
Abstract
Resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in non small cell lung cancer (NSCLC) is mediated by two major mechanisms namely secondary mutation T790M in EGFR and cMET amplification. Other molecular mediators which contribute towards TKI resistance include the activation of compensatory growth signaling, epithelial mesenchymal transition and microRNAs regulating EGFR and cMET levels. In this chapter, we have included the major mechanisms which contribute towards EGFR TKI resistance in NSCLC. Several therapeutic approaches to overcome TKI resistance are also presented which include second and third generation EGFR TKI inhibitors and cMET inhibitors. Further, the rationale to utilize the combination therapies to simultaneously target EGFR and other major oncogene addictive pathway such as ERBB2 and AXL kinase is outlined. Another promising approach to overcome TKI resistance is to potentiate EGFR protein for degradation. These studies will best be utilized when we can identify the oncogene addictions in an individual patient and tailor the therapy/therapies accordingly for the maximum benefits.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
EGFR Activating (Drug Sensitive) Mutations
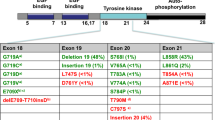
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase which belongs to the EGFR family, consisting of four members: EGFR, ERBB2, ERBB3, and ERBB4. Under physiological conditions, binding of ligands (e.g., epidermal growth factor, transforming growth factor-alpha, amphiregulin) activate the tyrosine kinase activity of EGFR via homo- or heterodimerization with EGFR family members [1]. In non small cell lung cancer (NSCLC), mutations in EGFR occur in exons encoding the ATP-binding pocket of the kinase domain (exons 18–21). In a cohort of nearly 1200 patients harboring EGFR mutations which are linked to clinical outcomes, more than 145 different types of nucleotide changes have been reported within the EGFR kinase domain [2]. However, the most clinically relevant and extensively studied drug-sensitive mutations are deletions in exon 19 that eliminate a common amino acid motif (LREA) and point mutations in exon 21 that lead to a substitution of arginine for leucine at position 858 (L858R). Together, these two classes of mutations account for approximately 85 % of EGFR mutations in NSCLC. They are constitutively active and oncogenic [3, 4] due to the disruption of autoinhibitory interactions in EGFR [5]. Biochemical studies indicate that these mutants preferentially bind to first generation tyrosine kinase inhibitors (TKIs) like gefitinib and erlotinib over ATP, which account for the dramatic response of patients harboring these mutations to TKIs [5, 6]. Other potential drug-sensitive mutations occur at much lower frequency: G719 × (3 %), L861 × (2 %), [2] and exon 19 insertions (1 %) [7]. The former two were associated with drug sensitivity in the original reports on EGFR mutations [8, 9], whereas the exon 19 insertions were recently reported as drug sensitive [7]. The rarity of clinical data associated with these less frequent mutants has made it more difficult to determine how drug sensitive they are in patients, however new data are emerging.
Mechanisms of Resistance to TKIs
T790M Mutation in EGFR
Despite initial response to EGFR first generation TKIs, patients with mutant EGFR NSCLC experience disease progression within 12 months of treatment [10]. The most common mechanism of acquired resistance is the emergence of a secondary mutation in exon 20, T790M, within the catalytic cleft of EGFR. T790M mutations are detected in approximately 50 % of NSCLCs that become resistant to first-generation EGFR TKIs [11]. The T790M mutation was identified in the germline of a family predisposed to NSCLC, indicating an important role in NSCLC genetic susceptibility [12]. An analysis of pretreatment biopsies from NSCLC patients with EGFR mutations who subsequently received erlotinib reported that the incidence of double EGFR mutations (L858R or exon 19 deletion as well as T790M) was 35 % (45 of 129) with no difference in the initial response to erlotinib (63.6 % versus 72.3 %) in patients with or without T790M mutations. However, those patients showed lower progression free survival where T790M mutation was present [13]. These findings suggest that the T790M mutation may be present in some patients prior to TKI therapy and may be selected during therapy because of the treatment resistance associated with the mutation, suggesting the possibility of intrinsic resistance in these patients. Initially, steric hindrance of TKIs by the “gatekeeper” T790M mutation has been hypothesized as the basis for T790M induced TKI resistance. Furthermore, the presence of T790M mutation increases the ATP affinity of the oncogenic L858R mutant by approximately fivefold. Therefore, enhanced ATP affinity reduces the ability of reversible TKIs such as gefitinib and erlotinib to effectively compete with ATP binding. These factors lead to a dramatically reduced potency of TKIs in the setting of the L858R and T790M double mutation [6].
