
Abstract
Organic cation transporter OCTs and multidrug and toxin extrusion (MATE) are involved in pharmacokinetics of various drugs. In the renal proximal epithelia, OCT2 mediates uptake of the drugs such as metformin or cimetidine at basolateral membrane, and MATE1 and MATE2-K mediated the secretion of cationic and zwitterionic drugs at brush-border membrane. In the liver, OCT1 are expressed on the sinusoidal membrane and MATE1 are expressed on the canalicular membrane. These transporters mediate the biliary excretion of drugs. The change of these transporter activities, caused by genetic alteration or drug–drug interaction, affected the pharmacokinetics of substrates. Inhibitors of the transporters reduce the biliary or urinary secretion of substrate drugs. In addition, OCTs and MATEs are involved in adverse drug reactions. For example, it was considered that renal toxicities of platinum agents cisplatine or oxaliplatin were affected by substrate specificities of renal OCT2 and MATEs. OCTs and MATEs play important roles for drug efficacies and toxicities especially in the liver and the kidney.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
Membrane transporters play pharmacokinetic important roles in various tissues, such as the brain, intestine, liver and kidney. The liver and kidney are the main organs responsible for the excretion of drugs and their metabolites. Renal excretion consists of glomerular filtration, tubular secretion and re-absorption. Organic cation transporters are involved in tubular secretion [1, 2]. The tubular secretion of drugs is mediated by a two-step membrane transport process [3] involving the basolateral membrane and the brush-border membrane. Organic cation transporters (OCTs) are expressed on the basolateral membranes of proximal tubular cells, and multidrug and toxin extrusion (MATE) proteins are expressed on the brush-border membrane. On the other hand, in the liver, OCTs are expressed on the sinusoidal membrane and MATEs are expressed on the canalicular membrane. These transporters are involved in the biliary excretion of cationic compounds.
General information about OCTs and MATEs was presented in the introduction Chap. 1 by G. Ciarimboli. This chapter highlights the pharmacological and toxicological significance of the organic cation transporters SLC22 (OCTs) and SLC47 (MATEs), specifically in the kidney and the liver. In addition, the membrane transport of victim drugs (substrate) by OCTs and MATEs can be inhibited by perpetrators (inhibitor), which is the one of the main mechanisms of drug–drug interaction in the kidney and the liver.
Pharmacokinetic Significance of Organic Cation Transporter SLC22 (OCTs) [4–14]
The transport characteristics of OCTs are similar in various species. OCT1, 2 and 3 mediated the facilitated transport of a broad range of structurally diverse organic cations and are inhibited by many additional compounds, which are not transported [5, 15]. The molecular size of the typical OCT substrate is under 500, and the OCTs mediate the bidirectional transport of small hydrophilic compounds. OCTs transport organic cations and weak bases, which are positively charged at physiological pH, in an electrogenic manner. In addition to these cations, zwitterionic compounds may also be the substrates transported by OCTs. The transported substrates and non-transported inhibitors of individual OCT transporters overlap broadly (Table 3.1).
OCT1 were originally isolated from rat kidney [50] and using knockout mice [51] it was demonstrated that OCT1 is an influential transporter in the renal secretion of organic cations in rodents. In contrast to rodents, OCT1 expression levels are quite low in the human kidney [52]. There are also species difference in the tissue distribution and expression level of OCT1. One of main organs, in which OCT1 mediates the membrane transports of drugs in humans, is the liver [53]. Here, OCT1 is expressed on the sinusoidal membranes of hepatocytes around the central veins. Therefore, considering its hepatic localization, OCT1 plays pivotal roles in the uptake of drugs by hepatocytes. The substrate specificities of OCTs, including OCT1, are reviewed extensively elsewhere [5]. For several drugs, which inhibit OCTs but are not transported, a highly inhibitory potency for OCT1 was observed compared with hOCT2 or hOCT3. These drugs include the glutamate receptor antagonist phencyclidine, the histamine receptors antagonists diphenylhydramine and ranitidine, the muscarinic acetylcholine receptor antagonist atropine, and the antidepressant desipramine. Some cations that are transported by OCT2 and OCT3 (e.g., epinephrine, norepinephrine, and histamine) are not transported by hOCT1.
OCT2 was cloned after OCT1 from the rat kidney and was later also cloned in human [17, 43, 54]. OCT2 is highly expressed in the rodent and human kidney. In the human kidney in particular, OCT2 is the most highly expressed among cation transporters, and OCT2 protein is expressed in all segments of proximal tubules [52, 55]. Hence, OCT2 is thought to uptake organic cations at the basolateral membrane and play pivotal roles in the tubular secretion of organic cations. OCT2 and OCT1 shares various substrate cations [5]. For example, OCT2 transports MPP, TEA, quinine, and metformin with similar Km values as OCT1 and transports acetylcholine with an approximately fourfold lower Km value compared with OCT1 [5, 15]. Uptake by OCT2 has also been demonstrated for choline; the neurotransmitters dopamine, norepinephrine, epinephrine, serotonin, histamine, agmatine; the glutamate receptor antagonists amantadine and memantine; the histamine H2 receptor antagonists cimetidine, famotidine and ranitidine; the cytostatic cisplatin; and the antihypertensive drug debrisoquine.
