
Abstract
Sleep is an orchestrated neurochemical process involving different biological systems. Many theories have attempted to explain the biological meaning of sleep including a primary restorative function, a central role in reinforcement, and consolidation of memory, an important place for thermoregulatory processes. Despite the lack of a general consensus on its purposes, the brain mechanisms controlling sleep and wakefulness as well as the associated changes in cardiovascular and respiratory function are largely known. The autonomic nervous system is of paramount importance in the regulation of the cardiovascular function during sleep, and it can also mediate cardiovascular events occurring during sleep as a result of cardiovascular diseases or primary sleep disorders such as sleep-disordered breathing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Sleep is an orchestrated neurochemical process involving different biological systems. Many theories have attempted to explain the biological meaning of sleep including a primary restorative function, a central role in reinforcement, and consolidation of memory, and an important place for thermoregulatory processes. Despite the lack of a general consensus on its purposes, the brain mechanisms controlling sleep and wakefulness as well as the associated changes in cardiovascular and respiratory function are largely known. The autonomic nervous system is of paramount importance in the regulation of the cardiovascular function during sleep, and it can also mediate cardiovascular events occurring during sleep as a result of cardiovascular diseases or primary sleep disorders such as sleep-disordered breathing.
The aim of this chapter is to review the autonomic effects of normal sleep and of sleep-related breathing abnormalities, the relationship of the autonomic nervous system to arrhythmogenesis and sudden death, and the role of the autonomic nervous system in mediating cardiovascular consequences of sleep apnea in heart failure.
1 The Cardiovascular System and Sleep Architecture
Polysomnography is the standard tool used to study the overall sleep process and diagnose sleep disorders. It involves the recording of the electroencephalogram (EEG), the electro-oculogram (EOG), and the electromyogram (EMG). Based on these signals, sleep is divided into two strikingly distinct states: non-rapid eye movement (NREM) sleep and rapid eye movement (REM) sleep. NREM sleep is subdivided further into light sleep (NREM1 and NREM2 or N1 and N2) and deep sleep (NREM3 or N3, sometimes referred to as slow wave sleep). NREM and REM occur in a cycle that is repeated four to six times every night, each cycle lasting approximately 90–110 min.
A normal sleep cycle begins with light N1 sleep, during which the theta waves (4–7 Hz) replace the alpha rhythm (8–13 Hz) of wakefulness. As sleep approaches, muscle activity, which is highest during wakefulness, starts to decrease. With transition to N2, sleep spindles and K complexes appear in the EEG, thus signaling the increasing depth of sleep. Sleep spindles are bursts of rapid, rhythmic brain wave activity in the range 12–15 Hz, while K complexes are slow (>4 Hz), large waves that stand out from the background with a negative than a positive deflection. In N3, the EEG pattern is characterized by synchronized high-amplitude (>75 μV), slow (0.5–3 Hz) delta waves. Slow wave sleep is associated with no eye movements and with a higher arousal threshold and a further decline in muscle tone than during lighter sleep. In contrast with the synchronized activity of NREM sleep, REM sleep is characterized by a very intense and desynchronized EEG activity (similar to wakefulness) coupled with a complete loss of muscle tone, bursts of rapid eye movements, and irregularities in respiration and heart rate.
It has to be underscored, however, that this sequence of sleep stages (N1, N2, N3, REM) undergoes some changes as sleep progresses from cycle to cycle. In the first cycle, sleep begins in N1 and progresses into N2 and N3. Afterward, N2 sleep is repeated before entering REM sleep. Once REM sleep is over, there is usually a return to N2 sleep. Thus, N1 sleep accounts for <5 % of total sleep as it usually occurs at sleep onset and as a transitional state across the night. N2 sleep occurs constantly through the night and represents about 45–55 % of sleep. N3 sleep, which represents about 15–25 % of total sleep time, is predominant in the first half of the night. The first cycle of REM sleep usually lasts only a short amount of time and becomes progressively longer at each new sleep cycle. REM sleep can last up to an hour as sleep progresses, amounting to 20–25 % of total sleep time [1].