cMET Amplification
Amplification of Hepatocyte growth factor receptor (HGFR/cMET), a receptor tyrosine kinase, was detected in up to 20 % of NSCLC patients that developed acquired resistance to gefitinib or erlotinib. Although, cMET amplification can coexist with the EGFR T790M mutation, approximately 60 % of MET amplification is independent of T790M mutation [14, 15]. cMET amplification was originally identified in a laboratory model of gefitinib resistance using HCC827 human EGFR mutant NSCLC cells. In this gefitinib resistant model, cells with EGFR TKI resistance developed dependency on cMET signaling to activate phospho AKT through ERBB3-mediated activation of PI3K signaling in the presence of EGFR TKIs [15]. Additionally, the cMET ligand hepatocyte growth factor (HGF) is also shown to induce gefitinib resistance through activation of cMET-PI3K signaling [16]. As seen in case of T790M mutations, cMET amplification was also observed at a low frequency in NSCLC patients prior to treatment and was associated with the development of acquired resistance to EGFR TKIs [17]. Together these findings suggest that EGFR TKI treatment may select for preexisting cells with cMET amplification to develop EGFR TKI resistance.
Epithelial Mesenchymal Transition (EMT)
Another mechanism which is shown to confer the resistance to TKI is an increase in EMT in NSCLC [10, 18]. In one study, the erlotinib resistant HCC4006 cells were shown to acquire mesenchymal phenotype and exhibited significant down regulation of E-cadherin [19]. EMT in response to TKI conferring resistance was mediated by TGF Beta and IL-6 axis. Other studies suggest the involvement of ERK2 signaling in TGF Beta mediated EMT in non-transformed cells [20–24]. In these tumors, ERK2 amplification was speculated to be responsible for EMT and TKI resistance [25]. In a recent study, authors utilized tumor xenografts with acquired resistance to erlotinib and found alterations in the expression of several genes that are established biomarkers of erlotinib resistance. For example in resistant tumors, elevated levels of COL6A1 (encoding a type IV collagen), HMGA1 and HMGA2 and reduced levels of keratin genes were found. Importantly, a critical mediator of EMT, AXL kinase was found to be dramatically induced by erlotinib resistance [26]. AXL kinase is a tyrosine protein kinase receptor UFO, which is involved in stimulation of cell proliferation [27]. In the TKI resistant tumors, up regulation of AXL kinase was found to be the second most prevalent mechanism by which resistance occurs followed by T790M mutation. Activation of AXL kinase occurred due to its over-expression as well as up regulation of its ligand GAS6 in the setting of resistant tumors. The up regulation of AXL kinase activity was a part of EMT associated transcriptional program and Vimentin was involved in it. In this study, AXL-overexpressing HCC827 erlotinib resistant cells showed increased migration and adhesion, the properties associated with EMT and the metastatic behavior of tumor cells. These findings are consistent with previous studies showing that over-expression of AXL is associated with increased metastasis in several types of cancers [28]. These findings also advocate that activation of multiple pathways involved in EMT may promote resistance to EGFR TKIs downstream of AXL upregulation. It is well studied that AXL can drive the growth of cancer cells through activation of several oncogenic pathways [28–30]. It will be important to determine the degree to which AXL activation may cooperate with other genetic and genomic alterations to induce resistance to EGFR inhibitors and other molecularly targeted therapies in NSCLC. The loss of E-cadherin can also be mediated by upregulation of Cyclooxygenase 2 (COX-2) metabolite, Prostaglandin E2 (PGE2) [31–33]. PGE2 induces rapid ERK phosphorylayion, reduce E-cadherin levels and upregulate the transcriptional repressors zinc-finger E-box binding homeobox 1 (ZEB-1) and zinc-finger factor Snail homologue 1 (Snail) in NSCLC. COX-2 inhibitors were shown to reverse these effects. Therefore, PGE2 or other inflammatory cytokines in the tumor microenvironment may contribute to EGFR TKI resistance in NSCLC by suppressing E-cadherin expression. These findings also provide a strong rationale for simultaneously targeting EGFR and COX-2 for lung cancer treatment.
Epigenetic Mechanisms
The involvement of specific micro RNAs (miRNAs) in regulating the expression of EGFR and cMET receptor tyrosine kinases and consequently, the metastatic behavior and gefitinib resistance in NSCLC is also reported [34]. miR-221 and miR-222 is shown to be regulated by c-MET and miR-30b, miR-30c, miR-221 and miR222 are regulated by both EGFR and cMET. In response to gefitinib treatment, miR-30b, miR-30c, miR-221 and miR222 get down regulated and consequently, Apoptotic protease activating factor 1 (APAF-1) and B cell lymphoma 2 interacting mediator of cell death (BIM) get up regulated in TKI sensitive cells. In gefitinib resistant cells, the levels of these four miRNAs did not decrease suggesting for their involvement in TKI resistance. Further, cMET inhibitors down regulate miR-30b, miR-30c, miR-221 and miR222 in TKI resistant cells. Taken together, these findings indicate that modulation of miR-30b, miR-30c, miR-221 and miR222 could have therapeutic implications to sensitize TKI resistant tumors. miR-221 and miR-222 has also been shown to regulated Phosphatase and Tensin homolog (PTEN) expression which might contribute towards TKI resistance [35–37]. Loss of PTEN is shown to aberrantly activate EGFR and c-MET signaling.