In contrast to OCT1 and OCT2, OCT3 is expressed ubiquitously [30, 56]. OCT3 mRNA was detected at high levels in the aorta, skeletal muscle, prostate, adrenal gland, salivary gland, liver, placenta, and fetal lung. OCT3 also transports substrates, such as MPP, with similar Km values as OCT1 and OCT2, whereas a much higher Km value was measured for the translocation of TEA by OCT3 compared with OCT1 and OCT2 [5]. Amantadine, memantine, phenylcyclidine, clonidine, diphenylhydramine, atropine, procainamide and cocaine were observed to inhibit OCT3 with much lower affinity compared to OCT1 and OCT2. High-affinity inhibitors of hOCT3 include disprocynium, decynium, and corticosterone.
Multidrug and Toxin Extrusion (MATE) Proteins [4–6, 57–60]
MATE1 was isolated as a mammalian orthologue of the bacterial multidrug and toxin extrusion family, which confers the multidrug resistance [28]. Human MATE1 and MATE2 were identified in 2005, followed by the isolation of the kidney-specific multidrug and toxin extrusion isoform MATE2-K [21]. In humans, the MATE1 is strongly expressed in liver, kidney and skeletal muscle and was also detected in the heart. [21, 28] MATE1 was localizes to the brush-border membrane of renal proximal tubules in the kidney [21, 55] and to the luminal membranes of bile canalicular epithelial cells in the liver. TEA uptake by MATE1 was independent of the membrane potential and the extracellular sodium concentration. Since MATE1-mediated uptake of TEA was stimulated by opposite-directed proton gradients, MATE1 is supposed to be a H+/cation antiporter as suggested in the TEA uptake by renal brush-border membrane vesicles. TEA uptake by MATE1 is inhibited by a large variety of organic cations such as MPP, serotonin, cimetidine, quinidine, and verapamil, suggesting that, much like OCTs, substrate recognition by MATE1 is polyspecific [22].
MATE2-K is highly expressed in the human kidney and is localized to the brush-border membrane in proximal tubules [21, 55]. When expressed in HEK293 cells, MATE2-K behaves as a H+ gradient-dependent TEA antiporter. MATE2-K is considered to be the active MATE2 variant in the human kidney. MATE2-K is a polyspecific H+/cation antiporter that transports TEA, cimetidine, MPP, procainamide, metformin and NMN, creatinine, guanidine, quinidine, thiamine, and verapamil.
MATE1 and MATE2-K transport typical organic cations such as TEA, cimetidine, MPP, and the antiarrhythmic drug procainamide. All of these compounds were demonstrated to be substrates for H+/organic cation antiporters characterized in brush-border membrane vesicles. Zwitterionic β-lactam antibiotics such as cephalexin and cephradine are effectively transported by MATE1 [32], and the results were also consistent with the transport experiments performed using renal brush-border membrane vesicles. A platinum anticancer agent, oxaliplatin, was a better substrate for MATE2-K rather than for MATE1. The pharmacokinetic and toxicokinetic significance of oxaliplatin transport is discussed below in the section “Renal Toxicity of Platinum Agents”. With a few exceptions, the substrate specificities of MATE1 and MATE2-K are generally similar.
Pharmacokinetic Roles of Organic Cation Transporters in the Liver [5, 53, 61–63]
Many endogenous or exogenous organic compounds, which are positively charged at physiological pH, are handled in the liver. Many of these compounds are highly hydrophilic, and therefore cannot passively diffuse across the plasma membrane. Hence, it has been considered that the membrane transport system is thought to be necessary for the translocation of these compounds. Based on their structural characteristics, organic cations are classified into two categories. Type I organic cations are small and highly hydrophilic, usually below 500 Da. Several quaternary ammonium compounds, such as TEA and MPP+, are considered as typical type I cations. Type II organic cations are bulky and less hydrophilic, and are often polyvalent compounds. d-tubocurarine and quinine are the typical type II cations.
OCT1, as well as other OCT isoforms, translocates organic cations in an electrogenic manner that is independent of Na+ or H+ gradients. OCT1 is localizes to the sinusoidal membrane of bile canalicular epithelial cells, and mediates the uptake of endogenous and xenobiotic cations into hepatocytes, which is the first step in the excretion or detoxification of many drugs. Importantly, OCT1 transports metformin, a biguanide antidiabetic agent that is widely used to treat type 2 diabetes. Metformin is a hydrophilic organic cation and OCT1 is responsible for the uptake of metformin into the hepatocytes. The main pharmacological activity of metformin is to decrease gluconeogenesis in the liver, which reduces blood glucose levels. Therefore, the transport activity of OCT1 in the sinusoidal membrane is important for the clinical efficacy of metformin. Another example of drug transported by OCT1 is lamivudine, a cytidine analog whose active metabolites prevent hepatitis B virus replication in the liver. Lamivudine is efficiently taken into hepatocytes by OCT1.
O-Desmethyltramadol is an opioid analgesic and the main active metabolite of tramadol, which is demethylated by CYP2D6 in the liver. O-Desmethyltramadol, but not tramadol, is transported by OCT1. Volunteers with loss-of-function OCT1 polymorphisms were reported to have significantly higher plasma concentrations of O-desmethyltramadol and significantly prolonged miosis, a surrogate marker of opioidergic effects . Interindividual differences observed in the OCT1 expression and/or activity levels affect pharmacokinetics of O-desmethyltramadol and thus the efficacy of tramadol treatment [62].
