-2 suppression after first- or second-line trastuzumab-based chemotherapy combination, will clarify whether dual blockage in maintenance will achieve better results in metastatic setting. Another ongoing phase III study, NCT00667251, which completed patient accrual, is comparing taxane plus trastuzumab or lapatinib combination therapy in untreated HER-2(+) MBC.
In the neoadjuvant setting, although lapatinib/trastuzumab combination therapy significantly improved pCR compared with trastuzumab alone (51.3 % vs. 29.5 %) in the NEO-ALTTO study [9], a head-to-head comparison of trastuzumab and lapatinib with chemotherapy combination therapy in the GeparQuinto study showed that trastuzumab achieved better pCR compared with lapatinib (31.75 % vs. 21.7 %, respectively) [10].. In the randomized phase II CHERLOB trial, preoperative taxane and anthracycline chemotherapy in combination with trastuzumab, lapatinib, or both was evaluated in stage II–IIIA breast cancer patients. The pCR rate was 28 %, 32 %, and 48 % in the trastuzumab, lapatinib, and combination arms, respectively [11]. The present data confirmed that the combination of these agents results in better pCR, whereas single-agent trastuzumab appears to be superior to single-agent lapatinib.
In tumors positive for HER-2 and hormone receptor (HR), inhibiting both the HER-2 and ER pathways might be a more reasonable option. There is crosstalk between these pathways. In lapatinib-exposed cells, continuous inhibition of the PI3K/Akt pathway can lead to upregulation of the transcription factor FOX03A, which can then increase ER signaling [12]. Two large randomized trials evaluated aromatose inhibition (AI) and anti-HER-2 therapy combinations. In postmenopausal hormone receptor- and HER-2-positive breast cancer patients, lapatinib in combination with letrozole achieved a significantly better median PFS (8.2 months vs. 3 months), ORR (28 % vs. 15 %), and CBR (48 % vs. 29 %) compared with letrozole alone [13]. In the TAnDEM study, trastuzumab and anastrozole combination therapy versus anastrozole alone showed a significantly superior median PFS (4.8 months vs. 2.4 months) and ORR (20.3 % vs. 6.8 %) in metastatic breast cancer [14]. In an ongoing study, NCT01160211, participants are being recruited to compare AI in combination with lapatinib, trastuzumab, or both for the treatment of hormone receptor-positive, HER-2-positive metastatic breast cancer.
Pertuzumab
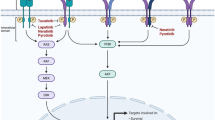
Pertuzumab is a recombinant humanized monoclonal antibody that binds to the HER-2 extracellular subdomain II and prevents HER-2 dimerization. Unlike trastuzumab, which is effective on HER-2 homodimers, pertuzumab can affect HER-2/EGFR and HER-2/HER-3 interactions (Fig. 36.1). In preclinical studies using pertuzumab as a single agent and in combination with trastuzumab, its activity was confirmed [15]. A subsequent phase II study evaluated the role of pertuzumab–trastuzumab combination therapy in HER-2(+) metastatic breast cancer patients who progressed on prior trastuzumab therapy. The objective response rate (ORR) and CBR were 24 % and 50 %, respectively. The median progression free-survival was 5.5 months [16]. The subsequent phase III CLEOPATRA study compared the efficacy of docetaxel, trastuzumab, and pertuzumab combination therapy with docetaxel, trastuzumab, and placebo in HER-2(+) metastatic breast cancer patients. The majority of the group (90 %) had not previously received an anti-HER-2 agent, and triple combination therapy showed significant improvements in the median PFS and ORR (19 months vs. 12 months; 80 % vs. 69 %, respectively) [17]. Based on this study, pertuzumab received FDA approval in June 2012 for use in the first-line setting as a combination therapy. The final OS results were presented at the 2014 ESMO Congress in Madrid, Spain. At last follow-up, the median OS was 56.5 versus 40.8 months in the pertuzumab–trastuzumab and trastuzumab only plus chemotherapy arms, respectively.

Therapies targeting HER signaling
The role of pertuzumab in second-line setting is not clear. As there is no present study comparing T-DM1 with pertuzumab combinations, the best option for the initial setting in MBC has not been clearly defined.
In neoadjuvant studies, the NEOSPHERE trial randomized operable, locally advanced, or inflammatory HER-2-positive breast cancers to four different treatment groups: (A) docetaxel and trastuzumab; (B) docetaxel, trastuzumab, and pertuzumab; (C) pertuzumab and trastuzumab; and (D) docetaxel and pertuzumab [18]. The pCRs of the four groups were 29 %, 45.8 %, 16.8 %, and 24 %, respectively. In this study, the triple combination showed superior pCR, but a subgroup of arm C also appeared to benefit from dual blockage without chemotherapy. The randomized phase II TRYPHAENA trial compared pertuzumab and trastuzumab (HP) with or without an anthracycline-based chemotherapy regimen in neoadjuvant setting. Fluorouracil, epirubicine, and cyclophosphamide (FEC)–HP followed by docetaxel (D)–HP, FEC followed by D–HP, and docetaxel–carboplatin (DC) with HP were the three treatment arms. The pCR rates were 62 %, 57 %, and 66 % in these three groups, respectively; no significant differences were found [19].