Targeting the Resistance Mechanisms
Tyrosine Kinase Inhibitors
To overcome the TKI resistance mediated by first generation TKIs, the second and third generation EGFR inhibitors are being developed which are outlined in Table 1.
Second Generations Inhibitors
The development of drugs that bind irreversibly to ERBB family members and/or inhibit multiple targets simultaneously, are being investigated to treat NSCLCs that are resistant to first-generation EGFR TKIs [11]. Unlike reversible TKIs, irreversible TKIs contain a reactive Michael-acceptor group that binds covalently with Cys797 present at the ATP-binding cleft of mutant EGFR. This approach provides greater presence at the ATP site and overcoming the competition with ATP [6, 38]. The ability of an irreversible TKI to overcome resistance was demonstrated in vitro in mutant EGFR cell lines [39]. Several investigational irreversible multitargeted HER family TKIs (Table 1) are being evaluated in patients with NSCLC. These include neratinib or HKI-272 (Wyeth, which was acquired by Pfizer in 2009, New London, CT), PF00299804 (Pfizer), and afatinib or BIBW 2992 (Boehringer Ingelheim, Ingelheim, Germany).
Neratinib (HKI-272)
Neratinib , an irreversible ERBB family inhibitor that targets EGFR/ERBB1, ERBB2, and ERBB4 [40, 41] (Table 1), was evaluated in a phase I trial of patients with advanced solid tumors [42]. Of 14 evaluable patients with NSCLC, stable disease (SD) for 24 weeks was observed in six (43 %) patients. Despite preclinical data suggesting a role for neratinib in overcoming resistance mediated by T790M [39], no patients with a known T790M mutation responded in another study. Based on overall results, neratinib is no longer in development for NSCLC (http://www.clinicaltrials.gov), although it is being investigated in ERBB-2 positive breast cancer [43].
PF00299804
PF00299804, an irreversible ERBB family inhibitor that targets EGFR/ERBB1, ERBB3, and ERBB4 [44] (Table 1), has demonstrated preclinical activity in gefitinib-resistant NSCLC models both in vitro and in vivo [45]. In a phase I/II trial of PF00299804 in patients with NSCLC who progressed following one or two prior chemotherapy regimens and erlotinib [46], 36 patients with adenocarcinoma and five patients with non-adenocarcinoma histology were evaluated for efficacy. Among patients with adenocarcinoma, 67 % had a clinical benefit (response), and among those with non-adenocarcinoma histology, the clinical benefit rate was 40 %.
Afatinib (BIBW 2992)
Afatinib is an oral irreversible ERBB family inhibitor that targets EGFR/ ERBB1, ERBB2 [47], and ERBB4 with preclinical data supporting a role in overcoming resistance to reversible EGFR TKIs [47]. Afatinib has been studied in multiple phase I clinical trials [47–52]. Three patients with NSCLC experienced PRs lasting 24, 18, and 34 months; their tumors were found to have mutations in EGFR, although none had received prior EGFR TKI treatment. Two additional patients (one with NSCLC and one with esophageal cancer) had unconfirmed partial responses (PRs). One of the NSCLC patients with an activating exon 19 mutation who had a PR was initially treated with afatinib (10 mg/day) but subsequently progressed and developed brain metastases. By investigator assessment, the objective relative risk (RR), disease control rate (DCR), median progression free survival (PFS) interval, and median OS time were 60 %, 86 %, 14 months, and 24 months, respectively, for all patients [49]. The objective RR, DCR, and median PFS were 59 %, 83 %, and 16.1 months, respectively, for patients with L858R mutations and 69 %, 93 %, and 13.7 months, respectively, for patients with exon 19 deletions.
Third Generation (T790M EGFR Specific) Tyrosine Kinase Inhibitors
Despite the initial promise, the 4-anilinoquinazoline core structure that is common to the clinically available irreversible inhibitors, the second generation TKIs, do not show specificity towards T790M EGFR compared with wild-type EGFR. New structurally distinct irreversible ERBB family inhibitors, such as the pyrimidine-based inhibitors described were recently introduced by Zhou et al. [53]. This study screened a library of compounds to identify agents that inhibited growth of gefitinib-resistant and gefitinib-sensitive cell lines without producing toxicity in mutant KRAS cells at high concentrations. One such compound, WZ4002, is an irreversible inhibitor with chemical properties that favor 100-fold greater binding to the T790M mutant. WZ4002 also demonstrated a 100-fold weaker binding to wild-type EGFR than with neratinib and other quinazoline-based second generation EGFR inhibitors. Additionally, WZ4002 inhibited L858R/T790M EGFR kinase activity more potently than wild-type EGFR protein activity, whereas the opposite was true for neratinib and gefitinib. Such findings indicate that the concept of irreversible ERBB family inhibition is a very promising and may yet provide a solution to the problem of acquired resistance.
