
Abstract
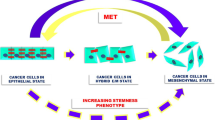
The progression of a cancer cell into a metastatic entity contributes to more than 90 % of cancer related deaths. Therefore, the prevention and treatment of metastasis is an unmet clinical need. Epithelial to mesenchymal transition (EMT) is an evolutionary conserved developmental program, which is induced during cancer progression and contributes to metastatic colonization. EMT endows metastatic properties upon cancer cells by enhancing mobility, invasion, and resistance to apoptotic stimuli. Furthermore, EMT-derived tumor cells acquire stem cell properties and exhibit therapeutic resistance. The disseminated tumor cells recruited to distant organs are suggested to subsequently undergo an EMT reversion through mesenchymal to epithelial transition (MET), necessary for efficient colonization and macrometastasis. A major focus of cancer research is to determine the cellular and molecular mechanisms underlying EMT/MET in tumor invasion, dissemination and metastasis. In this chapter, we will focus on the contribution of the EMT signaling pathways in lung cancer progression, cancer stem cells and acquired resistance to EGFR tyrosine kinase inhibitors and chemotherapy. We will also discuss the potential of targeting EMT pathways as an attractive strategy for the treatment of lung cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Epithelial Mesenchymal Transition in Cancer: Overview
Epithelial–mesenchymal transition (EMT), an evolutionarily conserved process, is essential for embryonic development, gastrulation, neural crest formation, and organ development [1]. EMT has been established as an important step in tissue repair, organ fibrosis, and cancer progression [2–4]. EMT is a dynamic and reversible process, during which epithelial cells transition from polarized, cobblestone-like cells to migratory, spindle-shaped mesenchymal cells. In addition to morphological changes , cells undergoing EMT also exhibit changes at the molecular level by losing expression of epithelial markers such as E-cadherin, ZO-1 and occludin, and gaining expression of mesenchymal markers including N-cadherin, vimentin, and fibronectin . Several signaling pathways regulate EMT including TGFs, BMPs, FGF, EGF, HGF, Wnt/beta-catenin and Notch, in which both transcriptional and post-transcriptional processes are involved [5, 6].
The similarities between transcriptional and epigenetic regulatory pathways in developmental and pathological EMTs suggest that the developmental EMT program is hijacked during tumor invasion and metastasis [3, 7–10]. To identify key EMT molecular pathways that govern the metastatic process, many studies have focused on cell-based experimental models. These studies have shown that EMT confers tumor cells with invasive and metastatic abilities, resistance to therapies, as well as cancer stem cell (CSC) phenotypes that have a major impact on cancer progression [11]. Consistent with the demonstration that EMT activators such as Twist, can induce EMT and breast CSC phenotypes [12, 13], enrichment of CSC/EMT signatures in residual tumors remaining after neoadjuvant chemotherapy was demonstrated [14]. However, a recent study showed that the homeobox factor “paired-related homeobox transcription factor 1” (Prrx1) is an EMT inducer conferring migratory and invasive properties. However, in contrast to other EMT-activators, Prrx1 suppresses CSC phenotypes [15]. This study suggests that unlike the classical EMT transcription factors, Prrx1 contributes to metastasis by uncoupling stemness from EMT.
While cancer cell intrinsic EMT signaling pathways have been well elucidated, the contribution of the tumor microenvironment (TME) in providing EMT activating signals to the cancer cells have only recently been investigated [16, 17]. Several paracrine and autocrine signals trigger induction of EMT resulting in mesenchymal and CSC states in cancer [18–20]. Following EMT, the disseminated mesenchymal cells undergo mesenchymal to epithelial transition (MET) at the site of metastasis [1, 21, 22].
The clinical relevance of EMT has been an area of long standing controversy, mainly due to the lack of evidence of EMT in clinical carcinomas and metastasis [23–26]. More recent efforts have been directed towards demonstrating EMT directly in vivo in mice and humans, and until now the direct role of EMT in vivo has remained elusive. In this chapter, we will focus on EMT in cancer progression, with emphasis on lung cancer, and discuss opportunities for novel anti-EMT therapeutic approaches.
2 Epithelial Mesenchymal Transition in Physiological Processes and Cancer
Three types of EMT have been proposed [27]. Type 1 EMT describes the transition of cells into the mesenchyme during embryogenesis and organ development, and does not involve pathological events [1]. Type 2 EMT is important for wound healing, tissue repair and organ fibrosis, where inflammatory cells produce EMT-inducing factors including TGFβ, PDGF, FGF and Matrix metalloproteinases (MMPs), which induce EMT in normal epithelial cells leading to extensive organ fibrosis [27, 28]. Type 3 EMT is associated with cancer progression and metastases [3, 8, 9]. In addition, following primary EMT, mesenchymal cells are capable of reversing back to epithelial phenotypes through mesenchymal to epithelial transition (MET), which is critical for organ formation including kidney organogenesis and somitogenesis [3, 29]. In cancer, histological analysis has revealed morphological similarities between primary tumors and their metastatic lesions [29], and it has been reported that E-cadherin levels are elevated in lymph node metastases relative to matched primary tumor samples. These data suggest that EMT in primary tumors may be followed by MET at distant metastatic sites [30, 31]. Consistent with these correlative clinical findings, recent studies have demonstrated that re-differentiation of disseminated tumor cells in the metastatic site through MET is critical for colonization [21, 24, 32]. The involvement of EMT in cancer progression is widely recognized; however, the potential role of MET is unclear, and constitutes an area of intense investigation.
3 Epithelial Mesenchymal Transition in Primary Tumor and Metastatic Dissemination
Since the first description of EMT in cancer progression, EMT has been inherently related to metastasis [27, 33]. Accumulating evidence from in vitro experiments have shown that EMT represents a major mechanism for tumor cells to acquire critical metastatic features including enhanced mobility, invasion, and resistance to apoptotic stimuli. Furthermore, as a result of EMT, tumor cells acquire chemo-resistance and exhibit increased potential for initiating secondary tumors [34]. More importantly, EMT has also been implicated in conferring CSC properties [12, 35], a rare subpopulation of cancer cells with capacity of self-renewal, regeneration and differentiation into diverse types of cancer cells.