The serotonin receptor antagonists, ondansetron and tropisetron, are used to treat nausea and vomiting caused by chemotherapy. These compounds are taken up into hepatocytes by OCT1 and are subsequently metabolized and inactivated by CYP enzymes, mainly CYP2D6. This pathway is a major detoxification route for ondansetron and tropisetron. OCT1 plays a crucial role in the first-pass effect through the liver and hence in the bioavailability of other cationic drugs such as amantadine, levodopa and pramipexole; cimetidine, ciprofloxacin and other fluoroquinolones, furamidine and pentamidine; lamotrigine, sulpiride, and zalcitabine. Most type II organic cations, which are typically hydrophobic, bulky, and polyvalent, such as atropine, decynium-22, prazosin, quinine, and d-tubocurarine, are inhibitors but not substrate of OCT1. Some exceptions include the clinically used type II organic cations quinidine, pancuronium, and rocuronium, which are all transported by OCT1.
MATE1 mediates the excretion of organic cations across the biliary membrane into the bile. MATE1 deficiency in the biliary membrane will results in increased intracellular concentrations of cationic drugs that are substrates of OCT1. This may lead to the increased hepatotoxicity of drugs. Hume et al. developed a new positron emission tomography probe, 11C-labelled metformin, to study hepatobiliary transport mediated by MATE1 [63]. This probe may be useful for non-invasively studies aimed at evaluating the in vivo function of hepatobiliary transport and drug–drug interactions, mediated by MATE1 in clinical investigations. Toyama et al. [64] reported that MATE1 dysfunction caused a marked elevation in the metformin concentration in the liver and led to lactic acidosis, suggesting that this homozygous MATE1 variant could be one of the risk factors for metformin-induced lactic acidosis.
Pharmacokinetic Roles of Organic Cation Transporters in the Kidney [1, 4, 57–59]
The kidney is one of the main organs responsible for the excretion of drugs and xenobiotics . Renal excretion is one of the determinants for the pharmacokinetics of cationic drugs and consists of three steps; glomerular filtration, tubular secretion and re-absorption. The secretion of xenobiotics is an important physiological function of the renal proximal tubules . Drugs are actively secreted via two distinct systems at the brush-border and basolateral membranes of tubular epithelial cells. Transport studies using isolated membrane vesicles and cultured renal epithelial cells characterized two distinct classes of organic cation transporters . Electrogenic transporter is facilitated by an internal negative membrane potential at the basolateral membranes and electroneutral transporter is driven by the transmembrane H+ gradient (H+/organic cation antiporter) at the brush-border membranes, which is dominated by a H+/organic cation antiport process. A prototype substrate, TEA, has been used for the functional characterization of these organic cation transport systems in the kidney. There are two SLC protein families involved in cationic drug secretion, including OCT2 at the basolateral membrane and MATEs at the brush-border membrane. OCT2 and MATEs can transport a variety of structurally unrelated organic cations.
The substrate specificity, membrane localization and driving forces indicated that OCT2 and MATE1 or MATE2-K coordinate the tubular secretion of cationic drugs from blood to urine. Double-transfected Madin-Darby canine kidney cells have been used as an in vitro model for the vectorial transport of cationic drugs across human epithelial cells [65]. In these cells, OCTs are expressed on the basolateral membrane and MATEs are on the apical membrane. Indeed, TEA was transported unidirectionally from the basolateral to apical side of the membrane in these double transfectants. In addition to TEA, the clear directional transport of procainamide and quinidine was also shown in the double transfectants. Procainamide and quinidine are actively secreted via the renal tubules into the urine suggesting that these drugs are substrates of membrane transporters. However, due to the lipophilicity of these compounds, it was difficult to detect the uptake of procainamide and quinidine by OCT- or MATE-expressing cells. Therefore, the double-transfected cells overcome the technical limitations of prior uptake experiments. These cells are convenient for in vitro examination to clarify the renal tubular secretion of cationic drugs in humans. Furthermore, OCT2 and MATEs in the proximal tubule are the sites of clinically important drug–drug interactions. For example, therapeutic doses of cimetidine decrease the renal elimination of procainamide. Double-transfected cells are thus also useful for the examining of drug–drug interactions. Details of drug–drug interactions are described below in section “Drug–Drug Interactions of Cationic Compounds in the Kidney”.
OCT2 may be involved in the renal excretion of a variety of drugs , such as the neurotransmitters dopamine, epinephrine and serotonin; agonists and antagonists of various receptors; various blockers of ion channels and transporters; and various other drugs, including a variety of psychoactive compounds. For example, OCT2 is important for the renal excretion of metformin, a biguanide that is used in the treatment of type-II diabetes and in polycystic ovary syndrome. Metformin is almost entirely excreted into the urine without being metabolized, but a portion of metformin is also excreted in bile. Metformin is mainly eliminated by glomerular filtration and tubular secretion, and OCT2, MATE1 and MATE2-K are involved in the tubular secretion of metformin. Lactic acidosis is a fatal adverse effect of metformin and its occurrence cannot be predicted in patients presymptomatically. Decreased renal excretion of metformin may lead to increased plasma levels. Increased concentration of metformin in the liver may lead to the excessive inhibition of mitochondrial respiratory enzymes and may cause lactic acidosis. Reduced OCT2 and/or MATEs activity in vivo, for example, through inhibition by the concomitant drugs or through renal impairments, may increase the risk of lactic acidosis.