cMET Inhibitors
Multiple agents that inhibit the cMET signaling at various points have been studied. HGF-competitive analogs, such as NK4, have shown inhibitory activity in various cancer cell lines, [54–59]. Other compounds such as decoy cMET and the isolated Sema domain of cMET have the ability to simultaneously bind to both the ligand HGF and the receptor cMET [60, 61]. These agents have shown inhibition of cMET signaling in preclinical studies. Studies with specific antibodies against HGF/cMET have also shown encouraging results. Monoclonal antibodies against cMET, such as OA-5D5 and DN30, have been shown to cause tumor-cell growth inhibition [62–64]. In addition, monoclonal antibodies against HGF have also been developed (L2G7 and AMG102) and validated in several preclinical studies [65, 66]. Another way to inhibit the cMET pathway is through competitors for the ATP binding site in the TK domain of cMET. This type of inhibition is carried by the small molecule inhibitors such as PHA665752, ARQ 197, SGX523, JNJ-38877605, EXEL-2880, XL-184, MGCD265, MK2461, crizotinib (PF-02341066), K252a and MP470 (Table 2). Engelman et al. exposed the gefitinib resistant cells to the cMET inhibitor PHA665752, and restored tumor sensitivity to gefitinib by reducing EGFR phosphorylation and inducing apoptosis [15, 67]. In the model of immnodeficient mice with enhanced tumor growth due to increased production of HGF, the MET-specific small-molecule kinase inhibitor SGX523 partially inhibits the HGF-dependent growth of lung, breast and pancreatic tumors [68]. So far, the most relevant compound developed in this arena is crizotinib (PF02341066), which has been recently approved by the US FDA for the treatment of EML4/ALK mutant NSCLC tumors, targets cMET and ALK receptor TK. In a panel of different tumor cell lines, crizotinib inhibited phosphorylation of wild-type cMET. In lung carcinoma cells, crizotinib inhibited HGF-stimulated cell migration and invasion [69].
Strategic Combination Therapies
In selected cases combination therapies have been utilized, specifically to inhibit the compensatory pathway which is activated and mediates TKI resistance.
Combining EGFR and c-MET Inhibitors
For tumors harboring cMET amplification as a determinant to cause TKI resistance, targeting cMET receptor in combination to EGFR inhibitors are likely to predict a better response compared with individual targeting of EGFR. Antibodies targeting the cMET ligand, antibodies targeting MET itself and small molecules inhibitors against cMET are the therapeutic options for these combination therapies. The simultaneous inhibition of EGFR and cMET was shown to suppress the proliferation of cells and anti-tumor efficacy in mice in HCC827 cells which develop gefitinib resistance [15, 17]. Another study was carried out in NCI-H820 cells which naturally harbor EGFR T790M mutation as well as cMET amplification. In this study, small molecule cMET inhibition or knockdown of cMET along with EGFR inhibition suppressed the compensatory ERBB3 signaling and compromised cell viability [14]. From these studies, it was not clear whether T790M EGFR and cMET amplification co-occur in the same cell and whether the cell type is dependent on both of these factors. To address these issues, Xu et al. carried out a study, where they developed mouse model of adenocarcinoma harboring both T790M EGFR and cMET. In this study treatment with individual inhibitors of EGFR and cMET was un-affective and the combinatorial targeting of both these receptors caused significant tumor regression. Importantly, this study strengthened the notion that in tumors harboring both T790M and cMET amplifications, both these lesions are drivers for growth [70].
EGFR and ERBB2 Inhibitors
The rationale of combining EGFR and ERBB2 inhibitors is via various molecular interactions across their downstream signaling pathways. It is known that the ligand independent activation of EGFR can be mediated by ERBB2 amplification. Over-expression and amplification of ERBB2 decreased the degradation of EGFR and increases its recycling to the cell membrane [71]. For simultaneous targeting of EGFR and ERBB2, two classes of inhibitors have been developed: agents which bind reversibly and those that bind irreversibly (covalently) to the ATP binding site in the tyrosine kinase domain in EGFR and ERBB2. Due to the mechanisms governing the resistance to reversible TKI, irreversible inhibitors targeting both EGFR and ERBB2 are likely to be better therapeutic choices. Irreversible TKI such as BIBW 2992 is shown to inhibit the autophosphorylation of EGFR and ERBB2. This agent was more than 100-fold potent that gefitinib against cells harboring the T790M+L858R mutation [47]. Another irreversible inhibitor HKI-272 caused dramatic tumor regression in mice model of TKI resistance [72]. In a Phase I clinical trial of BIBW2992, out of 26 patients with adenocarcinoma, 2 showed partial response. Another Phase II single arm clinical trial using BIBW2992, recently reported partial response in 43 patients out of 67 in mutation positive patients. The disease control rate was 96 % and a median progression free survival was 10.2 months. A clinical trial utilizing HKI-272 showed stable disease in 42 % of 16 NSCLC patients previously treated with gefitinib.