Substantial changes in cardiovascular function occur during sleep being mediated by the autonomic nervous system. However, it has to be emphasized that cardiovascular activity during sleep may not only be a consequence of sleep mechanisms, but may also reflect the influence of the circadian system [2]. Under normal circumstances, there is a fall in blood pressure and heart rate during NREM sleep as compared to relaxed wakefulness [3]. Blood pressure and heart rate may also be lower in slow wave sleep than stage 2 [4, 5]. During REM sleep, blood pressure undergoes frequent marked oscillations and heart rate becomes very variable. This behavior has led to contradictory results in the literature whenever mean values have been reported. Actually, both blood pressure and heart rate can further decrease during tonic REM sleep as compared to NREM, while they transiently increase to levels similar to wakefulness in association with phasic REM events [6].
2 The Autonomic Nervous System During Sleep
The autonomic nervous system with its two branches, the sympathetic and the parasympathetic systems, working in a delicately tuned, yet opposing fashion, provides adaptation of the cardiovascular system to the various activities of daily life. While the sympathetic system increases heart rate, myocardial contractility, and peripheral resistance, the parasympathetic system slows heart rate with a limited effect on cardiac contractility. It is worth noting that not only the sinus node but also the electrophysiological properties of the entire heart are modulated by these antagonistic influences [7]. The autonomic outflow to the heart and the peripheral circulation is regulated by cardiovascular reflexes, among which the arterial baroreflexes represent the primary mechanism for homeostatic control of arterial pressure and the maintenance of the optimal perfusion of critical organs. The baroreflex control of circulatory homeostasis occurs on a negative feedback basis. Generally, an increase in the loading condition of the baroreceptors by a rise in systemic arterial pressure increases their discharge to the cardiovascular control center in the brainstem that initiates appropriate compensatory responses, including an increase in the vagal outflow and a decrease in the sympathetic outflow to the heart and blood vessels. Conversely, an opposite response is elicited when the arterial baroreceptors are unloaded by a decrease in systemic arterial pressure.
It is worth to underscore that high brain centers continuously modulate baroreceptor responses and cardiovascular reflexes according to behavioral and physiological conditions. In particular, the sleep-dependent changes in cardiovascular function result from the integration between cardiovascular reflexes and central command that are specific to each sleep stage [8].
The first description that sleep alters the function of the arterial baroreceptors (i.e., that during sleep the baroreceptors do not follow the operating logic of wakefulness) dates back to 1969 when Smyth et al. [9] reported the occurrence of a nocturnal reduction of heart rate and arterial pressure as well as an increased gain of the reflex arc in the majority of subjects under study (Fig. 8.1), thereby suggesting that “fluctuations in the level of arousal might in turn alter the activity of baroreceptor reflexes and of other autonomic mechanisms.”

Baroreflex changes with sleep in one subject. Each line represents one baroreflex sensitivity estimation. A average systolic pressure awake, both before and after sleep, S average systolic pressure during slow wave sleep (Reproduced with permission from Ref. [12])
Although the neural mechanisms involved in the complex integration between sleep stages and cardiovascular reflexes are not completely known, the available evidence indicates that NREM sleep is characterized by a progressive decrease in neural sympathetic nerve activity [10] with an accompanying parasympathetic predominance [6, 11], and a downward resetting of the arterial baroreceptor reflex [12, 13]. The relative autonomic stability of NREM sleep is, however, interrupted by those burst of sympathetic nerve activity that are observed on the occurrence of arousals [10]. The resulting phasic increase in arterial pressure does not evoke the bradycardic response which would be expected due to the arterial baroreflex, but rather an increase in heart rate.
A marked sympathetic activation and instability in blood pressure and heart rate, likely of central origin, also characterize phasic REM sleep, during which sudden bursts of sympathetic nerve traffic, about twice the level seen during wakefulness, are documented [10]. As these increases in sympathetic activity are interspersed between the predominantly parasympathetic activity of tonic REM periods, this sleep stage as a whole appears to induce complex fluctuations in autonomic function.