To clarify the pharmacokinetic role of MATE1 in vivo, Tsuda et al. carried out the targeted disruption of the murine Mate1 gene [66]. After a single intravenous administration of metformin, the area under the blood concentration-time curve of metformin in Mate1-knockout mice showed a twofold increase. The urinary excretion of metformin after intravenous administration was also significantly decreased in Mate1-knockout mice. The report demonstrated an essential role for MATE1 in the systemic clearance of metformin . [66] Toyama et al. reported that significantly higher blood lactate levels and lower pH and HCO3- levels were observed in Mate1-knockout mice 7 days after metformin administration in the drinking water [64, 67]. In the knockout mice, dysfunctional Mate1 caused a remarkable elevation in the concentration of metformin in the liver and led to lactic acidosis. These results suggested that homozygous but not heterozygous MATE1 variants are risk factors for metformin-induced lactic acidosis.
Choi et al. characterized genetic variants of MATE2-K and determined their association with the response to metformin. [68] Four nonsynonymous variants and four variants in the MATE2-K basal promoter region were identified from ethnically diverse populations. Two nonsynonymous variants, including c.485C>T (Pro162Leu) and c.1177G>A (Gly393Arg), were shown to be associated with significantly lower metformin uptake and a reduction in protein levels when expressed in HEK293 cells. MATE2-K basal promoter haplotypes containing the most common variant, g.−130G>A (>26 % allele frequency), were associated with significantly increase in promoter activity and reduced binding to the transcriptional repressor myeloid zinc finger 1. Diabetic patients who were homozygous for g.−130A had significantly poorer responses to metformin treatment than carriers of the reference allele when assessed for the relative change in glycated hemoglobin (HbA1c). Choi et al. suggested that MATE2-K plays a role in the antidiabetic response to metformin and that the next challenge in pharmacogenomic research is to improve the outcome for patients through this pathway.
Tzvetkov et al. examined the effects of genetic polymorphisms in OCT1, OCT2, OCT3, OCTN1, and MATE1 on the pharmacokinetics of metformin in healthy male Caucasians [69]. Low-activity genotypes of OCT1 were related to an increase in the renal clearance of metformin. It is possible that OCT1 dysfunction indirectly affects the renal clearance of organic cations in humans.
Drug–Drug Interactions of Cationic Compounds in the Kidney [23, 39, 65, 70]
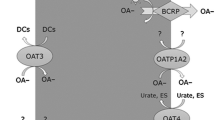
Drug–drug interactions are among the serious disturbances that render pharmacotherapies unsuccessful. In renal excretion, cationic perpetrators can reduce the renal secretion of victim drugs, resulting in severe adverse reactions due to elevations in the plasma concentrations. OCTs and MATEs are independent sites for drug–drug interactions between cationic drugs (Fig. 3.1). Perpetrator drugs exhibit considerable differences in the inhibitory potency with respect to MATE and OCT2 (Table 3.1). For example, pyrimethamine markedly inhibits MATEs, whereas it negligibly inhibited OCT transport activity at its clinically relevant dose.

Scheme of drug–drug interaction at renal proximal epithelia
Cimetidine has been well known to reduce the renal excretion of other cationic drugs. For example, the excretion of procainamide was inhibited by concomitant cimetidine administration. The drug interactions between cimetidine and metformin were assessed using double-transfected kidney cells that stably expressed OCT2 and MATE1 as an in vitro model of proximal tubular epithelial cells [20]. The results from the investigation of the blood concentrations at clinical doses suggested that apical MATE1 is the site of drug interactions whereby cimetidine inhibits other cationic compounds in proximal tubular epithelial cells in clinical situations. Similar conclusions were reached by other groups. Recently, Ito et al. concluded that competitive OCT2 inhibition is unlikely to underlie the drug–drug interaction caused by cimetidine in the renal elimination of cationic drugs and that the competitive inhibition of MATEs by cimetidine is likely to be important in vivo at clinical doses [23, 70]. The conclusion based on the reason that cimetidine interacts with higher affinity with MATEs than with OCT1.
Kusuhara et al. conducted a microdose study of metformin to investigate the predictability of drug–drug interactions at the therapeutic doses [39]. Healthy subjects received microdoses and therapeutic doses of metformin, along with a potent and competitive MATE inhibitor, pyrimethamine, in a crossover fashion. Pyrimethamine significantly reduced the renal clearance of metformin and caused significant increases in the plasma concentrations of metformin. Pyrimethamine also increased the plasma concentration of creatinine, which is an endogenous substrate secreted by proximal tubules. They considered that the drug–drug interaction was attributed to the inhibition of MATE proteins by pyrimethamine. Pyrimethamine is considered to be a useful tool as an in vivo inhibitor of MATE proteins. Ito et al. reported the usefulness of N-methylnicotinamide as an endogeneous probe for evaluating drug–drug interactions involving MATE1 and MATE2-K. NMN is an endogenous substrate of MATE1 and MATE2-K, as well as OCT2. NMN uptake by human brush-border membrane vesicles with proton gradients was reported to be saturable and completely inhibited by low concentrations of pyrimethamine. NMN has been suggested for use as an internal probe to evaluate drug–drug interactions in renal tubular secretion.