EGFR and EMT Inhibitors
To overcome the EMT associated with TKI resistance several mediators have been targeted [73]. Due to the role of ERK1/2 in EMT, ERK1/2 blockade by U0126 led to a suppression of TGF-Beta mediated EMT in NSCLC, restored epithelial phenotype of these cells and sensitized the TKI resistant cells in combination with gefitinib [74]. E-cadherin re-expression is also utilized to target EMT in overcoming TKI resistance. ZEB1 is known to down regulate E-cadherin and promote EMT. ZEB1 was shown to be inhibited by utilizing Histone deacetylase inhibitors. In this study, the combination of HDAC inhibitors with erlotinib led to the reversal of TKI resistance in HCC4006ER cells [19]. Since the up regulation of AXL kinase was shown to be a critical determinant of TKI resistance in NSCLC, targeting of AXL kinase sensitized HCC827ER cells. In this study, knockdown of AXL kinase in HCC827 parental cells did not affect survival, however, the knockdown decreased the survival of HCC827ER cells, suggesting the specific role of AXL kinase in mediating erlotinib resistance in these cells. Small molecule inhibitors of AXL kinase, MP-470 and XL-880 [28] in combination with erlotinib also decreased the viability of HCC827ER cells. The process of EMT is also governed by inflammatory signals [75]. The inflammatory enzyme COX-2 is frequently over-expressed in a variety of malignancies [76]. COX2 plays an important role in conferring malignant and metastatic phenotypes and its over-expression is involved in therapy resistance [77]. For instance, COX-2 over-expression in NSCLC is associated with apoptosis resistance, [78] angiogenesis, [79, 80] and metastasis [81, 82]. Most of these effects are mediated by prostaglandin E2 (PGE2). PGE2 and other inflammatory cytokines in the tumor microenvironment may contribute to TKI resistance by downregulating the levels of E-cadherin. These findings suggest a strong rationale of combining COX inhibitors with TKI to overcome TKI resistance [83]. However the clinical trials combining COX2 inhibitors with EGFR inhibitors did not show additional benefits compared with the individual treatment of EGFR inhibitor. It seems possible that in these trials sufficient dose of COX2 inhibitor was not used to inhibit maximum COX2 activity [84, 85].
EGFR and mTOR Pathway Inhibitors
The rationale for combining EGFR inhibitors with mTOR inhibitors was based on studies suggesting an important survival function of mTOR-AKT axis as downstream effector of EGFR signaling [86]. The ability of the irreversible EGFR inhibitor HKI-272 and rapamycin combination to promote more effective suppression of EGFR signaling to S6 and AKT kinases was demonstrated both in cultured NSCLC cell lines harboring double mutant EGFR alleles as well as in lung tumors in TL mice. The investigators suggest that HKI-272 may not sufficiently overcome the biochemical drug resistance conferred by T790M and that further suppression of an essential AKT-mTOR signal downstream of EGFR is required to achieve a therapeutic response [87].
Promoting Oncogene Degradation
An interesting aspect by which TKI resistance has been targeted is via EGFR degradation. It is now well accepted that EGFR degradation is superior in causing cancer cell cyto-toxicity compared to simply inhibiting its tyrosine kinase activity. Weihua et al. reported that knockdown of EGFR caused autophagy and inhibition of EGFR only caused a transient cell cycle arrest [88]. Other findings in head and neck tumor models suggest that promoting EGFR degradation may lead to cell death [89, 90]. Chemotherapeutic agents such as gemcitabine and cisplatin caused EGFR degradation which correlated with cell death. The importance of targeting EGFR for degradation to overcome TKI resistance was shown by knocking down EGFR in several EGFR mutant cell lines. In this study, knockdown of EGFR caused a decrease in survival of EGFR dependent cells including NCI-H1975 cells harboring EGFR-T790M mutation [91]. This approach was utilized recently to develop a peptide based therapy which caused EGFR degradation in TKI resistant cells harboring T790M mutant EGFR. The peptide, named as Disruptin was shown to decrease survival of TKI resistant NCI-H1975 cells by disrupting EGFR-Heat shock protein 90 interaction, inhibiting EGFR homodimerization and promoting EGFR degradation [92].