In light of the lengthening of REM cycles as sleep progresses, it has been hypothesized that REM sleep represents a vulnerable state for the cardiovascular system that can set the stage for nocturnal arrhythmias and cardiovascular events [10, 14, 15]. The fluctuations in autonomic activity across sleep stages and their impact on arrhythmia susceptibility may become particularly dangerous in the context of underlying cardiovascular diseases that are often associated with a deranged sympatho-vagal balance [16]. It has been described that after myocardial infarction, the loss in the capability of the vagal activity to physiologically increase during sleep results in a condition of relative sympathetic dominance even during NREM sleep, normally described as a condition at low risk for lethal events [17]. However, it is the sympathetic surge of the normal morning transition from sleep to wakefulness that has been more consistently implicated in the early morning peak in the occurrence of sudden death and implanted defibrillator discharges [18, 19].
3 The Respiratory System During Sleep and Sleep Apnea
Falling asleep removes postural muscle tone, voluntary respiratory control, and the wakefulness stimulus for breathing, leaving metabolic demand as the primary determinant of minute ventilation [20].
The sensors for chemical stimuli include the central and peripheral chemoreceptors. With increasing hypoxemia and hypercapnia, the carotid bodies send afferent impulses to the medullary centers to increase ventilation. Similarly, with hypercapnia, linear changes in ventilation occur in response to central chemoreceptor stimulation. The control of ventilation is organized as a negative feedback system to maintain the PaCO2 at approximately 40 mmHg during wakefulness. Three main factors determine the stability of this control system [21, 22]: controller gain (the ventilatory response to changes in arterial blood gas tensions), plant gain (the responsiveness of changes in arterial blood gases to changes in ventilation), and system delays (the combined effect of circulation time from the lungs to chemoreceptors and of phase shifts resulting from mixing and diffusion processes in the lungs, heart, vasculature, and brain tissues). In general, both the hypoxic and hypercapnic ventilatory responses – that represent the important elements of the controller gain in the respiratory control loop – decline during NREM sleep and decline further with REM sleep [23]. Thus, the transition from wakefulness to NREM sleep is accompanied by a decrease in ventilation (largely through a decrease in tidal volume) associated with an increase in upper airway resistance. As a result, the sleep eupneic PaCO2 increases by 2–7 mmHg, and oxygen saturation decreases by 1–3 %. While reductions in PaCO2 during wakefulness have no or limited effects on the breathing pattern, in NREM sleep small transient reductions in PaCO2 (even only to the waking levels) result in significant apnea [24]. Breathing during REM sleep is characteristically more rapid than during NREM sleep (despite muscle atonia) as a consequence of an excitatory drive to breath. Periods of hyperventilation are interspersed with period of more regular respiration and apneas of varying length.
In most healthy subjects of any age, the loss of wakefulness influences on neurochemical control of breathing and airway patency during sleep is of minor physiological consequences. In some other healthy subjects, and even more in individuals with cardiac abnormalities, these changes predispose to respiratory abnormalities during sleep, generally referred to as sleep-disordered breathing. Commonly, sleep-disordered breathing is divided into “central,” denoting an absence or marked reduction in central motor drive to respiratory muscle and “obstructive” events which are comprised of respiratory efforts against a narrowed or closed upper airway.
4 Sleep Apnea in Heart Failure
Both obstructive sleep apnea (OSA) and central sleep apnea (CSA) are a common comorbidity in patients with heart failure (independently of reduced or preserved left ventricular function) (Figs. 8.2 and 8.3), occurring approximately in half of them despite optimal pharmacological therapy [25–27].

A 3-min polysomnogram of a patient with heart failure revealing three obstructive apneas during stage 2 NREM sleep and REM sleep. Note the futile respiratory efforts associated with hypoxemia (min SpO2, 87 %) terminated by an abrupt arousal. Abbreviations: F3-A2, F4-A1, C3-A2, C4-A1, O1-A2, O2-A1, electroencephalograms, EOG electro-oculogram, EMG chin electromyogram, ECG electrocardiogram, Therm thermistor, Thor thorax, Abd abdomen

A 3-min polysomnogram of a patient with heart failure revealing three central apneas during stage 2 NREM sleep. Abbreviations as in Fig. 8.2
Irrespective of their origin, central or obstructive, such disorders typically manifest as a cyclic waxing and waning of tidal volume [28, 29], a pattern commonly referred to as periodic breathing [28].