T-DM1
T-DM1 is an antibody drug conjugate comprising trastuzumab and an antimitotic agent derivative maytansine that directly targets HER-2expressing cells [20] (Fig. 36.1). FDA approved the drug in February 2013 in consequence of EMILIA study results. This phase III study enrolled 978 HER-2(+) MBC patients treated previously with trastuzumab and taxane therapy. The patients were randomized to T-DM1 (3.6 mg/kg IV, D1) OR capecitabine (1,000 mg/m2 orally twice daily, days 1–14) and lapatinib (1,250 mg orally once daily) combination therapy given every 3 weeks. Significant improvements in the median PFS (10 months vs. 6 months, respectively), OS (31 months vs. 25 months, respectively), and ORR were achieved (44 % vs. 31 %, respectively). In a recent publication evaluating the patient-reported outcomes of the EMILIA study, greater efficacy and tolerability of T-DM 1 were revealed [21].
The THERESA phase 3 trial compared T-DM1 with the physician’s treatment of choice in patients with progressive disease following treatment with two or more HER-2-directed regimens for MBC, and this trial revealed that PFS was significantly improved with T-DM1 (median PFS 6.2 months vs. 3.3 months) [22].
Although the approval of the drug is restricted to patients who progressed after trastuzumab therapy, recent studies may confirm the role of T-DM1 in the first-line setting. A recently published phase II study in HER-2(+) MBC/locally advanced patients showed that in the first-line setting, T-DM1 produced a superior median PFS (14.2 months. vs. 9.2 months, respectively) and ORR (64.2 % vs. 58 %, respectively) compared with docetaxel and trastuzumab combination therapy [23]. The phase III MARIANNE study (NCT01120184) completed patient recruitment to compare T-DM1 plus pertuzumab, T-DM1 plus placebo, and trastuzumab plus taxane in HER-2(+) MBC patients without initial therapy in a locally advanced or metastatic setting, but the final results have not been presented. Additionally, an ongoing study, NCT00829166, is comparing the safety and efficacy of T-DM1 with capecitabine plus lapatinib. These studies will identify the therapeutic settings of the new anti-HER-2 drugs and also provide knowledge regarding whether combination therapy will improve the survival endpoints.
These agents mentioned above target HER-1, HER-2, and HER-3 in their dimerization and activation steps; however, various cascades are involved downstream of the ErbB family before the proliferation signals reach the nucleus.
Neratinib
Neratinib is an oral covalent drug that irreversibly inhibits the ATP-binding active site of the ErbB family [24] (Fig. 36.1). In a phase II study, advanced HER-2-positive breast cancer patients received oral neratinib 240 mg once daily. The median PFS was 22.3 or 38.6 weeks for patients with prior trastuzumab therapy or with no prior trastuzumab therapy, respectively. The ORR was 24 % among patients with prior trastuzumab treatment and 56 % in the trastuzumab-naive cohort [25]. In another study comparing neratinib monotherapy versus lapatinib plus capecitabine, the median PFS was 4.5 versus 6.8 months in the neratinib (240 mg/day) arm versus the combination arm. The ORRs were 29 % and 41 %, respectively. The noninferiority of neratinib could not be demonstrated in this study. Another study evaluated neratinib (240 mg/day) and capecitabine combination therapy (1,500 mg/m2/day) in HER-2(+) breast cancer. The ORRs for patients who had received prior lapatinib treatment and for lapatinib-naive patients were 57 % and 64 %, respectively. The median PFS was superior in the lapatinib-naive group (40.3 vs. 35.9 weeks) [26].
Lessons gained from phase II studies show that neratinib mainly improved responses and survival rates in anti-HER-2-naive patients. Based on the positive results in the metastatic setting, the efficacy of the drug in neoadjuvant and adjuvant settings is being investigated in ongoing trials.
The I-SPY 2 trial evaluated neratinib in combination with weekly paclitaxel with or without trastuzumab followed by doxorubicin and cyclophosphamide (AC) as neoadjuvant therapy for women with HER-2-positive locally advanced breast cancer. The final results were presented at AACR 2014 as an oral presentation. Neratinib plus chemotherapy achieved a pathological complete response rate of 56 % versus 33 % for patients treated with chemotherapy alone [27].
Additionally, an adjuvant study, the ExteNET trial, evaluated neratinib versus placebo after the completion of 1 year of standard trastuzumab therapy in 2,821 patients with HER-2-positive breast cancer. Adjuvant treatment with neratinib extended DFS by 33 % compared with the placebo. The full results have not been presented at a scientific meeting.
The main side effect of neratinib is diarrhea, which is usually of the secretory type without mucositis. Antidiarrheal therapy, including loperamide, can overcome this adverse event.