Conclusions and Future Perspectives
The main signaling pathways contributing towards TKI resistance and the therapeutic approaches to rationally target them are summarized in Fig. 1. In order to better target the EGFR TKI resistance, it will be critical to understand the driver oncogene mediating resistance in a specific patient. The prescreening of patients for the driver mutations and amplifications need to be carried out and the personalized therapies need to be tailored depending upon the driver oncogene. For example, in patients with AXL kinase amplification or COX2 over-expression, AXL or COX2 inhibitors need to be combined with EGFR TKI. Several preclinical studies suggest the re-emission of resistance due to another compensatory mechanism in response to individual therapies. As for instance, in case of irreversible EGFR inhibitors, resistance develops due to downstream signaling mediators. A more effective approach would be to combine the first generation EGFR inhibitor with the second or preferably third generation TKIs. Another promising approach is to target EGFR for degradation, which accounts for inhibition of other functions of EGFR apart from its tyrosine kinase activity which are important for cancer cell survival. These elusive functions of EGFR might be governing the activation of compensatory pathway or downstream signaling mediators imparting TKI resistance. Since the majority of NSCLC patients which harbor TKI sensitive mutations also contain T790M mutation, combining the T790M specific third generation inhibitors in combination with first generation inhibitors as a combined therapy could be a preferred therapeutic choice for these tumors. The Holy Grail to maximize the benefit of each NSCLC patient with EGFR activating mutation is to identify the oncogene addiction/s along with EGFR and provide a tailored combination therapy at the beginning of treatment before the resistance develops.

Major signaling pathways which contribute towards the resistance to EGFR TKIs. In addition to EGFR family members, ERBB2, ERBB3 and ERBB4, AXL and cMET kinases have been reported to mediate EGFR TKI resistance in NSCLC patients. These compensatory pathways can be co-targeted by small molecule inhibitors and antibodies in combination with EGFR TKIs to overcome resistance as shown in the figure
References
Schlessinger J (2002) Ligand-induced, receptor mediated dimerization and activation of EGF receptor. Cell 110:669–672
Horn L, Chen H, Lovly CM et al (2011) DIRECT: DNA-mutation inventory to refine and enhance cancer treatment—a catalogue of clinically relevant somatic mutations in lung cancer. J Clin Oncol 29(suppl; abstr 7575):494s
Greulich H, Chen TH, Feng W et al (2005) Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2, e313
Politi K, Zakowski MF, Fan PD et al (2006) Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 20:1496–1510
Yun CH, Boggon TJ, Li Y et al (2007) Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11:217–227
Yun CH, Mengwasser KE, Toms AV et al (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 105:2070–2075
He M, Capelletti M, Nafa K et al (2012) EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res 18:1790–1797
Lynch TJ, Bell DW, Sordella R et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
Paez JG, Janne PA, Lee JC et al (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Sequist LV, Waltman B, Dias-Santagata D et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3:26–75
Sharma SV, Bell DW, Settleman J et al (2007) Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7:169–181
Bell DW, Gore I, Okimoto RA et al (2005) Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 37:1315–1316
Rosell R, Molina MA, Costa C et al (2011) Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced nonsmall-cell lung cancer patients with EGFR. Clin Cancer Res 17(5):1160–1168
Bean J, Brennan C, Shih JY et al (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104:20932–20937
Engelman JA, Zejnullahu K, Mitsudomi T et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
Yano S, Wang W, Li Q et al (2008) Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 68:9479–9487
Turke AB, Zejnullahu K, Wu YL et al (2010) Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17:77–88
Vuoriluoto K, Haugen H, Kiviluoto S et al (2011) Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 30:1436–1448
Suda K, Tomizawa K, Fujii M et al (2011) Epithelial to mesenchymal transition in an epidermal growth factor receptor–mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol 6:1152–1161
Yao Z, Fenoglio S, Gao DC et al (2010) TGF beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A 107:15535–15540
Grande M, Franzen A, Karlsson JO et al (2002) Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci 115:4227–4236
Xie L, Law BK, Chytil AM et al (2004) Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6:603–610
Zavadil J, Bitzer M, Liang D et al (2001) Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci U S A 98:6686–6691
Shin S, Dimitri CA, Yoon SO et al (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif dependent signaling events. Mol Cell 38:114–127
Ercan D, Xu C, Yanagita M et al (2012) Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov 2:934–947
Zhang Z, Lee JC, Lin L et al (2012) Activation of the AXL kinase causes resistance to EGFR targeted therapy in lung cancer. Nat Genet 44:852–862