Using an apnea-hypopnea index (AHI, the number of apneas or hypopneas per hour of sleep) ≥15/h as a diagnostic threshold, the prevalence of OSA and CSA in patients with chronic stable heart failure and reduced ejection fraction ranges from 15 % to 26 % [25–27] and from 21 % to 38 % [25–27], respectively.
As for the relative distribution of OSA and CSA, data from previous investigations range from 28 % vs. 72 % [25, 26, 30] to 55 % vs. 45 % [27], respectively. However, it has to be underscored that many of these patients experience during the same night varying degrees of both central and obstructive sleep apneas [31]. Compared to patients without sleep apnea or OSA, patients with CSA have a worse hemodynamic profile and higher NYHA class [32].
It is recognized that heart failure contributes to the development of CSA and OSA by several mechanisms. First, in patients with heart failure, the ventilatory control system may become unstable and give rise to self-sustaining ventilatory oscillations owing to a long circulatory delay and an increased loop gain brought about, respectively, by impaired hemodynamics and augmented chemosensitivity [33, 34]. Second, the presence of a narrowed CO2 reserve enhances the susceptibility to central apneas [35]. Third, overnight rostral fluid displacement from the legs may cause fluid accumulation in the neck and lungs, thereby predisposing to the occurrence of OSA (due to the increased collapsibility of upper airways) and CSA (due to an increase in pulmonary congestion leading to worsening of the lung function and to stimulation of pulmonary vagal receptors, both of which would promote ventilatory instability) [36, 37].
The hallmark of sleep-disordered breathing – being central or obstructive in nature – is the triggering of intermittent episodes of hypoxemia and hypercapnia and of arousals which cause fragmentation of physiological sleep [38]. These events take place in repetitive cycles during sleep and induce the activation of a number of pathophysiological consequences including sympathetic activation, endothelial dysfunction, oxidative stress, metabolic dysregulation, and inflammation [39, 40], which have been shown to impact on the clinical outcome [41, 42]. Interestingly, in heart failure patients, disordered breathing also occurs during day time, possibly contributing to poor prognosis [43].
5 Sleep Apnea and the Autonomic Nervous System in Heart Failure
Autonomic cardiovascular control may be importantly implicated as a potential link between sleep-disordered breathing and its pathophysiological consequences. Autonomic responses to apnea are complex and mainly involve chemoreflex and baroreflex regulation.
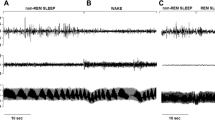
In OSA patients, several studies have demonstrated that recurrent nocturnal apneas are followed by sympathetic activation [44] and impaired baroreflex control [45] that persist during daytime wakefulness [44, 46], thus leading to a chronic dysfunction in autonomic balance (Fig. 8.4). Interestingly, the OSA-induced baroreflex dysfunction can be partly reversed following effective continuous positive airway (CPAP) treatment [47].

Recording of sympathetic neural activity (SNA), respiration (RESP), and intra-arterial blood pressure (BP) in the same subject when awake, with obstructive sleep apnea during REM sleep and with elimination of obstructive apnea by CPAP therapy during REM sleep. SNA is very high during wakefulness, but increases even further secondary to obstructive sleep apnea during REM. Blood pressure increases from 130/65 when awake to 256/110 at the end of the apnea. Elimination of apneas by CPAP results in decreased nerve activity and prevents BP surges during REM sleep (Reproduced with permission from Ref. [44])
It is currently thought that in patients with heart failure, sleep-disordered breathing causes cyclical increases in sympathetic nerve activity which results from the complex interplay of several mechanisms, including: (i) the reflex cardiac response to cyclical stimulation of chemoreceptors by hypoxia and hypercapnia, (ii) central effects of the cyclical increase in inspiratory drive and of arousals from sleep on sympathetic outflow, and (iii) the elimination of reflex inhibition of sympathetic outflow by pulmonary stretch receptors during obstructive apneas [37, 48, 49].