Afatinib
Afatinib is an oral small molecule inhibitor of the ErbB receptor family that covalently binds and irreversibly blocks ErbB family members (Fig. 36.1). In advanced HER-2(+) breast cancer patients after trastuzumab failure, afatinib 50 mg/day was given to patients once daily until progression occurred. SD was the best response in 37 % of patients, and 46 % achieved a clinical benefit. The median PFS and OS were 15.1 and 61 weeks, respectively [28]. According to a previous study, the activity appeared to be limited in HER-2(−) patients [29]. A phase 3 randomized study of afatinib in trastuzumab-resistant metastatic breast cancer was halted early due to unfavorable risk–benefit analysis [30].
Dual blocking of tyrosine kinases, pan-HER blocking agents, and drug-conjugated anti-HER-2 agents are being studied in early-phase clinical studies to explain the exact role of these agents in the near future (Table 36.1).
EGFR Inhibitors
EGFR (HER-1) is a member of the ErbB family that enhances tumorigenicity in breast cancer and is also associated with poorer survival and resistance to hormonal therapy [31, 32]. EGFR is not only related to ER(+) tumors but is also overexpressed in basal-like breast cancers [33]. The small molecule tyrosine kinase inhibitor gefitinib is being investigated in combination with endocrine therapy in hormone receptor-positive tumors, whereas cetuximab is being evaluated in triple-negative patients.
Gefitinib
Gefitinib is a small molecule tyrosine kinase inhibitor that inhibits downstream signaling pathways activated by phosphorylation. The efficacy of the drug could not be demonstrated in monotherapy in taxane- and anthracycline-pretreated metastatic breast cancer patients [34] but was shown to be a reasonable option in the neoadjuvant setting with anastrozole combination therapy in ER(+) and EGFR(+) tumors [35]. A phase II study in the advanced breast cancer setting demonstrated that paclitaxel and carboplatin combined with gefitinib (250 mg/day orally) achieved CR (10.3 %), PR (44.1 %), and SD (30.9 %) in a patient group [36]. In another study, first-line therapy in MBC with gefitinib and docetaxel revealed an ORR of 54 % with better PR and CR in an ER(+) versus ER(−) group (70 % vs. 21 %) [37]. Although various chemotherapeutic combinations had acceptable toxicity profiles, adding gefitinib to chemotherapy as well as to trastuzumab did not achieve a significant improvement in response rates or survival [38–40]. These results carried the drug to be used in a combination with hormonal therapy options.
According to the present data, the addition of gefitinib to anastrozole treatment had no additional clinical effect in a neoadjuvant setting [41]; however, the same combination is associated with improved PFS (17.4 vs. 8.4 months) and CBR (49 % vs. 34 %) compared with anastrozole alone in a metastatic setting [42]. A recent study comparing anastrozole plus gefitinib versus fulvestrant plus gefitinib in postmenopausal HR(+) MBC showed that both combinations have similar clinical benefit rates (44.1 % vs. 41 %, respectively), median PFS (5.3 vs. 5.2 months, respectively), and OS (30.3 vs. 23.9 months, respectively). However, the clinical benefit rates of both combinations are not clearly superior to gefitinib or endocrine therapy alone. Because EGFR expression is related to endocrine resistance, it is rational to hypothesize that gefitinib plus endocrine therapy might overcome hormonal therapy resistance. Additionally, in a phase II study, two patient groups with initial hormonal therapy received gefitinib. Stratum 1 included women with newly diagnosed metastases or who had recurred 1 year after stopping adjuvant therapy with tamoxifen. Stratum 2 involved patients with recurrent disease during or after AI adjuvant therapy or those who progressed after first-line hormonotherapy with AI in a metastatic setting. Patients were randomized to receive tamoxifen plus gefitinib (250 mg/day orally) versus tamoxifen plus placebo. The median PFS (10.9 vs. 8.8 months) was better in the combination arm in Stratum 1. No objective responses were detected in Stratum 2 with combination therapy [43].
These conflicting results reflect that the present data are not sufficient to identify the exact ideal setting of this agent. We believe that in the future, gefitinib can be a part of therapeutic options in HR (+) MBC patients in the initial setting to delay the development of hormone resistance.
Cetuximab
Cetuximab is an epidermal growth factor (EGF) antagonist that specifically binds to EGFR on both normal and tumor cells. The binding of cetuximab to EGFR blocks the phosphorylation and activation of receptor-associated kinases. Signal transduction through EGFR activates k-ras; however, mutant k-ras protein is constitutively active and does not depend on EGFR regulation. The majority of cetuximab studies included triple-negative breast cancers (TNBCs) because they have high EGFR expression. A phase II study evaluating weekly irinotecan/carboplatin with or without cetuximab in patients with metastatic breast cancer showed antitumor activity but also showed significant associated toxicity [44]. The TBCRC001 study evaluated cetuximab and carboplatin combination therapy in metastatic TNBCs; the combination clinical benefit ratio is higher (27 % vs. 10 %), whereas the median PFS was only 2 months in all study groups due to rapid disease progression. BALI-1, the largest EGFR trial in metastatic TNBC, compared cetuximab with cetuximab and cisplatin combination therapies. With combination therapy, the reduction in the risk of progression was 32.5 %, and PFS was longer in the cetuximab arm (3.7 months vs. 1.5 months, HR: 0.67, p = 0.03); no significant improvement was found in OS [45].