Gjerdrum C, Tiron C, Høiby T et al (2010) Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A 107(3):1124–1129
Linger RM, Keating AK, Earp HS et al (2010) Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets 14:1073–1090
Keating AK, Kim GK, Jones AE et al (2010) Inhibition of Mer and Axl receptor tyrosine kinases in astrocytoma cells leads to increased apoptosis and improved chemosensitivity. Mol Cancer Ther 9:1298–1307
Tai KY, Shieh YS, Lee CS et al (2008) Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-κB and Brg-1. Oncogene 27:4044–4055
Charuworn B, Dohadwala M, Krysan K et al (2006) Inflammation-mediated promotion of EMT in NSCLC: IL-1b mediates a MEK/Erk- and JNK/SAPK-dependent downregulation of E-cadherin. Proc Am Thorac Soc 3:D96
Dohadwala M, Yang SC, Luo J et al (2006) Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E2 induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res 66:5338–5345
Krysan K, Lee JM, Dohadwala M et al (2008) Inflammation, epithelial to mesenchymal transition, and epidermal growth factor receptor tyrosine kinase inhibitor resistance. J Thorac Oncol 3:107–110
Garofalo M, Di Leva G, Romano G et al (2009) MiR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 16:498–509
Garofalo M, Romano G, Di Leva G (2012) EGFR and MET receptor tyrosine kinase–altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med 18:74–82
Meng F, Henson R, Wehbe-Janek H et al (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133:647–658
Zhang JG, Wang JJ, Zhao F et al (2010) MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin Chim Acta 411:846–852
Suda K, Onozato R, Yatabe Y et al (2009) EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol 4:1–4
Kwak EL, Sordella R, Bell DW et al (2005) Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 102:7665–7670
Bose P, Ozer H (2009) Neratinib: an oral, irreversible dual EGFR/HER2 inhibitor for breast and nonsmall cell lung cancer. Expert Opin Investig Drugs 18:1735–1751
Rabindran SK, Discafani CM, Rosfjord EC et al (2004) Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res 64:3958–3965
Wong KK, Fracasso PM, Bukowski RM et al (2009) A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res 15:2552–2558
Burstein HJ, Sun Y, Dirix LY et al (2010) Neratinib, can irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol 28:1301–1307
Janne PA, Boss DS, Camidge DR et al (2011) Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin Cancer Res 17:1131–1139
Engelman JA, Zejnullahu K, Gale CM et al (2007) PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 67:11924–11932
Janne PA, Reckamp K, Koczywas M et al (2009) A phase 2 trial of PF-00299804 (PF299), an oral irreversible HER tyrosine kinase inhibitor (TKI), in patients (pts) with advanced NSCLC after failure of prior chemotherapy and erlotinib: preliminary efficacy and safety results. J Thorac Oncol 4(suppl 1):S293–S294
Li D, Ambrogio L, Shimamura T et al (2008) BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27:4702–4711
Eskens FA, Mom CH, Planting AS et al (2008) A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer 98:80–85
Awada A, Dumez H, Wolter P et al (2009) A phase I dose finding study of the 3-day administration of BIBW 2992, an irreversible dual EGFR/HER2 inhibitor, in combination with 3-weekly docetaxel in patients with advanced solid tumors. Presented at the 45th annual meeting of the American Society of Clinical Oncology, Orlando, FL
Stavridi F, Kristeleit R, Forster M (2009) Activity of BIBW 2992, an oral irreversible EGFR/HER2 dual kinase inhibitor, in combination with weekly paclitaxel in non-small cell lung cancer. J Thorac Oncol 4(suppl 1):S444
Vermorken JB, Machiels JH, Rottey S et al (2010) Phase Ib study evaluating the combination of BIBW 2992 with two different standard chemotherapy regimens, cisplatin/paclitaxel (PT) and cisplatin/5-FU (PF), in patients with advanced solid tumors. J Clin Oncol 28, e13521
Yap TA, Vidal L, Adam J et al (2010) Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol 28:3965–3972
Zhou W, Ercan D, Chen L et al (2009) Novel mutant selective EGFR kinase inhibitors against EGFR T790M. Nature 462:1070–1074
Hiscox S, Parr C, Nakamura T et al (2000) Inhibition of HGF/SF induced breast cancer cell motility and invasion by the HGF/SF variant, NK4. Breast Cancer Res Treat 59(3):245–254
Tomioka D, Maehara N, Kuba K et al (2001) Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res 61(20):7518–7524
Wen J, Matsumoto K, Taniura N et al (2004) Hepatic gene expression of NK4, an HGF-antagonist/angiogenesis inhibitor, suppresses liver metastasis and invasive growth of colon cancer in mice. Cancer Gene Ther 11(6):419–430
Heideman DA, van Beusechem VW, Bloemena E et al (2004) Suppression of tumor growth, invasion and angiogenesis of human gastric cancer by adenovirus-mediated expression of NK4. J Gene Med 6(3):317–327
Wen J, Matsumoto K, Taniura N et al (2007) Inhibition of colon cancer growth and metastasis by NK4 gene repetitive delivery in mice. Biochem Biophys Res Commun 358(1):117–123
Suzuki Y, Sakai K, Ueki J et al (2010) Inhibition of Met/HGF receptor and angiogenesis by NK4 leads to suppression of tumor growth and migration in malignant pleural mesothelioma. Int J Cancer 127(8):1948–1957
Michieli P, Mazzone M, Basilico C et al (2004) Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell 6:61–73
Kong-Beltran M, Stamos J, Wickramasinghe D (2004) The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 6:75–84