In patients with heart failure, the sympathetic activation that accompanies sleep-disordered breathing may significantly add to the already existing autonomic dysfunction that characterizes heart failure [50], thus contributing to the progression of the disease. Not only awake sympathetic activity – as assessed by microneurography – was significantly higher in patients with heart failure and either obstructive or central sleep apnea [51], but also baroreceptor sensitivity was found to be significantly more depressed when compared to patients with similar cardiac function but without breathing disorders [52].
6 Sleep Apnea and Sudden Death in Heart Failure
It has been previously shown how sleep apnea can be the cause of a derangement in autonomic cardiovascular control resulting in sympathetic overactivity [44, 45]. Moreover, the autonomic effects of arousals that can occur hundreds of times per night in patients with sleep-disordered breathing, also provide the milieu for an increased arrhythmia susceptibility. Clinical data support an increased risk for nocturnal arrhythmia and sudden death in patients with sleep-disordered breathing. In the Sleep Heart Health Study, not only the prevalence of nonsustained ventricular tachycardia was significantly higher in subjects with sleep-disordered breathing as compared to those without [53], but also the relative risk of the occurrence of an arrhythmia was markedly increased within 90 s after a respiratory disturbance in subjects with sleep-disordered breathing [54]. A retrospective analysis on death certificates of patients known to have OSA demonstrated a shift to nighttime (from midnight to 6 am) in the circadian distribution of sudden death as compared to the usual early morning peak of subjects without OSA in the general population [55]. In a large study on 10,701 consecutive adults undergoing a diagnostic polysomnogram who were followed up to 15 years, OSA predicted incident sudden cardiac death, and the magnitude of risk was predicted by multiple parameters characterizing OSA severity [56].
The role of the autonomic nervous system in triggering cardiac arrhythmias might be particularly relevant with regard to arrhythmic events taking place in patients with heart failure and sleep-disordered breathing. Several studies in patients with heart failure reported that the presence of both obstructive and central sleep apnea poses an independent risk factor for the occurrence of malignant arrhythmias documented by implantable cardioverter defibrillator therapies [57, 58]. In a study analyzing the circadian pattern, a striking increase in the incidence of ventricular arrhythmias occurring during the sleep period as opposed to periods of wakefulness was observed in patients with sleep-disordered breathing [59] (Fig. 8.5). Finally, a major impairment of the autonomic control has also been documented in patients with SDB and ventricular arrhythmias suggesting that sympathetic overactivity and vagal withdrawal are important contributing mechanisms [60].

Ventricular arrhythmias in patients with (right) and those without (left) sleep-disordered breathing according to the time of day. Error bars represent mean and 95 % confidence interval. Each circle represents the number of events in one patient. AHI apnea-hypopnea index (Reproduced with permission from Ref. [59])
7 Conclusions
The autonomic nervous system is deeply involved in the mechanisms mediating the cardiovascular effects of sleep within its different stages. Autonomic cardiac control during sleep is characterized by a vagal predominance for most of the time that is interrupted by bursts of sympathetic activity during arousals in NREM sleep and even more during the phasic REM sleep periods. The autonomic fluctuation during sleep associated with the lasting sympathetic activation that occurs in many cardiovascular diseases, particularly heart failure, sets the stage for an increased susceptibility to life-threatening arrhythmias. Sleep apneas, which are a common comorbidity in patients with heart failure, further complicate the picture of autonomic dysregulation and the risk of cardiac events in these subjects by eliciting a further sympathetic activation mostly due to the associated oxygen desaturation and recurrent arousals. Further understanding of the pathophysiological pathways linking sleep disorders to cardiac events is however important to improve clinical management and reduce cardiovascular risk.
References
Carskadon MA, Dement WC. Normal human sleep: an overview. In: Principle and practice of sleep medicine. 5th ed. St Louis: Elsevier Saunders; 2011. p. 17–26.
Trinder J, Waloszek J, Woods MJ, Jordan AS. Sleep and cardiovascular regulation. Pflugers Arch. 2012;463:161–8.
Coote JH. Respiratory and circulatory control during sleep. J Exp Biol. 1982;100:223–44.
George CF, Kryger MH. Sleep and control of heart rate. Clin Chest Med. 1985;6:595–601.