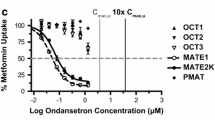
Minematu and Giacomini reported interactions of tyrosine kinase inhibitors with OCTs and MATEs [48]. In their report, IC50 values were estimated for eight TKIs (imatinib, dasatinib, nilotinib, gefitinib, erlotinib, sunitinib, lapatinib, and sorafenib) on metformin transport by OCT1, OCT2, OCT3, MATE1 and MATE2-K. The estimated IC50 values were comparable to the maximum clinical concentrations of unbound TKIs in plasma. Imatinib, nilotinib, gefitinib, and erlotinib exerted selectively potent inhibitory effects on MATE1, OCT3, MATE2-K, and OCT1, respectively. Major metabolites of several TKIs showed IC50 values similar to those for unmodified TKIs. TKIs may therefore possibly affect the disposition, efficacy, and toxicity of drugs that are substrates of these transporters.
The International Transporter Consortium (ITC), which includes members from academia, industry, and the US Food and Drug Administration (FDA), was formed in 2007. The ITC aimed to determine which transporters are determinants of pharmacokinetics, to discuss method for characterizing drug-transporter interactions, and to propose preclinical and clinical studies of transporter-mediated drug interactions for drug development. In 2010, the ITC published a white paper and the white paper identified seven transporters involved in clinical drug–drug interactions. OCTs were included among these transporters of white paper. The white paper recommended that these transporters should be studied in vitro to determine the potential of clinical drug–drug interactions. The European Medicines Agency also included recommendations on transporters in its published drug interaction guidelines. Based on the second ITC transporter workshop in 2012, the ITC identified additional transporters of emerging importance in pharmacokinetics, drug interference with the transport of endogenous compounds, and drug–drug interactions in humans. MATEs were added because of their importance in the excretion of organic cations into bile and urine. A comparison of the concentration of an inhibitor and its inhibitory potency toward a transporter predicts the likelihood of clinical drug–drug interactions [71, 72].
Renal Toxicity of Platinum Agents [58, 73, 74]
Due to the vast renal blood flow and accumulation via uptake mechanisms, the kidney experiences far greater exposure to the xenobiotics than other organs. Therefore, the cytotoxic effects of these drugs easily damage the renal cells. Platinum-based drugs are anticancer agents and are used individually or in combination with other antitumor and/or radiation therapies for the many human malignancies. Platinum-based chemotherapies have widely been used to treat solid tumors since the 1970s. However, renal impairment induced by cisplatin , the first of these compounds and typical platinum antitumor agent, is severe and limits cisplatin based chemotherapy.
Cisplatin, carboplatin, oxaliplatin, and nedaplatin are currently used to treat solid tumors. Of these drugs, only cisplatin induces nephrotoxicity with a higher accumulation in the kidney. Many groups have reported differential transport of platinum compounds by OCT2 [75]. OCT2 significantly increases the accumulation of oxaliplatin, ormaplatin, tetraplatin, transplatin and cisplatin but not carboplatin and nedaplatin [36]. Furthermore, OCT2-mediated oxaliplatin and cisplatin accumulation was time and concentration dependent, and OCT2 expression enhanced the sensitivity to oxaliplatin and cisplatin cytotoxicity. Many studies indicated that a kidney-specific OCT2 was the determining factor in cisplatin-induced nephrotoxicity, mediating the renal uptake of cisplatin. In contrast, the low-nephrotoxic platinum agents, carboplatin and nedaplatin, are not transported by OCT2. However, whereas oxaliplatin was revealed to be a good substrate for OCT2, it is not nephrotoxic. MATEs have been postulated to protect against oxaliplatin-induced nephrotoxicity by mediating the efflux of this agent from intracellular compartments. Marked transport of oxaliplatin by MATE2-K has been observed. MATE2-K may mediate the efflux of oxaliplatin from renal epithelial cells and protect these cells from platinum toxicity. These results clearly demonstrate the relationship between the renal pharmacokinetics and nephrotoxicity of platinum agents.
The nephrotoxicity of platinum agents is related to MATE and OCT (Fig. 3.2) [58]. Nakamura et al. investigated the role of MATE1 in the nephrotoxicity of cisplatin both in vivo and in vitro [74]. The intraperitoneal administration of cisplatin significantly reduced lifespan of Mate1-knockout mice compared with wild-type mice. The plasma concentration and renal accumulation of cisplatin were also higher in the knockout mice . Furthermore, the combination of pyrimethamine with cisplatin elevated serum creatinine and BUN levels. MATE1 is thus thought to mediate the translocation of cisplatin and is involved in attenuating cisplatin-induced nephrotoxicity via disposition of cisplatin.