Martens T, Schmidt NO, Eckerich C et al (2006) A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 12:6144–6152
Jin H, Yang R, Zheng Z et al (2008) MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res 68(11):4360–4368
Petrelli A, Circosta P, Granziero L et al (2006) Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc Natl Acad Sci U S A 103:5090–5095
Kim KJ, Wang L, Su YC et al (2006) Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clin Cancer Res 12:1292–1298
Jun HT, Sun J, Rex K (2007) AMG 102, a fully human anti-hepatocyte growth factor/ scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U-87 MG cells and xenografts. Clin Cancer Res 13:6735–6742
Yoshida T, Okamoto I, Okamoto W et al (2010) Effects of Src inhibitors on cell growth and epidermal growth factor receptor and Met signaling in gefitinib-resistant non-small cell lung cancer cells with acquired Met amplification. Cancer Sci 101(1):167–172
Zhang YW, Staal B, Essenburg C et al (2010) Met kinase inhibitor SGX523 synergizes with epidermal growth factor receptor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth. Cancer Res 70(17):6880–6890
Zou HY, Li Q, Lee JH et al (2007) An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res 67:4408–4417
Xu L, Kikuchi E, Xu C et al (2012) Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res 72(13):3302–3311
Pinkas-Kramarski R, Soussan L, Waterman H et al (1996) Diversification of Neu differentiation factor and epidermal growth factor receptor signaling by combinatorial receptor interactions. EMBO J 15:2452–2467
Ji H, Li D, Chen L et al (2006) The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 9:485–495
Witta SE, Gemmill RM, Hirsch FR et al (2006) Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 66:944–950
Buonato JM, Lazzara MJ (2013) Transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res
Huber MA, Kraut N, Beug H (2005) Molecular requirements for epithelial mesenchymal transition during tumor progression. Curr Opin Cell Biol 17:548–558
Soslow RA, Dannenberg AJ, Rush D et al (2000) COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 89:2637–2645
Riedl K, Krysan K, Pold M et al (2004) Multifaceted roles of cyclooxygenase-2 in lung cancer. Drug Resist Updat 7:169–184
Krysan K, Dalwadi H, Sharma S et al (2004) Cyclooxygenase 2-dependent expression of survivin is critical for apoptosis resistance in non-small cell lung cancer. Cancer Res 64:6359–6362
Pold M, Zhu LX, Sharma S et al (2004) Cyclooxygenase-2-dependent expression of angiogenic cxc chemokines ena-78/cxc ligand (cxcl) 5 and interleukin-8/cxcl8 in human non-small cell lung cancer. Cancer Res 64:1853–1860
Leahy KM, Koki AT, Masferrer JL (2000) Role of cyclooxygenases in angiogenesis. Curr Med Chem 7:1163–1170
Dohadwala M, Luo J, Zhu L et al (2001) Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem 276:20809–20812
Dohadwala M, Batra RK, Luo J et al (2002) Autocrine/paracrine prostaglandin E2 production by non-small cell lung cancer cells regulates matrixmetalloproteinase-2 and CD44 in cyclooxygenase-2-dependent invasion. J Biol Chem 277:50828–50833
Dannenberg AJ, Lippman SM, Mann JR et al (2005) Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol 23:254–266
Gadgeel SM, Ruckdeschel JC, Heath EI et al (2007) Phase II study of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFRTKI), and celecoxib, a cyclooxygenase-2 (COX-2) inhibitor, in patients with platinum refractory non-small cell lung cancer (NSCLC). J Thorac Oncol 2:299–305
O’Byrne KJ, Danson S, Dunlop D et al (2007) Combination therapy with gefitinib and rofecoxib in patients with platinum-pretreated relapsed non small-cell lung cancer. J Clin Oncol 25:3266–3273
Adjei AA (2006) Novel combinations based on epidermal growth factor receptor inhibition. Clin Cancer Res 12:4446–4450
Li D, Shimamura T, Ji H et al (2007) Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell 12(1):81–93
Weihua Z, Tsan R, Huang WC et al (2008) Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 13(5):385–393
Ahsan A, Hiniker SM, Ramanand SG et al (2010) Role of epidermal growth factor receptor degradation in cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res 70(7):2862–2869
Feng FY, Varambally S, Tomlins SA et al (2007) Role of epidermal growth factor receptor degradation in gemcitabine-mediated cytotoxicity. Oncogene 26(23):3431–3439
Chen G, Kronenberger P, Teugels E et al (2012) Targeting the epidermal growth factor receptor in non-small cell lung cancer cells: the effect of combining RNA interference with tyrosine kinase inhibitors or cetuximab. BMC Med 10:28
Ahsan A, Ray D, Ramanand SG et al (2013) Destabilization of the epidermal growth factor receptor (EGFR) by a peptide that inhibits EGFR binding to heat shock protein 90 and receptor dimerization. J Biol Chem 288(37):26879–26886
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ahsan, A. (2016). Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors and Therapeutic Approaches: An Update. In: Ahmad, A., Gadgeel, S. (eds) Lung Cancer and Personalized Medicine. Advances in Experimental Medicine and Biology, vol 893. Springer, Cham. https://doi.org/10.1007/978-3-319-24223-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-24223-1_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24221-7
Online ISBN: 978-3-319-24223-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)