Carrington MJ, Barbieri R, Colrain IM, Crowley KE, Kim Y, Trinder J. Changes in cardiovascular function during the sleep onset period in young adults. J Appl Physiol. 2005;98:468–76.
Zemaityte D, Varoneckas G, Sokolov E. Heart rhythm control during sleep. Psychophysiology. 1984;21:279–89.
Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. 2014;114:1004–21.
Silvani A. Physiological sleep-dependent changes in arterial blood pressure: central autonomic commands and baroreflex control. Clin Exp Pharmacol Physiol. 2008;35:987–94.
Smyth HS, Sleight P, Pickering GW. Reflex regulation of arterial pressure during sleep in man. A quantitative method of assessing baroreflex sensitivity. Circ Res. 1969;24:109–21.
Somers VK, Dyken ME, Mark AL, Abboud FM. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med. 1993;328:303–7.
Furlan R, Guzzetti S, Crivellaro W, Dassi S, Tinelli M, Baselli G, Cerutti S, Lombardi F, Pagani M, Malliani A. Continuous 24-hour assessment of the neural regulation of systemic arterial pressure and RR variabilities in ambulant subjects. Circulation. 1990;81:537–47.
Bristow JD, Honour AJ, Pickering TG, Sleight P. Cardiovascular and respiratory changes during sleep in normal and hypertensive subjects. Cardiovasc Res. 1969;3:476–85.
Parati G, Di Rienzo M, Bertinieri G, Pomidossi G, Casadei R, Groppelli A, Pedotti A, Zanchetti A, Mancia G. Evaluation of the baroreceptor-heart rate reflex by 24-hour intra-arterial blood pressure monitoring in humans. Hypertension. 1988;12:214–22.
Mancia G. Autonomic modulation of the cardiovascular system during sleep. N Engl J Med. 1993;328:347–9.
Verrier RL, Josephson ME. Impact of sleep on arrhythmogenesis. Circ Arrhythm Electrophysiol. 2009;2:450–9.
La Rovere MT, Pinna GD, Hohnloser SH, Marcus FI, Mortara A, Nohara R, ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators, et al. Baroreflex sensitivity and heart rate variability in the identification of patients at risk for life-threatening arrhythmias. Implications for clinical trials. Circulation. 2001;103:2072–7.
Vanoli E, Adamson PB, Ba-Lin, Pinna GD, Lazzara R, Orr WC. Heart rate variability during specific sleep stages. A comparison of healthy subjects with patients after myocardial infarction. Circulation. 1995;91:1918–22.
Willich SN, Goldberg RJ, Maclure M, Perriello L, Muller JE. Increased onset of sudden cardiac death in the first three hours after awakening. Am J Cardiol. 1992;70:65–8.
Englund A, Behrens S, Wegscheider K, Rowland E. Circadian variation of malignant ventricular arrhythmias in patients with ischemic and nonischemic heart disease after cardioverter defibrillator implantation. European 7219 Jewel Investigators. J Am Coll Cardiol. 1999;34:1560–8.
Sowho M, Amatoury J, Kirkness JP, Patil SP. Sleep and respiratory physiology in adults. Clin Chest Med. 2014;35:469–81.
Khoo MC, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol. 1982;53:644–59.
Cherniack NS, Longobardo GS. Mathematical models of periodic breathing and their usefulness in understanding cardiovascular and respiratory disorders. Exp Physiol. 2006;91:295–305.
Kara T, Narkiewicz K, Somers VK. Chemoreflexes-physiology and clinical implications. Acta Physiol Scand. 2003;177:377–84.
Dempsey JA, Smith CA, Przybylowski T, Chenuel B, Xie A, Nakayama H, Skatrud JB. The ventilatory responsiveness to CO(2) below eupnoea as a determinant of ventilatory stability in sleep. J Physiol. 2004;560(Pt 1):1–11.
Oldenburg O, Lamp B, Faber L, Teschler H, Horstkotte D, Topfer V. Sleep-disordered breathing in patients with symptomatic heart failure: a contemporary study of prevalence in and characteristics of 700 patients. Eur J Heart Fail. 2007;9:251–7.