In addition to these two agents, erlotinib was shown to have minimal activity in unselected previously treated women [46] and limited activity when combined with bevacizumab in MBC after first- or second-line chemotherapy [47]. However, preliminary evidence of anticancer activity was observed with trastuzumab combination therapy [48].
Among the EGFR inhibitors, cetuximab and gefitinib appear to be the most promising drugs according to recent data.
Targeting the PI3K Pathway in Breast Cancer to Overcome TKI Resistance
Phosphoinositide 3-kinases (PI3Ks) are a family of lipid kinases. They function as dimeric enzymes and comprise catalytic (p110 α, β, ɣ, and δ) and regulatory subunits (p85) in their structures. After a growth factor or a ligand binds to its tyrosine kinase receptor, the inhibitory effect of p85 on p110 is removed, and PI3K is activated. The activated kinases phosphorylate phosphatidylinositol bisphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3), which recruits proteins such as Akt and PDK1 to cellular membranes [49]. Phosphatase and tensin homologue deleted on chromosome ten (PTEN) acts as a catalytic antagonist of PI3K by hydrolyzing PIP3 to PIP2. Class 1 PI3Ks comprise the major subgroup, which is found to be involved in cancer. PI3K mutational activation or overexpression or PTEN inactivation by genetic or epigenetic alterations result in enhanced PI3K signaling. The majority of mutations are in PIK3CA in three hotspots within the p110α catalytic subunit and are gain-of-function mutations. Two of these mutations are helical, and one is on the kinase domain of p110α.
A recent paper published in Nature highlighted the genomic and proteomic features of breast cancer subtypes and showed that PIK3CA mutation was more common in luminal tumors, whereas PTEN mutation/loss was most common in basal-like breast cancers [50]. PIK3CA mutations were found in 49 %, 32 %, 7 %, and 42 % of luminal A, luminal B, basal-like, and HER-2(+) patients, respectively, whereas PTEN mutations/losses were found in 13 %, 24 %, 35 %, and 19 %, respectively.
Previous studies confirmed that PI3KCA mutations could confer favorable clinical outcomes. Because luminal A–B tumors have more frequent mutations with slower disease progression, especially in luminal A tumors, these mutations may be associated with less aggressive disease. However, these mutations are also associated with trastuzumab and lapatinib resistance in HER-2-positive breast cancer and to hormonal therapy resistance in HR-positive tumors by directly inducing ER transcription [51, 52]. Retrospective analyses in HER-2(+) MBC showed that tumors with PIK3CA mutations or PTEN loss are associated with low trastuzumab and lapatinib efficacy and also suggest that anti-HER-2 drug-resistant tumors may still benefit from PI3K inhibitors [53, 54]. In contrast, PTEN-deficient HER-2-positive cells still have upstream input from HER-2; therefore, dual blockage might be effective in this patient group [55].
PI3K pathway inhibitors can be divided into subgroups according to their targets: (1) pan-PI3K inhibitors; (2) mTOR inhibitors; (3) Akt inhibitors; and (4) PI3K/mTOR dual inhibitors. Regarding pan-PI3K inhibitors, phase 1 dose escalation study results in solid tumors have recently been published but will not be mentioned here because phase 2 study results are still pending [56, 57].
mTOR Inhibitors
mTOR is one of the major mediators of cell growth; it acts primarily via two downstream messengers, P70-S6 kinase 1 and 4E-BP1, which exert their activity at the translational level. Because PI3K/Akt/mTOR pathway activation was shown to contribute to trastuzumab and hormonal therapy resistance in previous studies, the addition of mTOR inhibitors to chemotherapy and hormonal therapy options was performed in an attempt to delay resistance in this patient group [58, 59].
Regarding the first studies initiated with temsirolimus, a phase II study exploring the combination of letrozole and temsirolimus compared with letrozole alone showed a longer median PFS in the combination group, but a subsequent phase III study was stopped early due to toxicity issues [60, 61].
Everolimus, with its improved toxicity profile, became the major agent being evaluated in this setting. Everolimus did not achieve a good objective ORR as a monotherapy [62]. However, in HER-2(+) MBC patients, everolimus in combination with paclitaxel plus trastuzumab or vinorelbine with trastuzumab demonstrated efficacy in trastuzumab-pretreated patients [63, 64]. In a phase I/II study, HER-2(+) MBC patients who progressed on trastuzumab-based therapy received everolimus in combination with trastuzumab. Among 47 patients, the combination of everolimus and trastuzumab resulted in PR in seven patients (15 %) and persistent SD (lasting 6 months or longer) in nine patients (19 %), translating to a clinical benefit rate of 34 %. The median PFS was 4.1 months. This study suggests that everolimus may have promising activity in trastuzumab-pretreated patients not receiving cytotoxic chemotherapy [65].