Renal transport of platinum agents at renal proximal tubules
Li et al. also reported corresponding results showing that ondansetron enhanced cisplatin-induced nephrotoxicity by inhibiting MATEs as well as pyrimethamine [42]. In cells that stably express OCT2 and MATEs, ondansetron was shown to inhibit both OCT2 and MATEs. Ondansetron significantly increased the renal accumulation of cisplatin and induced more severe pathohistological damage . Increased serum levels of creatinine and BUN, as well as changes in two molecular biomarkers of kidney injury, were indicative of cisplatin-induced nephrotoxicity in ondansetron-treated mice. Therefore, the potent inhibition of MATEs likely enhances the nephrotoxicity associated with cisplatin treatment. The potential nephrotoxic effects of combining the chemotherapeutic cisplatin with MATE inhibitors such as the antiemetic 5-hydroxytryptamine-3 receptor antagonists, should be investigated in patients.
However, whether the efficacy and/or toxicity of platinum agents are influenced by OCT or MATE transport activity in vivo has remained controversial. OCT2 mRNA expression in clinical ovarian cancer specimens was low and was not correlated with the treatment outcomes of platinum-based regimens. OCT2 is a critical determinant in the uptake and cytotoxicity of various platinum compounds, particularly oxaliplatin in vitro. However, the effects of OCT2 expression on the results of chemotherapy should be carefully considered. Sprowl et al. reported the influence of the OCT2 inhibitor cimetidine on the antitumor efficacy and systemic clearance of cisplatin [76]. In their reports, cimetidine affected the uptake and cytotoxicity of cisplatin in cultured ovarian cancer cells with highly OCT2 expression. In contrast, the antitumor efficacy of cisplatin in mice bearing luciferase-tagged IGROV-1 xenografts was unaffected by cimetidine. Data obtained from 18 patients receiving cisplatin in a randomized crossover study with or without cimetidine revealed that cimetidine did not alter exposure to unbound cisplatin, a marker of antitumor efficacy. Iwata et al. reported the effects of genetic variants of OCT2 and MATE1 on cisplatin-induced adverse events in patients [77]. They concluded that the 808G>T SNP in OCT2 ameliorated CDDP-induced nephrotoxicity without altering disposition, whereas the rs2289669 G>A SNP in MATE1 had no effect on CDDP toxicity. These results support the future clinical exploration of OCT2 and MATEs inhibitors as specific modifiers of cisplatin-induced nephrotoxicity .
In addition to the kidney, Sprowl et al. reported that oxaliplatin-induced neurotoxicity is also dependent on OCT2 [78]. Peripheral neurotoxicity is one of dose-limiting factors in the clinical use of oxaliplatin. OCT2 was found to be expressed on dorsal root ganglia cells in the nervous system, where oxaliplatin is known to accumulate. The cellular uptake of oxaliplatin was stimulated by the overexpression of human OCT2, and DNA platination and oxaliplatin-induced cytotoxicity were increased. Furthermore, genetic or pharmacologic knockout of Oct2 protected mice from acute oxaliplatin-induced neurotoxicity. These findings provide a rationale for the development of targeted approaches to mitigate this debilitating toxicity.
Summary
Among organic cation transporters OCT2, MATE1 and MATE2-K play pivotal roles in the renal tubular handling of various drugs. On the other hand, OCT1 and MATE1 are among the determinants of biliary excretion as well as hepatic detoxification. These transporters are the sites of drug–drug interaction, and differences in substrate affinities between OCTs and MATEs lead to different outcomes for each DDI. In addition, drug-induced kidney impairments were affected by OCT2 and MATE1 or MATE2-K activity. Information on these transporters is critical for the developing effective and safe pharmacotherapies.
References
Inui K, Masuda S, Saito H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000;58:944–58.
Inui K, Okuda M. Cellular and molecular mechanisms of renal tubular secretion of organic anions and cations. Clin Exp Nephrol. 1998;2:100–8.
Pritchard JB, Miller DS. Mechanisms mediating renal secretion of organic anions and cations. Physiol Rev. 1993;73:765–96.
Motohashi H, Inui K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013;15:581–8.
Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–51.
Yonezawa A, Inui K. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol. 2011;164:1817–25.
Jonker JW, Schinkel AH. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J Pharmacol Exp Ther. 2004;308:2–9.
Burckhardt G, Wolff NA. Structure of renal organic anion and cation transporters. Am J Physiol Renal Physiol. 2000;278:F853–66.
Koepsell H. Organic cation transporters in intestine, kidney, liver, and brain. Annu Rev Physiol. 1998;60:243–66.
Dresser MJ, Leabman MK, Giacomini KM. Transporters involved in the elimination of drugs in the kidney: organic anion transporters and organic cation transporters. J Pharm Sci. 2001;90:397–421.
Koepsell H, Schmitt BM, Gorboulev V. Organic cation transporters. Rev Physiol Biochem Pharmacol. 2003;150:36–90.
Koepsell H. Polyspecific organic cation transporters: their functions and interactions with drugs. Trends Pharmacol Sci. 2004;25:375–81.
Koepsell H, Endou H. The SLC22 drug transporter family. Pflugers Arch. 2004;447:666–76.
Wright SH, Dantzler WH. Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev. 2004;84:987–1049.