With the identification of a mesenchymal phenotype in the highly malignant breast CSCs, research focus has recently progressed towards understanding the role of EMT in metastasis in vivo. Using intravital imaging approaches, it was shown that single breast cancer cells gained mobility for hematogenous metastasis by activating EMT-promoting TGFβ-Smad2/3 signaling [36]. Indeed, EMT was also observed during metastasis in spontaneous tumor models in mice, where disseminated tumor cells in the lungs of MMTV-PyMT transgenic mice expressed a mesenchymal marker, FSP1, suggesting involvement of EMT in tumor dissemination [37]. Using a squamous cell carcinoma mouse model, activation of EMT-inducing transcription factor Twist was sufficient to promote carcinoma cells to undergo EMT and disseminate into blood circulation [38]. However, at the distant sites, turning off Twist1 to allow reversion of EMT was essential for disseminated tumor cells to proliferate and form overt metastases. Direct evidence of EMT has also been shown in a K-Ras mediated spontaneous pancreatic tumor model, which develops liver metastases [39]. Remarkably, EMT-positive cells were found in primary lesions, in the circulation, and as single cell deposits in the liver at a very early stage of primary tumor development, even before malignancy could be detected by rigorous histologic analysis. These post-EMT tumor cells gained expression of typical mesenchymal markers including fibronectin, Zeb1 and FSP1 and lost expression of E-cadherin. Importantly, the post-EMT tumor cells represent the majority of metastatic tumor cells that seeded the metastatic liver. However, more rigorous lineage tracing approaches are being developed to actually demonstrate the process of EMT in vivo. For example, using an EMT-lineage tracing strategy of mesenchymal specific (FSP1) Cre mediated β-galactosidase activity, Trimboli et al. compared the incidence of EMT events in three different oncogene-driven breast tumor models [40]. Significantly, post-EMT tumor cells were detected in the Myc-driven tumors, but not in the PyMT- or Neu-driven tumors. Notably, lung metastases were formed in almost all MMTV-PyMT and MMTV-neu mice, but not in MMTV-myc animals, suggesting that the contribution of EMT in metastasis may be tumor type specific. It is also possible that the β-galactosidase activity was not sensitive enough to monitor the relatively rare EMT events, and that better EMT-lineage tracing systems are required to clarify the biological contributions of post-EMT tumor cells in metastasis.
4 Epithelial Mesenchymal Transition in Lung Cancer
Lung cancer is a global public health problem with an estimated 1.3 million new cases each year [41]. In the United States, approximately 226,160 new cases of lung cancer are diagnosed per year with over 160,000 deaths. Despite advances in treatment options, including minimally invasive surgical resection, stereotactic radiation, and novel chemotherapeutic regimens, the 5-year survival rate in NSCLC remains only at approximately 15 %. Available targeted therapies such as EGFR tyrosine kinase inhibitors (TKIs, erlotinib and gefitinib) and EML4-ALK inhibitor (crizotinib) benefit only 15–20 % of NSCLC patients who carry specific drug-sensitive mutations. Even in these patients, acquired resistance is a major impediment to a durable therapeutic response [42–44]. Notably, EMT has been implicated in mediating resistance to therapy in lung cancer. A growing body of evidence supports the role of EMT in the progression of many cancers [2], and transcriptional factors and microRNAs involved in the EMT process have been identified in a number of signaling pathways. However, the role of EMT in lung cancer has not been extensively characterized.
4.1 Epithelial Mesenchymal Transition and Prognosis in Lung Cancer
Several studies have suggested an association between EMT factors including E-cadherin, hypoxia inducible factor 1α (HIF-1α), twist, snail and poor prognosis in lung cancer [45]. Notably, expression of Twist, Slug, and Foxc2 was an independent predictor of recurrence-free and overall survival in stage I NSCLC [46]. Analysis of archived tissue from primary human lung tumors, brain metastases and adjacent bronchial epithelial specimens showed high expression of EMT associated markers in progressing primary lung cancer specimens, particularly in squamous cell carcinoma [47]. Compared to primary NSCLC, brain metastases showed decreased EMT phenotype expression, consistent with the notion that disseminated tumor cells undergo MET at the site of metastasis [1, 21]. It was suggested that overexpression of Forkhead box M1 (FOXM1) , a member of the Fox family of transcriptional factors, may have prognostic value for patients with NSCLC, and FOXM1 was shown to promote metastasis by inducing EMT through activation of the AKT/p70S6K pathway [48].
In NSCLC, invasive tumor growth is accompanied by desmoplastic stroma reaction and concomitant upregulation of EMT markers at the invasive front [49]. Previously, an analysis of surgically resected 533 NSCLC specimens by immunohistochemistry showed that EMT proteins periostin, versican and elastin confer prognostic value [50, 51]. Clinically relevant EMT biomarkers with significant prognostic value in lung adenocarcinoma were identified recently [52]. In this study, analysis of the secretome from a TGF-β induced model of EMT by mass spectrometry unraveled a 97-gene EMT signature with positive correlations to lymph node metastasis, advanced tumor stage and histological grade. Moreover, a refined 20-gene signature predicted survival of both adenocarcinoma and squamous carcinoma patients. Increased expression of BRF2, a RNA polymerase II transcription factor was significantly associated with the poor prognosis of NSCLC patients by virtue of promoting EMT [53]. In another study, downregulation of BRAF activated non-coding RNA promoted EMT, which was associated with poor prognosis in NSCLC [54]. Importantly, in some studies, survival data related to the EMT profile is lacking.
4.2 Epithelial Mesenchymal Transition and Lung Cancer Progression
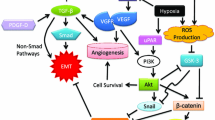
The association of EMT and cancer progression has been shown in several types of cancer, including breast cancer, prostate cancer, pancreatic cancer and hepatocellular carcinoma. However, the role of EMT in lung cancer has not been extensively studied, and the role of EMT in the pathogenesis of several lung disorders is currently intensely debated. More recently, a number of signaling pathways and biomarkers have been implicated in EMT-induced lung cancer progression (Fig. 1).

Schema depicting potential EMT pathways in lung cancer
EMT is orchestrated by several signaling pathways, including TGF-β/Smad and IL-6/JAK/STAT3 (signal transducer and activator of transcription 3) signaling. The JAK/STAT3 pathway was required for TGF-β-induced EMT and cancer cell migration and invasion via upregulation of p-Smad3 and Snail, and the IL-6/JAK/STAT3 and TGF-β/Smad signaling synergistically enhanced EMT in lung carcinomas [55]. In another study, activation of peroxisome proliferator-activated receptor-gamma (PPAR-γ) inhibited TGF-β-induced EMT in lung cancer cells and prevented metastasis by antagonizing Smad3 function [56]. TGF-β1-induced EMT in lung cancer cells resulted in the acquisition of a mesenchymal profile associated with elevated levels of stem cell markers [57–59]. In a related study, TGF-β1-induced EMT in lung cancer cells upregulated Neuropilin (NRP)-2, the high-affinity receptor for SEMA3F [60]. Notably, NRP2 blocked invasive potential of tumor xenografts and reversed TGF-β1-mediated growth inhibition. In NSCLC, Snail was shown to regulate Nanog during EMT via the Smad1/Akt/GSK3β signaling pathway [61].