In HR-positive tumors, because endocrine therapy resistance is associated with PI3K/Akt/mTOR pathway activation, the combination of everolimus and hormonal therapy is a rational option to overcome or delay endocrine resistance. The benefit of everolimus plus exemestane was shown in the BOLERO-2 trial in 724 patients who progressed on anastrozole. Patients were randomly assigned to receive either exemestane plus everolimus or exemestane plus placebo [66]. The final study results, with a median 18-month follow-up, show that the median PFS remained significantly longer with everolimus plus exemestane irrespective of age or metastasis region [investigator review: 7.8 versus 3.2 months, respectively; hazard ratio = 0.45 (95 % confidence interval 0.38–0.54); log-rank P < 0.0001; central review: 11.0 versus 4.1 months, respectively; hazard ratio = 0.38 (95 % confidence interval 0.31–0.48); log-rank P < 0.0001) [67].
The noninterventional BRAWO study including HR (+) and HER-2(−) MBC patients treated with everolimus and exemestane showed that the median PFS was 8.0 months in the everolimus and exemestane group, with more favorable results in the first-line treatment (PFS:10 months) group; these data were presented at the ESMO 2014 Congress in Spain [68].
In the phase II GINECO study, 111 HR(+), HER-2(−) MBC patients previously treated with aromatase inhibitors were randomly selected to receive tamoxifen alone or tamoxifen in combination with everolimus (10 mg/day). The CBR (61.1 % vs. 42.1 %) and TTP (8.6 months vs. 4.5 months) were significantly improved in the combination group, and the risk of death was reduced by 55 % with tamoxifen plus everolimus versus tamoxifen alone (HR, 0.45; 95 % CI, 0.24–0.81) [69]. When patients were stratified according to primary or secondary hormone resistance, TTP was more improved in secondary hormone-resistant patients who received combination therapy compared with those who received tamoxifen alone (17.5 months vs. 5 months, respectively), whereas TTP was only slightly improved by combination therapy in primary hormone-resistant patients (5.4 months vs. 3.9 months, respectively).
In a phase II study, ER(+) MBC patients who failed AI therapy within 6 months were randomized to receive everolimus 10 mg/day in combination with intramuscular fulvestrant (500 mg D1, 250 mg D14, 250 mg D28, or 250 mg once a month). Although the final results of the study have not been presented, a CBR of 55 % and TTP of 8.6 months were achieved by combination therapy [70].
The present data show that everolimus combined with hormonotherapy might be an ideal therapeutic option in secondary hormone-resistant patients.
The BOLERO-3 trial evaluated the combination of everolimus, vinorelbine, and trastuzumab in women with HER-2-positive, trastuzumab-resistant advanced breast carcinoma who had previously received taxane therapy. Eligible patients were randomly assigned to daily everolimus (5 mg/day) plus weekly trastuzumab (2 mg/kg) and vinorelbine (25 mg/m2) or to placebo plus trastuzumab plus vinorelbine in 3-week cycles [71]. The study revealed that the addition of everolimus to trastuzumab plus vinorelbine prolongs PFS in the patient group [the median PFS was 7.00 months (95 % CI 6.74–8.18) with everolimus and 5.78 months (5.49–6.90) with placebo (hazard ratio 0.78 [95 % CI 0.65–0.95]; p = 0.0067)].
An ongoing study, the BOLERO-1 trial (NCT00876395), is evaluating everolimus in combination with trastuzumab and paclitaxel in the first-line setting; the final results have not yet been released.
Everolimus has also evaluated in the neoadjuvant setting in combination with letrozole. Newly diagnosed ER (+) localized breast cancer patients were randomized to receive letrozole 2.5 mg/day plus placebo or letrozole plus everolimus 10 mg/day before surgery. The ORR was found to be 59 % and 68 % in the letrozole and combination arms, respectively [72].
Upon blocking mTOR with everolimus, compensatory Akt activation occurs. Baselga et al. explained in a recent review that this situation was due to reduced S6 following mTOR inhibition and claimed that reduced S6 could not suppress signaling of IGF-1R via supression of IRS-1 anymore. Activated IGF-1R increase PI3K signaling [49].
PI3K Inhibitors
Clinical trials with PI3K inhibitors are ongoing and still in early phases (Table 36.2). Among these inhibitors, XL-765 is a dual mTOR (TORC 1 and 2) and PI3K inhibitor, and XL-147 is a selective inhibitor of PI3K with a potent inhibitory effect on the class I PI3K family. Both agents were designed to be orally administered. Preliminary data from the NCT01082068 trial confirmed that both PI3K inhibitors can be safely combined with letrozole. The phase II studies will clarify whether dual PI3K and mTOR inhibition is better than PI3K inhibition alone.