Urakami Y, Okuda M, Masuda S, Akazawa M, Saito H, Inui K. Distinct characteristics of organic cation transporters, OCT1 and OCT2, in the basolateral membrane of renal tubules. Pharm Res. 2001;18:1528–34.
Muller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem Pharmacol. 2005;70:1851–60.
Gorboulev V, Ulzheimer JC, Akhoundova A, et al. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–81.
Hayer-Zillgen M, Bruss M, Bonisch H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br J Pharmacol. 2002;136:829–36.
Zhang L, Schaner ME, Giacomini KM. Functional characterization of an organic cation transporter (hOCT1) in a transiently transfected human cell line (HeLa). J Pharmacol Exp Ther. 1998;286:354–61.
Tsuda M, Terada T, Ueba M, et al. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J Pharmacol Exp Ther. 2009;329:185–91.
Masuda S, Terada T, Yonezawa A, et al. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17:2127–35.
Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem Pharmacol. 2007;74:359–71.
Ito S, Kusuhara H, Yokochi M, et al. Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug-drug interactions caused by cimetidine in the kidney. J Pharmacol Exp Ther. 2012;340:393–403.
Suhre WM, Ekins S, Chang C, Swaan PW, Wright SH. Molecular determinants of substrate/inhibitor binding to the human and rabbit renal organic cation transporters hOCT2 and rbOCT2. Mol Pharmacol. 2005;67:1067–77.
Tahara H, Kusuhara H, Endou H, et al. A species difference in the transport activities of H2 receptor antagonists by rat and human renal organic anion and cation transporters. J Pharmacol Exp Ther. 2005;315:337–45.
Busch AE, Karbach U, Miska D, et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol Pharmacol. 1998;54:342–52.
Amphoux A, Vialou V, Drescher E, et al. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology. 2006;50:941–52.
Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A. 2005;102:17923–8.
Lips KS, Volk C, Schmitt BM, et al. Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am J Respir Cell Mol Biol. 2005;33:79–88.
Wu X, Huang W, Ganapathy ME, et al. Structure, function, and regional distribution of the organic cation transporter OCT3 in the kidney. Am J Physiol Renal Physiol. 2000;279:F449–58.
Diao L, Shu Y, Polli JE. Uptake of pramipexole by human organic cation transporters. Mol Pharm. 2010;7:1342–7.
Watanabe S, Tsuda M, Terada T, Katsura T, Inui K. Reduced renal clearance of a zwitterionic substrate cephalexin in MATE1-deficient mice. J Pharmacol Exp Ther. 2010;334:651–6.
Mulgaonkar A, Venitz J, Grundemann D, Sweet DH. Human organic cation transporters 1 (SLC22A1), 2 (SLC22A2), and 3 (SLC22A3) as disposition pathways for fluoroquinolone antimicrobials. Antimicrob Agents Chemother. 2013;57:2705–11.
Jung N, Lehmann C, Rubbert A, et al. Relevance of the organic cation transporters 1 and 2 for antiretroviral drug therapy in human immunodeficiency virus infection. Drug Metab Dispos. 2008;36:1616–23.
Muller F, Pontones CA, Renner B, et al. N1-methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: studies on the metformin-trimethoprim interaction. Eur J Clin Pharmacol. 2015;71:85–94.
Zhang S, Lovejoy KS, Shima JE, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–57.
Takeda M, Khamdang S, Narikawa S, et al. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp Ther. 2002;300:918–24.
Kralj E, Zakelj S, Trontelj J, Roskar R, Cernelc P, Kristl A. Absorption and elimination of imatinib through the rat intestine in vitro. Int J Pharm. 2014;460:144–9.
Kusuhara H, Ito S, Kumagai Y, et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89:837–44.
Dresser MJ, Xiao G, Leabman MK, Gray AT, Giacomini KM. Interactions of n-tetraalkylammonium compounds and biguanides with a human renal organic cation transporter (hOCT2). Pharm Res. 2002;19:1244–7.
Kimura N, Masuda S, Tanihara Y, et al. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005;20:379–86.
Li Q, Guo D, Dong Z, et al. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs). Toxicol Appl Pharmacol. 2013;273:100–9.
Zhang L, Dresser MJ, Gray AT, Yost SC, Terashita S, Giacomini KM. Cloning and functional expression of a human liver organic cation transporter. Mol Pharmacol. 1997;51:913–21.
Sata R, Ohtani H, Tsujimoto M, et al. Functional analysis of organic cation transporter 3 expressed in human placenta. J Pharmacol Exp Ther. 2005;315:888–95.
Zhang L, Gorset W, Washington CB, Blaschke TF, Kroetz DL, Giacomini KM. Interactions of HIV protease inhibitors with a human organic cation transporter in a mammalian expression system. Drug Metab Dispos. 2000;28:329–34.
Ciarimboli G, Ludwig T, Lang D, et al. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005;167:1477–84.
Li Q, Sai Y, Kato Y, Muraoka H, Tamai I, Tsuji A. Transporter-mediated renal handling of nafamostat mesilate. J Pharm Sci. 2004;93:262–72.
Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10:531–9.
Tanihara Y, Masuda S, Katsura T, Inui K. Protective effect of concomitant administration of imatinib on cisplatin-induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem Pharmacol. 2009;78:1263–71.
Grundemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–52.
Jonker JW, Wagenaar E, Mol CA, et al. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (Oct1 [Slc22a1]) gene. Mol Cell Biol. 2001;21:5471–7.
Motohashi H, Sakurai Y, Saito H, et al. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol. 2002;13:866–74.
Lozano E, Herraez E, Briz O, et al. Role of the plasma membrane transporter of organic cations OCT1 and its genetic variants in modern liver pharmacology. Biomed Res Int. 2013;2013:692071.
Okuda M, Saito H, Urakami Y, Takano M, Inui K. cDNA cloning and functional expression of a novel rat kidney organic cation transporter, OCT2. Biochem Biophys Res Commun. 1996;224:500–7.
Motohashi H, Nakao Y, Masuda S, et al. Precise comparison of protein localization among OCT, OAT, and MATE in human kidney. J Pharm Sci. 2013;102:3302–8.
Kekuda R, Prasad PD, Wu X, et al. Cloning and functional characterization of a potential-sensitive, polyspecific organic cation transporter (OCT3) most abundantly expressed in placenta. J Biol Chem. 1998;273:15971–9.
Motohashi H, Inui K. Multidrug and toxin extrusion family SLC47: physiological, pharmacokinetic and toxicokinetic importance of MATE1 and MATE2-K. Mol Aspects Med. 2013;34:661–8.
Yonezawa A, Inui K. Organic cation transporter OCT/SLC22A and H+/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem Pharmacol. 2011;81:563–8.
Terada T, Inui K. Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A). Biochem Pharmacol. 2008;75:1689–96.
Damme K, Nies AT, Schaeffeler E, Schwab M. Mammalian MATE (SLC47A) transport proteins: impact on efflux of endogenous substrates and xenobiotics. Drug Metab Rev. 2011;43:499–523.
Chu X, Korzekwa K, Elsby R, et al. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther. 2013;94:126–41.
Tzvetkov MV, Saadatmand AR, Lotsch J, Tegeder I, Stingl JC, Brockmoller J. Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther. 2011;90:143–50.
Hume WE, Shingaki T, Takashima T, et al. The synthesis and biodistribution of [11C]metformin as a PET probe to study hepatobiliary transport mediated by the multi-drug and toxin extrusion transporter 1 (MATE1) in vivo. Bioorg Med Chem. 2013;21:7584–90.
Toyama K, Yonezawa A, Masuda S, et al. Loss of multidrug and toxin extrusion 1 (MATE1) is associated with metformin-induced lactic acidosis. Br J Pharmacol. 2012;166:1183–91.
Sato T, Masuda S, Yonezawa A, Tanihara Y, Katsura T, Inui K. Transcellular transport of organic cations in double-transfected MDCK cells expressing human organic cation transporters hOCT1/hMATE1 and hOCT2/hMATE1. Biochem Pharmacol. 2008;76:894–903.
Tsuda M, Terada T, Mizuno T, Katsura T, Shimakura J, Inui K. Targeted disruption of the multidrug and toxin extrusion 1 (mate1) gene in mice reduces renal secretion of metformin. Mol Pharmacol. 2009;75:1280–6.
Toyama K, Yonezawa A, Tsuda M, et al. Heterozygous variants of multidrug and toxin extrusions (MATE1 and MATE2-K) have little influence on the disposition of metformin in diabetic patients. Pharmacogenet Genomics. 2010;20:135–8.
Choi JH, Yee SW, Ramirez AH, et al. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90:674–84.
Tzvetkov MV, Vormfelde SV, Balen D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. 2009;86:299–306.
Ito S, Kusuhara H, Kumagai Y, et al. N-methylnicotinamide is an endogenous probe for evaluation of drug-drug interactions involving multidrug and toxin extrusions (MATE1 and MATE2-K). Clin Pharmacol Ther. 2012;92:635–41.
Hillgren KM, Keppler D, Zur AA, et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94:52–63.
International Transporter C, Giacomini KM, Huang SM, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36.
Sprowl JA, Ness RA, Sparreboom A. Polymorphic transporters and platinum pharmacodynamics. Drug Metab Pharmacokinet. 2013;28:19–27.
Nakamura T, Yonezawa A, Hashimoto S, Katsura T, Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80:1762–7.
Burger H, Zoumaro-Djayoon A, Boersma AW, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2). Br J Pharmacol. 2010;159:898–908.
Sprowl JA, van Doorn L, Hu S, et al. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: influence on antitumor efficacy and systemic clearance. Clin Pharmacol Ther. 2013;94:585–92.
Iwata K, Aizawa K, Kamitsu S, et al. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16:843–51.
Sprowl JA, Ciarimboli G, Lancaster CS, et al. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc Natl Acad Sci U S A. 2013;110:11199–204.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Motohashi, H., Inui, Ki. (2016). Pharmacological and Toxicological Significance of the Organic Cation Transporters OCT and MATE: Drug Disposition, Interaction and Toxicity. In: Ciarimboli, G., Gautron, S., Schlatter, E. (eds) Organic Cation Transporters. Springer, Cham. https://doi.org/10.1007/978-3-319-23793-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-23793-0_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23792-3
Online ISBN: 978-3-319-23793-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)