Notch-1 signaling is critical in lung development and disease [62, 63], and has been shown to promote EMT [64]. It has been demonstrated that blocking Notch-1 signaling by Hey-1 or Jagged1 knockdown or a γ-secretase inhibitor (GSI) attenuates EMT [65]. Radiation-induced Notch-1 overexpression promoted survival and EMT in NSCLC via miR-34a [66]. In this context, induction of miR-34a decreased the expression of Notch-1 and its downstream targets including Hes-1, Cyclin D1, Survivin and Bcl-2 and blocked proliferation and invasion in NSCLC cells [67]. Analysis of the Kras (G12D)-driven NSCLC mouse model showed that conditional Notch1 and Notch2 receptor deletion revealed opposing roles in NSCLC progression [68]. In another study, transcriptional factors Notch2 and Six1 induced EMT and conferred malignant phenotypes to lung adenocarcinomas [69].
MicroRNAs have been shown to contribute to EMT in NSCLC. miR-132 suppressed the migration and invasion of NSCLC cells through targeting ZEB2 [70]. Expression of miR-149, downregulated in lung cancer, was inversely correlated with invasive and EMT phenotypes in NSCLC cells [71]. miR-149 targeted Forkhead box M1 (FOXM1) , and FOXM1 was involved in the EMT induced by TGF-β1. miR-200s have recently been shown to inhibit EMT and promote MET by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2 [72–75]. The observation that the miR-200 family enforces the epithelial phenotype and inhibits EMT and invasion in vitro suggests that these miRNAs are likely to suppress metastasis. Recently, it was shown that, while re-differentiation induced by expression of miR-200 is required for metastatic colonization in a lung tumor xenograft model, miR-200 also directly targets SEC23A, which stimulates the secretion of metastasis-suppressive proteins [32]. Interestingly, cancer cells established from a mouse model of lung adenocarcinoma, driven by oncogenic K-Ras and loss of function p53 mutations, display epithelial plasticity [76], and undergo EMT following TGF-β exposure, which is dependent on downregulation of mir-200 with concomitant stabilization of ZEB1 expression . Ceppi and colleagues have shown that miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype, and that lower mir-200c levels were associated with poor grade of differentiation and higher metastatic potential in NSCLC patients [77]. In another study, immortalized human bronchial epithelial cells (HBECs) exposed to tobacco carcinogens exhibited EMT and stem-like features associated with miR-200 and miR-205. Notably, EMT was driven both by chromatin remodeling and promoter DNA methylation [78]. Some studies have reported conflicting roles of miR-200s in metastatic progression [76, 79, 80], possibly invalidating the therapeutic utility of miR-200s. Furthermore, it remains unclear whether metastasis-related functions of the miR-200s are mediated entirely or only partially through the ZEB–E-cadherin axis.
Osteopontin (OPN) , a prognostic marker in NSCLC [81, 82], through integrin αVβ3, activated the FAK, PI3K, Akt, ERK and NF-kB pathways, contributing to the migration of lung cancer cells [83]. Similarly, a role of pituitary tumor transforming gene (PTTG) , in regulating EMT by inducing expression of integrin αVβ3 and adhesion-complex proteins (FAK) in lung cancer cells was shown [84]. Zyxin was identified as a novel functional target and effector of TGF-β/Smad3 signaling that regulates lung cancer cell motility and EMT via Integrin α5β1 [85].
Inflammation is an important contributor of lung carcinogenesis. The inflammatory component of the TME has been shown to stimulate EMT in lung cancer by contributing to hypoxia, angiogenesis and differential regulation of miRNAs [86, 87]. Paracrine and autocrine contribution of signaling molecules in inducing EMT in lung cancer has been documented. For example, a role for IL-27 in regulating EMT and angiogenesis through modulation of the STAT pathways in human NSCLC was demonstrated [88]. Similarly, COX-2-dependent pathways via modulation of transcriptional repressors of E-cadherin, ZEB1 and Snail regulated EMT in NSCLC [89].
Although association between cigarette smoking and lung cancer is well documented, the molecular mechanisms underlying cigarette smoke-induced EMT processes that are critical for the progression and metastasis of lung cancer are not well understood. Cigarette smoking was shown to induce the repression of E-cadherin via transcription factors LEF-1 and Slug-mediated recruitment of histone deacetylase, HDAC [90]. In another study, cigarette smoke induced EMT through Rac1/Smad2 and Rac1/PI3K/Akt signaling pathways in pulmonary epithelial cells [91].
MMPs that degrade components of the extracellular matrix have been shown to induce EMT. MMP-3, MMP-7, and MMP-28 induce EMT in human A549 lung adenocarcinoma cells [92–94]. Recently MMP-induced upregulation of Rac1b contributed to EMT in a transgenic mice model of lung cancer [95].
4.3 Epithelial Mesenchymal Transition and Drug Resistance in Lung Cancer
Drug resistance constitutes a major challenge for the successful treatment of cancer patients. Cancer therapy is often associated with two major forms of drug resistance—de novo or acquired. Patients who are initially refractory to therapy display intrinsic or “de novo” drug resistance. Patients that initially respond to therapy typically relapse as a consequence of “acquired” drug resistance. EMT has been associated with resistance to chemotherapy, EGFR inhibitors, and other targeted drugs in cancers of the lung [96–98], bladder [99], head and neck [100], pancreas [101], and breast [102]. Intriguingly, EMT can trigger reversion to a CSC-like phenotype [12, 35], providing an association between EMT, CSCs and drug resistance.
In NSCLC , despite the initial response, patients with EGFR-mutant NSCLC eventually develop acquired resistance to EGFR TKIs. The EGFR-T790M secondary mutation is responsible for approximately half of acquired resistance cases, while MET amplification has been associated with acquired resistance in about 5–15 % of NSCLCs [43, 103]. Accumulating evidence suggests that reversible epigenetic changes that emerge during acquired drug resistance reflect changes in the differentiation state of the tumor, which is likely to reflect EMT and the emergence of chemoresistant cells with stem cell-like features [104, 105]. Notably, gefitinib inhibited invasive phenotype and EMT in drug-resistant NSCLC cells with MET amplification [106].
Overcoming de novo and acquired resistance to drug therapy remains a challenge in the clinical management of NSCLC, and approaches to reverse or inhibit EMT as a strategy for drug sensitization are being considered. For example, Buonato and colleagues showed that ERK 1/2 signaling maintained a mesenchymal phenotype in NSCLC cells, and prolonged exposure to MEK or ERK inhibitors restored epithelial phenotypes and overcome resistance to EGFR-targeted therapy [107]. Consistent with these observations, simultaneous EGFR and MEK inhibition are being considered in gastric cancer [108] and pancreatic cancer cells [109], and current clinical trials are evaluating erlotinib combined with MEK inhibitors in NSCLC.