Vazir A, Hastings PC, Dayer M, McIntyre HF, Henein MY, Poole-Wilson PA, Cowie MR, Morrell MJ, Simonds AK. A high prevalence of sleep disordered breathing in men with mild symptomatic chronic heart failure due to left ventricular systolic dysfunction. Eur J Heart Fail. 2007;9:243–50.
Yumino D, Wang H, Floras JS, Newton GE, Mak S, Ruttanaumpawan P, Parker JD, Bradley TD. Prevalence and physiological predictors of sleep apnea in patients with heart failure and systolic dysfunction. J Card Fail. 2009;15:279–85.
Cherniack NS. Apnea and periodic breathing during sleep. N Engl J Med. 1999;341:985–7.
Ryan CM, Bradley TD. Periodicity of obstructive sleep apnea in patients with and without heart failure. Chest. 2005;127:536–42.
Pinna GD, Robbi E, Pizza F, Taurino AE, Pronzato C, La Rovere MT, Maestri R. Can cardiorespiratory polygraphy replace portable polysomnography in the assessment of sleep-disordered breathing in heart failure patients? Sleep Breath. 2014;18:475–82.
Tkacova R, Niroumand M, Lorenzi-Filho G, Bradley TD. Overnight shift from obstructive to central apneas in patients with heart failure: role of PCO2 and circulatory delay. Circulation. 2001;103:238–43.
Solin P, Bergin P, Richardson M, Kaye DM, Walters EH, Naughton MT. Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation. 1999;99:1574–9.
Pinna GD, Maestri R, Mortara A, La Rovere MT, Fanfulla F, Sleight P. Periodic breathing in heart failure patients: testing the hypothesis of instability of the chemoreflex loop. J Appl Physiol. 2000;89:2147–57.
Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–200.
Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med. 2002;165:1245–50.
Yumino D, Redolfi S, Ruttanaumpawan P, Su MC, Smith S, Newton GE, Mak S, Bradley TD. Nocturnal rostral fluid shift: a unifying concept for the pathogenesis of obstructive and central sleep apnea in men with heart failure. Circulation. 2010;121:1598–605.
Kasai T, Arcand J, Allard JP, Mak S, Azevedo ER, Newton GE, Bradley TD. Obstructive sleep apnea and heart failure: pathophysiologic and therapeutic implications. J Am Coll Cardiol. 2011;57:119–27.
Pinna GD, Robbi E, Pizza F, Caporotondi A, La Rovere MT, Maestri R. Sleep-wake fluctuations and respiratory events during Cheyne-Stokes respiration in patients with heart failure. J Sleep Res. 2014;23:347–57.
Sánchez-de-la-Torre M, Campos-Rodriguez F, Barbé F. Obstructive sleep apnoea and cardiovascular disease. Lancet Respir Med. 2013;1:61–72.
Floras JS. Sleep apnea and cardiovascular risk. J Cardiol. 2014;63:3–8.
Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction, and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol. 2007;49:2028–34.
Jilek C, Krenn M, Sebah D, Obermeier R, Braune A, Kehl V, Schroll S, Montalvan S, Riegger GA, Pfeifer M, Arzt M. Prognostic impact of sleep disordered breathing and its treatment in heart failure: an observational study. Eur J Heart Fail. 2011;13:68–75.
La Rovere MT, Pinna GD, Maestri R, Robbi E, Mortara A, Fanfulla F, Febo O, Sleight P. Clinical relevance of short-term day-time breathing disorders in chronic heart failure patients. Eur J Heart Fail. 2007;9:949–54.
Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–904.
Parati G, Di Rienzo M, Bonsignore MR, Insalaco G, Marrone O, Castiglioni P, Bonsignore G, Mancia G. Autonomic cardiac regulation in obstructive sleep apnea syndrome: evidence from spontaneous baroreflex analysis during sleep. J Hypertens. 1997;15:1621–6.
Carlson JT, Hedner JA, Sellgren J, Elam M, Wallin BG. Depressed baroreflex sensitivity in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 1996;154:1490–6.