Concluding Remarks
Targeted therapies in breast cancer have had remarkable effects on patient survival since the first representative drug, trastuzumab, was used in HER-2(+) breast cancer. However, patients develop resistance to these drugs during the treatment period. This is mainly associated with cancer cells finding alternative pathways to maintain proliferative signaling. To delay the development of resistance to these therapies, combined modalities targeting different steps of the signaling cascade have been investigated. The main obstacle to this approach is tumor heterogeneity; because of this, we cannot use simple standard analytical techniques to predict the driving pathway in the tumor that should be blocked. Genomic analyses in recent decades have also confirmed this heterogeneity and revealed that by analyzing tumor characteristics, individualized therapy can be performed for each patient. This direction is also reflected in the ongoing trial protocols, which mainly include patients with demonstrated mutations amenable to treatment with the target drug. The future studies should not only confirm the efficacy of targeted combinations but also stratify the selected patient group for each developed drug.
References
Saxena R, Dwivedi A. ErbB family receptor inhibitors as therapeutic agents in breast cancer: current status and future clinical perspective. Med Res Rev. 2012;32(1):166–215. Review.
Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J Clin Oncol. 2011;2(2):80–93.
Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–9.
Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–38. Research Support, Non-U.S. Gov’t.
Di Leo A, Gomez HL, Aziz Z, Zvirbule Z, Bines J, Arbushites MC, et al. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26(34):5544–52. Clinical Trial, Phase III Multicenter Study Randomized Controlled Trial Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t.
Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, Romieu CG, et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat. 2008;112(3):533–43. Clinical Trial, Phase III Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(7):1124–30. Clinical Trial, Phase III Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G, et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 study. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(21):2585–92. Clinical Trial, Phase III Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–40. Clinical Trial, Phase III Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Untch M, Loibl S, Bischoff J, Eidtmann H, Kaufmann M, Blohmer JU, et al. Lapatinib versus trastuzumab in combination with neoadjuvant anthracycline-taxane-based chemotherapy (GeparQuinto, GBG 44): a randomised phase 3 trial. Lancet Oncol. 2012;13(2):135–44. Clinical Trial, Phase III Comparative Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Guarneri V, Frassoldati A, Piacentini F, Jovic G, Giovannelli S, Oliva C, et al. Preoperative chemotherapy plus lapatinib or trastuzumab or both in HER2-positive operable breast cancer (CHERLOB Trial). Clin Breast Cancer. April 2008. Vol 8 No. 2 192–194. Clinical Trial, Phase II Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Xia W, Bacus S, Husain I, Liu L, Zhao S, Liu Z, et al. Resistance to ErbB2 tyrosine kinase inhibitors in breast cancer is mediated by calcium-dependent activation of RelA. Mol Cancer Ther. 2010;9(2):292–9. Research Support, Non-U.S. Gov’t.
Johnston S, Pippen Jr J, Pivot X, Lichinitser M, Sadeghi S, Dieras V, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27(33):5538–46. Clinical Trial, Phase III Comparative Study Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27(33):5529–37. Clinical Trial, Phase III Comparative Study Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Brockhoff G, Heckel B, Schmidt-Bruecken E, Plander M, Hofstaedter F, Vollmann A, et al. Differential impact of cetuximab, pertuzumab and trastuzumab on BT474 and SK-BR-3 breast cancer cell proliferation. Cell Prolif. 2007;40(4):488–507. Research Support, Non-U.S. Gov’t.
Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(7):1138–44. Clinical Trial, Phase II Multicenter Study Research Support, Non-U.S. Gov’t.
Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–19. Comparative Study Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32. Clinical Trial, Phase II Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Schneeweiss A, Chia S, Hickish T, Harvey V, Hegg R, Tausch C, et al. Pertuzumab and trastuzumab in combination with an anthracycline-containing or an anthracycline-free standard chemotherapy in the neoadjuvant treatment of HER2-positive breast cancer (TRYPHAENA). Eur J Cancer. 2012;48:S96–S.
Isakoff SJ, Baselga J. Trastuzumab-DM1: building a chemotherapy-free road in the treatment of human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29(4):351–4. Comment Editorial.
Welslau M, Dieras V, Sohn JH, Hurvitz SA, Lalla D, Fang L, et al. Patient-reported outcomes from EMILIA, a randomized phase 3 study of trastuzumab emtansine (T-DM1) versus capecitabine and lapatinib in human epidermal growth factor receptor 2-positive locally advanced or metastatic breast cancer. Cancer. 2014;120(5):642–51. Clinical Trial, Phase III Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, et al. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(7):689–99. Clinical Trial, Phase III Comparative Study Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, et al. Phase II randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(9):1157–63.
Lopez-Tarruella S, Jerez Y, Marquez-Rodas I, Martin M. Neratinib (HKI-272) in the treatment of breast cancer. Future Oncol. 2012;8(6):671–81. Review.
Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(8):1301–7. Clinical Trial, Phase II Comparative Study Multicenter Study Research Support, Non-U.S. Gov’t.
Saura C, Martin M, Moroose R, Harb W, Liem K, Arena F, et al. Safety of neratinib (HKI-272) in combination with capecitabine in patients with solid tumors: a phase 1/2 study. Cancer Res. 2009;69(24):801S–2.