In an attempt to explain resistance to EGFR TKIs, Sordella and colleagues have uncovered the existence of a subpopulation of lung cancer cells that are intrinsically resistant to erlotinib and display EMT phenotypes. These cells by virtue of secreting elevated amounts of TGF-β and IL-6 resisted Tarceva treatment independently of the EGFR pathway [110]. In a previous study, lung adenocarcinomas harboring EGFR mutations were shown to exhibit upregulated IL-6 which activated the gp130/JAK/STAT3 pathway [111]. In this context, Varmus and colleagues showed that inducible expression of EGFR kinase domain–activating mutations targeted to the lung epithelium gave rise to adenocarcinomas containing pSTAT3 and pAKT, demonstrating an association between this oncogene and activated STAT3 [112]. Interestingly, metformin that suppress the IL-6/STAT3 pathway mediated EMT, and sensitized EGFR-TKI-resistant human lung cancer cells to erlotinib or gefitinib [113]. In another study, the expression of Ras-related nuclear protein (Ran) GTPase was elevated in invasive NSCLC. Ran induced EMT and enhanced invasion in NSCLC cells through the activation of PI3K-AKT signaling [114].
In EGFR-TKI resistant lung cancer, activated Notch-1 was found to promote EMT associated with increased Snail and Vimentin expression, suggesting that gefitinib resistance was secondary to Notch-activated EMT [115]. Consistent with this observation, cisplatin was shown to induce the enrichment of multidrug resistant CD133+ CSCs by the activation of Notch signaling [116]. Consistent with this observation, Notch pathway activity identified cells with CSC-like properties and correlated with worse survival in human lung adenocarcinoma [117]. High Notch activity has also been shown to induce radiation resistance in NSCLC [118]. The Hedgehog (Hh) pathway is implicated in lung squamous cell carcinomas (SCC). Notably, activated Hh signaling was shown to regulate metastasis through EMT, and the Shh/Gli pathway was implicated in SCC recurrence, metastasis and resistance to chemotherapy [119]. In NSCLC, TGF-β1-mediated upregulation of shh induced EMT in NSCLC cells [120], and conferred resistance to EGFR-TKIs [121]. Importantly, both genetic and pharmacological inhibition of the Hh pathway reversed the EMT phenotype and improved the therapeutic efficacy of EGFR-TKIs [121].
The miR-134/487b/655 cluster was shown to regulate TGF-β1-induced EMT and induced resistance to gefitinib by targeting MAGI2 (membrane-associated guanylate kinase, WW, and PDZ domain-containing protein 2) in which suppression subsequently caused loss of PTEN stability in lung cancer cells [122].
Platinum-based chemotherapy is the standard first-line approach for the treatment of NSCLC, but recurrence occurs in most patients [123]. Novel combination of chemotherapeutic agents have enhanced the overall median survival of NSCLC patients [124]. However, chemoresistance of tumor cells continues to be a challenge in the management of NSCLCs. Tumor cells often show initial sensitivity to chemotherapeutic drugs, but acquired resistance develops during the treatment, leading to tumor recurrence and further tumor progression. Analysis of cisplatin resistant lung cancer cells showed acquisition of the EMT phenotype, decreased connexin43 (Cx43) expression, and increased capability of invasion and migration [125]. In a related study, resistance of lung cancer cells to docetaxel was associated with EMT, and inhibition of ZEB1 reversed EMT and chemoresistance [126]. Integrinβ1 induced EGFR TKI resistance in NSCLC tumors was associated with an EMT phenotype. [127].
5 Therapeutic Potential of Targeting Epithelial Mesenchymal Transition in Lung Cancer
In lung cancer, EMT has been associated with key tumorigenic properties including increased invasion, angiogenesis and metastasis. Mechanistic insights on how EMT affects signaling pathways contributing to carcinogenesis is necessary to develop effective therapeutics. A number of signaling pathways including notch, wnt, hedgehog and PI3K-AKT, have been implicated in EMT. Furthermore, a growing body of evidence suggests that epithelial cells are more likely to initially respond to therapy, and that EMT confers acquisition of therapeutic resistance. As such, EMT, CSCs, and drug resistance have been described as an emerging axis of evil in cancer [128]. Targeting EMT has been considered a promising strategy against lung cancer, as it would provide novel translational and clinical studies for the benefit of advanced stage cancer patients with metastatic disease [129]. NSCLCs resistant to EGFR TKIs have been shown to downregulate EGFR and increase expression of platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), and AXL [130].
EMT is currently being investigated as a therapeutic target for overcoming drug resistance in lung cancer. For example, HGF-mediated activation of Met receptor induced EMT conferred an aggressive phenotype and induced chemoresistance in preclinical models. Notably, treatment with Met inhibitor resensitized cells to chemotherapy [131]. These findings have clinical relevance, as human NSCLC specimens expressing mesenchymal markers were associated with Met activation, predicted worse survival, and were upregulated in chemorefractory disease . These results support the rationale for Met inhibitor and chemotherapy-centered clinical trials, and suggest that the selection of SCLC patients based on mesenchymal biomarkers in combination with Met expression may be a superior alternative for clinical trials of Met inhibitors plus chemotherapy. Similarly, in drug-resistant NSCLC cells with MET amplification, gefitinib was shown to inhibit invasive phenotype and EMT [106].
ERK1/2 signaling was shown to maintain a mesenchymal phenotype in NSCLC cells associated with resistance to EGFR-TKIs. Prolonged exposure to MEK or ERK inhibitors restored epithelial phenotypes and overcame resistance of NSCLC to EGFR-targeted therapy [107]. For example, combination treatment with gefitinib and MEK inhibitors was effective in the treatment of gefitinib-resistant lung adenocarcinoma cells harboring EGFR mutations [132]. Indeed, current clinical trials have begun to evaluate erlotinib in combination with MEK inhibitors in NSCLC (NCT01229150). Similarly, the IL-6/STAT3 pathway-mediated EMT is also being exploited in EGFR-TKI-resistant NSCLC. Suppression of this pathway with metformin sensitized resistant lung cancer cells to erlotinib or gefitinib [113]. Metformin also inhibited IL-6-induced EMT and lung adenocarcinoma growth and metastasis [133].
A 76-gene EMT signature was found to predict resistance to EGFR and PI3K/Akt inhibitors, and AXL (a member of the RTK family), was identified as a potential therapeutic target for overcoming EGFR inhibitor resistance associated with the mesenchymal phenotype [134]. In this context, activated phospho-AXL was detected in 59.8 % of adenocarcinoma cases examined and correlated significantly with larger tumor size and with overall survival of the patients [135]. A recent study has shown that EMT rewires the mechanism of PI3K pathway activation -dependent proliferation in NSCLC cells [136, 137]. In epithelial cells, autocrine ERBB3 activation maintained PI3K signaling; however EMT altered the proliferative potential of cells by modulating ERBB3 expression.