Bonsignore MR, Parati G, Insalaco G, Marrone O, Castiglioni P, Romano S, Di Rienzo M, Mancia G, Bonsignore G. Continuous positive airway pressure treatment improves baroreflex control of heart rate during sleep in severe obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 2002;166:279–86.
van de Borne P, Oren R, Abouassaly C, Anderson E, Somers VK. Effect of Cheyne-Stokes respiration on muscle sympathetic nerve activity in severe congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1998;81:432–6.
Yumino D, Bradley TD. Central sleep apnea and Cheyne-Stokes respiration. Proc Am Thorac Soc. 2008;5:226–36.
La Rovere MT, Pinna GD, Maestri R, Robbi E, Caporotondi A, Guazzotti G, Sleight P. Prognostic implications of baroreflex sensitivity in heart failure patients in the beta-blocking era. J Am Coll Cardiol. 2009;53:193–9.
Spaak J, Egri ZJ, Kubo T, Yu E, Ando S, Kaneko Y, Usui K, Bradley TD, Floras JS. Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension. 2005;46:1327–32.
Ueno LM, Drager LF, Rodrigues AC, Rondon MU, Mathias Jr W, Krieger EM, Júnior RF, Negrão CE, Lorenzi-Filho G. Day-night pattern of autonomic nervous system modulation in patients with heart failure with and without sleep apnea. Int J Cardiol. 2011;148:53–8.
Mehra R, Benjamin EJ, Shahar E, Gottlieb DJ, Nawabit R, Kirchner HL, Sahadevan J, Redline S. Sleep heart health study. Association of nocturnal arrhythmias with sleep-disordered breathing: the sleep heart health study. Am J Respir Crit Care Med. 2006;173:910–6.
Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med. 2005;352:1206–14.
Gami AS, Olson EJ, Shen WK, Wright RS, Ballman KV, Hodge DO, Herges RM, Howard DE, Somers VK. Obstructive sleep apnea and the risk of sudden cardiac death: a longitudinal study of 10,701 adults. J Am Coll Cardiol. 2013;62:610–6.
Monahan K, Storfer-Isser A, Mehra R, Shahar E, Mittleman M, Rottman J, Punjabi N, Sanders M, Quan SF, Resnick H, Redline S. Triggering of nocturnal arrhythmias by sleep-disordered breathing events. J Am Coll Cardiol. 2009;54:1797–804.
Bitter T, Westerheide N, Prinz C, Hossain MS, Vogt J, Langer C, Horstkotte D, Oldenburg O. Cheyne-Stokes respiration and obstructive sleep apnoea are independent risk factors for malignant ventricular arrhythmias requiring appropriate cardioverter-defibrillator therapies in patients with congestive heart failure. Eur Heart J. 2011;32:61–74.
Kreuz J, Skowasch D, Horlbeck F, Atzinger C, Schrickel JW, Lorenzen H, Nickenig G, Schwab JO. Usefulness of sleep-disordered breathing to predict occurrence of appropriate and inappropriate implantable-cardioverter defibrillator therapy in patients with implantable cardioverter-defibrillator for primary prevention of sudden cardiac death. Am J Cardiol. 2013;111:1319–23.
Zeidan-Shwiri T, Aronson D, Atalla K, Blich M, Suleiman M, Marai I, Gepstein L, Lavie L, Lavie P, Boulos M. Circadian pattern of life-threatening ventricular arrhythmia in patients with sleep-disordered breathing and implantable cardioverter-defibrillators. Heart Rhythm. 2011;8:657–62.
Yamada S, Suzuki H, Kamioka M, Suzuki S, Kamiyama Y, Yoshihisa A, Saitoh S, Takeishi Y. Sleep-disordered breathing increases risk for fatal ventricular arrhythmias in patients with chronic heart failure. Circ J. 2013;77:1466–73.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
La Rovere, M.T., Pinna, G.D. (2016). Whispering During Sleep: Autonomic Signaling During Sleep, Sleep Apnea, and Sudden Death. In: Gronda, E., Vanoli, E., Costea, A. (eds) Heart Failure Management: The Neural Pathways. Springer, Cham. https://doi.org/10.1007/978-3-319-24993-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-24993-3_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24991-9
Online ISBN: 978-3-319-24993-3
eBook Packages: MedicineMedicine (R0)