Park J, Liu M, Yee D, et al. Neratinib plus standard neoadjuvant chemotherapy for high-risk breast cancer: efficacy results from the I-SPY trial. 2014 AACR Annual Meeting 2014.
Lin NU, Winer EP, Wheatley D, Carey LA, Houston S, Mendelson D, et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res Treat. 2012;133(3):1057–65. Clinical Trial, Phase II Multicenter Study Research Support, Non-U.S. Gov’t.
Schuler M, Awada A, Harter P, Canon JL, Possinger K, Schmidt M, et al. A phase II trial to assess efficacy and safety of afatinib in extensively pretreated patients with HER2-negative metastatic breast cancer. Breast Cancer Res Treat. 2012;134(3):1149–59. Clinical Trial, Phase II Multicenter Study Research Support, Non-U.S. Gov’t.
Hurvitz SA, Shatsky R, Harbeck N. Afatinib in the treatment of breast cancer. Expert Opin Investig Drugs. 2014;23(7):1039–47.
Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. 2000;60(11):3081–7. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.
Nicholson RI, McClelland RA, Gee JM, Manning DL, Cannon P, Robertson JF, et al. Epidermal growth factor receptor expression in breast cancer: association with response to endocrine therapy. Breast Cancer Res Treat. 1994;29(1):117–25. Clinical Trial Comparative Study Research Support, Non-U.S. Gov’t.
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2004;10(16):5367–74. Research Support, U.S. Gov’t, P.H.S.
von Minckwitz G, Jonat W, Fasching P, du Bois A, Kleeberg U, Luck HJ, et al. A multicentre phase II study on gefitinib in taxane- and anthracycline-pretreated metastatic breast cancer. Breast Cancer Res Treat. 2005;89(2):165–72. Clinical Trial Clinical Trial, Phase II Multicenter Study Research Support, Non-U.S. Gov’t.
Polychronis A, Sinnett HD, Hadjiminas D, Singhal H, Mansi JL, Shivapatham D, et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial. Lancet Oncol. 2005;6(6):383–91. Clinical Trial Clinical Trial, Phase II Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Fountzilas G, Pectasides D, Kalogera-Fountzila A, Skarlos D, Kalofonos HP, Papadimitriou C, et al. Paclitaxel and carboplatin as first-line chemotherapy combined with gefitinib (IRESSA) in patients with advanced breast cancer: a phase I/II study conducted by the Hellenic Cooperative Oncology Group. Breast Cancer Res Treat. 2005;92(1):1–9. Clinical Trial Clinical Trial, Phase I Clinical Trial, Phase II Multicenter Study Research Support, Non-U.S. Gov’t.
Ciardiello F, Troiani T, Caputo F, De Laurentiis M, Tortora G, Palmieri G, et al. Phase II study of gefitinib in combination with docetaxel as first-line therapy in metastatic breast cancer. Br J Cancer. 2006;94(11):1604–9. Clinical Trial, Phase II Multicenter Study.
Dennison SK, Jacobs SA, Wilson JW, Seeger J, Cescon TP, Raymond JM, et al. A phase II clinical trial of ZD1839 (Iressa) in combination with docetaxel as first-line treatment in patients with advanced breast cancer. Investig New Drugs. 2007;25(6):545–51. Clinical Trial, Phase II Multicenter Study Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t.
Arteaga CL, O’Neill A, Moulder SL, Pins M, Sparano JA, Sledge GW, et al. A phase I-II study of combined blockade of the ErbB receptor network with trastuzumab and gefitinib in patients with HER2 (ErbB2)-overexpressing metastatic breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14(19):6277–83. Clinical Trial, Phase I Clinical Trial, Phase II Research Support, N.I.H., Extramural.
Gioulbasanis I, Saridaki Z, Kalykaki A, Vamvakas L, Kalbakis K, Ignatiadis M, et al. Gefitinib in combination with gemcitabine and vinorelbine in patients with metastatic breast cancer pre-treated with taxane and anthracycline chemotherapy: a phase I/II trial. Anticancer Res. 2008;28(5B):3019–25. Clinical Trial, Phase I Clinical Trial, Phase II Research Support, Non-U.S. Gov’t.
Smith IE, Walsh G, Skene A, Llombart A, Mayordomo JI, Detre S, et al. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(25):3816–22. Clinical Trial, Phase II Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Cristofanilli M, Valero V, Mangalik A, Royce M, Rabinowitz I, Arena FP, et al. Phase II, randomized trial to compare anastrozole combined with gefitinib or placebo in postmenopausal women with hormone receptor-positive metastatic breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16(6):1904–14. Clinical Trial, Phase II Comparative Study Multicenter Study Randomized Controlled Trial Research Support, N.I.H., Extramural.
Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, Underhill C, et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17(5):1147–59. Clinical Trial, Phase II Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
O’Shaughnessy J, Weckstein DJ, Vukelja SJ, McIntyre K, Krekow L, Holmes FA, et al. Preliminary results of a randomized phase II study of weekly irinotecan/carboplatin with or without cetuximab in patients with metastatic breast cancer. Breast Cancer Res Treat. 2007;106:S32–3.
Baselga J, Gomez P, Awada A, Greil R, Braga S, Climent MA, et al. The addition of cetuximab to cisplatin ıncreases overall response rate (orr) and progressıon-free survıval (pfs) ın metastatıc trıple-negatıve breast cancer (tnbc): results of a randomızed phase II study (Bali-1). Ann Oncol. 2010;21:96.
Dickler MN, Cobleigh MA, Miller KD, Klein PM, Winer EP. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res Treat. 2009;115(1):115–21. Clinical Trial, Phase II Multicenter Study.
Dickler MN, Rugo HS, Eberle CA, Brogi E, Caravelli JF, Panageas KS, et al. A phase II trial of erlotinib in combination with bevacizumab in patients with metastatic breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2008;14(23):7878–83. Clinical Trial, Phase II Multicenter Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t.
Britten CD, Finn RS, Bosserman LD, Wong SG, Press MF, Malik M, et al. A phase I/II trial of trastuzumab plus erlotinib in metastatic HER2-positive breast cancer: a dual ErbB targeted approach. Clin Breast Cancer. 2009;9(1):16–22. Clinical Trial, Phase I Clinical Trial, Phase II Research Support, N.I.H., Extramural.
Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16 Suppl 1:12–9. Research Support, Non-U.S. Gov’t Review.
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–60. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t.
Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(16):5049–59. Research Support, Non-U.S. Gov’t.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–30. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t.
Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, et al. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer. 2011;11:248. Clinical Trial Research Support, Non-U.S. Gov’t.
Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177(4):1647–56. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res BCR. 2011;13(6):224. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review.
Ando Y, Inada-Inoue M, Mitsuma A, Yoshino T, Ohtsu A, Suenaga N, et al. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105(3):347–53. Clinical Trial, Phase I Research Support, Non-U.S. Gov’t.
Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Brana I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res Off J Am Assoc Cancer Res. 2014;20(1):233–45. Clinical Trial, Phase I Research Support, Non-U.S. Gov’t.
Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19(53):6102–14. Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.Research Support, U.S. Gov’t, P.H.S. Review.
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. Clinical Trial Multicenter Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t.
Baselga J, Fumoleau P, Gil M, Colomer R, Roche H, Cortes-Funes H, et al. Phase II, 3-arm study of CCI-779 in combination with letrozole in postmenopausal women with locally advanced or metastatic breast cancer: preliminary results. J Clin Oncol. 2004;22(14):13S–S.
Chow LWC, Sun Y, Jassem J, Baselga J, Hayes DF, Wolff AC, et al. Phase 3 study of temsirolimus with letrozole or letrozole alone in postmenopausal women with locally advanced or metastatic breast cancer. Breast Cancer Res Treat. 2006;100:S286–S.
Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27(27):4536–41. Clinical Trial, Phase II Comparative Study Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Andre F, Campone M, O’Regan R, Manlius C, Massacesi C, Sahmoud T, et al. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(34):5110–5. Clinical Trial, Phase I Multicenter Study Research Support, Non-U.S. Gov’t.
Jerusalem G, Fasolo A, Dieras V, Cardoso F, Bergh J, Vittori L, et al. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2011;125(2):447–55. Clinical Trial, Phase I Multicenter Study Research Support, Non-U.S. Gov’t.
Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29(23):3126–32. Clinical Trial, Phase IClinical Trial, Phase II Multicenter Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t.
Beaver JA, Park BH. The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol. 2012;8(6):651–7. Clinical Trial, Phase III Randomized Controlled Trial.
Yardley DA, Noguchi S, Pritchard KI, Burris 3rd HA, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30(10):870–84. Clinical Trial, Phase III Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Lueftner D, Schuetz F, Grischke EM, Fasching PA. Breast cancer treatment with everolimus and exemestane for ER+ women: results of the first interim analysis of the noninterventional trial BRAWO. Ann Oncol. 2014;25(Supplement 5):v1–41.
Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(22):2718–24. Clinical Trial, Phase II Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Barnett CM. Everolimus: targeted therapy on the horizon for the treatment of breast cancer. Pharmacotherapy. 2012;32(4):383–96. Research Support, Non-U.S. Gov’t Review.
Andre F, Gianni L. BOLERO-3 results: pharmacological activity or pharmacokinetic effect? – authors’ reply. Lancet Oncol. 2014;15(8):e304–5. Comment Letter.
Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27(16):2630–7. Clinical Trial, Phase II Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cakar, B., Göker, E. (2016). Tyrosine Kinase Inhibitors. In: Aydiner, A., İgci, A., Soran, A. (eds) Breast Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-26012-9_36
Download citation
DOI: https://doi.org/10.1007/978-3-319-26012-9_36
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26010-5
Online ISBN: 978-3-319-26012-9
eBook Packages: MedicineMedicine (R0)