The CXCR4/CXCL12 axis contributes to the pathology of NSCLC, and targeting this axis has been considered as a potential therapeutic approach for the treatment of NSCLC [138]. Importantly, elevated CXCR4 levels were observed in NSCLC cells high in self-renewal capacity and increased chemotherapeutic resistance [139]. Inhibition of CXCR4 suppressed the self renewal capacity of NSCLC cells [140], and a previous study had shown that the transcription factor 5T4 via CXCR4 may induce EMT and increase migration of NSCLC [141]. The therapeutic potential of CXCR4/CXCL12 axis is being considered for cancer treatment [142–144]. EMT-induced CSC phenotypes have been implicated in resistance to cisplatin, as cisplatin-treated patients with lung cancer showed enrichment of CD133+ stem cells due to activated Notch signaling, suggesting that blocking Notch signaling may reduce the recurrence of NSCLCs [116]. Similarly, the AKT/β-catenin/Snail signaling pathway has been associated with CSC-like properties and EMT features in NSCLC cells, implying the therapeutic potential of this pathway for the treatment of NSCLC [145].
6 Future Perspectives
Current EMT research efforts are directed towards understanding the interplay of multiple regulatory networks that contribute to the conversion of an epithelial tumor cell to a mesenchymal state resulting in acquisition of various acquired capabilities such as resistance to anoikis, oncogene-induced senescence, and resistance to apoptosis/chemotherapy and CSC properties. Various transcriptional and post-transcriptional processes have been identified; however, the mechanisms by which these pathways are interconnected during cancer progression are not completely understood. A variety of contextual paracrine and autocrine signaling factors that maintain mesenchymal and CSC phenotypes have begun to emerge [20], and recent studies have implicated the contribution of chromatin modification as a mechanism to attain widespread changes in gene expression that accompany the EMT process [9]. In lung cancer, EMT is associated with metastatic progression, resistance to EGFR inhibitors, chemotherapy, and other targeted drugs [96–98]. Acquired resistance to the EGFR inhibitor erlotinib resulted from the selection and expansion of a mesenchymal subpopulation [110], and restoring E-cadherin expression in mesenchymal-like NSCLC cells potentiated sensitivity to EGFR inhibitors [146] suggesting that a treatment approach eliciting a mesenchymal to epithelial transition (MET) may be useful for expanding the efficacy of EGFR inhibitors. In addition, growing evidence for AXL-mediated EGFR inhibitor resistance has been linked to EMT [147]. EMT regulators are being considered as potential molecular biomarkers and therapeutic targets for developing multi-targeted strategies for improving current cancer therapies and preventing disease relapse. For example, TGF-β has been shown to induce EMT in NSCLC [57], and clinical benefits of TGF-β signaling inhibitors is being considered [148]. Consistent with this notion, IN-1130, a novel inhibitor of TGF-β type I receptor, was shown to impair breast cancer lung metastasis through inhibition of EMT [149]. Similarly, Wnt signaling has emerged as a critical pathway in lung carcinogenesis, and Wnt pathway antagonists are being explored in NSCLC [150, 151]. Given that EMT contributes to resistance of EGFR-TKIs, inhibition of EMT constitutes a critical therapeutic strategy for overcoming to EGFR-TKis resistance in lung cancer.
Despite the significant and rapid progress in the EMT field, several issues have still remained unresolved. For example, circulating tumor cell (CTC) number in metastatic cancer patients is being considered as prognostic markers consistent with enhanced cell migration and invasion via loss of adhesion, a feature of EMT. Evidence of prognostic significance of CTC number emerged from a study of resectable NSCLC, demonstrating an association between increased CTC number and shorter disease free survival [152]. A hybrid EMT phenotype of CTCs was also demonstrated in patients with metastatic NSCLC [153]. In light of these studies, multiplex analysis and further detailed exploration of metastatic potential and EMT in CTCs is now warranted in a larger patient cohort.
Finally, the role of EMT in cancer progression has been a topic of debate in the scientific community mainly due to paucity of robust in vivo data demonstrating the importance of EMT in tumorigenesis. Furthermore, the clinical relevance of EMT is often questionable due to the lack of evidence of EMT in clinical carcinomas and metastasis [23, 25] [24, 26]. More recent efforts are directed towards EMT demonstration directly in vivo using lineage tracing approaches and live intravital microscopy imaging. These analyses may establish a more direct role of EMT in vivo during tumorigenesis.
References
Lim J, Thiery JP (2012) Epithelial-mesenchymal transitions: insights from development. Development 139:3471–3486
Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW (2007) Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol 213:374–383
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890
Yang J, Weinberg RA (2008) Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14:818–829
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15:178–196
Said NA, Williams ED (2011) Growth factors in induction of epithelial-mesenchymal transition and metastasis. Cells Tissues Organs 193:85–97
De Craene B, Berx G (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13:97–110
Nieto MA (2011) The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol 27:347–376
Tam WL, Weinberg RA (2013) The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 19:1438–1449
Thomson S, Petti F, Sujka-Kwok I, Mercado P, Bean J, Monaghan M, Seymour SL, Argast GM, Epstein DM, Haley JD (2011) A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis 28:137–155
Dave B, Mittal V, Tan NM, Chang JC (2012) Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res 14:202
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3, e2888
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI et al (2009) Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 106:13820–13825
Ocaña OH, Córcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA (2012) Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22:709–724
Gao D, Mittal V (2012) Tumor microenvironment regulates epithelial-mesenchymal transitions in metastasis. Expert Rev Anticancer Ther 12:857–859
Gao D, Vahdat LT, Wong S, Chang JC, Mittal V (2012) Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res 72:4883–4889
Katoh Y, Katoh M (2008) Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int J Mol Med 22:271–275
Moustakas A, Heldin CH (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 98:1512–1520
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL et al (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145:926–940
Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, Sadik H, Argani P, Wagner P, Vahdat LT et al (2012) Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res 72:1384–1394
Sheng W, Wang G, La Pierre DP, Wen J, Deng Z, Wong CK, Lee DY, Yang BB (2006) Versican mediates mesenchymal-epithelial transition. Mol Biol Cell 17:2009–2020
Bastid J (2012) EMT in carcinoma progression and dissemination: facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev 31:277–283
Brabletz T (2012) To differentiate or not–routes towards metastasis. Nat Rev Cancer 12:425–436
Ledford H (2011) Cancer theory faces doubts. Nature 472:273
Tarin D, Thompson EW, Newgreen DF (2005) The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 65:5996–6000, discussion 6000–5991
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428
Zeisberg M, Kalluri R (2013) Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 304:C216–C225
Chaffer CL, Thompson EW, Williams ED (2007) Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 185:7–19
Jeschke U, Mylonas I, Kuhn C, Shabani N, Kunert-Keil C, Schindlbeck C, Gerber B, Friese K (2007) Expression of E-cadherin in human ductal breast cancer carcinoma in situ, invasive carcinomas, their lymph node metastases, their distant metastases, carcinomas with recurrence and in recurrence. Anticancer Res 27:1969–1974
Park D, Kåresen R, Axcrona U, Noren T, Sauer T (2007) Expression pattern of adhesion molecules (E-cadherin, alpha-, beta-, gamma-catenin and claudin-7), their influence on survival in primary breast carcinoma, and their corresponding axillary lymph node metastasis. APMIS 115:52–65
Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y et al (2011) Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17:1101–1108
Scheel C, Weinberg RA (2011) Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer 129:2310–2314
Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C et al (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 69:4116–4124
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265–273
Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E (2009) Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 11:1287–1296
Xue C, Plieth D, Venkov C, Xu C, Neilson EG (2003) The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res 63:3386–3394
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J (2012) Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22:725–736
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH et al (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148:349–361
Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, Creasap N, Rosol TJ, Robinson ML, Eng C et al (2008) Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res 68:937–945
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62:10–29
Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H et al (2010) EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 363:1734–1739
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2, e73
Xiao D, He J (2010) Epithelial mesenchymal transition and lung cancer. J Thorac Dis 2:154–159
Jiang W, Pang XG, Wang Q, Shen YX, Chen XK, Xi JJ (2012) Prognostic role of Twist, Slug, and Foxc2 expression in stage I non-small-cell lung cancer after curative resection. Clin Lung Cancer 13:280–287
Prudkin L, Liu DD, Ozburn NC, Sun M, Behrens C, Tang X, Brown KC, Bekele BN, Moran C, Wistuba II (2009) Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod Pathol 22:668–678
Kong FF, Qu ZQ, Yuan HH, Wang JY, Zhao M, Guo YH, Shi J, Gong XD, Zhu YL, Liu F et al (2014) Overexpression of FOXM1 is associated with EMT and is a predictor of poor prognosis in non-small cell lung cancer. Oncol Rep 31:2660–2668
Soltermann A (2012) Epithelial-mesenchymal transition in non-small cell lung cancer. Pathologe 33(suppl 2):311–317
Morra L, Moch H (2011) Periostin expression and epithelial-mesenchymal transition in cancer: a review and an update. Virchows Arch 459:465–475
Soltermann A, Tischler V, Arbogast S, Braun J, Probst-Hensch N, Weder W, Moch H, Kristiansen G (2008) Prognostic significance of epithelial-mesenchymal and mesenchymal-epithelial transition protein expression in non-small cell lung cancer. Clin Cancer Res 14:7430–7437
Reka AK, Chen G, Jones RC, Amunugama R, Kim S, Karnovsky A, Standiford TJ, Beer DG, Omenn GS, Keshamouni VG (2014) Epithelial-mesenchymal transition-associated secretory phenotype predicts survival in lung cancer patients. Carcinogenesis 35:1292–1300
Tian Y, Lu M, Yue W, Li L, Li S, Gao C, Si L, Qi L, Hu W, Tian H (2014) TFIIB-related factor 2 is associated with poor prognosis of nonsmall cell lung cancer patients through promoting tumor epithelial-mesenchymal transition. Biomed Res Int 2014:530786
Sun M, Liu XH, Wang KM, Nie FQ, Kong R, Yang JS, Xia R, Xu TP, Jin FY, Liu ZJ et al (2014) Downregulation of BRAF activated non-coding RNA is associated with poor prognosis for non-small cell lung cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Mol Cancer 13:68
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu Z, Zhao J, Zhang HT (2014) JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int J Oncol 44:1643–1651
Reka AK, Kurapati H, Narala VR, Bommer G, Chen J, Standiford TJ, Keshamouni VG (2010) Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol Cancer Ther 9:3221–3232
Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N, Rocco G (2011) Epithelial to mesenchymal transition by TGFβ-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One 6, e21548
Pirozzi G, Tirino V, Camerlingo R, La Rocca A, Martucci N, Scognamiglio G, Franco R, Cantile M, Normanno N, Rocco G (2013) Prognostic value of cancer stem cells, epithelial-mesenchymal transition and circulating tumor cells in lung cancer. Oncol Rep 29:1763–1768
Tirino V, Camerlingo R, Bifulco K, Irollo E, Montella R, Paino F, Sessa G, Carriero MV, Normanno N, Rocco G et al (2013) TGF-β1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell Death Dis 4, e620
Nasarre P, Gemmill RM, Potiron VA, Roche J, Lu X, Barón AE, Korch C, Garrett-Mayer E, Lagana A, Howe PH et al (2013) Neuropilin-2 Is upregulated in lung cancer cells during TGF-β1-induced epithelial-mesenchymal transition. Cancer Res 73:7111–7121
Liu CW, Li CH, Peng YJ, Cheng YW, Chen HW, Liao PL, Kang JJ, Yeng MH (2014) Snail regulates Nanog status during the epithelial-mesenchymal transition via the Smad1/Akt/GSK3β signaling pathway in non-small-cell lung cancer. Oncotarget 5:3880–3894
Westhoff B, Colaluca IN, D’Ario G, Donzelli M, Tosoni D, Volorio S, Pelosi G, Spaggiari L, Mazzarol G, Viale G et al (2009) Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A 106:22293–22298
Xu K, Moghal N, Egan SE (2012) Notch signaling in lung development and disease. Adv Exp Med Biol 727:89–98
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008) Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A 105:6392–6397
Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP (2004) Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J 23:1155–1165
Kang J, Kim E, Kim W, Seong KM, Youn H, Kim JW, Kim J, Youn B (2013) Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J Biol Chem 288:27343–27357
Ji X, Wang Z, Geamanu A, Goja A, Sarkar FH, Gupta SV (2012) Delta-tocotrienol suppresses Notch-1 pathway by upregulating miR-34a in nonsmall cell lung cancer cells. Int J Cancer 131:2668–2677
Baumgart A, Mazur PK, Anton M, Rudelius M, Schwamborn K, Feuchtinger A, Behnke K, Walch A, Braren R, Peschel C et al (2015) Opposing role of Notch1 and Notch2 in a Kras(G12D)-driven murine non-small cell lung cancer model. Oncogene 34:578–588
Mimae T, Okada M, Hagiyama M, Miyata Y, Tsutani Y, Inoue T, Murakami Y, Ito A (2012) Upregulation of notch2 and six1 is associated with progression of early-stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clin Cancer Res 18:945–955
You J, Li Y, Fang N, Liu B, Zu L, Chang R, Li X, Zhou Q (2014) MiR-132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS One 9, e91827
Ke Y, Zhao W, Xiong J, Cao R (2013) miR-149 inhibits non-small-cell lung cancer cells EMT by targeting FOXM1. Biochem Res Int 2013:506731
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9:582–589
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914
Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907
Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A et al (2009) Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev 23:2140–2151
Ceppi P, Mudduluru G, Kumarswamy R, Rapa I, Scagliotti GV, Papotti M, Allgayer H (2010) Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol Cancer Res 8:1207–1216
Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA, Tessema M, Leng S, Belinsky SA (2011) EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res 71:3087–3097
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J (2009) miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One 4:e7181
Olson P, Lu J, Zhang H, Shai A, Chun MG, Wang Y, Libutti SK, Nakakura EK, Golub TR, Hanahan D (2009) MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev 23:2152–2165
Boldrini L, Donati V, Dell’Omodarme M, Prati MC, Faviana P, Camacci T, Lucchi M, Mussi A, Santoro M, Basolo F et al (2005) Prognostic significance of osteopontin expression in early-stage non-small-cell lung cancer. Br J Cancer 93:453–457
Donati V, Boldrini L, Dell’Omodarme M, Prati MC, Faviana P, Camacci T, Lucchi M, Mussi A, Santoro M, Basolo F et al (2005) Osteopontin expression and prognostic significance in non-small cell lung cancer. Clin Cancer Res 11:6459–6465
Fong YC, Liu SC, Huang CY, Li TM, Hsu SF, Kao ST, Tsai FJ, Chen WC, Chen CY, Tang CH (2009) Osteopontin increases lung cancer cells migration via activation of the alphavbeta3 integrin/FAK/Akt and NF-kappaB-dependent pathway. Lung Cancer 64:263–270
Shah PP, Fong MY, Kakar SS (2012) PTTG induces EMT through integrin αVβ3-focal adhesion kinase signaling in lung cancer cells. Oncogene 31:3124–3135
Mise N, Savai R, Yu H, Schwarz J, Kaminski N, Eickelberg O (2012) Zyxin is a transforming growth factor-β (TGF-β)/Smad3 target gene that regulates lung cancer cell motility via integrin α5β1. J Biol Chem 287:31393–31405
Heinrich EL, Walser TC, Krysan K, Liclican EL, Grant JL, Rodriguez NL, Dubinett SM (2012) The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron 5:5–18
Krysan K, Lee JM, Dohadwala M, Gardner BK, Reckamp KL, Garon E, St John M, Sharma S, Dubinett SM (2008) Inflammation, epithelial to mesenchymal transition, and epidermal growth factor receptor tyrosine kinase inhibitor resistance. J Thorac Oncol 3:107–110
Kachroo P, Lee MH, Zhang L, Baratelli F, Lee G, Srivastava MK, Wang G, Walser TC, Krysan K, Sharma S et al (2013) IL-27 inhibits epithelial-mesenchymal transition and angiogenic factor production in a STAT1-dominant pathway in human non-small cell lung cancer. J Exp Clin Cancer Res 32:97
Dohadwala M, Yang SC, Luo J, Sharma S, Batra RK, Huang M, Lin Y, Goodglick L, Krysan K, Fishbein MC et al (2006) Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res 66:5338–5345
Nagathihalli NS, Massion PP, Gonzalez AL, Lu P, Datta PK (2012) Smoking induces epithelial-to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin. Mol Cancer Ther 11:2362–2372
Shen HJ, Sun YH, Zhang SJ, Jiang JX, Dong XW, Jia YL, Shen J, Guan Y, Zhang LH, Li FF et al (2014) Cigarette smoke-induced alveolar epithelial-mesenchymal transition is mediated by Rac1 activation. Biochim Biophys Acta 1840:1838–1849
Illman SA, Lehti K, Keski-Oja J, Lohi J (2006) Epilysin (MMP-28) induces TGF-beta mediated epithelial to mesenchymal transition in lung carcinoma cells. J Cell Sci 119:3856–3865
McGuire JK, Li Q, Parks WC (2003) Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol 162:1831–1843
Radisky DC, Przybylo JA (2008) Matrix metalloproteinase-induced fibrosis and malignancy in breast and lung. Proc Am Thorac Soc 5:316–322
Stallings-Mann ML, Waldmann J, Zhang Y, Miller E, Gauthier ML, Visscher DW, Downey GP, Radisky ES, Fields AP, Radisky DC (2012) Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci Transl Med 4:142ra195
Garofalo M, Romano G, Di Leva G, Nuovo G, Jeon YJ, Ngankeu A, Sun J, Lovat F, Alder H, Condorelli G et al (2012) EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med 18:74–82
Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD (2008) Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis 25:843–854
Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S et al (2005) Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 11:8686–8698
Shrader M, Pino MS, Brown G, Black P, Adam L, Bar-Eli M, Dinney CP, McConkey DJ (2007) Molecular correlates of gefitinib responsiveness in human bladder cancer cells. Mol Cancer Ther 6:277–285
Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA, Raben D (2007) Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther 6:1683–1691
Yin T, Wang C, Liu T, Zhao G, Zha Y, Yang M (2007) Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J Surg Res 141:196–203
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC et al (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 100:672–679
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S et al (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104:20932–20937
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA et al (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141:69–80
Voulgari A, Pintzas A (2009) Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta 1796:75–90
La Monica S, Caffarra C, Saccani F, Galvani E, Galetti M, Fumarola C, Bonelli M, Cavazzoni A, Cretella D, Sirangelo R et al (2013) Gefitinib inhibits invasive phenotype and epithelial-mesenchymal transition in drug-resistant NSCLC cells with MET amplification. PLoS One 8, e78656
Buonato JM, Lazzara MJ (2014) ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res 74:309–319
Yoon YK, Kim HP, Han SW, Hur HS, Oh DY, Im SA, Bang YJ, Kim TY (2009) Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Ther 8:2526–2536
Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H (2011) Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res 17:2744–2756
Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V et al (2010) TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A 107:15535–15540
Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B et al (2007) Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 117:3846–3856
Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE (2006) Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 20:1496–1510
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H, Li K, Chen H, Sun F, Yang Z et al (2014) Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res 20:2714–2726
Ning J, Liu W, Zhang J, Lang Y, Xu S (2014) Ran GTPase induces EMT and enhances invasion in non-small cell lung cancer cells through activation of PI3K-AKT pathway. Oncol Res 21:67–72
Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, Zhao LP, Tian Y (2012) Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem 113:1501–1513
Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, Lai TC, Lee CH, Hsiao YW, Lu J et al (2013) Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res 73:406–416
Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS (2013) Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res 19:1972–1980
Theys J, Yahyanejad S, Habets R, Span P, Dubois L, Paesmans K, Kattenbeld B, Cleutjens J, Groot AJ, Schuurbiers OC et al (2013) High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiother Oncol 108:440–445
Yue D, Li H, Che J, Zhang Y, Tseng HH, Jin JQ, Luh TM, Giroux-Leprieur E, Mo M, Zheng Q et al (2014) Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J Exp Clin Cancer Res 33:34
Maitah MY, Ali S, Ahmad A, Gadgeel S, Sarkar FH (2011) Up-regulation of sonic hedgehog contributes to TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS One 6, e16068
Ahmad A, Maitah MY, Ginnebaugh KR, Li Y, Bao B, Gadgeel SM, Sarkar FH (2013) Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J Hematol Oncol 6:77
Kitamura K, Seike M, Okano T, Matsuda K, Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K, Gemma A (2014) MiR-134/487b/655 cluster regulates TGF-β-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther 13:444–453
Thatcher N, Faivre-Finn C, Blackhall F, Anderson H, Lorigan P (2005) Sequential platinum-based chemotherapy-thoracic radiotherapy in early stage non-small cell lung cancer. Clin Cancer Res 11:5051s–5056s
Socinski MA (2004) Clinical issues in the management of non-small-cell lung cancer and the role of platinum-based therapy. Clin Lung Cancer 5:274–289
Yu M, Zhang C, Li L, Dong S, Zhang N, Tong X (2014) Cx43 reverses the resistance of A549 lung adenocarcinoma cells to cisplatin by inhibiting EMT. Oncol Rep 31:2751–2758
Ren J, Chen Y, Song H, Chen L, Wang R (2013) Inhibition of ZEB1 reverses EMT and chemoresistance in docetaxel-resistant human lung adenocarcinoma cell line. J Cell Biochem 114:1395–1403
Ju L, Zhou C (2013) Integrin beta 1 enhances the epithelial-mesenchymal transition in association with gefitinib resistance of non-small cell lung cancer. Cancer Biomark 13:329–336
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29:4741–4751
Ginnebaugh KR, Ahmad A, Sarkar FH (2014) The therapeutic potential of targeting the epithelial-mesenchymal transition in cancer. Expert Opin Ther Targets 18:731–745
Niederst MJ, Engelman JA (2013) Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal 6:re6
Cañadas I, Rojo F, Taus Á, Arpí O, Arumí-Uría M, Pijuan L, Menéndez S, Zazo S, Dómine M, Salido M et al (2014) Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res 20:938–950
Huang MH, Lee JH, Chang YJ, Tsai HH, Lin YL, Lin AM, Yang JC (2013) MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol Oncol 7:112–120
Zhao Z, Cheng X, Wang Y, Han R, Li L, Xiang T, He L, Long H, Zhu B, He Y (2014) Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS One 9, e95884
Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK et al (2013) An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19:279–290
Iida S, Miki Y, Suzuki T, Mori K, Saito M, Niikawa H, Kondo T, Yamada-Okabe H, Sasano H (2014) Activation of AXL and antitumor effects of a monoclonal antibody to AXL in lung adenocarcinoma. Anticancer Res 34:1821–1827
Niederst MJ, Benes CH (2014) EMT twists the road to PI3K. Cancer Discov 4:149–151
Salt MB, Bandyopadhyay S, McCormick F (2014) Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov 4:186–199
Wald O, Shapira OM, Izhar U (2013) CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic roles and therapeutic potential. Theranostics 3:26–33
Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S et al (2009) Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A 106:16281–16286
Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB, Ko YG, Lee JS, Lee SJ, Lee JC, Park MJ (2013) Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 32:209–221
Damelin M, Geles KG, Follettie MT, Yuan P, Baxter M, Golas J, DiJoseph JF, Karnoub M, Huang S, Diesl V et al (2011) Delineation of a cellular hierarchy in lung cancer reveals an oncofetal antigen expressed on tumor-initiating cells. Cancer Res 71:4236–4246
Fahham D, Weiss ID, Abraham M, Beider K, Hanna W, Shlomai Z, Eizenberg O, Zamir G, Izhar U, Shapira OM et al (2012) In vitro and in vivo therapeutic efficacy of CXCR4 antagonist BKT140 against human non-small cell lung cancer. J Thorac Cardiovasc Surg 144:1167–1175.e1161
Otani Y, Kijima T, Kohmo S, Oishi S, Minami T, Nagatomo I, Takahashi R, Hirata H, Suzuki M, Inoue K et al (2012) Suppression of metastases of small cell lung cancer cells in mice by a peptidic CXCR4 inhibitor TF14016. FEBS Lett 586:3639–3644
Peled A, Wald O, Burger J (2012) Development of novel CXCR4-based therapeutics. Expert Opin Investig Drugs 21:341–353
Wang H, Zhang G, Zhang H, Zhang F, Zhou B, Ning F, Wang HS, Cai SH, Du J (2014) Acquisition of epithelial-mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur J Pharmacol 723:156–166
Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M et al (2006) Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 66:944–950
Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK et al (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44:852–860
Nagaraj NS, Datta PK (2010) Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs 19:77–91
Park CY, Min KN, Son JY, Park SY, Nam JS, Kim DK, Sheen YY (2014) An novel inhibitor of TGF-β type I receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition. Cancer Lett 351:72–80
Stewart DJ (2014) Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst 106:djt356
Stewart DJ, Chang DW, Ye Y, Spitz M, Lu C, Shu X, Wampfler JA, Marks RS, Garces YI, Yang P et al (2014) Wnt signaling pathway pharmacogenetics in non-small cell lung cancer. Pharmacogenomics J 14:509–522
Hofman V, Ilie MI, Long E, Selva E, Bonnetaud C, Molina T, Vénissac N, Mouroux J, Vielh P, Hofman P (2011) Detection of circulating tumor cells as a prognostic factor in patients undergoing radical surgery for non-small-cell lung carcinoma: comparison of the efficacy of the Cell Search Assay™ and the isolation by size of epithelial tumor cell method. Int J Cancer 129:1651–1660
Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, Farace F (2011) Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer 105:1338–1341
Acknowledgments
We thank Sharrell Lee for reading the manuscript. VM is supported by NIH grants and by Cornell Center on the Microenvironment and Metastasis through Award Number U54CA143876 from the National Cancer Institute, and the Neuberger Berman Lung Cancer Center. The authors apologize for studies that could not be included due to space limitations.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mittal, V. (2016). Epithelial Mesenchymal Transition in Aggressive Lung Cancers. In: Ahmad, A., Gadgeel, S. (eds) Lung Cancer and Personalized Medicine: Novel Therapies and Clinical Management. Advances in Experimental Medicine and Biology, vol 890. Springer, Cham. https://doi.org/10.1007/978-3-319-24932-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-24932-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24931-5
Online ISBN: 978-3-319-24932-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)